

Abstract
Curcumin (Cur) has various pharmacological activities, including anti‐inflammatory, antiapoptotic and anticancer effects. However, there is no report on the effect of Cur on endothelial cell fibrosis. This study was designed to investigate the effect and mechanism of Cur on endothelial cell fibrosis. An endothelial cell fibrosis model was established by using transforming growth factor (TGF) induction. Proliferation assays, qRT‐PCR, western blotting and immunostaining were performed to investigate the effects and mechanism of Cur on endothelial cell fibrosis. We found that in human umbilical vein endothelial cells (HUVECs), TGF‐β1 treatment significantly decreased the expression of nuclear factor erythroid‐2‐related factor 2 (NRF‐2), dimethylarginine dimethylaminohydrolase‐1 (DDAH1), and VE‐cadherin, the secretion of cellular nitric oxide (NO) and the activity of nitrous oxide synthase (NOS), while asymmetric dimethylarginine (ADMA) and the release of inflammatory factors were elevated. Immunofluorescence showed decreased CD31 and increased α‐smooth muscle actin (α‐SMA). Overexpression of NRF‐2 significantly attenuated the effects of TGF‐β1, while downregulation of DDAH1 potently counteracted the effect of NRF‐2. In addition, ADMA treatment resulted in similar results to those of TGF‐β1, and Cur significantly attenuated the effect of TGF‐β1, accompanied by increased VE‐cadherin, DDAH1 and NRF‐2 and decreased matrix metalloproteinase‐9 (MMP‐9) and extracellular regulated protein kinases 1/2 (ERK1/2) phosphorylation. The NRF‐2 inhibitor ML385 had the opposite effect as that of Cur. These results demonstrated that Cur inhibits TGF‐β1‐induced endothelial‐to‐mesenchymal transition (EndMT) by stimulating DDAH1 expression via the NRF‐2 pathway, thus attenuating endothelial cell fibrosis.
Keywords: curcumin, endothelial‐to‐mesenchymal transition (EndMT), NRF‐2‐DDAH‐ADMA‐NO, TGF‐β1
1. INTRODUCTION
The endothelial‐to‐mesenchymal transition (EndMT) is a complex biological process that is one of the mechanisms that generates myofibroblasts in fibrotic tissues or organs. 1 , 2 , 3 , 4 The main characteristics of the EndMT are the loss of adhesion and polarity between endothelial cells, transformation to mesenchymal cells, acquisition of a mesenchymal phenotype, and an increase in cell migration and collagen secretion. 5 The EndMT refers to the gradual loss of endothelial cell‐specific markers such as CD31 and VE‐cadherin and the gradual decrease in endothelial cell junctions, 6 , 7 , 8 as well as the acquisition of a mesenchymal or myofibroblastic phenotype and expression of mesenchymal cell products such as α‐smooth muscle actin (α‐SMA) and enhanced collagen synthesis, migration, and secretion of a large amount of extracellular matrix and collagen, leading to myocardial fibrosis. 9 , 10 Studies have reported that in addition to fibroblasts and myofibroblasts, endothelial cells can transdifferentiate to mesenchymal cells through the EndMT process, further leading to endothelial dysfunction. 11
Fibroblasts and myofibroblasts are key fibroblastic effector cells. 12 , 13 Recent studies have shown that the EndMT process in organ fibrosis is one of the important sources of fibroblasts and is one of the important mechanisms of fibrosis development. 14 , 15 Studies have shown that abnormal EndMT is a key mechanism in the development of fibrosis in multiple tissues or organs in humans. 8 , 16 , 17 Studies have shown that transforming growth factor‐β1 (TGF‐β1) is a potent factor in inducing the EndMT process. 18 , 19 Therefore, in this study, the EndMT was used as an entry point, TGF‐β1 was used to induce human umbilical vein endothelial cells (HUVECs) to establish an EndMT model. Actively searching for and discovering innovative drugs that can interfere with and regulate the EndMT may be an effective way to prevent and treat myocardial fibrosis.
Acute and chronic inflammation often leads to fibrosis. 20 Inflammation can damage epithelial cells (usually vascular endothelial cells), and excessive release of inflammatory mediators promotes the development of organ or tissue fibrosis. 21 Studies have shown that cardiac fibrosis is a chronic inflammatory process, and inflammatory stimuli accelerate the progression of cardiac fibrosis through the EndMT. 22
Asymmetric dimethylarginine (ADMA) is an endogenous nitric oxide (NO) synthesis inhibitor and promotes the occurrence of cardiovascular disease. Dimethylarginine dimethylaminohydrolase (DDAH) is a metabolic enzyme of ADMA. Studies have found that increased expression of DDAH inhibits matrix metalloproteinase‐9 (MMP9), ADMA, epidermal growth factor receptor (EGFR) and extracellular regulated protein kinases (ERK) signalling pathways, while TGF‐β1 inhibits the expression and activity of DDAH, thereby inducing ADMA. 23 , 24 In addition, studies have shown that TGF‐β1 significantly inhibits the expression and activity of nuclear factor erythroid‐2‐related factor 2 (NRF‐2), which is a potential therapeutic target for liver fibrosis. 25
Curcumin (Cur) is a polyphenolic substance that is naturally extracted from the rhizome of turmeric plants and has antifibrotic, anti‐inflammatory, antioxidant and hypolipidaemic effects. 26 , 27 However, it is not clear whether curcumin inhibits myocardial fibrosis induced by the EndMT. Therefore, this study aimed to investigate whether curcumin attenuates TGF‐β1‐induced fibrosis in HUVECs by inhibiting the EndMT process via regulating the NRF2‐DDAH1‐ADMA‐NO pathway.
2. RESULTS
2.1. Involvement of the NRF‐2/DDAH1/ADMA pathway in TGF‐β1‐induced EndMT in human umbilical vein endothelial cells (HUVECs)
First, the involvement of the NRF‐2/DDAH1/ADMA pathway in TGF‐β1‐induced EndMT in HUVECs was investigated. As shown in Figure 1A‐D, TGF‐β1 significantly decreased NRF‐2 (Figure 1A) and DAAH1 (Figure 1B) expression (P < .01) and inhibited NO levels (Figure 1C) and nitric oxide synthase (NOS) (Figure 1D) activity (P < .01), whereas IL‐1β (Figure 1E), TNF‐α (Figure 1F) and ADMA (Figure 1G) levels were significantly increased. Western blotting showed that TGF‐β1 significantly decreased the expression of VE‐cadherin, DDAH and NRF‐2, whereas vimentin was increased (Figure 1H, P < .01). Immunofluorescence staining showed an obvious decrease in CD31 (Figure 1I) fluorescence in the nuclei of the TGF‐β1‐induced group, while α‐SMA (Figure 1J) fluorescence was significantly enhanced. In addition, overexpression of NRF‐2 potently counteracted the effect of TGF‐β1 (P < .01), while downregulation of DDAH1 had the opposite effect to NRF‐2 overexpression (P < .01). The cell morphology of EndMT process was showed in Figure S1 and the efficiency of NRF‐2 overexpression and DDAH1 downregulation is shown in Figure S2. These results indicated that the NRF‐2/DDAH1/ADMA pathway may be involved in TGF‐β1‐induced EndMT in HUVECs.
Figure 1.
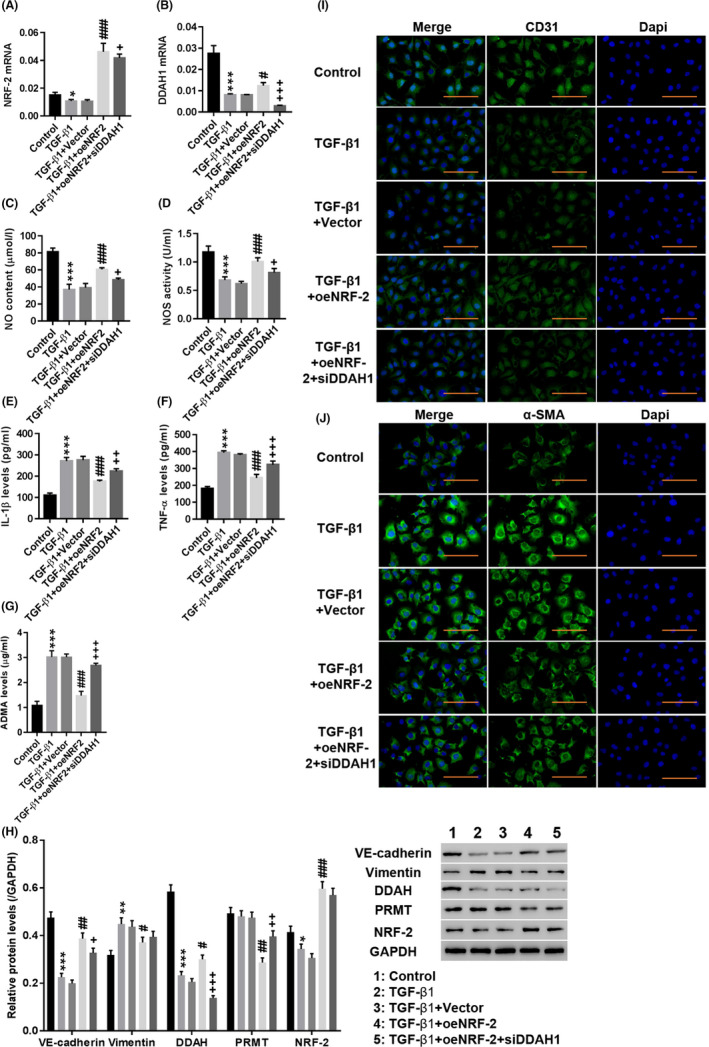
Involvement of the NRF‐2/DDAH1/ADMA pathway in TGF‐induced EndMT in human umbilical vein endothelial cells (HUVECs). HUVECs were treated with medium (Control), TGF‐β1, TGF‐β1 + Vector, TGF‐β1 + oeNRF‐2 and TGF‐β1 + oeNRF‐2 + siDDAH1. A, B, The mRNA expression of NRF‐2 and DDAH1 was detected. C–G, The levels of NO (C), NOS (D), IL‐1β (E), TNF‐α (F) and ADMA (G) were detected. H, The protein levels of VE‐cadherin, vimentin, DDAH, PRMT and NRF‐2 were detected. I, J, Immunofluorescence detection of CD31 (I) and α‐SMA (J) at a magnification of 200 ×. *P < .05, **P < .01, ***P < .001 vs Control, siNC or Vector; #
P < .05, ##
P < .01, and ###
P < .001 vs TGF + Vector; +
P < .05, ++
P < .01, and +++
P < .001 vs TGF + oeNRF‐2. (H)  , Control;
, Control;  , TGF‐β1;
, TGF‐β1;  , TGF‐β1 + Vector;
, TGF‐β1 + Vector;  , TGF‐β1 + oeNRF2;
, TGF‐β1 + oeNRF2;  , TGF‐β1 + oeNRF2 + siDDAH1
, TGF‐β1 + oeNRF2 + siDDAH1
2.2. ADMA induced the EndMT in HUVECs
CCK‐8 assays were performed to detect cell viability. The results are shown in Figure S3. Treatment with ADMA significantly increased the cell viability in a time‐ and dose‐dependent manner compared with that of the control group. Therefore, 72‐hour treatment with 10 and 20 μmol/L ADMA were selected for subsequent testing.
High‐performance liquid phase analysis showed that treatment with both 10 and 20 μmol/L ADMA significantly increased the level of ADMA in HUVECs, and TGF‐β1 also increased ADMA levels, but the effect of ADMA treatment was stronger than that of TGF‐β1 (Figure 2A). Moreover, treatment with ADMA significantly decreased the NO content (Figure 2B) and NOS activity (Figure 2C) (P < .01), and TGF‐β1 had a similar effect (P < .01). In contrast, the levels of IL‐1β (Figure 1D) and TNF‐α (Figure 1E) were significantly increased by ADMA (P < .01), and TGF‐β1 also increased the levels of TNF‐α and IL‐1β (P < .01). Western blotting showed that the expression of vimentin and DDAH was significantly increased by ADMA (P < .01), and TGF‐β1 had a similar effect (P < .01), while ADMA had no significant effect on the protein expression levels of VE‐cadherin or PRMT (Figure 2F). Immunofluorescence staining showed that ADMA and TGF‐β1 significantly weakened CD31 fluorescence in HUVECs (Figure 2G), while α‐SMA fluorescence was significantly enhanced (Figure 2H). These results indicated that ADMA induced the EndMT in HUVECs, which was consistent with the effect of TGF‐β1.
Figure 2.
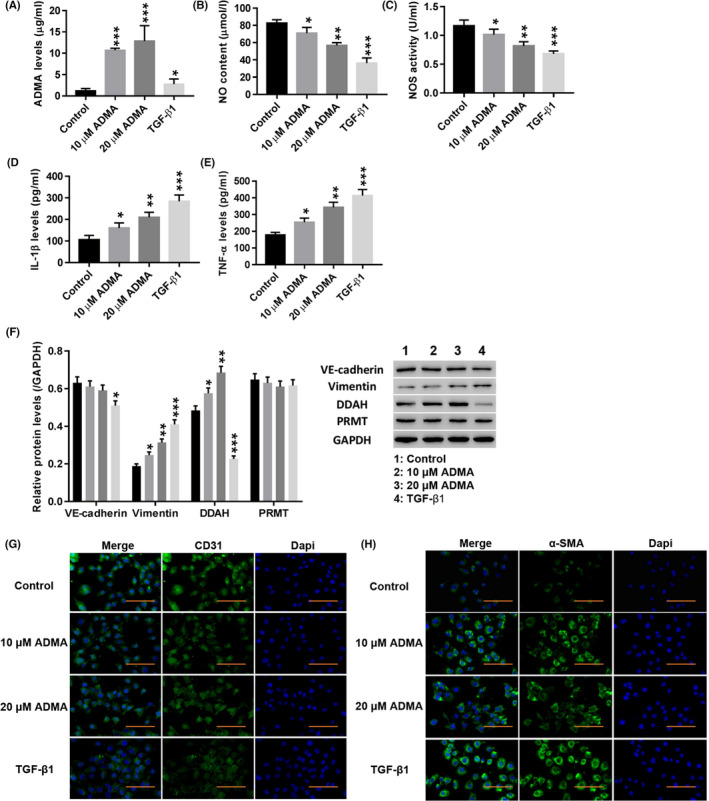
ADMA induced the EndMT in HUVECs. HUVECs were treated with TGF‐β1 (10 µg/L; AF‐100‐21C‐10; PeproTech) and ADMA (5, 10, 20 μmol/L; D4268; Sigma). A–E, The levels of ADMA (A), NO (B), NOS (C), IL‐1β (D) and TNF‐α (E) were detected. F, The protein levels of VE‐cadherin, vimentin, DDAH and PRMT were detected. G, H, Immunofluorescence detection of CD31 (G) and α‐SMA (H) at a magnification of 200×. *P < .05, **P < .01, and ***P < .001 vs Control. (F)  , Control;
, Control;  , 10 μmol/L ADMA;
, 10 μmol/L ADMA;  , 20 μmol/L ADMA;
, 20 μmol/L ADMA;  , TGF‐β1
, TGF‐β1
2.3. Curcumin (Cur) inhibited TGF‐β1‐induced EndMT, and the ERK pathway may be involved in this regulation
Next, the effect of Cur (Figure S4 showed that 5 and 10 µmol/L Cur has no toxic effect on cells) on TGF‐β1‐induced EndMT and ERK1/2 pathways was examined. Figure 3A‐D shows that treatment with Cur significantly reduced TGF‐β1‐induced IL‐1β (Figure 3A) and TNF‐α (Figure 3B) levels, while DDAH1 (Figure 3C) and NRF‐2 (Figure 3D) were significantly increased (P < .01). Overexpression of NRF‐2 potently counteracted the effects of TGF‐β1 in HUVECs (P < .01). Moreover, TGF‐β1 significantly induced the expression of EGFR and MMP9 (P < .01), which was potently counteracted by Cur treatment (Figure 3E,F). In addition, treatment with Cur significantly reduced TGF‐β1‐induced expression of vimentin, p‐ERK1/2 and MMP9 in HUVECs, while VE‐cadherin, DDAH and NRF‐2 were significantly increased (Figure 3G,H) (P < .01). Immunofluorescence staining showed that Cur significantly enhanced CD31 (Figure 3I) fluorescence in HUVECs compared to that of the TGF‐β1 group, while α‐SMA (Figure 3J) fluorescence was decreased. Overexpression of NRF‐2 had a similar effect as that of Cur. These results indicated that Cur inhibited TGF‐β1‐induced EndMT and ERK1/2 pathway activation.
Figure 3.
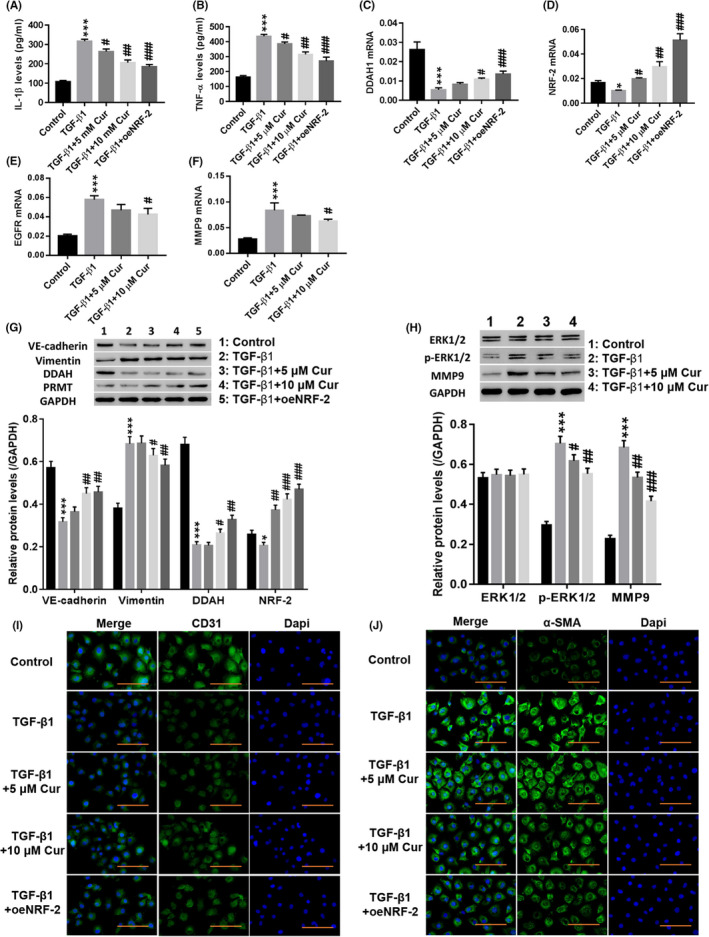
Curcumin (Cur) inhibited TGF‐β1‐induced EndMT, and the ERK pathway may be involved in this regulation. HUVECs were treated with TGF‐β1 (10 µg/L), TGF‐β1 (10 µg/L) + 5 µmol/L Cur (C1386; Sigma), TGF‐β1 (10 µg/L) + 10 µmol/L Cur and TGF‐β1 (10 µg/L) + oeNRF‐2 for 48 h. A, B, IL‐1β (A) and TNF‐α (B) levels were detected. C–F, DDAH1 (C), NRF‐2 (D), EGFR (E) and MMP9 (F) mRNA expression was detected. G, Protein levels of VE‐cadherin, vimentin, DDAH and PRMT were detected. H, Protein levels of ERK1/2, p‐ERK1/2 and MMP9 were detected. I, J, Immunofluorescence detection of CD31 (I) and α‐SMA (J) at a magnification of 200×. *P < .05 and ***P < .001 vs Control, siNC or Vector; #
P < .05, ##
P < .01, and ###
P < .001 vs TGF‐β1. (G)  , Control;
, Control;  , TGF‐β1;
, TGF‐β1;  , TGF‐β1 + 5 μmol/L Cur;
, TGF‐β1 + 5 μmol/L Cur;  , TGF‐β1 + 10 μmol/L Cur;
, TGF‐β1 + 10 μmol/L Cur;  , TGF‐β1 + oeNRF‐2. (H)
, TGF‐β1 + oeNRF‐2. (H)  , Control;
, Control;  , TGF‐β1;
, TGF‐β1;  , TGF‐β1 + 5 μmol/L Cur;
, TGF‐β1 + 5 μmol/L Cur;  , TGF‐β1 + 10 μmol/L Cur
, TGF‐β1 + 10 μmol/L Cur
2.4. Cur inhibited TGF‐β1‐induced EndMT by stimulating NRF‐2 and DDAH1 to degrade ADMA
Different concentrations of the NRF‐2 inhibitor ML385 were used to measure cell proliferation at different times. Figure 4A shows that ML385 increased cell viability in a dose‐ and time‐dependent manner. Therefore, 72‐hour treatment with 2.5 μmol/L ML385 was selected for subsequent testing.
Figure 4.
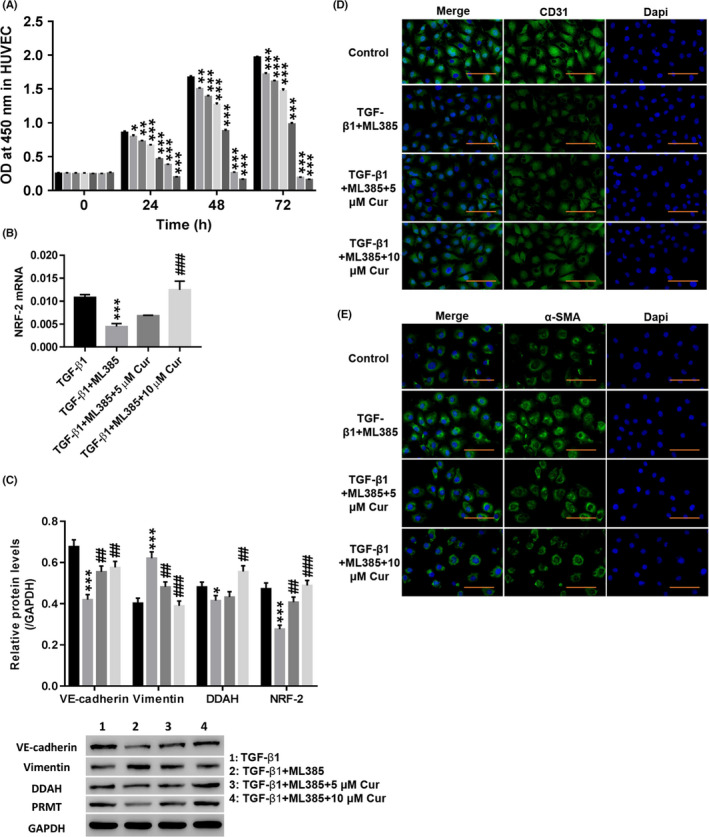
Cur inhibited TGF‐β1‐induced EndMT by stimulating NRF‐2 and DDAH to degrade ADMA. A, After treatment with different concentrations (0, 0.5, 1, 2.5, 5, 10, or 20 μmol/L) of ML385 (NRF‐2 inhibitor; SML1833; Sigma), the cell proliferation was detected at 0, 24, 48 and 72 h. HUVECs were treated with TGF‐β1 (10 µg/L), TGF‐β1 (10 µg/L) + 2.5 μmol/L ML385, TGF‐β1 (10 µg/L) + 2.5 μmol/L ML385 + 5 μmol/L Cur, and TGF‐β1 (10 µg/L) + 2.5 μmol/L ML385 + 10 μmol/L Cur for 48 h. B, NRF‐2 mRNA expression was detected. C, Protein levels of VE‐cadherin, vimentin, DDAH and NRF‐2 were detected. D, E, Immunofluorescence detection of CD31 (D) and α‐SMA (E) at a magnification of 200×. *P < .05, **P < .01, and ***P < .001 vs 0 μmol/L ML385 or TGF‐β1; ##
P < .01 and ###
P < .001 vs TGF‐β1 + ML385. (A)  , 0 μmol/L ML385;
, 0 μmol/L ML385;  , 0.5 μmol/L ML385;
, 0.5 μmol/L ML385;  , 1 μmol/L ML385;
, 1 μmol/L ML385;  , 2.5 μmol/L ML385;
, 2.5 μmol/L ML385;  , 5 μmol/L ML385;
, 5 μmol/L ML385;  , 10 μmol/L ML385;
, 10 μmol/L ML385;  , 20 μmol/L ML385;
, 20 μmol/L ML385;  , TGF‐β1;
, TGF‐β1;  , TGF‐β1 + ML385;
, TGF‐β1 + ML385;  , TGF‐β1 + ML385 + 5 μmol/L Cur;
, TGF‐β1 + ML385 + 5 μmol/L Cur;  , TGF‐β1 + ML385 + 10 μmol/L Cur
, TGF‐β1 + ML385 + 10 μmol/L Cur
As shown in Figure 4B,C, the NRF‐2 inhibitor ML385 significantly inhibited TGF‐β1‐induced expression of VE‐cadherin, DDAH and NRF‐2, while vimentin was increased (P < .01). In addition, the NRF‐2 inhibitor ML385 significantly weakened TGF‐β1‐induced CD31 (Figure 4D) fluorescence, while the α‐SMA (Figure 4E) fluorescence was enhanced. Treatment with Cur potently counteracted the effects of the NRF‐2 inhibitor ML385 (P < .01). The above results indicated that Cur stimulated NRF‐2 and DDAH1 to promote the degradation of ADMA and thereby inhibit TGF‐β1‐induced EndMT.
3. DISCUSSION
Heart failure is a series of clinical syndromes caused by various cardiovascular diseases, such as coronary heart disease, cardiomyopathy, hypertension, and diabetic myocarditis, that causes changes in the structure and function of the normal myocardium, resulting in low ventricular ejection or filling and an inability to meet the metabolic needs of the body and a high morbidity and mortality. 28 , 29 The progression of most cardiovascular diseases leads to heart failure, which is the leading cause of death from cardiovascular disease. 30 Myocardial fibrosis is one of the main components of ventricular remodelling and is the main physiological and pathological basis of heart failure. Under pathological conditions, such as increased circulatory pressure and myocardial necrosis, myocardial fibrosis activates fibroblasts or myofibroblasts to secrete a large amount of extracellular matrix, leading to an imbalance in the metabolism of the extracellular matrix. 31 , 32 When collagen production and mesenchymal cell products are greater than degradation, a large amount of collagen is deposited in the interstitium of myocardial cells, leading to myocardial fibrosis, myocardial contraction, and diastolic and electrophysiological damage. It has been reported that myocardial fibrosis is a common pathological feature of end‐stage heart failure due to various causes. 33 Therefore, myocardial fibrosis is an important target for the treatment of heart failure.
Recent studies have found that abnormal EndMT promotes the progression of myocardial fibrosis. 8 , 17 Therefore, studying the mechanism of the EndMT and finding effective drugs to block this process will help to provide potential clinical treatments for preventing and treating myocardial fibrosis and heart failure. Abnormal EndMT refers to the process in which endothelial cells gradually lose their original morphology, phenotype and function and obtain the morphology, phenotype and function of mesenchymal cells such as fibroblasts or myofibroblasts. 15 , 34 The expression of VE‐cadherin, a specific marker of endothelial cells, is gradually weakened or lost, and the myofibroblast‐specific marker α‐SMA is expressed instead. 35 Studies have shown that many regulatory factors are involved in the EndMT, of which TGF‐β1 is considered to be the most potent factor in the EndMT and myocardial fibrosis. Therefore, upregulation of TGF‐β1 expression is both the mechanism of the EndMT and one of the signs indicating the possible existence of abnormal EndMT. 36 NRF‐2 plays a protective role in fibrosis by regulating antioxidant enzyme activity and downstream gene expression. 37 As a new high‐risk factor for certain diseases, DDAH1 has recently received extensive attention in cardiovascular and renal diseases, but little research has been done on the role of DDAH1 in fibrosis. 38 , 39 In this study, TGF‐β1 was used to induce the EndMT in HUVECs. We found that TGF‐β1‐induced EndMT was potently counteracted by NRF‐2 overexpression, while downregulation of DDAH1 counteracted the effect of NRF‐2. We hypothesized that NRF‐2 inhibits TGF‐β1‐induced EndMT by regulating the expression of ADMA via regulating DDAH1.
Asymmetric dimethylarginine can inhibit NOS activity and reduces NO production. DDAH1 affects NOS activity by regulating ADMA levels in vivo, thereby regulating NO production in vivo. 40 , 41 Studies have shown that NO is closely related to the occurrence and development of fibrosis. 42 These findings are consistent with our results that, similar to TGF‐β1, ADMA decreased NO secretion, decreased NOS activity, and increased inflammatory factor release. Immunofluorescence analysis showed decreased CD31 and increased α‐SMA. Therefore, ADMA induced the EndMT at the cellular level.
Curcumin is a fat‐soluble polyphenolic compound that is extracted from the rhizome of traditional Chinese medicines such as turmeric and other plants (such as turmeric, medlar, and calamus). 43 Studies have shown that curcumin effectively inhibits the malignant behaviour of cancer cells and has broad applications in the treatment of cancer. 44 , 45 Curcumin also has many functions such as antibacterial, anti‐inflammatory, analgesic and wound healing properties, and its immunomodulatory potential in many inflammatory disease models has been demonstrated. 46 , 47 Moreover, studies have shown that curcumin protects against myocardial fibrosis through various mechanisms. For example, curcumin inhibits myocardial fibrosis by inhibiting the activation of MMP‐9, TIMP‐1 and fibroblasts. 48 , 49 Whether curcumin inhibits myocardial fibrosis by inhibiting abnormal EndMT remains unclear. In this study, curcumin alleviates TGF‐β1‐induced EndMT, accompanied by decreased release of extracellular matrix and inflammatory factors, and inactivation of ERK1/2 signalling. Overexpression of NRF‐2 had a similar effect to that of Cur. These results indicated that curcumin may modulate the EndMT by stimulating the NRF‐2 pathway.
In this study, we demonstrated that curcumin inhibits TGF‐β1‐induced EndMT by regulating the NRF2–DDAH–ADMA–NO signalling pathway, thus attenuating endothelial cell fibrosis. Targeting the EndMT is potential new strategy for the prevention and treatment of fibrosis.
4. METHODS
4.1. Culture of human umbilical vein endothelial cells (HUVECs)
The HUVECs were purchased from Changsha YRGene Co. (NC006) and were cultured in DMEM (SH30243.01; HyClone) containing 10% fetal bovine serum (10% FBS + heparin 0.1 mg/mL + ECGS 10 μL/mL + DMEM) (16000‐044; Gibco) and a 1% mixture of penicillin and streptomycin (P1400‐100; Solarbio) at 37°C and 5% CO2.
4.2. Configuration of curcumin solution
The curcumin dry powder (C1386; Sigma) was weighed with an electronic balance, added to dimethyl sulfoxide (DMSO) and stored in a refrigerator at 4°C. The above medium was used to dilute the solution to the required concentration with a final concentration of DMSO in the medium of <0.2%.
4.3. Lentivirus construction and cell transfection
The coding DNA sequence (CDS) region of the NRF2 gene containing the restriction site (upstream EcoRI, downstream BamHI) was synthesized. The CDS was ligated into the pLVX‐Puro vector, followed by transformation and plasmid extraction. Lentiviral packaging was performed by lipofection (Lipofectamine 2000; Invitrogen) in 293T cells. 293T cells were seeded into 6‐well plates for transfection, and vector DNA and liposomes were transfected at a ratio of 1:2 as follows: 1000 ng of pLVX‐Puro‐NRF2, 100 ng of psPAX2, and 900 ng of pMD2G. After 4 hours of culture, the transfection medium was replaced with complete medium. Supernatants containing virus were collected at 48 and 72 hours.
4.4. Interference of DDAH1
Three siRNAs targeting the DDAH1 gene (siDDAH1‐1, siDDAH1‐2, and siDDAH1‐3) were designed, and then the cells were transfected with the three siDDAH1 constructs by lipofection (Lipofectamine 2000; Invitrogen). The sequences of the three siDDAH1 constructs are listed in Table 1.
Table 1.
Interference sequences of DDAH1
| siDDAH‐1 (407‐425) |
Sense: 5′‐GGAGGAAGGAGGUUGACAUUU‐3′; Antisenes: 5′‐AUGUCAACCUCCUUCCUCCUU‐3′ |
| siDDAH‐2 (835‐853) |
Sense: 5′‐GAGUAUCCAGAAAGUGCAAUU‐3′; Antisense: 5′‐UUGCACUUUCUGGAUACUCUU‐3′ |
| siDDAH‐3 (1192‐1210) |
Sense: 5′‐CCUAGAAGAUACAGAGCUAUU‐3′; Antisense: 5′‐UAGCUCUGUAUCUUCUAGGUU‐3′ |
| siNC |
Sense: 5′‐CAGUACUUUUGUGUAGUACAA‐3′; Antisense: 5′‐UUGUACUACACAAAAGUACUG‐3′ |
4.5. RNA purification and qRT‐PCR
Total RNA in cells was extracted by using TRIzol reagent (Invitrogen), followed by verification of RNA quantity and quality. After reverse transcription by using a reverse transcription kit (#K1622; Fermentas), qRT‐PCR was performed by using a SYBR Green PCR kit (#K0223; Thermo) on a ViiATM 7 real‐time PCR system (Life Technologies) using the following parameters: 95°C for 10 min, followed by 40 cycles of 95°C for 15 seconds and 60°C for 45 seconds, and ending with 95°C for 15 seconds. GAPDH and U6 were used as internal references. The 2−ΔΔCT method was used to determine relative gene expression levels. Three replicate analyses were performed for each sample. The specific qRT‐PCR experimental methods were performed as previously described. 50 The sequences of the primers used are listed in Table 2.
Table 2.
Sequences of primers
| NRF‐2 |
Forward: 5′‐GAGGTTTCTTCGGCTACGTTTC‐3′; Reverse: 5′‐TGGTAGTCTCAACCAGCTTGTC‐3′ |
| DDAH1 |
Forward: 5′‐AACTACACAGAGGCACATC‐3′; Reverse: 5′‐ACTGGAAATACGGTGAGTC‐3′ |
| MMP9 |
Forward: 5′‐GTGGCACCACCACAACATCAC‐3′; Reverse: 5′‐CGCGACACCAAACTGGATGAC‐3′ |
| EGFR |
Forward: 5′‐TTCGGCACGGTGTATAAG‐3′; Reverse: 5′‐TTGTGTTCCCGGACATAG‐3′ |
| GAPDH |
Forward: 5′‐AATCCCATCACCATCTTC‐3′; Reverse: 5′‐AGGCTGTTGTCATACTTC‐3′ |
4.6. Western blot analysis
Total protein was extracted, and the protein concentration was quantified using a BCA protein assay kit. The proteins were separated by 10% SDS‐PAGE, followed by semidry transfer to polyvinylidene fluoride (PVDF) membranes (HATF00010; Millipore). After blocking in 5% skim milk (BYL40422; BD Biosciences) for 1 hour at room temperature, the membrane was incubated with anti‐DDAH1 (1:1000; Ab180599; Abcam), anti‐NRF‐2 (1:1000; Ab62352; Abcam), anti‐VE‐cadherin (1:1000; Ab166715; Abcam), anti‐PRMT (1:1000; Ab190892; Abcam), anti‐Vimentin (1:1000; ab8925; Abcam), anti‐ERK1/2 (1:1000; #4695; Cell Signaling Technology [CST]), anti‐p‐ERK1/2 (1:2000; #4370; CST), anti‐MMP9 (1:1000; Ab76003; Abcam) and anti‐GAPDH (1:2000; #5174; CST) at 4°C overnight. Then, a horseradish peroxidase‐conjugated secondary antibody (1:1000; Beyotime) was added. After developed by using a chemiluminescent reagent (WBKLS0100; Millipore) for 5 minutes, the protein bands were visualized on an ECL imaging system (Tanon‐5200; Tanon). Western blot analysis was performed as previously described. 51
4.7. ELISA
Human interleukin 1β (IL‐1β) and human tumor necrosis factor alpha (TNF‐α) levels were measured using an enzyme‐linked immunosorbent assay (ELISA) kit.
4.8. Biochemical testing
After treatment, the cells were collected. A nitric oxide (NO) kit (a012; Nanjing Institute of Bioengineering) and a NOS test kit (a014‐1; Nanjing Institute of Bioengineering) were used according to the manufacturer's instructions. Briefly, the corresponding reagents were added and mixed well, followed by 60 minutes in a water bath at 37°C. After vortexing well for 30 seconds, the solution was incubated for 10 minutes at room temperature. Following centrifugation for 10 minutes at 2500 g/min, the supernatant was obtained, and the absorbance was detected at 550 nm, which was used to calculate the NO content and NOS activity.
4.9. Cell proliferation
The DMEM was discarded, and the cells in the 96‐well plate were randomly divided into different groups. Then, 1, 5, 10, 15, or 20 μmol/L ADMA medium was added to the cells for further culture. The cells were cultured for different times (0, 24, 48, or 72 hours), and then 10 μL of Cell Counting Kit‐8 (CCK‐8; CP002; SAB) solution was added to the culture plate. The plate was incubated for 1 hour in a 5% CO2 incubator. The optical density (OD) value at 450 nm was measured to compare cell growth activity.
4.10. Immunofluorescence staining
The cells were washed three times with 3% bovine serum albumin (BSA) in PBS, placed in PBS containing 4% PFA, and incubated for 1 hour. Then, the cells were incubated in PBS containing 0.5% Triton X‐100 for permeabilization. Specific experimental methods were performed as previously described. 52 Anti‐CD31 (ab24590; Abcam) and anti‐α‐SMA (ab124964; Abcam) primary antibodies and goat anti‐rabbit (A0423; Beyotime) and goat anti‐mouse (A0428; Beyotime) Alexa Fluor 488 secondary antibodies were used for this study.
4.11. ADMA detection
The ADMA detection was performed as previously described. Approximately 10 mg of ADMA was accurately weighed and placed in a 10 mL volumetric flask, and then a standard stock solution with a concentration of 1 mg/mL was prepared. Next, 450 μL of methanol was added to 50 μL of the culture supernatant and vortexed for 1 minute. After centrifuged at high speed for 15 minutes (28 000 g, 4°C), 200 μL of the supernatant was obtained in a sample bottle, followed by detection of ADMA levels by liquid chromatography as previously described. 53 The ADMA peak shape was good, the separation was complete, and there was no impurity peak interference. 53 The regression equation Y = 310470X−5673.6, R 2 = 0.9998, using the peak area as the ordinate and ADMA mass as the abscissa, were obtained by computer regression.
4.12. Statistical methods
The data were analyzed by using spss19.0 statistical software (IBM Coprporation, Armonk, NY, USA). The data are shown as the mean ± standard deviation (mean ± SD). Multigroup data analysis was based on one‐way ANOVA. A LSD test was used for subsequent analysis. A value of P < .05 indicated a significant difference.
CONFLICTS OF INTEREST
All authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Zhigang Guo designed this projected and wrote the manuscript; Xiao Chen performed the experiments; Xuliang Chen, Xiangxiang Shi, Zhan Gao analyzed the data and edited diagrams. All authors have contributed to, read, and agreed upon the final contents of the manuscript for submission.
Supporting information
FigS1
FigS2
FigS3
FigS4
FigS5
Chen X, Chen X, Shi X, Gao Z, Guo Z. Curcumin attenuates endothelial cell fibrosis through inhibiting endothelial–interstitial transformation. Clin Exp Pharmacol Physiol. 2020;47:1182–1192. 10.1111/1440-1681.13271
Xiao Chen is first author.
REFERENCES
- 1. Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial‐to‐mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952. [DOI] [PubMed] [Google Scholar]
- 2. Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial‐to‐mesenchymal transition. J Am Soc Nephrol. 2008;19:2282‐2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Potenta S, Zeisberg E, Kalluri R. The role of endothelial‐to‐mesenchymal transition in cancer progression. Br J Cancer. 2008;99:1375‐1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li J, Qu X, Bertram JF. Endothelial‐myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin‐induced diabetic mice. Am J Pathol. 2009;175:1380‐1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maddaluno L, Rudini N, Cuttano R, et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature. 2013;498:492‐496. [DOI] [PubMed] [Google Scholar]
- 6. Piera‐Velazquez S, Li Z, Jimenez SA. Role of endothelial‐mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol. 2011;179:1074‐1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang Y, Wu X, Li Y, et al. Endothelial to mesenchymal transition contributes to arsenic‐trioxide‐induced cardiac fibrosis. Sci Rep. 2016;6:33787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hua JY, Zhang ZC, Jiang XH, He YZ, Chen P. [Relationship between endothelial‐to‐mesenchymal transition and cardiac fibrosis in acute viral myocarditis]. J Zhejiang Univ. 2012;41:298‐304. [PubMed] [Google Scholar]
- 9. Cipriani P, Liakouli V, Marrelli A, et al. AB0248 Epidermal growth factor‐like domain 7 (EGFL7) in skin of systemic sclerosis. Ann Rheum Dis. 2013;71:651. [Google Scholar]
- 10. Mihira H, Suzuki HI, Akatsu Y, et al. TGF‐β‐induced mesenchymal transition of MS‐1 endothelial cells requires Smad‐dependent cooperative activation of Rho signals and MRTF‐A. J Biochem. 2012;151:145‐156. [DOI] [PubMed] [Google Scholar]
- 11. Ciszewski WM, Sobierajska K, Wawro ME, et al. The ILK‐MMP9‐MRTF axis is crucial for EndMT differentiation of endothelial cells in a tumor microenvironment. Biochim Biophys Acta Mol Cell Res. 2017;1864:2283. [DOI] [PubMed] [Google Scholar]
- 12. Meran S, Steadman R. Fibroblasts and myofibroblasts in renal fibrosis. Int J Exp Pathol. 2011;92:158‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baum J, Duffy HS. Fibroblasts and myofibroblasts: what are we talking about? J Cardiovasc Pharmacol. 2011;57:376‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sharma V, Dogra N, Saikia U, Khullar M. Transcriptional regulation of EndMT in cardiac fibrosis: role of MRTF‐A and ATF3. Can J Physiol Pharmacol. 2017; 95:1263‐1270 [DOI] [PubMed] [Google Scholar]
- 15. Yu W, Liu Z, An S, et al. The endothelial‐mesenchymal transition (EndMT) and tissue regeneration. Curr Stem Cell Res Ther. 2014;9:196‐204. [DOI] [PubMed] [Google Scholar]
- 16. Cruz‐Solbes AS, Youker K. Epithelial to mesenchymal transition (EMT) and endothelial to mesenchymal transition (EndMT): role and implications in kidney fibrosis. Results Probl Cell Differ. 2017;60:345. [DOI] [PubMed] [Google Scholar]
- 17. Zhou H, Chen X, Chen L, et al. Anti‐fibrosis effect of scutellarin via inhibition of endothelial‐mesenchymal transition on isoprenaline‐induced myocardial fibrosis in rats. Molecules. 2014;19:15611‐15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shu Y, Liu YU, Li X, et al. Aspirin‐triggered resolvin D1 inhibits TGF‐β1‐induced EndMT through increasing the expression of Smad7 and is closely related to oxidative stress. Biomol Ther. 2016;24:132‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xiang Y, Zhang Y, Tang Y, Li Q. MALAT1 modulates TGF‐β1‐induced endothelial‐to‐mesenchymal transition through downregulation of miR‐145. Cell Physiol Biochem. 2017;42:357. [DOI] [PubMed] [Google Scholar]
- 20. Yang H‐Z, Wang J‐P, Mi SU, et al. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am J Pathol. 2012;180:275‐292. [DOI] [PubMed] [Google Scholar]
- 21. Ding S, Walton KL, Blue RE, Mcnaughton K, Magness ST, Lund PK. Mucosal healing and fibrosis after acute or chronic inflammation in wild type FVB‐N mice and C57BL6 procollagen α1(I)‐promoter‐GFP reporter mice. PLoS ONE. 2012;7:e42568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elliott WH, Tan Y, Li M, Tan W. High pulsatility flow promotes vascular fibrosis by triggering endothelial EndMT and fibroblast activation. Cell Mol Bioeng. 2015;8:285‐295. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23. Liu Z, Wang J, Xing W, Peng Y, Huang Y, Fan X. Role of DDAH/ADMA pathway in TGF‐β1‐mediated activation of hepatic stellate cells. Mol Med Rep. 2018;17:2549‐2556. [DOI] [PubMed] [Google Scholar]
- 24. Shahin NN, Abdelkader NF, Safar MM. A novel role of irbesartan in gastroprotection against indomethacin‐induced gastric injury in rats: targeting DDAH/ADMA and EGFR/ERK signaling. Sci Rep. 2018;8:4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wei X, Chen Y, Huang W. Ginsenoside Rg1 ameliorates liver fibrosis via suppressing epithelial to mesenchymal transition and reactive oxygen species production in vitro and in vivo. BioFactors. 2018. [DOI] [PubMed] [Google Scholar]
- 26. Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4:807‐818. [DOI] [PubMed] [Google Scholar]
- 27. Aggarwal BB, Harikumar KB. Potential therapeutic effects of curcumin, the anti‐inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int J Biochem Cell Biol. 2009;41:40‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Metra M, Teerlink JR. Heart failure. Lancet. 2017;390:1981‐1995. [DOI] [PubMed] [Google Scholar]
- 29. Von Haehling S, Ebner N, Dos Santos MR, Springer J, Anker SD. Muscle wasting and cachexia in heart failure: mechanisms and therapies. Nat Rev Cardiol. 2017;14:323. [DOI] [PubMed] [Google Scholar]
- 30. Kreuzer M, Kreisheimer M, Kandel M, Schnelzer M, Tschense A, Grosche B. Mortality from cardiovascular diseases in the German uranium miners cohort study, 1946–1998. Radiat Environ Biophys. 2006;45:159‐166. [DOI] [PubMed] [Google Scholar]
- 31. Espeland T, Lunde IG, H‐Amundsen B, Gullestad L, Aakhus S. Myocardial fibrosis. Tidsskr Nor Laegeforen. 2018;138. [DOI] [PubMed] [Google Scholar]
- 32. Chin CWL, Everett RJ, Kwiecinski J, et al. Myocardial fibrosis and cardiac decompensation in aortic stenosis. JACC Cardiovasc Imaging. 2017;10:1320‐1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Centurión OA, Alderete JF, Torales JM, García LB, Scavenius KE, Miño LM. Myocardial fibrosis as a pathway of prediction of ventricular arrhythmias and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. Crit Pathw Cardiol. 2019;18:89‐97. [DOI] [PubMed] [Google Scholar]
- 34. Xiao L, Kim DJ, Davis CL, et al. Tumor endothelial cells with distinct patterns of TGFβ‐driven endothelial‐to‐mesenchymal transition. Can Res. 2015;75:1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhu K, Pan QI, Jia L‐Q, et al. MiR‐302c inhibits tumor growth of hepatocellular carcinoma by suppressing the endothelial‐mesenchymal transition of endothelial cells. Sci Rep. 2014;4:5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cooley BC, Nevado J, Mellad J, et al. TGF‐Î2 signaling mediates endothelial‐to‐mesenchymal transition (EndMT) during vein graft remodeling. Sci Transl Med. 2014;6:227ra234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nguyen T, Nioi P, Pickett CB. The Nrf2‐antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu X, Fassett J, Wei Y, Chen Y. Regulation of DDAH1 as a potential therapeutic target for treating cardiovascular diseases. Evid Based Complement Alternat Med. 2013;2013:619207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dowsett L, Piper S, Slaviero A, et al. Endothelial DDAH1 is an important regulator of angiogenesis but does not regulate vascular reactivity or hemodynamic homeostasis. Circulation. 2015;131:2217‐2225. [DOI] [PubMed] [Google Scholar]
- 40. Goette A, Hammwöhner M, Bukowska A, et al. The impact of rapid atrial pacing on ADMA and endothelial NOS. Int J Cardiol. 2012;154:141‐146. [DOI] [PubMed] [Google Scholar]
- 41. Wang J‐H, Chen D, Zhang K‐Q, Zhang H, Fu Q. Effect of DDAH/ADMA/NOS regulation pathway on cavernae corporum cavernosorum rat penis of different age. Andrologia. 2016;48:262‐267. [DOI] [PubMed] [Google Scholar]
- 42. Grasemann H, Gärtig SS, Wiesemann HG, Teschler H, Konietzko N, Ratjen F. Effect of l ‐arginine infusion on airway NO in cystic fibrosis and primary ciliary dyskinesia syndrome. Eur Respir J. 2010;13:114‐118. [DOI] [PubMed] [Google Scholar]
- 43. Rezaee R, Momtazi AA, Monemi A, Sahebkar A. Curcumin: a potentially powerful tool to reverse cisplatin‐induced toxicity. Pharmacol Res. 2017;117:218‐227. [DOI] [PubMed] [Google Scholar]
- 44. Mirzaei H, Masoudifar A, Sahebkar A, et al. MicroRNA: a novel target of curcumin in cancer therapy. J Cell Physiol. 2018;233:3004‐3015. [DOI] [PubMed] [Google Scholar]
- 45. Lee J‐S, Wang T‐S, Lin MC, Lin W‐W, Yang J‐J. Inhibition of curcumin on ZAK alpha activity resultant in apoptosis and anchorage‐independent growth in cancer cells. Chin J Physiol. 2017;60:267‐274. [DOI] [PubMed] [Google Scholar]
- 46. Afrin R, Arumugam S, Rahman A, et al. Curcumin ameliorates liver damage and progression of NASH in NASH‐HCC mouse model possibly by modulating HMGB1‐NF‐κB translocation. Int Immunopharmacol. 2017;44:174‐182. [DOI] [PubMed] [Google Scholar]
- 47. Yang Z, Sun N, Cheng R, Zhao C, Liu J, Tian Z. Hybrid nanoparticles coated with hyaluronic acid lipoid for targeted co‐delivery of paclitaxel and curcumin to synergistically eliminate breast cancer stem cells. J Mater Chem B. 2017;5:6762‐6775. [DOI] [PubMed] [Google Scholar]
- 48. Xiao J, Sheng X, Zhang X, Guo M, Ji X. Curcumin protects against myocardial infarction‐induced cardiac fibrosis via SIRT1 activation in vivo and in vitro. Drug Des Devel Ther. 2016;2016:1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu CH, Zhao‐Fa HE, Liu CH, et al. Effects of curcumin on cardiac function and myocardial fibrosis in rats with acute myocardial infarction. Chin J Cardiovasc Rehab Med. 201524:407‐410 [Google Scholar]
- 50. Li T, Wang J, Lu M, Zhang T, Qu X, Wang Z. Selection and validation of appropriate reference genes for qRT‐PCR analysis inIsatis indigoticaFort. Front Plant Sci. 2017;8:1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sayre KR, Dodd RY, Tegtmeier G, Layug L, Alexander SS, Busch MP. False‐positive human immunodeficiency virus type 1 western blot tests in noninfected blood donors. Transfusion. 2010;36:45‐52. [DOI] [PubMed] [Google Scholar]
- 52. Donaldson JG. Immunofluorescence staining. Curr Protoc Cell Biol. 2015;69:431‐437 [DOI] [PubMed] [Google Scholar]
- 53. Maas R, Böger RH, Schwedhelm E, et al. Plasma Concentrations of Asymmetric Dimethylarginine (ADMA) in Colombian women with pre‐eclampsia. JAMA. 2004;291:823‐824. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FigS1
FigS2
FigS3
FigS4
FigS5