

Abstract
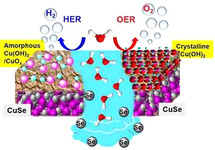
Electrochemical water splitting remains a frontier research topic in the quest to develop artificial photosynthetic systems by using noble metal‐free and sustainable catalysts. Herein, a highly crystalline CuSe has been employed as active electrodes for overall water splitting (OWS) in alkaline media. The pure‐phase klockmannite CuSe deposited on highly conducting nickel foam (NF) electrodes by electrophoretic deposition (EPD) displayed an overpotential of merely 297 mV for the reaction of oxygen evolution (OER) at a current density of 10 mA cm−2 whereas an overpotential of 162 mV was attained for the hydrogen evolution reaction (HER) at the same current density, superseding the Cu‐based as well as the state‐of‐the‐art RuO2 and IrO2 catalysts. The bifunctional behavior of the catalyst has successfully been utilized to fabricate an overall water‐splitting device, which exhibits a low cell voltage (1.68 V) with long‐term stability. Post‐catalytic analyses of the catalyst by ex‐situ microscopic, spectroscopic, and analytical methods confirm that under both OER and HER conditions, the crystalline and conductive CuSe behaves as an electro(pre)catalyst forming a highly reactive in situ crystalline Cu(OH)2 overlayer (electro(post)catalyst), which facilitates oxygen (O2) evolution, and an amorphous Cu(OH)2/CuOx active surface for hydrogen (H2) evolution. The present study demonstrates a distinct approach to produce highly active copper‐based catalysts starting from copper chalcogenides and could be used as a basis to enhance the performance in durable bifunctional overall water splitting.
Keywords: copper selenide, electrocatalysis, klockmannite, non-noble metal catalyst, overall water splitting
Transform to perform: A highly crystalline klockmannite CuSe has been used as an efficient bifunctional electro(pre)catalyst for the reaction of alkaline water splitting and, subsequently, its active structure at the anode and cathode has been uncovered.
Introduction
Rapid depletion of fossil fuels is driving the urgent requirement for alternative systems to harness energy.1 Splitting of water into oxygen (O2) and hydrogen (H2) is one of the promising ways to overcome the current energy crisis, and this process is vital in natural photosynthesis, which directly converts photon energy to chemical energy.2 In this regard, the development of artificial photosynthesis by mimicking nature's strategy has become an essential pathway for a clean and sustainable energy‐based society.3 However, the high thermodynamic barrier of the overall water splitting (OWS), which comprises two half‐cell reactions—oxygen evolution reaction (OER) at the anode and hydrogen evolution reaction (HER) at the cathode—makes this process extremely demanding.4 Therefore, the design of efficient bifunctional OER and HER catalysts to perform OWS remains a great challenge and deserves significant attention.4, 5 Currently, the state‐of‐the‐art electrocatalysts to perform water splitting (WS) mainly rely on the noble‐metal‐based materials (RuO2 and IrO2 for OER, and Pt for HER).6 However, the low abundance of these precious elements limits their wide practical application. Conversely, recent studies infer that higher OER or HER catalytic activity could be achieved by a subtle choice of Earth‐abundant transition‐metal (TM) based catalysts, particularly the first‐row TMs.7 However, only a few of them have been shown to mediate efficient bifunctionality for OWS.
On the other hand, copper is one of the most abundant metals and has immense biological significance.8 Moreover, high catalytic activity and conductivity of metallic copper and/or copper‐based materials have widely been used for organic transformations,9 photothermal,10 electrical and electronic devices.11 Apart from this, copper and copper oxide nanomaterials have also been employed as efficient CO2 reduction catalysts with profound catalytic efficacy.9, 12 Most recently, in a handful cases, copper‐based nanomaterials have also been applied as efficient electrocatalysts for OER.13 In this direction, copper pnictides and chalcogenides such as Cu3N, Cu3P, and Cu9S5 have gained considerable interest owing to their favorable OWS activity with low cell potential in alkaline electrolytes.14 Under electrochemical conditions, both Cu3N and Cu3P nanostructures act as electro(pre)catalysts. They undergo an in situ transformation to form the electro(post)catalyst CuOx overlayer, which efficiently accomplishes OER, HER, and OWS with remarkably low overpotential and cell potentials.14a In this context, copper chalcogenides are relatively less explored as OWS catalysts, and in the recent reports, Cu2S and Cu2Se have only been used as OER catalysts.15 Consequently, the lack of activity for HER in copper chalcogenides prompted us to investigate a copper‐based selenide (CuSe) as a potential bifunctional catalyst for OWS.15a, 15b Over the years, numerous TM selenides, mainly based on Fe, Co, and Ni have been uncovered as attractive bifunctional electrocatalysts in alkaline media.7h, 16 Although much is known about the electrical,11d electronic, and conducting11a properties of CuSe,11b, 11c, 17 its bifunctionality for OWS has never been documented.5c
Herein, we report, for the first time, a highly crystalline klockmannite CuSe as a bifunctional electro(pre)catalyst for the OWS reaction in alkaline electrolyte. CuSe has been synthesized from a high‐temperature calcination approach, and without altering its chemical identity, the material has been deposited on highly conducting electrode substrates (nickel foam, NF, and fluorinated tin oxide, FTO) through the electrophoretic deposition (EPD) technique for simultaneous oxidation and reduction of water to O2 and H2, respectively. Impressively, the recorded overpotentials at a current density of 10 mA cm−2 (297 mV for OER and 162 mV for HER) are among the best values reported for copper‐based materials. When both anode and cathode are fabricated by using CuSe deposited on NF as electro(pre)catalyst for OWS, a low cell potential (1.68 V) with a faradaic efficiency of near 100 % has been achieved. The detailed post‐OER and HER analyses indicate the formation of a crystalline Cu(OH)2 overlayer as an active phase for OER whereas an amorphous Cu(OH)2/CuOx overlayer on the CuSe behaves as the active catalyst for the HER . Additionally, the conductive CuSe core accelerates the electron transport between the active layer and the electrode substrate. As Cu‐based materials have a limited exposure for OER, HER, and OWS, the presented study opens a promising scope to explore new bifunctional catalytic systems based on copper.
Results and Discussion
CuSe crystalline particles were synthesized by the high‐temperature calcination of metallic copper and selenium powder in an inert atmosphere at a 900 °C (see the Experimental Section and the Supporting Information).13 High crystallinity and phase purity of the as‐prepared powder sample was confirmed by powder X‐ray diffraction (XRD) analysis. The obtained diffraction patterns were consistent with the klockmannite CuSe (ICSD‐82 331; Figure 1 a), and the crystal structure of the compound synthesized herein belongs to a hexagonal P63/mmc group with lattice parameters a=3.9428(1) Å and c=17.2574(7) Å.18 The arrangement of the Cu and Se atoms in the unit cell results in a layered‐type structure (Figure S1 a in the Supporting Information) where each CuII ion is surrounded by four Se atoms possessing a tetrahedral geometry with a Cu−Se distance of 2.384–2.435 Å and CuI ion is trigonal‐planar coordinated by Se atoms with Cu−Se distances of 2.276 Å. Two geometrically distinguishable (five‐ and four‐coordinate) Se atoms present in the layered framework, and each four‐coordinated Se atom is positioned in such a way that a Se−Se covalent type interaction occurs at a distance of 2.340 Å (Figure S1 b in the Supporting Information).18a
Figure 1.

Characterization of CuSe crystals. (a) Powder XRD pattern of the as‐prepared CuSe, which consists of a sharp diffraction pattern matching CuSe ICSD‐82331 (the crystal structure shown in the inset). (b) TEM images of the powder CuSe showing smaller crystals. (c) High‐resolution TEM image with atomic fringes matches with the (0 0 6) lattice plane of CuSe, and SAED pattern exhibiting diffraction rings of CuSe (inset). (d) SEM image of CuSe particle with (e, f) EDX mapping, which displays a homogeneous distribution of elements Cu (blue) and Se (orange).
Transmission electron microscopic (TEM) analysis of the as‐prepared CuSe revealed large particles (ca. 200×120 nm; Figure 1 b, Figure S2 in the Supporting Information). The high‐resolution TEM image of a crystal provided lattice fringes matching with the d(0 0 6) planes of CuSe with an inter‐planar distance of 0.288 nm (Figure 1 c). Selected area electron diffraction (SAED) analysis conducted on the particles displayed well‐defined diffraction rings which could be correlated to the Miller indices {(1 0 1), (0 0 6), (1 1 0), (1 1 6), (2 1 2), and (2 1 8)} of CuSe (Figure 1 c, inset). Scanning electron microscopic (SEM) images also illustrated the block morphology of the particles with varying sizes (Figure S3 in the Supporting Information). Elemental mapping of the particles confirmed the homogeneous distribution of Cu and Se (Figure 1 d–f) whereas energy‐dispersive X‐ray (EDX) analysis showed a Cu/Se ratio of approximately 1:1 (Figures S4, S5, and Table S1 in the Supporting Information). An identical elemental composition was further confirmed by inductively coupled plasma atomic emission spectroscopy (ICP‐AES) analysis of the powder sample (Table S1 in the Supporting Information).
To ascertain the elemental (oxidation) state of the CuSe material, X‐ray photoelectron spectroscopic (XPS) studies were performed with the as‐prepared powder. In the high‐resolution XPS, the Cu 2p envelope consisted of two peaks corresponding to Cu 2p3/2 and Cu 2p1/2 at the binding energies of 932.1 eV and 952.2 eV, respectively (Figure S6 a in the Supporting Information). The spin‐orbit coupling spacing value (2p3/2–2p1/2) of 20.1 eV and the detailed deconvolution indicated the presence of CuI with an appreciable amount of CuII (CuI/CuII 1:0.11 ratio, Figure S6 a in the Supporting Information). The CuI species belongs to the CuSe and CuII arises from the surface passivation owing to aerial oxidation. The presence of the latter species is consistent with earlier reports of Cu2−xSe and CuSe materials.11c, 18b, 19 The Se 3d envelope recorded with the powder sample showed an overlapped spin‐orbit component with two maxima for 3d5/2 and 3d3/2 peaks at 53.8 and 54.5 eV (Δ=0.7 eV). These energy values (after deconvolution) are consistent with the presence of SeII−, as shown for reported CuSe materials.11c, 18b, 19a Moreover, a weak loss feature at 56.1 eV and a weak peak corresponding to SeIV arising from surface passivation at 58.3 eV, as observed in other metal selenides (Figure S6 b in the Supporting Information).16c, 20
After an in‐depth microscopic and spectroscopic analysis, the ground CuSe was deposited on NF (a three‐dimensional and high surface area electrode substrate) through EPD. The as‐deposited films of CuSe on NF were further analyzed by microscopic and analytical techniques, which confirmed the excellent chemical stability of CuSe after EPD (Figures S7–S9 in the Supporting Information). The electrocatalytic properties of CuSe were measured and compared with that of CuO and Cu nanoparticles. A three‐electrode set‐up in 1 m aqueous KOH electrolyte where catalysts deposited on NF served as the working electrode was used. The linear sweep voltammetry (LSV) curve displayed a slow increase in current density with the applied potential (Figure 2 a) and reached up to 500 mA cm−2 at 1.7 V (vs. reversible hydrogen electrode, RHE), indicating catalytic oxidation of water. The overpotentials recorded for the OER with CuSe/NF were 297 mV and 382 mV at 10 mA cm−2 and 100 mA cm−2, respectively. A much lower current density resulted when CuO/NF and metallic Cu/NF were used as OER electrodes. The overpotential recorded with CuO/NF was 339 mV (at 10 mA cm−2), whereas an overpotential of 389 mV (at 10 mA cm−2) was achieved for Cu/NF. The bare NF measured in similar LSV conditions was almost inactive. Interestingly, the overpotential (297 mV) achieved with CuSe is slightly higher than the recently reported Cu2Se (270 mV), which could arise from the stability of Cu2Se under OER, the substrate effect (Cu substrate), different mass loading (5 mg), and the higher accessibility of Cu sites of Cu2Se at the surface.15b The mass normalized activity and TOF of CuSe/NF, CuO/NF, and Cu/NF is summarized in Figures S10 and S11 (in the Supporting Information), which further presented the superior activity of the CuSe/NF. Recent studies by the groups of Mayer, Brudvig, and Lin have highlighted that electrooxidation of water catalyzed by CuII complexes proceeds through an in situ formed active CuIII intermediate and, in all the cases, a similar redox feature was observed.21 By analogy to these studies, as well as other reported Cu‐based heterogeneous catalysts, it could be proposed that under applied potentials, CuIIIO(OH) species were formed at the surface to catalyze the OER.14a, 22
Figure 2.
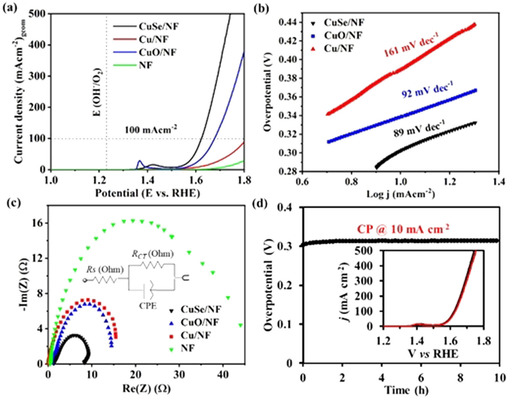
Electrochemical study of CuSe, CuO, and Cu on NF. (a) Polarization curves from LSV for OER, (b) Tafel plot, (c) Nyquist plot from EIS analysis, and (d) the chronopotentiometry (CP) measurements at a current density of 10 mA cm−2 (OER) for 10 h. (d, inset) LSV measured before (black) and after 10 h of CP (OER) experiment (red), exhibiting almost overlapped polarization curves demonstrating the remarkable stability of CuSe. The LSV and Tafel slopes were recorded at a scan rate of 1 mV s−1.
To understand the electrokinetics of the OER, Tafel slopes were calculated from the Tafel plot (Figure 2 b). For CuSe/NF, a Tafel slope of 89 mV dec−1 was attained, which was significantly lower than CuO/NF (92 mV dec−1) and Cu/NF (161 mV dec−1), indicating faster OER kinetics of CuSe/NF. To evaluate the electron transfer capacity, electrochemical impedance spectra (EIS) were recorded. The semicircular Nyquist plot depicts a considerably low charge‐transfer resistance (R ct) for CuSe/NF (Table S2 in the Supporting Information) compared with those of other materials, suggesting a facile electron transfer between the substrate electrode surface and CuSe catalyst providing better OER activity (Figure 2 c).23 The electrochemical double‐layer capacitance (C dl) was further determined by collecting CVs in a non‐faradic region (Figure S12 in the Supporting Information) and, consequently, the electrochemically active surface area (ECSA), which is proportional to the C dl was obtained.7b A C dl of 1.45 mF cm−2, calculated for CuSe/NF was higher than that of CuO/NF (1.0 mF cm−2) and Cu/NF (0.98 mF cm−2) as well as nine‐fold larger compared with bare NF (0.16 mF cm−2; Table S3 in the Supporting Information).24 A higher value of the ECSA normalized current density further pointed out the better intrinsic activity (Figure S13 in the Supporting Information) of CuSe/NF within the investigated materials. To assess the long‐term durability of CuSe/NF, a chronopotentiometry (CP) measurement was conducted, maintaining a current density of 10 mA cm−2, which substantiated the inherent stability of the catalyst in the long run (Figure 2 d). The sustainable activity of CuSe/NF was further demonstrated by measuring the LSV before and after OER CP catalysis (Figure 2 d, inset). Furthermore, the performance of CuSe/NF was compared with the benchmark RuO2/NF and IrO2/NF catalysts (Figure S14 in the Supporting Information), and the resulting overpotential of CuSe/NF was strikingly better than that of noble‐metal‐based catalysts, making it one of the most proficient OER catalysts designed from earth‐abundant first‐row transition metals. In addition, the catalytic OER activity achieved here is not only superior to well established copper oxide and/or hydroxides but also comparable to Cu9S5, Cu3N, Cu3P, and Cu2Se materials (Table S4 in the Supporting Information).14a, 14b, 14c, 14e, 14f, 15b Moreover, the overpotential of CuSe/NF even surpasses some of the Co, Fe, and Ni‐containing active electrocatalysts reported in the literature.7b, 25
Recent reports have revealed that metallic copper and copper‐based materials can act as potential reduction catalysts.9, 12, 26 Additionally, the bifunctional behavior of the previously reported Fe,7h Co,16c and Ni16b selenide materials towards WS inspired us to study HER with CuSe/NF films. Subsequently, the CuSe/NF catalyst was tested as a working electrode for HER in 1 m aqueous KOH solution in a similar fashion as that of OER. A graphite rod, instead of Pt as the reference electrode was used to rule out the influence of Pt (leaching and redeposition on the working electrode). From the LSV polarization curves, an overpotential of 162 mV was achieved for CuSe/NF at a current density of −10 mA cm−2 (Figure 3 a), whereas CuO/NF (245 mV) and Cu/NF (186 mV) recorded significantly high overpotentials at the same current density. Similar to OER, NF showed limited activity; however, Pt was the most active for HER among the tested catalysts. Notably, the overpotential of CuSe/NF is closely comparable to the recently reported copper and other transition metal‐based HER catalysts (Table S5 in the Supporting Information).27 The mass and ECSA normalized activity and TOF of CuSe/NF, CuO/NF, and Cu/NF are shown in Figures S15–S17 (in the Supporting Information). Further, a Tafel slope of 129 mV dec−1 resulted from CuSe/NF, which was lower than Cu/NF (136 mV dec−1) and CuO/NF (140 mV dec−1) displaying faster kinetics for HER catalysis (Figure 3 b). The CuSe/NF catalyst was tested under CP conditions at a constant current density −10 mA cm−2 for HER (Figure 3 c), and a marginal drop in overpotentials was observed, highlighting its superior stability.
Figure 3.
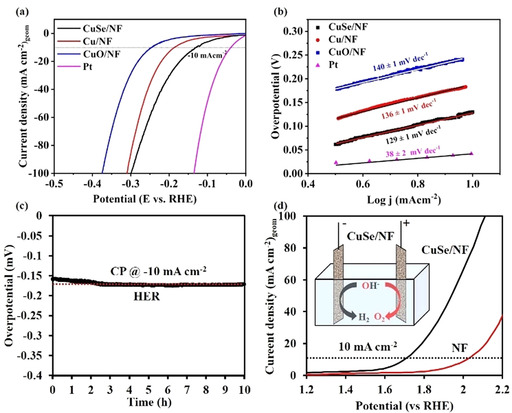
Electrochemical HER and OWS with CuSe/NF. (a) Polarization curves of HER of CuSe/NF compared with Cu/NF, CuO/NF, Pt, and bare NF. (b) Tafel plot (along with Tafel slopes) obtained from the LSV curves for HER measurements with CuSe, Cu, CuO, and Pt. (c) HER CP of CuSe at a constant current of −10 mA cm−2 in a three‐electrode set‐up. (d) Polarization curve of OWS in the two‐electrode set‐up (c, inset) using CuSe/NF as both cathode and anode.
It has been documented previously that the electrode support plays a crucial role in electrocatalysis.7f, 7g, 28 To verify this, CuSe, CuO, and Cu were electrophoretically deposited on FTO, characterized (Figures S18–S21 in the Supporting Information), and then measured similarly to that of the NF substrate in 1 m aqueous KOH electrolyte (Figures S22–S23 in the Supporting Information). Under the tested OER conditions, the overpotential of CuSe/FTO was found to be 380 mV at a current density of 10 mA cm−2, whereas CuO/FTO and Cu/FTO displayed an overpotential of 455 and 610 mV, respectively (Figure S22 in the Supporting Information). Similarly, CuSe/FTO also outperformed CuO/FTO and Cu/FTO in HER conditions (Figure S23 in the Supporting Information). Notably, the catalytic performance of CuSe for both OER and HER based on FTO followed the same trend as that of NF.
Inspired by the promising catalytic half‐cell activities for OER and HER, we designed an OWS device with a two‐electrode configuration by using CuSe/NF as both anode and cathode in 1 m aqueous KOH electrolyte at room temperature (Figure 3 d, inset and Figure S24 in the Supporting Information). The polarization curve in Figure 3 d shows the high performance of CuSe/NF, reaching a current density of 10 mA cm−2 with a requirement of cell voltage of 1.68 V whereas bare NF displayed a large cell voltage (>2 V). Notably, the catalytic OWS performance of CuSe/NF is clearly higher than the recently reported monometallic selenides, better than mono and bimetallic chalcogenide materials, and comparable with other highly active non‐noble based bifunctional electrocatalysts (Table S6 in the Supporting Information).14f, 15b, 16 The long‐term stability (CP) of this bifunctional catalyst was also tested in similar conditions at a current density of 10 mA cm−2, which demonstrated the superior durability of the electrodes (Figure S25 in the Supporting Information). To demonstrate the efficiency of the catalysts, an inverted (graduated) electrolyzer cell was constructed and the relative evolution of H2 to that of O2 gas was collected separately at atmospheric pressure (Figure S26 in the Supporting Information). The measured volume of the evolved O2 and H2 at the anode and cathode followed the theoretically predicated 1:2 ratio (Figure S27 in the Supporting Information). In a separate experiment, under a closed electrochemical cell, the evolved gases were further quantified by GC analysis, and that gave almost 90 % and 98 % faradaic efficiency for OER and HER, respectively (Table S7 in the Supporting Information).
After obtaining successful results for each half‐reaction of OER and HER, the structure of the active catalysts on the electrode was systematically investigated through various ex situ methods. Previous studies have confirmed that under alkaline electrochemical OER and HER operational conditions, the surface of non‐oxide materials such as TM chalcogenides, phosphides, phosphates, as well as intermetallic compounds, undergoes a rapid structural change forming a highly reactive metal oxide/hydroxide/oxyhydroxide.7g, 7h, 29 In this regard, we examined the CuSe electrodes separately after CP measurements (termed as OER CP and HER CP hereafter) by spectroscopic, microscopic, and analytical methods to gain detailed insights. The TEM measured after OER CP suggested a severe morphological and structural change compared with the as‐prepared material. The high‐resolution TEM images provided concrete evidence in support of the formation of crystalline Cu(OH)2 (Figure 4 a and Figure S28 in the Supporting Information) on the surface and were further evidenced by the well‐defined diffraction rings of Cu(OH)2 in the SAED pattern (Figure 4 a, inset, JCPDS 42‐746). The TEM also showed the remaining crystalline CuSe core identified by the lattice fringes matching with the d(1 0 2) planes with an inter‐planar distance of 0.301 nm. The elemental analysis (SEM and EDX) of the catalyst after OER CP exhibited a significant loss of Se from the catalyst surface and concomitant oxygen incorporation into the structure (Figures S29–S30 in the Supporting Information). This is in accordance with the formation of a layer‐type Cu(OH)2.30 The ratio of Cu and Se in the electrolyte solution was determined by ICP‐AES analysis, which also supported the loss of Se (≈40 %; Table S8 in the Supporting Information). However, under HER CP, TEM and SAED analyses showed the formation of an amorphous overlayer on the crystalline CuSe, which was predicted to be an amorphous copper oxide/hydroxide (Figure 4 b, Figure S31 in the Supporting Information). Elemental mapping (SEM and EDX) confirmed the presence of oxygen in the amorphous layer structure (Figures S32–S33 in the Supporting Information). ICP‐AES analysis of the electrolyte after HER indicated a similar extent of Se loss (Table S9 in the Supporting Information) in comparison to that observed for OER CP.
Figure 4.
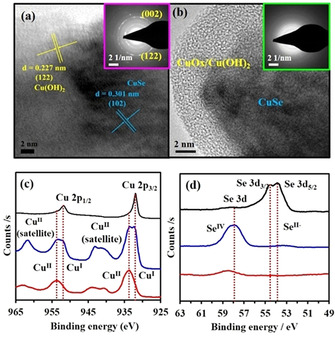
Post‐catalytic characterization of CuSe catalysts. (a) The high‐resolution TEM image of the catalysts after OER CP where the Bragg's planes match perfectly with Cu(OH)2 and the remaining CuSe core. The SAED (inset a) shows the diffraction rings matching to (0 0 2) and (1 2 2) from Cu(OH)2. (b) TEM image after HER CP displaying the formation of an amorphous CuOx layer surrounding the crystalline CuSe core. The SAED (b, inset) confirms the formation of an amorphous layer. Plots of XPS analysis for (c) Cu 2p and (d) Se 3d scans of the catalysts before (black), after OER CP (blue) and after HER CP (red).
To analyze the change in the surface structure, XPS was conducted after OER CP and HER CP and compared directly with the deposited CuSe electrode. The binding energy values of Cu 2p and Se 3d, in addition to their spin‐orbit coupling values (2p1/2–2p3/2), differ dramatically after CP for both OER and HER (Figure 4 c–d). In the deconvoluted Cu 2p spectrum recorded after OER CP (Figure 4 c, blue curve), two higher binding energy peaks (Cu 2p3/2 933.3 eV and Cu 2p1/2 953.7 eV) appeared with two major satellite peaks (at 944.8 and 961.7 eV) indicating a higher amount of CuII from Cu(OH)2 on the surface in comparison to the as‐prepared film, which is almost pure CuI (Figure 4 c, Figure S34 a in the Supporting Information). Despite the oxidation of CuI to CuII on the surface, peaks for CuI were still present (Cu 2p3/2 932.8 eV and Cu 2p1/2 952.2 eV), demonstrating that the CuSe core was preserved after OER (Figure S34 a in the Supporting Information).15b However, in the high‐resolution XPS of Se, two strongly overlapped Se 3d5/2 and 3d3/2 binding energies (Figure 4 d, black curve, and Figure S34 b in the Supporting Information) mostly disappeared after OER CP and a broad peak appeared at 57.8 eV confirming the presence of SeIV from SeOx, formed during the alkaline electrochemical condition by oxidation of Se (blue and red curves). Such oxidation of Cu and Se under OER in strongly alkaline conditions has already been well documented for copper selenides.11c, 16c, 18b, 19a, 20 Interestingly, the O 1s spectrum after OER CP displayed a sharp peak at 531.2 eV, suggesting the formation of Cu(OH)2 (Figure S34 c in the Supporting Information), in accordance with TEM and SAED results. On the other hand, similar to OER CP, slightly oxidized Cu 2p and completely oxidized Se 3d spectra were obtained after HER CP (Figure 4 c–d, red curves). However, the O 1s spectrum of HER CP showed two prominent peaks 529.2 and 531.3 eV, strongly supporting the formation of CuOx and Cu(OH)2 surface (Figure S35 c in the Supporting Information). Moreover, this observation can also be linked to the amorphous surface structure as displayed by TEM images (Figure 4 d and Figure S31 in the Supporting Information). Detailed comparison of the high‐resolution XPS spectra for Cu 2p, Se 3d, and O 1s of OER and HER CP has been provided in the Supporting Information (Figures S34–S35). From the detailed investigation, it was clear that during OER, a crystalline Cu(OH)2 overlayer behaves as the active catalyst whereas a mixed amorphous Cu(OH)2/CuOx surface boosts the HER activity. Also, the conductive CuSe core in both cases accelerates the electron mobility between the active catalyst to the electrode substrate, illustrating the dual benefit of the CuSe for electrocatalysis.7h, 14a, 25c, 31
The post‐(electro)catalytic characterization of CuSe confirmed the formation of two different active phases under OER and HER conditions. Based on the obtained results, the higher catalytic activity of CuSe for OER can be attributed to the formation of the in situ crystalline Cu(OH)2 overlayer, which could form CuIII species to facilitate O−O bond formation.14a, 14c, 14e, 22a, 22c, 31, 32 Alternatively, the amorphous Cu(OH)2/CuOx overlayer possibly creates a low‐valent Cu species to catalyze the reaction of HER, as known for other Cu‐based materials.13, 14c, 14d, 27a Moreover, it has also been shown that selenides have optimum bond strength to adsorb protons and can act as a base to accelerate deprotonation to furnish H2 evolution.7d, 7h, 16a, 16b As the core of the particle still contained CuSe, the possibility of having selenium on the surface cannot entirely be ruled out. Similarly, the loss of Se from the surface of CuSe in both OER and HER could provide abundant defects and disorders, which has often been shown to be beneficial for electrocatalysis.7g, 14a, 29b, 29c, 33 Most importantly, the conductive core of CuSe sandwiched between the surface layer and the electrode substrate, enhances the charge mobility for both OER and HER (as also shown by EIS).5a, 7h, 14a Finally, the large ECSA of CuSe furnishes the increased number of active sites, favoring the efficient adsorption and transfer of reactants to accelerate the electrochemical reaction.7f, 7g, 25c, 28b, 33, 34
Conclusions
CuSe crystalline particles have been synthesized by using a high‐temperature solid‐state synthetic approach. The CuSe nanostructure behaves as an effective electro(pre)catalyst in alkaline media, displaying considerably low overpotentials for both reactions of OER and HER. Under OER conditions, the as‐prepared CuSe generates an in situ crystalline Cu(OH)2 overlayer that acts as the active site to facilitate O−O bond formation, whereas in HER, an amorphous Cu(OH)2/CuOx shell is formed, which generates low‐valent Cu species (alongside Se) to efficiently adsorb protons to evolve H2. The superior conductivity of CuSe present at the core also plays a vital role by enhancing the charge transport between the active layer and the electrode surface for both OER and HER. Additionally, the loss of Se creates surface defects by increasing the active surface area to boost the catalytic activity of CuSe precatalyst. Keeping the activity and conductivity of the presented catalyst in mind, we have examined CuO and Cu for OER and HER, which validates our reasons for the higher activity of CuSe. Finally, the bifunctionality of CuSe has also been demonstrated by fabricating a two‐electrode alkaline electrolyzer for OWS, which only requires a cell voltage of 1.68 V to reach 10 mA cm−2. In the quest to design a novel catalyst relying on noble‐metal‐free and earth‐abundant sources, the present study opens up new opportunities to modulate the active and electronic structure of electrocatalysts for practical water electrolysis.
Experimental Section
General considerations and instrumentation
All synthetic procedures related to the preparation of CuSe were carried out under inert conditions by using standard Schlenk techniques or an MBraun inert atmosphere dry box containing an atmosphere of purified nitrogen. The commercial RuO2 (99 %), IrO2 (99 %), and copper(II) acetate monohydrate were purchased from Alfa Aesar. Nickel foam (NF) and fluorine‐doped tin oxide (FTO, resistivity 8–12 Ω sq−1) were obtained from Racemat BV and Sigma–Aldrich, respectively. Microscopic and spectroscopic characterizations and details of electrochemical measurements are provided in the Supporting Information.
Synthesis of CuSe
For the synthesis of the CuSe, all materials were handled in an argon atmosphere by using an argon‐filled glovebox (MBraun, H2O/O2 level <1.0 ppm) and other standard inert gas techniques. A binary CuSe compound was prepared from elemental copper and selenium in a stoichiometric ratio: Cu wire (5.62 mmol, purity 99.9 %, Chem Pur) and Se granules (5.62 mmol, purity 99.999 %, Chem Pur) were filled in double‐walled silica glass ampules and sealed by using a O2/H2 flame in argon atmosphere (0.6 atm). The ampule was heated in a muffle furnace (Nabertherm, P330 controller) to 500 °C at a rate of 3 K min−1, held at this temperature for 4 h, then heated to 900 °C at a rate of 3 K min−1, and held at this temperature for 5 h. In the next step, the sample was slowly cooled to 300 °C at a rate of 5 K min−1, held at this temperature for 20 h, and finally cooled down to room temperature at a rate of 10 K min−1. An air‐stable crystalline dark product was obtained from the reaction. The powder XRD pattern of the product shows the presence of the CuSe phase (see Figure 1 a).
Synthesis of Cu and CuO
Copper(II) acetate monohydrate (300 mg) was heated to 300 °C while the temperature was increased slowly with a heating rate of 5 °C min−1 and in the presence of a H2 flow (slow, 2–3 L h−1) and the temperature was maintained for 2 h. The resulting black powder sample was thoroughly washed with water and ethanol (3×50 mL) and dried at 60 °C to obtain 100 mg (yield >90 %) of phase pure Cu. Following a very similar procedure, CuO (yield >95 %) nanoparticles (NP) were obtained whereas the heating was done in presence of O2 flow.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC 2008/1–390540038—UniSysCat. The authors thank J. Niklas Haussmann (TU Berlin) for TEM and the group of Prof. Martin Lerch (TU Berlin) for PXRD measurements.
B. Chakraborty, R. Beltrán-Suito, V. Hlukhyy, J. Schmidt, P. W. Menezes, M. Driess, ChemSusChem 2020, 13, 3222.
Contributor Information
Dr. Prashanth W. Menezes, Email: prashanth.menezes@mailbox.tu-berlin.de.
Prof. Dr. Matthias Driess, Email: matthias.driess@tu-berlin.de.
References
- 1.
- 1a. Dresselhaus M. S., Thomas I. L., Nature 2001, 414, 332–337; [DOI] [PubMed] [Google Scholar]
- 1b. Qi J., Zhang W., Cao R., Adv. Energy Mater. 2018, 8, 1701620; [Google Scholar]
- 1c. Höök M., Tang X., Energy Policy 2013, 52, 797–809. [Google Scholar]
- 2.
- 2a. Gray H. B., Nat. Chem. 2009, 1, 7;21378780 [Google Scholar]
- 2b. Suen N.-T., Hung S.-F., Quan Q., Zhang N., Xu Y.-J., Chen H. M., Chem. Soc. Rev. 2017, 46, 337–365; [DOI] [PubMed] [Google Scholar]
- 2c. Song F., Bai L., Moysiadou A., Lee S., Hu C., Liardet L., Hu X., J. Am. Chem. Soc. 2018, 140, 7748–7759; [DOI] [PubMed] [Google Scholar]
- 2d. Li J., Güttinger R., Moré R., Song F., Wan W., Patzke G. R., Chem. Soc. Rev. 2017, 46, 6124–6147. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Faunce T. A., Lubitz W., Rutherford A. W., MacFarlane D., Moore G. F., Yang P., Nocera D. G., Moore T. A., Gregory D. H., Fukuzumi S., Yoon K. B., Armstrong F. A., Wasielewski M. R., Styring S., Energy Environ. Sci. 2013, 6, 695–698; [Google Scholar]
- 3b. Menezes P. W., Indra A., Schuster F., Schwarze M., Driess M., ChemPlusChem 2016, 81, 1068–1074; [DOI] [PubMed] [Google Scholar]
- 3c. Bozal-Ginesta C., Durrant J. R., Faraday Discuss. 2019, 215, 439–451; [DOI] [PubMed] [Google Scholar]
- 3d. Concepcion J. J., House R. L., Papanikolas J. M., Meyer T. J., Proc. Natl. Acad. Sci. USA 2012, 109, 15560–15564; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Long X., Qiu W., Wang Z., Wang Y., Yang S., Mater. Today Chem. 2019, 11, 16–28. [Google Scholar]
- 4. You B., Sun Y., Acc. Chem. Res. 2018, 51, 1571–1580. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Panda C., Menezes P. W., Driess M., Angew. Chem. Int. Ed. 2018, 57, 11130–11139; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11298–11308; [Google Scholar]
- 5b. Chakraborty B., Gan-Or G., Duan Y., Raula M., Weinstock I. A., Angew. Chem. Int. Ed. 2019, 58, 6584–6589; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 6656–6661; [Google Scholar]
- 5c. Chakraborty B., Gan-Or G., Raula M., Gadot E., Weinstock I. A., Nat. Commun. 2018, 9, 4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Reier T., Oezaslan M., Strasser P., ACS Catal. 2012, 2, 1765–1772; [Google Scholar]
- 6b. Jiao Y., Zheng Y., Jaroniec M., Qiao S. Z., Chem. Soc. Rev. 2015, 44, 2060–2086. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Roger I., Shipman M. A., Symes M. D., Nat. Rev. Chem. 2017, 1, 928; [Google Scholar]
- 7b. McCrory C. C. L., Jung S., Peters J. C., Jaramillo T. F., J. Am. Chem. Soc. 2013, 135, 16977–16987; [DOI] [PubMed] [Google Scholar]
- 7c. Hunter B. M., Gray H. B., Müller A. M., Chem. Rev. 2016, 116, 14120–14136; [DOI] [PubMed] [Google Scholar]
- 7d. Anantharaj S., Ede S. R., Sakthikumar K., Karthick K., Mishra S., Kundu S., ACS Catal. 2016, 6, 8069–8097; [Google Scholar]
- 7e. Dinh K. N., Liang Q., Du C.-F., Zhao J., Tok A. I. Y., Mao H., Yan Q., Nano Today 2019, 25, 99–121; [Google Scholar]
- 7f. Menezes P. W., Indra A., Zaharieva I., Walter C., Loos S., Hoffmann S., Schlögl R., Dau H., Driess M., Energy Environ. Sci. 2019, 12, 988–999; [Google Scholar]
- 7g. Menezes P. W., Panda C., Loos S., Bunschei-Bruns F., Walter C., Schwarze M., Deng X., Dau H., Driess M., Energy Environ. Sci. 2018, 11, 1287–1298; [Google Scholar]
- 7h. Panda C., Menezes P. W., Walter C., Yao S., Miehlich M. E., Gutkin V., Meyer K., Driess M., Angew. Chem. Int. Ed. 2017, 56, 10506–10510; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10642–10646; [Google Scholar]
- 7i. Yu F., Yu L., Mishra I. K., Yu Y., Ren Z. F., Zhou H. Q., Mater. Today Phys. 2018, 7, 121–138; [Google Scholar]
- 7j. Chakraborty B., Indra A., Menezes P. V., Driess M., Menezes P. W., Mater. Today Chem. 2020, 15, 100226. [Google Scholar]
- 8. Solomon E. I., Heppner D. E., Johnston E. M., Ginsbach J. W., Cirera J., Qayyum M., Kieber-Emmons M. T., Kjaergaard C. H., Hadt R. G., Tian L., Chem. Rev. 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gawande M. B., Goswami A., Felpin F.-X., Asefa T., Huang X., Silva R., Zou X., Zboril R., Varma R. S., Chem. Rev. 2016, 116, 3722–3811. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Shen K., Hua C., Liang Z., Wang Y., Sun H., Hu J., Zhang H., Li H., Jiang Z., Huang H., Wang P., Sun Z., Wahlström E., Lu Y., Song F., ACS Appl. Electron. Mater. 2019, 1, 1817–1824; [Google Scholar]
- 10b. Singh S. C., Li H., Yao C., Zhan Z., Yu W., Yu Z., Guo C., Nano Energy 2018, 51, 774–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Pazhamalai P., Krishnamoorthy K., Kim S. J., Int. J. Hydrogen Energy 2016, 41, 14830–14835; [Google Scholar]
- 11b. Gong J., Li J.-C., Yang J., Zhao S., Yang Z., Zhang K., Bao J., Pang H., Han M., ACS Appl. Mater. Interfaces 2018, 10, 38341–38349; [DOI] [PubMed] [Google Scholar]
- 11c. Choi J., Kang N., Yang H. Y., Kim H. J., Son S. U., Chem. Mater. 2010, 22, 3586–3588; [Google Scholar]
- 11d. Liu Y.-Q., Wang F.-X., Xiao Y., Peng H.-D., Zhong H.-J., Liu Z.-H., Pan G.-B., Sci. Rep. 2014, 4, 5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li C. W., Kanan M. W., J. Am. Chem. Soc. 2012, 134, 7231–7234. [DOI] [PubMed] [Google Scholar]
- 13. Liu X., Zheng H., Sun Z., Han A., Du P., ACS Catal. 2015, 5, 1530–1538. [Google Scholar]
- 14.
- 14a. Panda C., Menezes P. W., Zheng M., Orthmann S., Driess M., ACS Energy Lett. 2019, 4, 747–754; [Google Scholar]
- 14b. Han A., Zhang H., Yuan R., Ji H., Du P., ACS Appl. Mater. Interfaces 2017, 9, 2240–2248; [DOI] [PubMed] [Google Scholar]
- 14c. Tian J., Liu Q., Cheng N., Asiri A. M., Sun X., Angew. Chem. Int. Ed. 2014, 53, 9577–9581; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9731–9735; [Google Scholar]
- 14d. Hao J., Yang W., Huang Z., Zhang C., Adv. Mater. Interfaces 2016, 3, 1600236; [Google Scholar]
- 14e. Du J., Li F., Wang Y., Zhu Y., Sun L., ChemElectroChem 2018, 5, 2064–2068; [Google Scholar]
- 14f. Chakraborty B., Kalra S., Beltrán-Suito R., Das C., Hellmann T., Menezes P. W., Driess M., Chem. Asian J. 2020, 15, 852–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Li Q., Wang X., Tang K., Wang M., Wang C., Yan C., ACS Nano 2017, 11, 12230–12239; [DOI] [PubMed] [Google Scholar]
- 15b. Masud J., Liyanage W. P. R., Cao X., Saxena A., Nath M., ACS Appl. Energy Mater. 2018, 1, 4075–4083; [Google Scholar]
- 15c. Zhao X., Liu L., Zhang Y., Zhang H., Wang Y., Nanotechnology 2017, 28, 345402. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Tang C., Cheng N., Pu Z., Xing W., Sun X., Angew. Chem. Int. Ed. 2015, 54, 9351–9355; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9483–9487; [Google Scholar]
- 16b. Swesi A. T., Masud J., Liyanage W. P. R., Umapathi S., Bohannan E., Medvedeva J., Nath M., Sci. Rep. 2017, 7, 2401; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16c. Masud J., Swesi A. T., Liyanage W. P. R., Nath M., ACS Appl. Mater. Interfaces 2016, 8, 17292–17302. [DOI] [PubMed] [Google Scholar]
- 17. Zhou R., Huang Y., Zhou J., Niu H., Wan L., Li Y., Xu J., Xu J., Dalton Trans. 2018, 47, 16587–16595. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Milman V., Acta Crystallogr. Sect. B 2002, 58, 437–447; [DOI] [PubMed] [Google Scholar]
- 18b. Xie Y., Zheng X., Jiang X., Lu J., Zhu L., Inorg. Chem. 2002, 41, 387–392. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Xie Y., Riedinger A., Prato M., Casu A., Genovese A., Guardia P., Sottini S., Sangregorio C., Miszta K., Ghosh S., Pellegrino T., Manna L., J. Am. Chem. Soc. 2013, 135, 17630–17637; [DOI] [PubMed] [Google Scholar]
- 19b. Singh S. C., Peng Y., Rutledge J., Guo C., ACS Appl. Electron. Mater. 2019, 1, 1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wei C., Zhong D., Li D., Hao G., Liu G., Li J., Zhao Q., Int. J. Hydrogen Energy 2019, 44, 21979–31986. [Google Scholar]
- 21.
- 21a. Fisher K. J., Materna K. L., Mercado B. Q., Crabtree R. H., Brudvig G. W., ACS Catal. 2017, 7, 3384–3387; [Google Scholar]
- 21b. Zhang T., Wang C., Liu S., Wang J.-L., Lin W., J. Am. Chem. Soc. 2014, 136, 273–281; [DOI] [PubMed] [Google Scholar]
- 21c. Barnett S. M., Goldberg K. I., Mayer J. M., Nat. Chem. 2012, 4, 498–502. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Deng Y., Handoko A. D., Du Y., Xi S., Yeo B. S., ACS Catal. 2016, 6, 2473–2481; [Google Scholar]
- 22b. Liu X., Cui S., Qian M., Sun Z., Du P., Chem. Commun. 2016, 52, 5546–5549; [DOI] [PubMed] [Google Scholar]
- 22c. Cui S., Liu X., Sun Z., Du P., ACS Sustainable Chem. Eng. 2016, 4, 2593–2600. [Google Scholar]
- 23. Kang Q., Vernisse L., Remsing R. C., Thenuwara A. C., Shumlas S. L., McKendry I. G., Klein M. L., Borguet E., Zdilla M. J., Strongin D. R., J. Am. Chem. Soc. 2017, 139, 1863–1870. [DOI] [PubMed] [Google Scholar]
- 24. Yoon Y., Yan B., Surendranath Y., J. Am. Chem. Soc. 2018, 140, 2397–2400. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Rao Y., Wang Y., Ning H., Li P., Wu M., ACS Appl. Mater. Interfaces 2016, 8, 33601–33607; [DOI] [PubMed] [Google Scholar]
- 25b. Zhang X., Xu H., Li X., Li Y., Yang T., Liang Y., ACS Catal. 2016, 6, 580–588; [Google Scholar]
- 25c. Beltrán-Suito R., Menezes P. W., Driess M., J. Mater. Chem. A 2019, 7, 15749–15756. [Google Scholar]
- 26. Ray C., Lee S. C., Jin B., Chung K. Y., Guo S., Zhang S., Zhang K., Park J. H., Jun S. C., ChemElectroChem 2019, 6, 5014–5021. [Google Scholar]
- 27.
- 27a. Wei S., Qi K., Jin Z., Cao J., Zheng W., Chen H., Cui X., ACS Omega 2016, 1, 1367–1373; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Nichols E. M., Gallagher J. J., Liu C., Su Y., Resasco J., Yu Y., Sun Y., Yang P., Chang M. C. Y., Chang C. J., Proc. Natl. Acad. Sci. USA 2015, 112, 11461–11466; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27c. Sun Y., Liu C., Grauer D. C., Yano J., Long J. R., Yang P., Chang C. J., J. Am. Chem. Soc. 2013, 135, 17699–17702; [DOI] [PubMed] [Google Scholar]
- 27d. Son C. Y., Kwak I. H., Lim Y. R., Park J., Chem. Commun. 2016, 52, 2819–2822. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Menezes P. W., Indra A., Das C., Walter C., Göbel C., Gutkin V., Schmeißer D., Driess M., ACS Catal. 2017, 7, 103–109; [Google Scholar]
- 28b. Walter C., Menezes P. W., Orthmann S., Schuch J., Connor P., Kaiser B., Lerch M., Driess M., Angew. Chem. Int. Ed. 2018, 57, 698–702; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 706–710. [Google Scholar]
- 29.
- 29a. Indra A., Menezes P. W., Das C., Göbel C., Tallarida M., Schmeißer D., Driess M., J. Mater. Chem. A 2017, 5, 5171–5177; [Google Scholar]
- 29b. Menezes P. W., Panda C., Garai S., Walter C., Guiet A., Driess M., Angew. Chem. Int. Ed. 2018, 57, 15237–15242; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15457–15462; [Google Scholar]
- 29c. Menezes P. W., Walter C., Hausmann J. N., Beltran-Suito R., Schlesiger C., Praetz S., Verchenko V. Y., Shevelkov A., Driess M., Angew. Chem. Int. Ed. 2019, 58, 16569–16574; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 16722–16727. [Google Scholar]
- 30. Oswald H. R., Reller A., Schmalle H. W., Dubler E., Acta Crystallogr. Sect. C 1990, 46, 2279–2284. [Google Scholar]
- 31. Menezes P. W., Indra A., Littlewood P., Schwarze M., Göbel C., Schomäcker R., Driess M., ChemSusChem 2014, 7, 2202–2211. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Du J., Chen Z., Ye S., Wiley B. J., Meyer T. J., Angew. Chem. Int. Ed. 2015, 54, 2073–2078; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 2101–2106; [Google Scholar]
- 32b. Hou C.-C., Fu W.-F., Chen Y., ChemSusChem 2016, 9, 2069–2073. [DOI] [PubMed] [Google Scholar]
- 33. Menezes P. W., Panda C., Walter C., Schwarze M., Driess M., Adv. Funct. Mater. 2019, 29, 1808632. [Google Scholar]
- 34. Panda C., Menezes P. W., Yao S., Schmidt J., Walter C., Hausmann J. N., Driess M., J. Am. Chem. Soc. 2019, 141, 13306–13310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary