

Abstract
Mycenarubin C, a previously unknown red pyrroloquinoline alkaloid, was isolated from fruiting bodies of the mushroom Mycena rosea and its structure was elucidated mainly by NMR spectroscopy and mass spectrometry. Unlike mycenarubin A, the major pyrroloquinoline alkaloid in fruiting bodies of M. rosea, mycenarubin C, contains an eight‐membered ring with an additional C1 unit that is hitherto unprecedented for pyrroloquinoline alkaloids known in nature. Incubation of mycenarubin A with an excess of formaldehyde revealed that mycenarubin C was generated nearly quantitatively from mycenarubin A. An investigation into the formaldehyde content of fresh fruiting bodies of M. rosea showed the presence of considerable amounts of formaldehyde, with values of 5 μg per gram of fresh weight in fresh fruiting bodies. Although mycenarubin C did not show bioactivity against selected bacteria and fungi, formaldehyde inhibits the growth of the mycoparasite Spinellus fusiger at concentrations present in fruiting bodies of M. rosea. Therefore, formaldehyde might play an ecological role in the chemical defence of M. rosea against S. fusiger. In turn, S. fusiger produces gallic acid—presumably to detoxify formaldehyde by reaction of this aldehyde with amino acids and gallic acid to Mannich adducts.
Keywords: alkaloids, chemical ecology, formaldehyde, natural products, pyrroloquinolines
Natural protection: Mycenaubin C, a pyrroloquinoline alkaloid with an unprecedented core structure, was isolated from fruiting bodies of M. rosea. The alkaloid is generated from mycenarubin A and formaldehyde. The amounts of formaldehyde present in the fruiting bodies of M. rosea exhibit a fungistatic effect against the mycoparasite S. fusiger. In turn, S. fusiger is probably able to detoxify formaldehyde with gallic acid and amino acids to Mannich adducts.

Introduction
Mycena rosea (Bull.) Gramberg, the rosy bonnet (German name: Rosa Rettichhelmling), is a relatively small mushroom with a pinkish coloured cap (Figure 1) that is widespread in beech forests in Europe.1 Nevertheless, its fruiting bodies have not been well investigated, to date, for its secondary metabolite content. In 2007, an investigation of the coloured principles of the fruiting bodies revealed the presence of the previously unknown red pyrroloquinoline alkaloids mycenarubin A (1) and mycenarubin B.2 Pyrroloquinoline alkaloids were earlier considered to be typically “marine alkaloids”, because known representatives,3 such as the batzellines,4 damirones,5 discorhabdines,6 isobatzellines,7 and makaluvamines,8 were isolated from marine sources. However, meanwhile, a considerable number of different pyrroloquinoline alkaloids have been isolated from fruiting bodies of other Mycena species, for instance, from Mycena haematopus,9, 10 Mycena sanguinolenta,11 and Mycena pelianthina.12 Regarding volatile constituents, hexanal and hexan‐2‐one have been reported to be present in fruiting bodies of M. rosea.13
Figure 1.

A photograph of M. rosea and structures of mycenarubin A (1) and mycenarubin C (2).
In the course of a comparative metabolic screening for pyrroloquinoline alkaloids with different Mycena species, we found that 1 was not only present in M. rosea, but also in the fruiting bodies of several other Mycena species. Moreover, metabolic profiling revealed the presence of a previously undescribed, red, pyrroloquinoline alkaloid in fruiting bodies of M. rosea that we named mycenarubin C (2; Figure 1). The unusual structure of this pyrroloquinoline alkaloid, containing an eight‐membered ring system, inspired us to investigate the biosynthesis of this compound in more detail. Herein, we describe the structure elucidation of 2; its biosynthesis; and the occurrence of its biosynthetic precursor, formaldehyde, in fruiting bodies of M. rosea. Moreover, we show that formaldehyde protects the fruiting bodies of M. rosea, to some degree, from infestation with the mycoparasite Spinellus fusiger.
Results and Discussion
For metabolic profiling of fruiting bodies of selected Mycena species, one fruiting body of each species was extracted with methanol, and the resulting extract was subjected to HPLC‐UV and LC‐ESIMS and LC‐ESIMS/MS analysis. Mycenarubin A (1) not only occurred in M. rosea, but also in M. pelianthina, M. haematopus, Mycena renati, and M. sanguinolenta (Figure S1 in the Supporting Information). In the HPLC‐UV analysis of fresh and frozen fruiting bodies of M. rosea, apart from 1 and mycenarubin B, a third peak with a UV maximum of λ=360 nm was present, which corresponded to so far undescribed compound 2.
Structure elucidation of 2
To elucidate the structure of 2, fruiting bodies of M. rosea were extracted with methanol and purified by means of semipreparative HPLC on an RP‐18ec column. An amount of 100 g of freshly collected fruiting bodies yielded approximately 1 mg 2.
The UV/Vis spectrum of red compound 2 resembles that of 1 2 and exhibits three maxima at λ=249, 360, and 529 nm; this suggests the presence of a pyrroloquinoline core structure.
The HR‐(+)‐ESIMS spectrum showed an [M+H]+ ion at m/z 302.11356, consistent with the molecular formula C15H15N3O4 for 2, and thus, indicating that 2 contained one more carbon than that of 1. The presence of 15 carbon atoms in 2 was confirmed by means of 13C NMR spectroscopy. Because some proton signals were strongly broadened in the 1H NMR spectrum, if recorded at 300 K in D2O, all NMR spectra, except the 13C NMR spectrum, were recorded at 335 K; this is a temperature at which signal broadening is much lower. The 1H NMR spectrum, measured at 335 K in D2O, showed 12 nonexchangeable protons. Hence, compound 2 contains three exchangeable protons. The HSQC data showed signals attributed to five CH2 groups and two CH groups, which implied the presence of eight quaternary carbon atoms. Analysis of the spin systems in the COSY spectrum revealed the presence of CH2−CH2−CH2, CH−CH2, isolated CH2, and isolated CH fragments. A comparison of the NMR spectra of 2 with those of 1 revealed that the two compounds were closely related to each other because most carbon atoms and protons showed similar shift values in both compounds (Table 1 and Figures S6–S21). The major differences in the NMR spectra are an additional CH2 group in 2 and the fact that C‐6 is quaternary instead of a CH group. Moreover, in 2, there are only three exchangeable protons instead of four. The carbon chemical shift value of the CH2 group in position 14 indicates that it is flanked by two amino groups. HMBC correlations from the diastereotopic protons at H‐14 to C‐5a, C‐6, C‐7, and C‐12 reveal that C‐14 is connected through an NH group to the CH2−CH2−CH2 residue at C‐12 and to the pyrroloquinoline core at C‐6; thus a hexahydro‐1,5‐diazocine ring is formed (Figures 1 and 2). The presence of an NOE correlation between Ha‐10 and Hb‐14 suggests that in D2O at 335 K 2 occurs in the form of a predominantly pseudo‐boat–chair conformer (Figure 2); this is a conformer that is similar to that of the preferred boat–chair conformation present in cyclooctane.14
Table 1.
NMR spectroscopic data of 1 and 2.
|
No. |
2 |
1 2 |
||||
|---|---|---|---|---|---|---|
|
δ C (ppm)[a] |
δ H (ppm)[b] |
HMBC (H→C)[b,e] |
δ C (ppm)[c] |
δ H (ppm)[d] |
HMBC (H→C)[d,e] |
|
|
2 |
128.7 (CH) |
7.13 (s) |
2a, (7), (8), 8a, 8b |
127.4 (CH) |
6.95 (s) |
2a, 7, (8), 8a, 8b |
|
2a |
119.6 (qC) |
|
|
117.5 (qC) |
|
|
|
3 |
26.2 (CH2) |
3.233 (dd, J=16.7, 1.9 Hz; Ha) 3.18 (dd, J=16.7, 6.3 Hz; Hb) |
2, 2a, 4, 8b, 9, 10 2, 2a, 4, 8b, 9 |
25.5 (CH2) |
3.20 (d, J=16.6 Hz; Ha) 3.15 (dd, J=16.6, 5.0 Hz; Hb) |
2, 2a, 4, 8b, 9 (2), 2a, 4, (8b) |
|
4 |
72.8 (CH) |
4.32 (dd, J=6.3, 1.9 Hz) |
2a, 3, 5a, 9, 10 |
67.1 (CH) |
4.25 (d, J=5.0 Hz) |
2a, 3, 5a, 9, 10 |
|
5a |
159.4 (qC) |
|
|
157.7 (qC) |
|
|
|
6 |
97.7 (qC) |
|
|
93.9 (CH) |
5.36 (s) |
2, 7, (8), 8b |
|
7 |
181.9 (qC) |
|
|
180.8 (qC) |
|
|
|
8 |
171.4 (qC) |
|
|
172.6 (qC) |
|
|
|
8a |
126.4 (qC) |
|
|
125.5 (qC) |
|
|
|
8b |
127.8 (qC) |
|
|
126.2 (qC) |
|
|
|
9 |
178.7 (qC) |
|
|
177.9 (qC) |
|
|
|
10 |
54.0 (CH2) |
4.20 (ddm, J=14.7, 6.6 Hz; Ha) 3.98 (dm, J=14.7 Hz; Hb) |
|
49.9 (CH2) |
3.75 (ddd, J≈14, ≈7, ≈7 Hz; Ha) 3.32 (ddd, J=≈14, ≈7, ≈7 Hz; Hb) |
4, 5a, 11, 12 4, 5a, 11, 12 |
|
11 |
27.4 (CH2) |
2.28 (ddddd, J=15.4, 6.6, 5.9, 3.7, 3.3 Hz; Ha) 2.24 (ddddd, J=15.4, 9.8, 4.2, 4.2, 3.9 Hz; Hb) |
10, 12 10, 12 |
26.6 (CH2) |
2.06 (m, 2 H) |
10, 12 10, 12 |
|
12 |
41.1 (CH2) |
3.60 (ddd, J=13.1, 9.8, 3.7 Hz; Ha) 3.234 (ddd, J=13.1, 5.9, 3.9 Hz; Hb) |
10, 11, 14 10, 11, 14 |
38.5 (CH2) |
3.11 (m, 2 H) |
10, 11 |
|
14 |
42.5 (CH2) |
4.64 (d, J=15.3 Hz; Ha) 4.45 (d, J=15.3 Hz; Hb) |
5a, 6, 7, 12 5a, 6, 7 |
|||
[a] Recorded at 151 MHz in D2O at 330 K. [b] Recorded at 500 MHz in D2O at 335 K. [c] Recorded at 226 MHz in D2O at 300 K. [d] Recorded at 900 MHz in D2O at 300 K. [e] HMBC correlations, optimised for 6 Hz, are from proton(s) to the indicated carbon. Brackets indicate weak HMBC correlations.
Figure 2.

Selected HMBC (→) and NOE (↔) correlations of 2.
The structure of 2 is unique, since no other pyrroloquinoline alkaloids containing a hexahydro‐1,5‐diazocine or any other eight‐membered ring system are known in nature, to date.
The absolute configuration of 2 at position C‐4 was determined by comparison with the circular dichroism (CD) spectrum of 1. The CD spectra of both compounds resemble each other closely (Figure S24). Consequently, C‐4 in 2 is S configured.
Bioactivity of 2
If tested in agar diffusion assays at amounts up to 0.5 μmol (0.15 mg) per paper disk against selected gram‐negative bacteria, such as Azospirillum brasilense, Azovibrio restrictus, Azoarcus tolulyticus, and Escherichia coli, neither 1 nor 2 displayed any significant activity. Likewise, no activity was found for 2 against the gram‐positive bacteria Bacillus fastidiosus, Bacillus subtilis, Nocardioides simplex, Paenibacillus polymyxa, Sporosarcina pasteurii, and Staphylococcus capitis under the same conditions. Moreover, compounds 1 and 2 showed no activity in the agar diffusion assay against S. fusiger and Mucor hiemalis at amounts up to 0.5 μmol (0.15 mg) per paper disk. Further tests with 2 and other bacteria, fungi, and the nematode Caenorhabditis elegans were negative (Tables S1 and S2). Because mycenarubin D, differing from 1 only by the presence of a C=NH unit instead of a C=O unit at position 7, displayed considerable bioactivity;10 the presence of a C=NH unit at position 7 seems to be an important prerequisite for bioactivity of this type of pyrroloquinoline alkaloid.
Biosynthesis of 2 and formaldehyde
Compound 1 is distinguished from 2 by the presence of an additional C1 unit, which is used for the formation of the eight‐membered ring in 2. Consequently, compound 2 should be derived biosynthetically from 1. The latter is very likely to originate from tryptophan and S‐adenosylmethionine.2 We hypothesised that the additional C1 unit in 2 originated from formaldehyde because formaldehyde could react with the amino group of the side chain in 1, and thus, form an imine that could be attacked from the nucleophilic ring position at C‐6, forming the eight‐membered ring in 2. To test this hypothesis, both 1 obtained through total synthesis15 and that isolated from fruiting bodies of M. rosea were dissolved in two separate experiments in water, and an excess of formalin was added; this resulted in both cases in the quantitative conversion of 1 into 2. Moreover, the NMR spectroscopy data and CD spectra (Table S3 and Figure S25) of both synthetic and isolated 2 resembled each other very closely, and thus, confirmed the structure and absolute configuration of 2.
Because 2 is generated from 1 and formaldehyde, we have checked if formaldehyde occurs in free form in fresh fruiting bodies.
To detect formaldehyde and to quantify the amount of free formaldehyde produced in freshly harvested fruiting bodies of M. rosea, an aqueous crude extract of M. rosea was derivatised with O‐(2,3,4,5,6‐pentafluorobenzyl)‐hydroxylamine hydrochloride (PFBHA) and analysed through GC‐EIMS, according to a procedure published by the U.S. Environmental Protection Agency.16 A collection of fresh fruiting bodies of M. rosea contained an average formaldehyde concentration of (5.4±0.7) μg g−1 (Figure 3). Similar amounts of formaldehyde were detected in fruiting bodies of Mycena pura with (2.0±0.5) μg g−1, whereas in M. pelianthina nearly no formaldehyde was present. So far, only the formaldehyde content of a few edible mushrooms has been studied in detail, particularly in shiitake mushrooms (Lentinula edodes), from which formaldehyde concentrations as high as 110 to 494 μg g−1 have been reported.17, 18 Nevertheless, these high concentrations are a negligible health risk, if L. edodes is ingested in normal amounts.19
Figure 3.

Formaldehyde concentration in fresh fruiting bodies of M. pura, in fresh fruiting bodies of M. rosea, in fresh fruiting bodies of M. rosea infested with S. fusiger, in fruiting bodies of M. pura stored for 70 days at −32 °C, and in fruiting bodies of M. rosea stored for 72 days at −32 °C (Table S6).
In L. edodes, formaldehyde originates from lentinic acid,20 which is not present in M. rosea. Consequently, in M. rosea, formaldehyde is generated from other sources than that in L. edodes. Some fungi are known to be able to produce formaldehyde through oxidation of sarcosine (N‐methylglycine) or N,N‐dimethylglycine. For instance, Cylindrocarpon didymum M‐1 contains a sarcosine oxidase that oxidises sarcosine to glycine, formaldehyde, and H2O2.21 Similarly, formaldehyde might be generated in fruiting bodies of M. rosea. However, metabolic profiling through LC‐ESIMS analysis of a methanol/water crude extract of M. rosea showed no convincing evidence for the presence of sarcosine and dimethylglycine in M. rosea. Another source of formaldehyde could be methyl esterified pectins present in rotting plant material that the saprophytic basidiomycete M. rosea might degrade by a pectin methylesterase to pectin and methanol.22 The latter would then be oxidised to formaldehyde by a methanol oxidase23 or a methanol dehydrogenase.24 A feeding experiment with fruiting bodies of M. rosea and the injection of [D4]methanol (10 μL) into each fruiting body led to incorporation rates as high as 57 % at position 14 in 2 (Figure S37), suggesting that formaldehyde was effectively generated from methanol as a biosynthetic precursor compound (Scheme 1).
Scheme 1.
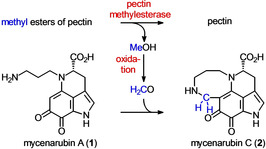
Hypothetical biosynthesis of 2.
Chemical defence mechanism of M. rosea against S. fusiger with formaldehyde and its inactivation by S. fusiger
Formaldehyde is a well‐known biocide that also is effective against various fungi and their spores.25 Therefore, we speculated that formaldehyde might play a role in the chemical defence of M. rosea.26, 27 Fruiting bodies of Mycena species, including M. rosea, are known to be infected at low temperatures and under humid conditions by the mycoparasite S. fusiger.28 To verify if formaldehyde exhibited a protective effect against this mycoparasite, we applied different concentrations of formaldehyde in agar diffusion assays to cultures of S. fusiger. For instance, an amount of 10 μg formaldehyde per test plate resulted, after an incubation time of 9 days, in an average inhibition zone of (1.5±0.5) cm, whereas a collection of fresh fruiting bodies of M. rosea contained a formaldehyde concentration of (5.4±0.7) μg g−1 and 5–10 times more total formaldehyde, because an average‐sized fruiting body of M. rosea usually has a weight between 5 and 10 g. Therefore, the amounts of formaldehyde present in M. rosea might protect fruiting bodies of M. rosea, at least to some degree, against S. fusiger. To test if fruiting bodies of M. rosea infected with S. fusiger reacted to attack with an increase of their formaldehyde content, we determined the formaldehyde content of infected fruiting bodies. The formaldehyde content in infested fruiting bodies was (4.2±0.7) μg g−1, which was similar to the formaldehyde content of non‐infested fruiting bodies. Therefore, formaldehyde seems to act in M. rosea as a constitutive chemical defence compound. To gain more insight into how S. fusiger was able to grow on fruiting bodies of M. rosea, metabolic profiling of the pertrimethylsilylated methanol/ethyl acetate (1:1, v/v) crude extract of S. fusiger was performed by means of GC‐MS; this revealed the presence of high amounts of gallic acid, which is a well‐known polyphenol with antioxidative and prooxidative properties,29, 30 that might protect S. fusiger from the harmful effects of formaldehyde (Figures S39 and S40). It is known that gallic acid reacts in living organisms with formaldehyde and amino acids to produce Mannich adducts, and thus, detoxify formaldehyde (Scheme 2).31
Scheme 2.

Hypothetical chemical defence of M. rosea against the mycoparasite S. fusiger with formaldehyde, and subsequent inactivation of this defence mechanism by the formation of Mannich adducts of formaldehyde with gallic acid and amino acids in S. fusiger.
Increase of the content of formaldehyde and 2 in fruiting bodies during long‐term storage at −32 °C
Upon using frozen fruiting bodies of M. rosea that had been stored for 72 days at −32 °C, we found that these contained (30.4±1.1) μg g−1 formaldehyde, which is significantly higher than that present in fresh fruiting bodies (Figure 3). Moreover, the amount of 2 was also significantly increased in fruiting bodies stored at −32 °C, and thus, suggested that formaldehyde was generated in frozen fruiting bodies during long‐term storage and partially reacted with 1 to 2. The ratio of the amount of 2 to that of the total amounts of 1 and 2 turned out to correlate well with the total amount of formaldehyde present in fruiting bodies of M. rosea (Figure 4).
Figure 4.

Correlation of the ratio [%] of the amount of 2 to the total amounts of 1 and 2 with the measured formaldehyde concentration. The relative amounts of 1 and 2 were determined by measuring the peak areas of 1 and 2 at λ=360 nm after HPLC separation.
Therefore, the amount of formaldehyde present in the fruiting bodies of M. rosea can also be deduced from the ratio of the amount of 2 to that of the total amounts of 1 and 2. The significant increase in the amount of formaldehyde in frozen organisms has also been reported for fish. In this case, trimethylamine is converted by bacteria into the corresponding N‐oxide, which is then oxidised to yield formaldehyde.32 A similar degradation reaction might also take place in frozen mushroom samples.
Consequently, meaningful experiments on the formaldehyde content and the content of 1 in fresh fruiting bodies could only be performed with fresh fruiting bodies during the short mushroom season in autumn. Similarly to M. rosea, the amount of formaldehyde in M. pura was significantly increased in frozen fruiting bodies of M. pura. Fruiting bodies of M. pura stored at −32 °C for 70 days contained (14.8±1.3) μg g−1 formaldehyde (Figure 3). Preliminary experiments with L. edodes also showed that the amount of formaldehyde dramatically increased in fruiting bodies of L. edodes both upon injury of fresh fruiting bodies and upon storage at −32 °C. Consequently, to obtain reliable results, care has to be taken when determining the formaldehyde content of any mushroom species.
Conclusions
From fruiting bodies of M. rosea, the previously undescribed pyrroloquinoline alkaloid 2 was isolated, its structure elucidated, and a total synthesis for the compound was developed. Compound 2 possesses a unique structure; so far, it is the only pyrroloquinoline alkaloid known in nature that contains an eight‐membered ring. The biosynthesis of 2 apparently proceeds by the reaction of the side‐chain amino group of 1 with formaldehyde, yielding an imine intermediate that forms an eight‐membered ring after nucleophilic attack from the pyrroloquinoline residue. It is remarkable that this ring‐closing reaction is only intramolecular and does not lead to polymers, even if the reaction is not supported by the presence of an enzyme.
Compound 2 did not show any antibacterial, antifungal, nematicidal, or cytotoxic activity against selected test organisms.
M. rosea not only uses formaldehyde for the biosynthesis of 2, but it also contains free formaldehyde. Formaldehyde is likely to be produced by the degradation of methyl esterified pectins, which are abundant in decaying plant material that is used by M. rosea as a nutrient. Probably, M. rosea hydrolyses methyl esterified pectins to the corresponding pectins and MeOH. Feeding experiments revealed that fruiting bodies of M. rosea very effectively converted methanol into formaldehyde, which was finally incorporated into 2 at C‐14.
According to agar diffusion assays with formaldehyde and the mycoparasite S. fusiger, the concentrations of formaldehyde present in M. rosea are sufficient to exhibit fungistatic activity against S. fusiger, and thus, protect the fruiting bodies, to some degree, from infestation with S. fusiger. Infested fruiting bodies do not produce more formaldehyde, and consequently, formaldehyde is a constitutive chemical defence agent of M. rosea. S. fusiger is presumably even able to protect itself, to some extent, from formaldehyde present in M. rosea by producing gallic acid in large quantities, by using gallic acid and amino acids to react with formaldehyde, and thus, produce Mannich adducts, which results in the detoxification of formaldehyde.
Experimental Section
General experimental procedures: The evaporation of organic solvents was performed under reduced pressure by using a rotary evaporator and in the case of H2O by freeze‐drying. Preparative HPLC was performed on Waters 590EF pumps equipped with an automated gradient controller 680 and a Knauer UV/Vis detector. CD: J‐715 spectropolarimeter (Jasco). NMR spectroscopy was performed on a Bruker DMX 900 spectrometer equipped with a TXI cryoprobe (1H at 900.13 MHz, 13C at 226.3 MHz), a Bruker Avance DRX‐600 spectrometer equipped with a TXI probe (1H at 600.22 MHz, 13C at 150.91 MHz), and a Bruker DMX 500 spectrometer equipped with a TXI probe (1H at 500.11 MHz, 13C at 125.74 MHz). 1H chemical shifts were calibrated by using the chemical shift of the HDO solvent signal corrected for the well‐known temperature dependency of water (δ H (HDO)=δ H 4.7476 ppm, δ H (HDO)=4.4346 ppm at 330 K, and δ H (HDO)=4.3861 ppm at 335 K).33 LC‐ESIMS spectra were obtained on an LCQ DecaXP Plus ESIMS spectrometer (Thermo Fisher Scientific Inc., USA). The spectrometer was operated in positive mode (0.625 spectra per s; mass range 50–1000). As a sheath gas, nitrogen was used (80 a.u.) and helium served as a collision gas. The spectrometer was equipped with a Hewlett–Packard HPLC system (Series 1100) composed of a degasser, two binary pumps, a diode array detector (DAD), and an autosampler (injection volume 10 μL). HR‐ESIMS spectra were obtained on an LTQ Orbitrap ESIMS spectrometer (Thermo Fisher Scientific). The spectrometer was operated in positive mode (1 spectrum per s; mass range 50–1000) with nitrogen as a sheath gas (6 arbitrary units) and helium as a collision gas. The HR‐ESIMS/MS spectra were obtained on an Impact II mass spectrometer (Bruker Daltonics). The spectrometer was operated in positive mode (mass range 50–1300) with nitrogen as a sheath gas and helium as a collision gas. The GC‐EIMS spectra for the formaldehyde measurements and for the identification of gallic acid were obtained on a Trace GC Ultra instrument with a Trace DSQ EIMS and an AS300 Autosampler (Thermo Finnigan, Thermo Fisher Scientific Inc., USA). Helium was used as a carrier gas with a constant flow rate of 1 mL min−1. Separations were performed on an Optima 5 ms Accent column (15 m×0.25 mm i.d., 0.25 μm film thickness). The mass spectrometer was operated in either positive mode for the full scan (1.9763 scans s−1; mass range 34–600 u, 1171.1 amu s−1) or in selected‐ion monitoring mode. Retention indices, R i, were determined according to a method reported by Kováts through injection of a sample (0.5 μL) of a standard mixture of saturated straight‐chain alkanes (C7–C30) in n‐hexane.34
Mushrooms: Fruiting bodies of M. rosea (leg. et det. P. Spiteller) were collected in September, October, and November of 2004 to 2018 in beech forests near Bayreuth, Breitbrunn, Leutstetten, Mühltal, Rieden, Starnberg, Wolfratshausen (Bavaria), and in Bad Fallingbostel (Lower Saxony), Germany. They were immediately frozen and stored at −35 °C. Voucher samples of M. rosea were deposited at the Institut für Organische und Analytische Chemie, Universität Bremen, Germany.
Fruiting bodies of M. haematopus, M. pelianthina, M. renati, and M. sanguinolenta (leg. et det. P. Spiteller) were collected in September and October of 2018 in beech forests near Mühltal (Bavaria), Germany. They were immediately frozen and stored at −35 °C. Voucher samples of M. haematopus, M. pelianthina, M. renati, and M. sanguinolenta were deposited at the Institut für Organische und Analytische Chemie, Universität Bremen, Germany.
Extraction for the metabolite screening of selected Mycena species: Frozen fruiting bodies of M. haematopus, M. pelianthina, M. renati, M. rosea, and M. sanguinolenta (each ca. 1 g) were extracted with H2O/MeCN (10 mL 50:50, v/v) for 20 min at 150 rpm and 25 °C. The solvents were evaporated in vacuo and then the residues were redissolved in H2O and filtered over a solid‐phase extract using an RP‐18ec cartridge. The three fractions—H2O, then H2O/MeCN (50:50, v/v), and then MeCN—were combined. The extracts were measured by using an analytical LC‐ESI‐(+)MS instrument. Separations were achieved with an RP‐18ec column (Nucleodur, 100 Å, 5 μm, 3×250 mm, Macherey–Nagel) using the following gradient: 10 min at 100 % H2O+0.1 % HOAc, then within 35 min linear to 100 % MeCN, flow rate 0.66 mL min−1. 1: t R=16.53 min; 2: t R=17.11 min (Figure S1).
Extraction and isolation of 2 for structure elucidation: Fresh fruiting bodies (100 g) were extracted first with MeOH (200 mL) for 10 min, followed by H2O/MeOH (100 mL 50:50, v/v) for another 10 min at 200 rpm and 25 °C. The solvents of the combined extract were evaporated and the resulting residue was redissolved in H2O (10 mL) before being centrifuged at 15000 rpm and 25 °C for 5 min. The supernatant was pre‐purified with an RP‐18ec cartridge by using H2O and then H2O/MeOH (50:50, v/v) as the eluent to obtain two fractions. The H2O/MeOH fraction contained 2 and was further purified by means of HPLC on a semipreparative RP‐18ec column (Nucleodur, 100 Å, 5 μm, 21×250 mm, Macherey–Nagel). For sample separation, the following gradient program was used: 5 min at 100 % H2O, then within 40 min linear to 100 % MeOH, flow rate 12 mL min−1; UV detection at 360 nm. A sample of 100 g of freshly collected fruiting bodies yielded 1 mg of 2. If frozen fruiting bodies stored for several months or even years were used, the yield of 2 was much higher and reached approximately 20 mg per 100 g of complete fruiting bodies. For analytical LC‐ESI‐(+)MS separations of 1 and 2, an RP‐18ec column (Nucleodur, 100 Å, 5 μm, 3×250 mm, Macherey–Nagel) and the following gradient program were used: 5 min at 100 % H2O, then within 40 min linear to 100 % MeOH, flow rate 0.66 mL min−1.
Mycenarubin A (1):2 Red solid; [α] =+669 (c=0.0055 in H2O);15 HPLCprep: t R=21.8 min; LC‐(+)‐ESIMS: t R=17.24 min; 1H NMR (900 MHz, D2O, 300 K): see Table 1; 13C NMR (226 MHz, D2O, 300 K): see Table 1; (+)‐ESI‐MS: m/z: 290 [M+H]+.
Mycenarubin C (2): Red solid; [α] =+433 (c=0.006 in H2O); HPLCprep: t R=23.6 min; LC‐(+)‐ESIMS: t R=18.68 min; 1H NMR (500 MHz, D2O, 335 K): see Table 1; 13C NMR (151 MHz, D2O, 330 K): see Table 1; UV/Vis (H2O): λ max(ϵ)=529 (2.90), 360 (4.10), 249 nm (4.21 mol−1 dm3 cm−1); CD (H2O): λ (Δϵ)=245 (−0.2), 272 (+0.1), 304 (+0.1), 362 (−0.4), 523 nm (+0.1 mol−1 dm3 cm−1); HRMS (ESI+): m/z calcd for C15H16N3O4: 302.11353 [M+H]+; found: 302.11356; HRMS2 (Figure S5, ESI+, precursor ion m/z (%) 302.11408 (63.7), 15 eV): m/z calcd for C14H13N2O4: 273.08698 [M+H−CH2=NH]+; found: 273.08753 (29.4); m/z calcd for C14H16N3O2: 258.12370 [M+H−CO2]+; found: 258.12425 (100); m/z calcd for C13H13N2O2: 229.09715 [M+H−CO2−CH2=NH]+; found: 229.09761 (14.0); m/z calcd for C12H11N2O2: 215.08150 [M+H−CO2−C2H5N]+; found: 215.08199 (22.5).
Biological tests with 2: For agar plate diffusion assays, 2 (0.5 μmol) in H2O (10 μL) was dropped onto paper discs, dried under sterile conditions, and placed in the middle of the plates inoculated with the organisms (A. tolulyticus, A. brasilense, A. restrictus, B. fastidiosus, B. subtilis, E. coli, N. simplex, P. polymyxa, S. pasteurii, S. capitis, M. hiemalis, and S. fusiger). The plates were incubated at 37 °C for 24 h for E. coli and B. subtilis. The other bacterial cultures were incubated for 24 h at 30 °C. M. hiemalis was incubated at 25 °C for 24 h and S. fusiger at 12 °C for 11 days.
The dilution assays in 96‐well microtiter plates were performed with 2 (0.066 μmol) against the following organisms: Schizosaccharomyces pombe, Pichia anomala, M. hiemalis, Rhodotorula glutinis, Micrococcus luteus, B. subtilis, E. coli, Mycolicibacterium smegmatis, Chromobacterium violaceum, and Pseudomonas aeruginosa for 24 h.35
Biofilm inhibition was tested against Candida albicans and Staphylococcus aureus under the same conditions.36, 37
Cytotoxicity was tested against the tumour cell lines L929 and KB3.1 at a concentration of 1 mg mL−1 of 2.38
The test for nematicidal activity against C. elegans was performed with concentrations up to 100 μg mL−1 in 24‐well microtiter plates for 20 °C for 18 h.39
Synthesis of 2 from 1 and formaldehyde: Enantiopure 1 obtained by total synthesis15 (2.0 mg, 6.9 μmol) was dissolved in H2O (1 mL), treated with an excess of formaldehyde (37 % aqueous solution, 5 μL, 69 μmol), and stirred for 2 h at room temperature. The solvent was evaporated and the reaction product was purified on a Sephadex LH20 column with H2O as eluent. Compound 2 (1.9 mg, 6.3 μmol) was obtained in 91 % yield. In the same way, compound 2 was synthesised from 1 isolated from M. rosea. However, in this case, product 2 was purified by semipreparative HPLC, as described for the isolation of 2.
Quantification of formaldehyde in fruiting bodies of Mycena species: To ensure a stable stock concentration, a standard solution of 0.1 mg/10 mL solvent was prepared for each reagent and diluted to the concentration needed. The stock standard solutions were stored at −35 °C. For calibration, formaldehyde spiking solutions were taken from a primary dilution standard and subsequently diluted to the specific concentration.
The water for calibration and aqueous extracts was LC‐MS grade (Sigma–Aldrich) and treated with 500 mg L−1 each of copper sulfate and ammonium sulfate, to prevent bacteria from emitting formaldehyde in decomposition processes.16 To minimise the background level of formaldehyde, the water used was treated with UV light (λ=366 nm) for at least 90 min.
For sample preparation, to a sample volume (20 mL) of calibration water or of an aqueous extract of M. rosea potassium hydrogen phthalate (200 mg) was added to adjust the pH of the solution to 4. To ascertain the quality of derivatisation, surrogate analyte, 2′,4′,5′‐trifluoroacetophenone (20 μL, 200 μg mL−1), which was unlikely to be found in mushrooms, was added to the solution. For calibration, calibration spiking solution (20 μL) was added. A freshly produced aqueous solution of PFBHA (1 mL, 7.5 mg mL−1) was added to the sample. Derivatisation was conducted at (35±2) °C for 2 h in an EPA‐certified 30 mL closed glass vessel. After derivatisation, the sample was allowed to cool to room temperature before the addition of three drops of concentrated sulfuric acid. A liquid–liquid extraction was carried out with n‐hexane (4 mL) containing the internal standard 1,2‐dibromopropane (800 μg L−1). The organic phase was washed with 0.2 n sulfuric acid (3 mL). Two GC samples (1.5 mL) were taken from the organic phase, one for direct measurement and the other was stored for subsequent verification. The injection volume was 1 μL.16
For GC‐MS analysis, the same temperature program as that described in EPA‐556 method was used.16 After holding the temperature at 50 °C for 1 min, a linear gradient (4 °C min−1) to 220 °C was used, followed by a second linear gradient (20 °C min−1) to 250 °C. Then, the temperature was kept at 250 °C for 10 min. For sample injection, a heated PTV injector (200 °C) in splitless injection mode was used. The mass spectrometer was operated in positive selected‐ion monitoring mode with SIM masses of 121, 123, and 181 and a scan time of 0.36 s per scan. By performing the measurements in SIM mode, a significant gain of resolution was achieved.40
The calibration was carried out with 10 calibration points for each calibration curve. The calibration points were chosen to be equidistant and in a repetition of n=6 non‐technical replicates.41 Since the range of operation for fresh fruiting bodies was significantly lower than that for stored fruiting bodies, two separate calibrations were conducted.
For fresh fruiting bodies, the chosen concentration range was 25 to 250 μg L−1, in equidistant steps of 25 μg L−1. The regression function obtained was y=7.853(±0.074)x, r 2=0.9993. The area under the curve and the concentrations were standardised to the values of the internal standard, 1,2‐dibrompropane. The residual standard deviation of the first calibration (s y1) was calculated to be 0.228457. For the first calibration, the standard deviation of variation (s xo) was 0.0291. This led to a coefficient of variation (CV) of 3.38 %. The Mandel fitting test was applied for the mathematical verification of linearity, resulting in F calcd=0.593 versus F tab(α=0.01; 1, n−3)=8.40, proving the linearity of the regression. The minimum detection level (MDL)41 was estimated to be 3.052×10−8 mol L−1 with a student's t value for the 99 % confidence level. The concentration estimated to be near the MDL, 2 to 3 times the noise level, was 5 μg L−1.
The limit of detection (LOD) and the limit of quantification (LOQ) were determined as described previously.42 Thus, LOD1 and LOQ1 were determined to be 2.2×10−7 and 4.4×10−7 mol L−1, respectively.
Experiments with regard to the recovery of the regression function showed an average of (91.5±5.6) % (n=6) by adding formaldehyde spiking solutions (75 μg L−1) to half of the samples already containing 50 μg L−1 formaldehyde. For stored fruiting bodies, the concentration range was found to be significantly higher, and therefore, the calibration had to be more widespread. This calibration ranged from 200 to 2000 μg L−1 in equidistant steps of 200 μg L−1. The second regression function obtained was y=14.371(±0.093)x−6.5073, r 2=0.9997. The area under the curve and the concentrations were standardised to the values of the internal standard, 1, 2‐dibromopropane. The residual standard deviation (s y1) was calculated to be 0.9534109. For the second calibration, the standard deviation of variation (s xo) was 0.06790676, which led to a CV of 0.99 %. The Mandel fitting test was applied for the mathematical verification of linearity, resulting in F calcd=0.795 versus F tab(α=0.01; 1, n−3)=8.40, proving the linearity of the regression. LOD2 and LOQ2 were calculated, as in calibration 1, to be 3.5×10−6 and 7.07×10−6 mol L−1, respectively. Both calibrations fitted the criteria of linear regression and could be used to measure the formaldehyde content in fruiting bodies. The measured values were given with their respective confidence interval (CI), which was calculated as shown in the Supporting Information.43
To include the possibility of matrix effects in the measured data, a matrix calibration was performed and compared with the second calibration. A systematic–proportional matrix effect was found in the differences of the slopes and included in the calculations as a correction factor. The values were removed from the comparison between matrix calibration and normal calibration. Matrix recovery experiments led to an average recovery of (89.3±0.2) %. Calibration curves and all formulae for the calculations are shown in the Supporting Information.
Feeding experiment of fruiting bodies of M. rosea with [D4]MeOH: The feeding experiments were performed in October 2007 and 2008 in a forest at Mühltal near Starnberg. Five young fruiting bodies of Mycena rosea were chosen for the experiments and [D4]MeOH (10 μL) was injected into each fruiting body. Three days later, the fruiting bodies were harvested. Then each fruiting body was extracted for 20 min with formaldehyde‐free MeOH (40 mL). The extract was filtered and the solvents were removed at 35 °C, and the residue was dissolved in MeOH and H2O (1:1, v/v, 2 mL) and pre‐purified on an RP‐18 cartridge. The extract was then subjected to LC‐HR‐(+)‐ESIMS analysis. The incorporation rate was calculated by determining the ratio of the peak area at m/z 304 to the sum of the peak areas at m/z 302 and 304, yielding an incorporation rate of 57 % into 2.
Agar diffusion assay with formaldehyde against mycelial cultures of S. fusiger: Ninety agar plates with BPM medium44 were inoculated with spores of S. fusiger obtained from previously grown cultures (CBS: 633.80) and incubated at 12 °C. After 3 days on each plate, signs of growth (first hyphae) were visible. For the tests, aqueous stock solutions with different formaldehyde concentrations (3.33, 16.7, 33.3, 50.0, 66.7, 83.3, 166, and 833 μmol mL−1) and a negative control with H2O were prepared. For a single test, 10 μL of the respective stock solution and negative control were dropped onto a paper disc (0.6 cm diameter) and placed on the centre of the plates with the growing cultures of S. fusiger. The plates were incubated for a further 9 days at 12 °C. After 9 days, the growth of S. fusiger was observed and the diameter of the inhibition zone was determined (Table S8). Each test was performed for each concentration and for the control 10 times in parallel.
Identification of gallic acid in cultures of S. fusiger: Three mycelial cultures of S. fusiger (CBS 633.80) grown on 9 cm agar plates on BPM medium44 for 23 days at 12 °C were removed from the plates and extracted with MeOH (25 mL). Then, the extract was filtered and the solvent was evaporated in vacuum. The residue was dissolved in ethyl acetate (1.2 mL) and dried with Na2SO4. After filtration and removal of the solvent, the residue was pertrimethylsilylated with N‐methyl‐N‐(trimethylsilyl)trifluoroacetamide (MSTFA, 50 μL) at 60 °C for 15 min. Then, the obtained solution (0.5 μL) was subjected to GC‐MS analysis by using a PTV injector at 200 in the split mode (split ratio: 1:30). For sample separation, the following temperature program was used: 50 °C for 1 min, then linear with 5 K min−1 to 300 °C, then isotherm for 10 min at 300 °C. Pertrimethylsilylated gallic acid: GC‐(+)‐EIMS: R i=2008; m/z (%): 458 (30), 443 (12), 399 (3), 369 (2), 355 (3), 311 (4), 281 (63), 253 (3) 237 (2), 207 (3), 193 (3), 179 (13), 147 (10), 133 (7), 73 (100), 45 (13).
Correlation of the ratio of the amount of 2 to the total amounts of 1 and 2 with the formaldehyde content: To evaluate the percentage of 2 in comparison to the sum of 1 and 2 in the same extracts from which the formaldehyde content was measured, half of the extract was dried by means of rotary evaporation and pre‐purified by using an RP‐18ec cartridge. Pigments 1 and 2 readily eluted from the cartridge with 1:1 (v/v) H2O/MeCN solvent. The obtained samples were analysed with an LC‐(+)‐ESI‐MS instrument equipped with a UV/Vis DAD. For sample separation, an RP‐18ec column (Nucleodur 250/3, 100‐5) was used with the following gradient program: 10 min at 100 % H2O+0.1 % HOAc, then within 40 min linear to 100 % MeCN, flow rate 0.66 mL min−1, detection at λ=360 nm. Because pigments 1 and 2 exhibited nearly the same extinction values at λ=360 nm, the relative peak areas of 1 and 2 could be used to determine the ratio of the amount of 2 to the total amounts of 1 and 2.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful to Prof. Dr. Michael Spiteller, to Dr. Marc Lamshöft (Institut für Umweltforschung, Universität Dortmund), to Dr. Thomas Dülcks and Dorit Kemken for the measurements of the HR‐(+)‐ESIMS spectra; to Dr. Wieland Willker and Johannes Stelten for their help with the measurements of the NMR spectra; to Prof. Dr. Horst Kessler (TU München) for providing access to the 900 MHz NMR spectrometer; to Prof. Dr. Dieter Spiteller (Universität Konstanz) for the measurement of the CD spectra; to Prof. Dr. Lucio Colombi Ciacchi and Dr. Monika Michaelis for assistance and providing access to their CD spectrometer; to Prof. Dr. Barbara Reinhold‐Hurek for providing three soil bacteria cultures; to Wera Collisi for performing cytotoxicity tests; and to the Deutsche Forschungsgemeinschaft (SP718/4‐1) for financial support.
R. Himstedt, S. Wagner, R. J. R. Jaeger, M.-L. Lieunang Watat, J. Backenköhler, Z. Rupcic, M. Stadler, P. Spiteller, ChemBioChem 2020, 21, 1613.
Dedicated to Professor Horst Kessler on the occasion of his 80th birthday
References
- 1. Robich G., Mycena d'Europa, Associazione Micologica Bresadola, Trento, 2003, pp. 123–127. [Google Scholar]
- 2. Peters S., Spiteller P., Eur. J. Org. Chem. 2007, 1571–1576. [Google Scholar]
- 3. Antunes E. M., Copp B. R., Davies-Coleman M. T., Samaai T., Nat. Prod. Rep. 2005, 22, 62–72. [DOI] [PubMed] [Google Scholar]
- 4. Sakemi S., Sun H. H., Jefford C. W., Bernardinelli G., Tetrahedron Lett. 1989, 30, 2517–2520. [Google Scholar]
- 5. Stierle D. B., Faulkner D. J., J. Nat. Prod. 1991, 54, 1131–1133. [DOI] [PubMed] [Google Scholar]
- 6. Perry N. B., Blunt J. W., McCombs J. D., Munro M. H. G., J. Org. Chem. 1986, 51, 5476–5478. [Google Scholar]
- 7. Sun H. H., Sakemi S., Burres N., McCarthy P., J. Org. Chem. 1990, 55, 4964–4966. [Google Scholar]
- 8. Radisky D. C., Radisky E. S., Barrows L. R., Copp B. R., Kramer R. A., Ireland C. M., J. Am. Chem. Soc. 1993, 115, 1632–1638. [Google Scholar]
- 9. Peters S., Jaeger R. J. R., Spiteller P., Eur. J. Org. Chem. 2008, 319–323. [Google Scholar]
- 10. Lohmann J. S., Wagner S., von Nussbaum M., Pulte A., Steglich W., Spiteller P., Chem. Eur. J. 2018, 24, 8609–8614. [DOI] [PubMed] [Google Scholar]
- 11. Peters S., Spiteller P., J. Nat. Prod. 2007, 70, 1274–1277. [DOI] [PubMed] [Google Scholar]
- 12. Pulte A., Wagner S., Kogler H., Spiteller P., J. Nat. Prod. 2016, 79, 873–878. [DOI] [PubMed] [Google Scholar]
- 13. Talou T., Breheret/Hulin-Bertraut S., Gaset A. in Frontiers of Flavour Science (Eds.: P. Schieberle, K.-H. Engel), Deutsche Forschungsanstalt für Lebensmittelchemie, Garching, 2000, pp. 46–50. [Google Scholar]
- 14. Dorofeeva O. V., Mastryukov V. S., Allinger N. L., Almenningen A., J. Phys. Chem. 1985, 89, 252–257. [Google Scholar]
- 15. Backenköhler J., Reck B., Plaumann M., Spiteller P., Eur. J. Org. Chem. 2018, 2806–2816. [Google Scholar]
- 16.J. W. Munch, D. J. Munch, S. D. Winslow, S. C. Wendelken, B. V. Pepich, Method 556, Determination of Carbonyl Compounds in Drinking Water by Pentafluorobenzylhydroxylamine Derivatization and Capillary Gas Chromatography with Electron Capture Detection, United States Environmental Protection Agency, Cincinnati, 1998, pp. 1–37.
- 17. Mason D. J., Sykes M. D., Panton S. W., Rippon E. H., Food Addit. Contam. 2004, 21, 1071–1082. [DOI] [PubMed] [Google Scholar]
- 18. Liu J.-F., Peng J.-F., Chi Y.-G., Jiang G.-B., Talanta 2005, 65, 705–709. [DOI] [PubMed] [Google Scholar]
- 19. Claeys W., Vlemincky C., Dubois A., Huyghebaert A., Hoefte M., Daenes P., Schiffers B., Food Addit. Contam. A 2009, 26, 1265–1272. [Google Scholar]
- 20. Kurashima Y., Tsuda M., Sugimura T., J. Agric. Food Chem. 1990, 38, 1945–1949. [Google Scholar]
- 21. Mori N., Sano M., Tani Y., Yamada H., Agric. BioI. Chem. 1980, 44, 1391–1397. [Google Scholar]
- 22. Massiot P., Perron V., Baron A., Drilleau J.-F., Lebensm.-Wiss. Technol. 1997, 30, 697–702. [Google Scholar]
- 23. de Oliveira B. V., Teixeira G. S., Reis O., Barau J. G., Teixeira P. J. P. L., do Rio M. C. S., Domingues R. R., Meinhardt L. W., Paes Lemec A. F., Rinconesa J., Pereira G. A. G., Fung. Gen. Biol. 2012, 49, 922–932. [DOI] [PubMed] [Google Scholar]
- 24. Reid M. F., Fewson C. A., Crit. Rev. Microbiol. 1994, 20, 13–56. [DOI] [PubMed] [Google Scholar]
- 25. Power E. G. M., Prog. Med. Chem. 1997, 34, 149–201. [Google Scholar]
- 26. Spiteller P., Chem. Eur. J. 2008, 14, 9100–9110. [DOI] [PubMed] [Google Scholar]
- 27. Spiteller P., Nat. Prod. Rep. 2015, 32, 971–993. [DOI] [PubMed] [Google Scholar]
- 28. Helfer W., Pilze auf Pilzfruchtkörpern, IHW-Verlag, Eching, 1991. [Google Scholar]
- 29. Aruoma O., Murcia A., Butler J., Halliwell B., J. Agric. Food Chem. 1993, 41, 1880–1885. [Google Scholar]
- 30. Haslam E., J. Nat. Prod. 1996, 59, 205–215. [DOI] [PubMed] [Google Scholar]
- 31. Jiang H.-P., Cai N., Ju X.-L., Huang J., Wang X., Rapid Commun. Mass Spectrom. 2018, 32, 2074–2080. [DOI] [PubMed] [Google Scholar]
- 32. Bianchi F., Careri M., Musci M., Mangia A., Food Chem. 2007, 100, 1049–1053. [Google Scholar]
- 33. Gottlieb H. E., Kotlyar V., Nudelman A., J. Org. Chem. 1997, 62, 7512–7515. [DOI] [PubMed] [Google Scholar]
- 34. Kováts E., Helv. Chim. Acta 1958, 41, 1915–1932. [Google Scholar]
- 35. Surup F., Mohr K. I., Jansen R., Stadler M., Phytochemistry 2013, 95, 252–258. [DOI] [PubMed] [Google Scholar]
- 36. Brambillaa L. Z. S., Endo E. H., Cortez D. A. G., Dias Filho B. P., Rev. Bras. Farmacogn. 2017, 27, 112–117. [Google Scholar]
- 37. Chepkirui C., Yuyama K. T., Wanga L. A., Decock C., Matasyoh J. C., Abraham W. R., Stadler M., J. Nat. Prod. 2018, 81, 778–784. [DOI] [PubMed] [Google Scholar]
- 38. Surup F., Halecker S., Nimtz M., Rodrigo S., Schulz B., Steinert M., Stadler M., Steroids 2018, 135, 92–97. [DOI] [PubMed] [Google Scholar]
- 39. Schrey H., Müller F. J., Harz P., Rupcic Z., Stadler M., Spiteller P., Phytochemistry 2019, 160, 85–91. [DOI] [PubMed] [Google Scholar]
- 40. Yu J., Jeffries H. E., Le Lacheur R. M., Environ. Sci. Technol. 1995, 29, 1923–1932. [DOI] [PubMed] [Google Scholar]
- 41.German Norm for Calibration: DIN 3840251A.
- 42. Glaser J. A., Foerst D. L., McKee G. D., Quave S. A., Budde W. L., Environ. Sci. Technol. 1981, 15, 1426–1435. [Google Scholar]
- 43. Funk W., Dammann V., Donnevert G., Qualitätssicherung in der Analytischen Chemie , 2nd ed., Wiley-VCH, Weinhein, 2005, pp. 1–50. [Google Scholar]
- 44. Benny G., Aliso 2008, 26, 37–61. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary