

Short abstract
Lessons from extinction can be used in trials designed to pursue a cure for cancer. When cancer cannot be cured, similar strategies may be unwise, and strategies that leverage the adaptations of cancer to therapy should be considered.
Keywords: adaptive therapy, clinical trial, Ewing sarcoma, evolution, extinction therapy, first strike, osteosarcoma, resistance, rhabdomyosarcoma, second strike
Patients and families affected by sarcomas often ask, “What is cancer?” Answers frequently portray cancer as mutant cells gone rogue with rapid, uncontrolled, and even zombie‐like growth. This analogy may be helpful for encouraging compliance during chemotherapy, when cure requires enduring significant toxicity. However, this “zombie” paradigm understates cancer's most lethal and sophisticated property—its ability to evolve.1, 2, 3, 4 In contrast, viewing cancer as an invasive and evolving species may provide a more accurate and accessible analogy. This model “normalizes” cancer cells by emphasizing their obedience to the same laws of ecology and evolution that govern all living systems. Within this framework, the Darwinian dynamics of evolution can be leveraged to improve understanding of tumor growth and resistance.
Normal mammalian cells do not evolve because their fates are determined by collective tissue controls, making their fitness (for a Glossary of Terms, see Table 1) identical to that of the host organism. Fundamentally, cancer is a shift in the order of natural selection from the host level to that of an individual cancer cell. This new, “self‐defined” fitness function of the cancer cell can be considered a speciation event.3 In becoming a singular unit of natural selection, a cancer cell must respond to external influences from the host environment. To understand malignancy as a complex, adapting system of cancer cells, we introduce key terms and emerging theories from evolutionary biology that may translate into novel clinical trials. Although evolutionary principles are applicable to many cancer types across adult and pediatric oncology, here, we apply these concepts to pediatric sarcoma, using an evolution‐inspired clinical trial in metastatic fusion‐positive rhabdomyosarcoma (FPRMS) as an illustration.
Table 1.
Example Hypotheses, Proposed Preclinical Research, and Implications for Translation to Clinical Trials from Evolutionary Inspired Therapies
| Term | Definition |
|---|---|
| Adaptive therapy | Using models and measures of the past and present state of the individual patient's cancer to anticipate and steer the ecological and evolutionary dynamics of the disease by adjusting the time course of therapy accordingly. An example would be competitive‐release adaptive therapy, whereby therapy(ies) moderate the evolution of resistance by maintaining sensitive cancer cell types that, in turn, can compete with and continue to suppress more resistance cell types. This can be accomplished by starting and stopping therapy at patient‐specific upper and lower bounds of tumor burden |
| Background extinction | The continuous and regular loss and/or replacement of individual species over time that can be seen in the fossil record. Such extinctions are seen as resulting from clade‐specific events associated with habitat destruction or change and shifts in community composition. It is likely that contributors include second strikes rather than a major first strike alone |
| Clade | A group of individuals, subpopulations, and/or species derived from 1 common ancestor, often distinguished by sharing some set of 1 or more derived characteristics. A cancer clade would be a lineage of cells descended from a common ancestral cell and possessing a common set of mutations, epigenes, and/or heritable phenotypes |
| Competitive release | When there are 2 or more competing species (or types of cancer cells) that exist together, the removal of 1 of the species will result in expansion and increases in the remaining species (or types). Often the types will be therapy‐resistant and therapy‐sensitive cancer cells |
| Cost of resistance | The fitness cost to a cancer cell of supporting or deploying resistance mechanisms. The cost may involve less access to or efficient use of resources, costly support structures, or costly upregulated metabolic pathways. Consequently, in the absence of therapy, resistant cancer cells will have a lower proliferation rate and/or survival rate than sensitive cells; thus the resistant population is initially outcompeted by sensitive cancer cells. After many lines of continuous therapy over long periods of time, resistant cancer cells can be expected to evolve mechanisms for minimizing their costs of resistance thus rendering them highly resistant and proliferative. Therefore, the difference in competitive ability between sensitive and resistant cell types to cytotoxic chemotherapy is likely greatest at diagnosis |
| Evolutionary rescue | When a species or clade evolves successful adaptations after a stressor (therapy) that otherwise would have driven the clade extinct. Evolutionary rescue is much less likely when the clade's population size is small and it has little heritable variation |
| Evolutionary triage | The eco‐evolutionary dynamics leading to adaptations in cancer cells as phenotypes that are less fit become replaced by those that are more fit given the context and circumstances. Natural selection acts as “creative destruction.” The observable cancer cells, genomically and phenotypically, belie all of those that had been but died off. The observable clades represent successful genotypic and phenotypic trajectories driven by past and current selection forces. The selection forces themselves are not static. Therapy imposes additional and changed selection forces |
| First strike | A large‐scale and high‐impact stressor (therapy) that drives a species or clade to extinction or to the brink of extinction. The dramatic decline in population sizes happens when the therapy directly kills or indirectly lowers fitness by disrupting the populations' environment and ecology |
| Fitness | The expected per capita growth rate, as proliferation and survival rates, of a cancer cell clade. Fitness will usually be highly context‐dependent and will include the microenvironment, densities, and frequencies of different cancer cell clades and therapy |
| Inverse problem approach | The process of using a set of observations to calculate or infer the causal factors that produced them |
| Mass extinction | The rapid and collective extinction of many diverse clades of species generally in response to a singular global or large‐scale, catastrophic event. Paleontologists recognize 5 major mass extinctions over the last 600 million years |
| Minimum viable population | The lower bound on a population's size below which it cannot survive and has a high likelihood of extinction. Sometimes, it is expressed as the population size at which the likelihood of persistence equals some probability over a specified amount of time. For cancer, it might the number of cells at which the probability of extinction becomes greater than 90% in 4 months |
| Second strike | A single event or sequences of subtle and relatively undramatic events that affect vulnerable populations and favor extinction |
Pediatric Sarcomas
Sarcomas, such as osteosarcoma, Ewing sarcoma, and rhabdomyosarcoma, collectively make up 10% of malignancies in children and young adults. Even when disease is clinically localized at presentation, surgery and/or radiation alone is usually insufficient for cure, as a majority of patients will relapse, frequently through development of distant metastases. Because there are no current ways to identify patients who can be cured by local control alone, chemotherapy is recommended for all patients. Cure rates increase up to 65% to 80% for localized disease with the addition of combination chemotherapy.5, 6, 7, 8, 9 Although there is a growing understanding of the genetic features distinguishing sarcoma cells from somatic cells, the most successful treatments target pathways common to tumor and normal cells alike, so‐called “never mutated pathways,” such as DNA synthesis and replication, topoisomerase‐mediated DNA repair, and microtubule function.2, 10, 11, 12, 13, 14, 15 Unfortunately, outcomes for metastatic pediatric sarcomas have changed little over the past 2 decades, and the prognosis for metastatic pediatric sarcomas remains dismal.16, 17, 18 The application of evolutionary concepts to pediatric sarcomas can deepen our understanding of treatment failure from resistance and may improve treatment strategies and trial designs for a patient population in which improved outcomes are urgently needed.
A Phenotypic View of Cancer Ontogeny
Cancer cells compete against each other in a dynamic environment. Their tumor ecosystems exhibit spatial and temporal fluctuations in blood‐borne nutrients, oxygen, growth factors, immune cells, and hormones.19, 20, 21 Even when considering 2 genetically identical cancer cells, differences between these cells in proximity to nutrients may promote different transcription rates of growth factors. Ultimately, this may affect the rate of progression through the cell cycle, leading to distinct rates of proliferation and mutational acquisition. Eventually, each clade (see Table 1) emerging from these 2 cells will have different adaptations for exploiting and avoiding specific microenvironmental opportunities and hazards.19, 20, 22, 23, 24 By the time a cancer becomes clinically apparent, cancer cells have transformed from a single clone into a diverse community of cell types evolving in response to a spatially and temporally heterogeneous tumor environment.25, 26 Theoretically, a 10‐gram cancer may contain the same order of magnitude of cancer cells as there are humans on earth, with tremendous diversity of phenotypes and environments.27
Evolutionary triage (defined in Table 1) continues beyond diagnosis. The cancer cells, through competition with each other and interactions with stromal and immune cells, see the progressive replacement of less fit phenotypes by those that are more fit. Cancer cells enjoy another advantage that aids in the development of successful resistance to therapies: they do not need to be unconditionally resistant. They only need to be more resistant than critical normal cells. This may explain why initial responses in certain solid tumors (notably rhabdomyosarcoma) do not predict eventual survival.28, 29, 30 The sensitivities of the dominant cancer cell populations dictate the initial response, but it is the ecology and evolution of the rare and more resistant populations that determine cure or relapse. The cost of resistance (defined in Table 1) becomes key to understanding recurrence after the initial responses to therapy. An important point for clinical translation is that, in general, the relative fitness of any given cancer phenotype can be estimated using an inverse problem approach (defined in Table 1) and thus does not require complete knowledge of each cancer clade. This explicit link allows for a simple generality—the observed abundance of each tumor subpopulation provides a reasonable estimate of its fitness.
Preclinical and precision medicine panel investigations focus on genomic changes in cancer cells rather than on genomic changes that do not happen; however, both mutations and wild‐type status are selected. Mutations suggest directional selection for promoting the progressive evolution of cancer. Targeting these changes has been useful and widely practiced. But genes and pathways that are always unchanged suggest stabilizing selection, in which the cancer cells pay a substantial fitness cost for altering these never‐mutated genes. These may provide promising and novel therapeutic options.2 Instead of targeting gains of a particular target in cancer on which somatic cells also rely (and thus inhibition is relatively toxic), one can envision characterizing wild‐type genes on which cancer cells rely more than somatic cells. Thus inhibition would be selectively toxic to the cancer cells. An example of such a target would be the CMG helicase, an essential complex for DNA replication.31
Mass Extinctions and First‐Strike Strategy
Curing pediatric sarcomas requires the extinction of dispersed and diverse cancer cell populations without destroying essential normal tissues. Although the task of curing sarcomas (especially high‐risk, metastatic sarcomas) is formidable, nature and human activity have unwittingly performed analogous tasks by driving numerous species to extinction.32
Most current frontline therapies for pediatric sarcomas rely solely on a first‐strike (defined in Table 1) strategy that primarily administers maximum tolerated doses (MTDs) of cytotoxic drugs in combination. This mimics the dynamics of a mass extinction (defined in Table 1), famously illustrated by the extinction of the dinosaurs after an asteroid impact. This first‐strike strategy is sometimes successful because natural selection does not prepare a population for events that have never happened. An asteroid impact that blocks photosynthesis for an extended time leads to calamitous changes in the food supply, leaving many otherwise well adapted species ill prepared for survival. The collapse of the population is too rapid to allow for an evolutionary rescue (defined in Table 1). In pediatric sarcomas, especially those with clinically localized disease, a large enough perturbation over a short interval is often a successful first strike, resulting in cure (Fig. 1A).2, 11, 12, 13 Unfortunately, MTD therapies often have narrow therapeutic windows and may result in considerable toxicities. This represents a central ecological and evolutionary problem in treating cancers: although cancer cells are a distinct, evolving population, they arose from the host's somatic cells and thus share many pathways.2 Despite the heavy burden of toxicity from MTD therapy, residual disease may persist from the survival of cancer cell subpopulations with greater intrinsic or environmental resistance.
Figure 1.
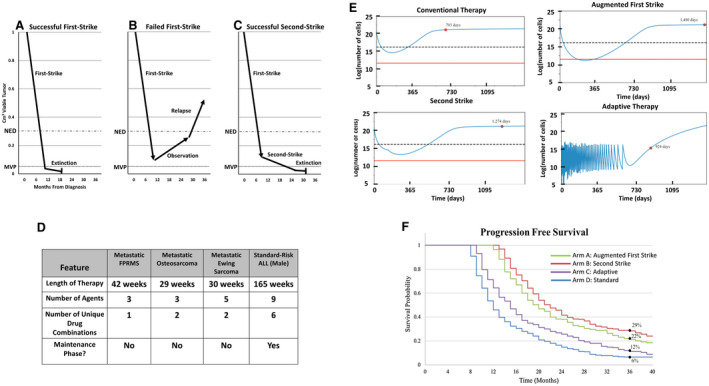
(A) First‐strike therapy reduces disease burden below the threshold of radiographic detection (no evidence of disease [NED]). (B) Minority resistant cell populations are below the NED level, but not the minimum viable population (MVP), driving eventual relapse. (C) With successful second‐strike therapy, the disease burden falls below the MVP, eventually causing extinction (cure). (D) Current sarcoma therapy based on North American protocols contains relatively few agents that are administered for 7 to 9 months. Acute lymphoblastic leukemia (ALL) therapy contains more agents, increased variation in multiagent combinations and dosing, and a prolonged maintenance phase. (E) Simulated in silico outcomes of model parameterized by a conventional chemotherapy setting of an event‐free survival (EFS) of 6% at 3 years are illustrated. Red dots represent EFS for a simulated patient. An augmented first‐strike strategy can affect extinction by reducing levels below a theoretical MVP (indicated by a red line). Second‐strike therapy delays progression but does not increase extinction, whereas adaptive therapy controls progression longer than conventional chemotherapy. A dashed line represents the threshold of detection by imaging. (F) A simulated cohort of patients is illustrated with random variability around key parameters presented as a Kaplan‐Meier curve. Importantly, this is based on a calibration of parameters that generates a 3‐year EFS of 6%. The data that accrue through a proposed clinical trial will update and improve model parameterization and model predictions. These baseline data show an example of how an evolutionary framework can improve therapeutic regimens.
Background Extinctions and Second‐Strike Strategies
An understanding of the dynamics of background extinctions (defined in Table 1) may help improve treatment strategies after partially successful first strikes. As an illustration of this evolutionary process, the North American passenger pigeon was once an abundant species with an estimated population of 4 billion. A series of seemingly independent and noncatastrophic insults led to its extinction. Habitat loss from deforestation caused an overconcentration of birds that invited over‐hunting, leaving remaining populations small and fragmented. Loss of genetic diversity, stochastic failures of local nut production, lack of safety from natural predators, and breakdowns in the social structure of breeding colonies were critical factors that drove extinction.33, 34 Although there was no single event that “cured” the Earth of the passenger pigeon, humans and nature unwittingly concocted a sequence of “treatments” that individually were minor but together were decisive. Once first strikes of deforestation and hunting reduced the birds to small, fragmented populations, a series of what would otherwise have been minor second strikes (defined in Table 1) pushed the passenger pigeon below its extinction threshold, or minimum viable population (MVP) (defined in Table 1).
Background extinctions such as the passenger pigeon may provide valuable lessons for curing pediatric sarcomas and other cancers. In pediatric sarcomas, the initial success of first‐strike systemic therapy over surgery alone and the inability to measure residual disease has limited the ability to refine therapeutic strategies before overt relapse. However, what should be done if the first strike does not cure (Fig. 1B)? Clearly, continued use of the first‐strike drug is evolutionarily unwise because the remaining cells are resistant. Here, using a second strike could further reduce the remaining population size, leading to extinction by reducing the number of individuals (cancer cells) in each fragmented subpopulation to below its MVP (Fig. 1C).35 Importantly, a good second‐strike agent need not be a good first‐strike agent, and vice versa.
The passenger pigeon likely had a high MVP because of its social breeding structure and reliance on safety in numbers from nonhuman predators. Less is known regarding the MVP size of cancer cell populations within patients. Although it is speculative, elements such as paracrine growth factors, cell‐cell interactions, or need for a critical mass of different populations may mean that different pediatric sarcomas have distinct MVPs. Currently, we do not have good tools for measuring disease burden below the detection limit of traditional radiology studies in pediatric sarcomas. Patients have no evidence of disease (NED) when we cannot see a mass on a computed tomography scan or a magnetic resonance image down to a few millimeters. The NED threshold must be above the MVP because many patients relapse after achieving NED (Fig. 1B). This highlights a critical research need for more sensitive measures of residual disease below NED. Preclinical experiments could help us better understand disease dynamics below NED thresholds. The emerging fields of peripheral blood tumor evaluations for circulating tumor cells and DNA, along with radiomics, may enhance our ability to follow cancer responses over time.36, 37, 38 Radiomics attempts to identify phenotypic properties within a tumor, independent of its size, which may accurately predict histopathology and future response or resistance to therapy. Examples include novel, radiolabeled pharmaceuticals, which bind to tumor‐specific antigens; the detection of microscopic changes in the tumor microenvironment, such as water diffusivity or tumor hypoxia; and the application of mathematical algorithms to images to predict histopathologic diagnosis and patient prognosis.39
Second strikes should be timed to occur around the time when the first strike has achieved its greatest effect, presumably at the point when the disease becomes clinically undetectable or at a measurable nadir (Fig. 1C). Ideally, second‐strike therapies should have modes of action that require different resistance strategies by the cancer cells than those needed for resistance to the first strike. Consider the treatment for standard‐risk pediatric acute lymphoblastic leukemia (ALL), which causes extinction of the cancer in >90% of patients (Fig. 1D). Here, the 1‐month first‐strike induction leads to at least a 3‐orders‐of‐magnitude reduction in leukemia cells. This is followed by years of sequential second strikes that vary in mechanism and intensity. The result is near certain extinction and patient cure.
Making sarcoma therapy more like ALL therapy is typically met with skepticism given that sarcomas offer fewer and less diverse drugs, less profound responses to any given drug, and greater difficulty in sampling tumor burden and composition over time. Nevertheless, these limitations, in theory, are quantifiable and can be modeled. Variables such as cell death, growth rates, adaptability, and heterogeneity can be parameterized and used to design preclinical experiments. Mathematical models of the ecological and evolutionary dynamics can evaluate therapy efficacies. The models aim to reflect key biologic processes. Prospectively, the models suggest hypotheses and make predictions. Their utility increases as more data and more types of data become available. With additional data, the models can be updated and refined, and the iterative processes continues. In the first step of model creation, we used known tumor biology, hypothesized resistance mechanisms, and a calibration such that current therapies yielded an expected 3‐year event‐free survival rate of 6% (Fig. 1E,F). For this particular calibration, first‐strike second‐strike therapy was overall most effective, although not for all patients. In simulating a broad range of calibrations, the 3 eco‐evolutionary therapies were always more successful than conventional therapy, and which was most effective varied. The next step establishes the iterative process of updating and refining the models and improving the in silico simulations of possible patient outcomes from treatment regimens (see Mathematical Model, below).
When Extinction Is Not Possible
The objective of the extinction therapy described above is to cure and render this section moot. Unfortunately, relapse occurs in 94% of patients with metastatic FPRMS within 3 years after diagnosis, which highlights an important issue.40 Through an evolutionary lens, an optimal treatment strategy for driving the cancer populations to extinction is different from the optimal therapy designed to maintain long‐term population control. Intensive therapy aimed at a very unlikely chance at cure may add toxicity and shorten lifespan by hastening the emergence of resistant disease. It may be important to design evolutionary therapies that contain rather than cure the cancer.
Heterogeneous populations of cancer cells cannot anticipate clinical interventions, but they are adept at adapting. An innovative approach to the treatment of an incurable cancer might be informed by approaches in agriculture. Led by pesticide manufacturers motivated to limit resistance to their products, the agricultural community adopted a strategy termed integrated pest management (IPM).41 IPM recognizes that continuous application of pesticides at maximal concentrations accelerates the emergence of resistant insect populations, causing competitive release, whereby loss of sensitive individuals permits resistant individuals to flourish (see Table 1, Fig. 2A).42 IPM applies pesticides judiciously, stopping applications while populations are declining and only reapplying upon reemergence to “pest levels.” The goal is to limit crop damage while retaining the sensitivity of the insects to the pesticides. Resistance most often comes at a cost. In the absence of the pesticide, sensitive individuals will outcompete resistant individuals.
Figure 2.
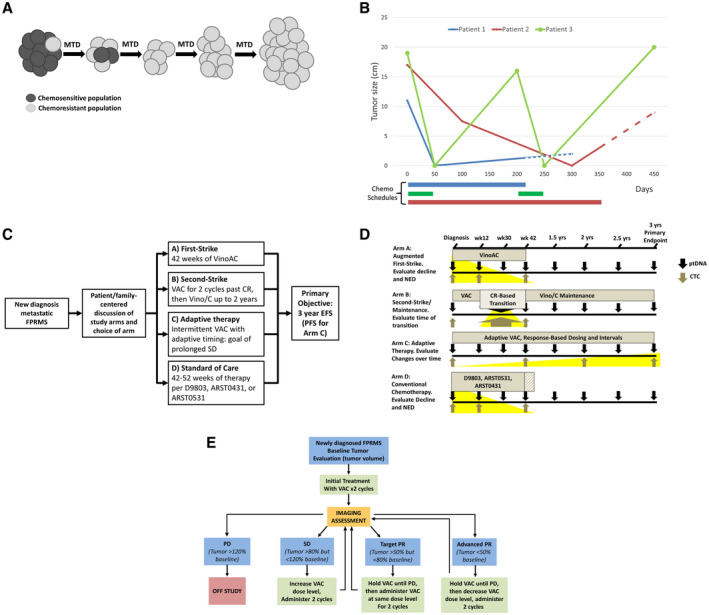
(A) Over time and under selection of continued first‐strike therapy at the maximum tolerated dose (MTD), minor resistant populations emerge, leading to relapse, termed competitive release (adapted from West et al, 201942). (B) An illustration of clinical courses compares MTD therapy with “adaptive” therapy in metastatic, fusion‐positive rhabdomyosarcoma (FPRMS). Data are from aggregate tumor measurements of 3 patients who demonstrated an initial complete response to therapy followed by subsequent progression, with the truncation of lines representing death from disease. Although patients 1 and 2 progressed on therapy and were refractory to second‐line therapies, patient 3 chose to stop therapy while in remission, demonstrating a second complete response and again stopping therapy. Dashed lines represent refractory disease in which continued cytotoxic therapy was delivered without observable benefit. Chemo indicates chemotherapy. (C) An open‐label, phase 2 study to assess the efficacy of 4 approaches to metastatic FPRMS is illustrated. The shared primary endpoint is 3‐year event‐free survival (EFS), with the exception of arm C (see E). ARST0431 indicates the Children's Oncology Group trial “High‐Dose Combination Chemotherapy and Radiation Therapy in Treating Patients With Newly Diagnosed Metastatic Rhabdomyosarcoma or Ectomesenchymoma (clinicaltrials.gov identifier NCT00354744); ARST0531, Children's Oncology Group trial “Combination Chemotherapy and Radiation Therapy in Treating Patients With Newly Diagnosed Rhabdomyosarcoma” (clinicaltrials.gov identifier NCT00354835); CR, complete response; D9803, Children's Oncology Group trial “Combination Chemotherapy in Treating Patients With Previously Untreated Rhabdomyosarcoma” (clincialtrials.gov identifier NCT00003985); PFS, progression‐free survival; VAC, vincristine, actinomycin‐D, and cyclophosphamide; Vino/C, vinorelbine and oral cyclophosphamide; VinoAC, vinorelbine, actinomycin‐D, and cyclophosphamide. (D) Plasma tumor DNA (ptDNA) and circulating tumor cells (CTCs) will be collected at specified intervals across all arms, with focused collections that may be of interest for a particular arm highlighted in yellow (ie, the rate of decline may be greater with an augmented first strike on arm A; circulating cell‐free ptDNA at a second‐strike transition point that may indicate whether different populations are being eliminated by a second strike in arm B; CTCs with single‐cell RNAseq to evaluate genetic changes over time in arm C with the shared primary endpoint of 3‐year EFS). NED indicates no evidence of disease. (E) A schema for adaptive therapy in metastatic FPRMS is illustrated in which therapy will be withheld when the tumor responds, corresponding with arm C (see C) of an upcoming, evolutionary inspired FPRMS trial. PD indicates progressive disease; PR, partial response; SD, stable disease.
Adaptive therapy (defined in Table 1) uses the concepts embodied in IPM and exploits the cost of resistance by applying treatment only until some predetermined response is observed in the dominant, sensitive population (Fig. 2B). Therapy is then withdrawn and not resumed until the tumor returns to a predetermined size. Therapy‐sensitive cells, which have a proliferative advantage over resistant cells, will regrow, and the same therapy can be used again, minimizing iatrogenic selection and expansion of resistant cells. Theoretical, preclinical, and clinical studies have now demonstrated that this strategy can delay the proliferation of resistant populations and prolong the time to progression while reducing the cumulative drug dose compared with continuous treatment.43, 44, 45 A clinical trial in prostate cancer used the testosterone production inhibitor abiraterone and patient‐specific dosing schedules. Therapy was withdrawn when a patient's prostate‐specific antigen level declined to 50% of the baseline value and then resumed once the level returned to baseline. Importantly, progression‐free survival on that trial increased even as the cumulative abiraterone dose delivered over time declined.44 Using adaptive therapy, clinicians can maintain the initial heterogeneity of the tumor, allowing subsequent treatment cycles to retain effectiveness.
Translating Evolutionary Concepts into Trials
Viewing pediatric sarcoma therapy through an evolutionary lens reveals opportunities to translate evolutionary strategies into innovative clinical trials (Fig. 2C). Although intensification of therapy has improved outcomes in localized Ewing sarcoma and in fusion‐negative rhabdomyosarcoma, outcomes for metastatic FPRMS remain dismal.8, 16, 40 Incorporating relapsed therapies for newly diagnosed patients provides a novel first‐strike opportunity. Because vinorelbine is among the most active agent in relapsed disease, incorporation of vinorelbine into frontline therapy may eliminate the population that leads to relapse after combined vincristine, actinomycin‐D, and cyclophosphamide (VAC) (Fig. 2C).46, 47 Another first‐strike intervention would be to time S‐phase–specific therapies to create “hypercompression.” After initial chemotherapy using VAC, irinotecan could be given starting on day 4 of a cycle, with the expectation that any cells entering S‐phase after VAC chemotherapy are VAC‐resistant but potentially sensitive to irinotecan. This concept finds support in trial data from timed sequential therapy in acute myeloid leukemia.48, 49, 50 Both of these strategies may add toxicity, but a prospective trial could evaluate whether that toxicity is justified by improved survival.
As for second strikes, a recent European trial of selected patients with high‐risk, localized rhabdomyosarcoma showed benefits of vinorelbine‐based maintenance therapy.51 In localized Ewing sarcoma, high‐dose chemotherapy and autologous stem‐cell rescue could be considered for poor histologic responders to neoadjuvant chemotherapy as an effective second strike.52 The optimal timing of transition from first strike to a second strike designed to push the population below its MVP is unknown, but the second strike is best implemented before the first strike has become ineffective. The continued investigation and optimization of emerging technologies such as circulating tumor cells and circulating cell‐free tumor DNA are critical (Fig. 2D).36, 37, 38 Borrowing from ALL, advancing the detection of residual disease from “remission” (<5% blasts) to negative minimal residual disease (<0.01% blasts) has been fundamental to trial design and continued improvements in outcomes.53 Importantly, there is no need to be rigid with the duration of the current conventional chemotherapy plans, especially in metastatic populations for which data on these durations and drug combinations are controversial and outcomes remain poor.16, 17
For metastatic FPRMS, we propose that the transition from first strike to a second‐strike maintenance with vinorelbine and cyclophosphamide should be patient‐specific and implemented within 6 weeks of achieving NED, as determined by conventional imaging (Fig. 2C). After establishing a trial design to evaluate second strikes, the scope of strategies can be widened to investigate schedules and combinations of agents with orthogonal mechanisms of action, such as habitat perturbation (eg, antiangiogenesis), introduction of predators (eg, immunotherapy), and disruption of cancer cell foraging (eg, metabolic and RAS pathway perturbation), as well as agents that target pathways important to minor populations, such as micrometastases.
In addition to extinction therapy, adaptive therapy for chemotherapy‐sensitive, metastatic FPRMS could be offered to patients. In this approach, we shift away from rigid strategies that ultimately lead to competitive release of resistant populations in the overwhelming majority of patients with metastatic FPRMS. The significant costs of resistance should be incorporated into therapeutic plans rather than iatrogenically selected.1, 4 The chemotherapeutically sensitive, dominant population must not be viewed entirely as the enemy to be eliminated but, rather, as a pest to be controlled. Accordingly, we propose VAC therapy to maintain tumor burden over time, with dose and interval adjustments based on patient‐specific responses (Fig. 2C,E). We are designing a clinical trial that will compare the arms mentioned above with a conventional‐care arm using previously published North American treatment strategies (Fig. 2C).
Conclusion
Cancer cells are subject to the same evolutionary principles that apply to all living systems. When a cure for pediatric sarcoma is an achievable outcome, observations from extinctions in nature provide a theoretical framework for optimizing the probability of a cure by either augmenting first strikes or following first strikes closely with diverse second strikes before disease progression. The application of these strategies may improve on current conventional care, which relies on fixed combination chemotherapies. We need to leverage imaging studies and eventually incorporate more sensitive assessments of residual tumor burden. Armed with this information, adjustments to therapy can prevent overtreatment after a successful first strike and the swifter initiation of second‐strike strategies in patients who need alternative therapy (Table 2).
Table 2.
Specific Adaptive Therapy Hypotheses and Preclinical Models
| Specific Hypotheses | Interpretation | Preclinical Models | Translation |
|---|---|---|---|
| Adaptive therapy can improve survival by delaying the emergence of resistant cells, aiming to maintain a population of sensitive cancer cells rather than to eradicate all cancer cells | Different strategies and dosing regimens are needed when seeking disease control as opposed to cure | Need to characterize responses to conventional therapy (scheduled, repetitive treatments at the maximum tolerated dose) versus adaptive therapy in preclinical models | For situations in which conventional therapy, despite an initial response, leads to virtually assured progression, consider adaptive therapy; this offers the potential to extend life with less toxicity (thus increased quality of life) but does forgo the possibility of cure |
| Therapies in minimal disease states can have effects even when there are no measurable effects on bulky tumor | Evaluate the benefit of treatment for minimal disease using circulating cancer cells/DNA instead of radiographic response | Identify a minimal residual disease model for solid tumors by measuring circulating tumor cells/DNA and seek to identify sensitivities of the minimal viable population (“bottleneck survivors”) | Explore dose and schedule of combinations |
| Consider agents in terms of reduction of metastases and clearance of established metastases for second‐strike agents, even when the patient has no evidence of disease | |||
| Second strikes should be used early for maximum effect | Evaluate the ideal timing and duration of second‐strike therapy | Explore the initiation of second strikes while tumor is shrinking, at nadir, and after nadir to determine the optimal timing for second strikes | Current convention is to continue the same regimen for a predetermined number of cycles independent of tumor response |
| Determine variable susceptibilities of subpopulations of tumor cells | We propose identifying the ideal time to discontinue initial therapy and implement a second‐strike regimen | ||
| Explore optimal durations of second strikes |
In contrast, when it is believed that a cure is unachievable, evolutionary principles suggest that optimal cancer treatment could move away from the conventional strategy of therapy at the MTD until progression. Although adaptive therapy would represent a major paradigm shift in pediatric oncology, this approach would exploit the chemotherapy‐sensitive population to prevent the emergence of resistant populations, optimizing tumor control with less toxicity. An upcoming trial in metastatic FPRMS will serve as an initial investigation of these evolutionary concepts by offering patients extinction therapy, adaptive therapy, or conventional chemotherapy. We hypothesize that both extinction therapy and adaptive therapy will provide clinical benefits and shape a more patient‐specific approach to pediatric sarcomas.
Mathematical Model
We construct a fitness‐generating function (G‐function) to mathematically model the population dynamics of tumor growth and evolution of phenotypic adaptations to promote resistance over time. Our G‐function assumes logistic tumor growth that is suppressed by multiple cancer drugs, resulting in per capita growth rate of a cell using specific, resistant strategies.
In the model, r is the growth rate of the cancer cell, K is the tumor‐carrying capacity, and x is the density of cells. represents cell death caused by specific drugs (eg, vinorelbine, dactinomycin, cyclophosphamide):
Where m is the death rate caused by drug exposure, k is the innate resistance that may be present before drug exposure, b is the benefit the cell gains by reducing sensitivity to the drug, and v is the evolving resistant strategy. For example, a mechanism of resistance to vinorelbine is the upregulation of p‐glycoprotein before drug exposure, v = 0, simplifying our cell death term to:
Without the evolution of resistance, cell death is only determined by drug toxicity and de novo resistance. Whereas v ∈ [0,1] and, as v increases, the effectiveness of the drug diminishes. The current model assumes that there is a unique resistant strategy, v, for every drug used during treatment. Large values of k and b denote a higher efficacy in the cell's resistant strategy.
The cost a cell incurs by exhibiting a resistant strategy is applied to the carrying capacity. The model proposes that resources or energy that would have been used to maintain a larger cell population are instead used to increase mechanisms of resistance. A cell evolves an optimal resistant strategy value based on the increasing fitness:
where σ characterizes evolutionary speed (how fast and often a genetic and/or phenotypic difference arises and sweeps through the population). How the population size changes over time is determined by:
The design of the model is flexible and can be easily adjusted for each trial arm. Parameters in the model, such as b and k, can be patient‐specific, are determined based on clinical data gained over time, and thus are iterative and likely to improve with more data. The model can compare the different trial arms for a given set of parameters in which the enhanced first‐strike arm leads to a cure, whereas the first‐strike/second‐strike and adaptive‐therapy arms extend progression‐free survival (Fig. 1E). If we randomly draw parameters from a fixed distribution, a Kaplan‐Meier curve can be generated (Fig. 1F). Which of these 3 therapies is best depends on specific parameter values.
Funding Support
This work was generously supported by the National Pediatric Cancer Foundation and by grant U54CA143970‐05 (Physical Science Oncology Network; “Cancer as a Complex Adaptive System”) from the National Institutes of Health/National Cancer Institute. Ryan Roberts is supported by a Career Development Award from the National Cancer Institute (K08 CA201638).
Conflict of Interest Disclosures
Damon R. Reed reports personal fees from Epizyme, LOXO, and Shire, outside the submitted work. The remaining authors made no disclosures.
References
- 1. Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12:487‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gatenby RA, Cunningham JJ, Brown JS. Evolutionary triage governs fitness in driver and passenger mutations and suggests targeting never mutations. Nat Commun. 2014;5:5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gatenby RA, Brown J. Mutations, evolution and the central role of a self‐defined fitness function in the initiation and progression of cancer. Biochim Biophys Acta. 2017;1867:162‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gatenby R, Brown J. The evolution and ecology of resistance in cancer therapy. Cold Spring Harb Perspect Med. 2018;8:a033415. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5. Bielack SS, Smeland S, Whelan JS, et al. Methotrexate, doxorubicin, and cisplatin (MAP) plus maintenance pegylated interferon alfa‐2b versus MAP alone in patients with resectable high‐grade osteosarcoma and good histologic response to preoperative MAP: first results of the EURAMOS‐1 good response randomized controlled trial. J Clin Oncol. 2015;33:2279‐2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gorlick R, Janeway K, Lessnick S, Randall RL, Marina N. Children's Oncology Group's 2013 blueprint for research: bone tumors. Pediatr Blood Cancer. 2013;60:1009‐1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marina NM, Smeland S, Bielack SS, et al. Comparison of MAPIE versus MAP in patients with a poor response to preoperative chemotherapy for newly diagnosed high‐grade osteosarcoma (EURAMOS‐1): an open‐label, international, randomised controlled trial. Lancet Oncol. 2016;17:1396‐1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Womer RB, West DC, Krailo MD, et al. Randomized controlled trial of interval‐compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol. 2012;30:4148‐4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hawkins DS, Spunt SL, Skapek SX. Children's Oncology Group's 2013 blueprint for research: soft tissue sarcomas. Pediatr Blood Cancer. 2013;60:1001‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reed DR, Hayashi M, Wagner L, et al. Treatment pathway of bone sarcoma in children, adolescents, and young adults. Cancer. 2017;123:2206‐2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hustu HO, Pinkel D, Pratt CB. Treatment of clinically localized Ewing's sarcoma with radiotherapy and combination chemotherapy. Cancer. 1972;30:1522‐1527. [DOI] [PubMed] [Google Scholar]
- 12. Link MP, Goorin AM, Miser AW, et al. The effect of adjuvant chemotherapy on relapse‐free survival in patients with osteosarcoma of the extremity. N Engl J Med. 1986;314:1600‐1606. [DOI] [PubMed] [Google Scholar]
- 13. Maurer HM, Moon T, Donaldson M, et al. The intergroup rhabdomyosarcoma study: a preliminary report. Cancer. 1977;40:2015‐2026. [DOI] [PubMed] [Google Scholar]
- 14. Hingorani P, Janeway K, Crompton BD, et al. Current state of pediatric sarcoma biology and opportunities for future discovery: a report from the Sarcoma Translational Research Workshop. Cancer Genet. 2016;209:182‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Whelan JS, Bielack SS, Marina N, et al. EURAMOS‐1, an international randomised study for osteosarcoma: results from pre‐randomisation treatment. Ann Oncol. 2015;26:407‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weigel BJ, Lyden E, Anderson JR, et al. Intensive multiagent therapy, including dose‐compressed cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, irinotecan, and radiation, in patients with high‐risk rhabdomyosarcoma: a report from the Children's Oncology Group. J Clin Oncol. 2016;34:117‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348:694‐701. [DOI] [PubMed] [Google Scholar]
- 18. Ebb D, Meyers P, Grier H, et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: a report from the Children's Oncology Group. J Clin Oncol. 2012;30:2545‐2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891‐899. [DOI] [PubMed] [Google Scholar]
- 20. Gillies RJ, Brown JS, Anderson ARA, Gatenby RA. Eco‐evolutionary causes and consequences of temporal changes in intratumoural blood flow. Nat Rev Cancer. 2018;18:576‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gillies RJ, Gatenby RA. Hypoxia and adaptive landscapes in the evolution of carcinogenesis. Cancer Metastasis Rev. 2007;26:311‐317. [DOI] [PubMed] [Google Scholar]
- 22. Verduzco D, Lloyd M, Xu L, et al. Intermittent hypoxia selects for genotypes and phenotypes that increase survival, invasion, and therapy resistance. PLoS One. 2015;10:e0120958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lloyd MC, Cunningham JJ, Bui MM, Gillies RJ, Brown JS, Gatenby RA. Darwinian dynamics of intratumoral heterogeneity: not solely random mutations but also variable environmental selection forces. Cancer Res. 2016;76:3136‐3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Estrella V, Chen T, Lloyd M, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013;73:1524‐1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stewart E, Federico SM, Chen X, et al. Orthotopic patient‐derived xenografts of paediatric solid tumours. Nature. 2017;549:96‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stewart E, Goshorn R, Bradley C, et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep. 2014;9:829‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Del Monte U. Does the cell number 10(9) still really fit one gram of tumor tissue? Cell Cycle. 2009;8:505‐506. [DOI] [PubMed] [Google Scholar]
- 28. Anderson ND, de Borja R, Young MD, et al. Rearrangement bursts generate canonical gene fusions in bone and soft tissue tumors. Science. 2018;361:eaam8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rosenberg AR, Anderson JR, Lyden E, et al. Early response as assessed by anatomic imaging does not predict failure‐free survival among patients with group III rhabdomyosarcoma: a report from the Children's Oncology Group. Eur J Cancer. 2014;50:816‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vaarwerk B, van der Lee JH, Breunis WB, et al. Prognostic relevance of early radiologic response to induction chemotherapy in pediatric rhabdomyosarcoma: a report from the International Society of Pediatric Oncology Malignant Mesenchymal Tumor 95 Study. Cancer. 2018;124:1016‐1024. [DOI] [PubMed] [Google Scholar]
- 31. Bryant VL, Elias RM, McCarthy SM, Yeatman TJ, Alexandrow MG. Suppression of reserve MCM complexes chemosensitizes to gemcitabine and 5‐fluorouracil. Mol Cancer Res. 2015;13:1296‐1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gatenby RA, Zhang J, Brown JS. First strike‐second strike strategies in metastatic cancer: lessons from the evolutionary dynamics of extinction. Cancer Res. 2019;79:3174‐3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murray GGR, Soares AER, Novak BJ, et al. Natural selection shaped the rise and fall of passenger pigeon genomic diversity. Science. 2017;358:951‐954. [DOI] [PubMed] [Google Scholar]
- 34. Hung CM, Shaner PJ, Zink RM, et al. Drastic population fluctuations explain the rapid extinction of the passenger pigeon. Proc Natl Acad Sci U S A. 2014;111:10636‐10641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frankham R, Bradshaw CJA, Brook BW. Genetics in conservation management: revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biol Conservation. 2014;170:56‐63. [Google Scholar]
- 36. Hayashi M, Zhu P, McCarty G, et al. Size‐based detection of sarcoma circulating tumor cells and cell clusters. Oncotarget. 2017;8:78965‐78977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hayashi M, Chu D, Meyer CF, et al. Highly personalized detection of minimal Ewing sarcoma disease burden from plasma tumor DNA. Cancer. 2016;122:3015‐3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Abbou SD, Shulman DS, DuBois SG, Crompton BD. Assessment of circulating tumor DNA in pediatric solid tumors: the promise of liquid biopsies. Pediatr Blood Cancer. 2019;66:e27595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gillies RJ, Kinahan PE, Hricak H. Radiomics: images are more than pictures, they are data. Radiology. 2016;278:563‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Malempati S, Weigel BJ, Chi YY, et al. The addition of cixutumumab or temozolomide to intensive multiagent chemotherapy is feasible but does not improve outcome for patients with metastatic rhabdomyosarcoma: a report from the Children's Oncology Group. Cancer. 2019;125:290‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gould F, Brown ZS, Kuzma J. Wicked evolution: can we address the sociobiological dilemma of pesticide resistance? Science. 2018;360:728‐732. [DOI] [PubMed] [Google Scholar]
- 42. West JB, Dinh MN, Brown JS, Zhang J, Anderson AR, Gatenby RA. Multidrug cancer therapy in metastatic castrate‐resistant prostate cancer: an evolution‐based strategy. Clin Cancer Res. 2019;25:4413‐4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Enriquez‐Navas PM, Kam Y, Das T, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8:327ra324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang J, Cunningham JJ, Brown JS, Gatenby RA. Integrating evolutionary dynamics into treatment of metastatic castrate‐resistant prostate cancer. Nat Commun. 2017;8:1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ibrahim‐Hashim A, Robertson‐Tessi M, Enriquez‐Navas PM, et al. Defining cancer subpopulations by adaptive strategies rather than molecular properties provides novel insights into intratumoral evolution. Cancer Res. 2017;77:2242‐2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kuttesch JF Jr, Krailo MD, Madden T, Johansen M, Bleyer A. Phase II evaluation of intravenous vinorelbine (Navelbine) in recurrent or refractory pediatric malignancies: a Children's Oncology Group study. Pediatr Blood Cancer. 2009;53:590‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mascarenhas L, Chi YY, Hingorani P, et al. Randomized phase II trial of bevacizumab or temsirolimus in combination with chemotherapy for first relapse rhabdomyosarcoma: a report from the Children's Oncology Group. J Clin Oncol. 2019;37:2866‐2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smith FO, Alonzo TA, Gerbing RB, Woods WG, Arceci RJ; Children's Cancer Group . Long‐term results of children with acute myeloid leukemia: a report of three consecutive phase III trials by the Children's Cancer Group: CCG 251, CCG 213 and CCG 2891. Leukemia. 2005;19:2054‐2062. [DOI] [PubMed] [Google Scholar]
- 49. Woods WG, Kobrinsky N, Buckley JD, et al. Timed‐sequential induction therapy improves postremission outcome in acute myeloid leukemia: a report from the Children's Cancer Group. Blood. 1996;87:4979‐4989. [PubMed] [Google Scholar]
- 50. Loeb DM, Bowers DC, Civin CI, Friedman AD. Intensive timed sequential remission induction chemotherapy with high‐dose cytarabine for childhood acute myeloid leukemia. Med Pediatr Oncol. 2001;37:365‐371. [DOI] [PubMed] [Google Scholar]
- 51. Bisogno G, De Salvo GL, Bergeron C, et al. Maintenance low‐dose chemotherapy in patients with high‐risk (HR) rhabdomyosarcoma (RMS): a report from the European Paediatric Soft Tissue Sarcoma Study Group (EpSSG) [abstract]. J Clin Oncol. 2018;36(18 suppl):LBA2. [Google Scholar]
- 52. Whelan J, Le Deley MC, Dirksen U, et al. High‐dose chemotherapy and blood autologous stem‐cell rescue compared with standard chemotherapy in localized high‐risk Ewing sarcoma: results of Euro‐E.W.I.N.G.99 and Ewing‐2008. J Clin Oncol. 2018;36:3110‐3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Coustan‐Smith E, Sancho J, Hancock ML, et al. Clinical importance of minimal residual disease in childhood acute lymphoblastic leukemia. Blood. 2000;96:2691‐2696. [PubMed] [Google Scholar]