

Abstract
Filamin C (FLNC) variants are associated with cardiac and muscular phenotypes. Originally, FLNC variants were described in myofibrillar myopathy (MFM) patients. Later, high‐throughput screening in cardiomyopathy cohorts determined a prominent role for FLNC in isolated hypertrophic and dilated cardiomyopathies (HCM and DCM). FLNC variants are now among the more prevalent causes of genetic DCM. FLNC‐associated DCM is associated with a malignant clinical course and a high risk of sudden cardiac death. The clinical spectrum of FLNC suggests different pathomechanisms related to variant types and their location in the gene. The appropriate functioning of FLNC is crucial for structural integrity and cell signaling of the sarcomere. The secondary protein structure of FLNC is critical to ensure this function. Truncating variants with subsequent haploinsufficiency are associated with DCM and cardiac arrhythmias. Interference with the dimerization and folding of the protein leads to aggregate formation detrimental for muscle function, as found in HCM and MFM. Variants associated with HCM are predominantly missense variants, which cluster in the ROD2 domain. This domain is important for binding to the sarcomere and to ensure appropriate cell signaling. We here review FLNC genotype–phenotype correlations based on available evidence.
Keywords: cardiomyopathy, filamin, FLNC, genotype–phenotype correlation, myopathy
The location of causative variants leading to the filaminopathies A are mapped onto FLNA protein monomers. Variants leading to ‘loss‐of‐function’ disorders (left monomer) and ‘gain‐of‐function’ disorders (right monomer) can clearly be seen to cluster. ‘Hotspot’ regions are marked with larger symbols. PH: periventricular heterotopia, CIPX: congenital intestinal pseudo‐obstruction, IMT: isolated macrothrombocytopenia, FCMPD: familial cardiac myxomatous polyvalvular dystrophy, OPD: otopalatodigital syndrome, FMD: frontometaphyseal dysplasia, CKCO: condition comprising contractures, keloid, cardiac defects and optic anomalies, MNS: Melnick‐Needles syndrome, DCD: digitocutaneous dysplasia, ABD: actin‐binding domain, CHD: calponin homology domain, H: hinge.
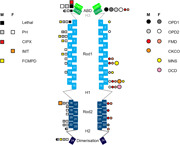
1. BACKGROUND
Similar to other filamins, Filamin C (FLNC) is a structural protein which has an actin‐binding domain (ABD) composed of two calponin homology (CH) domains, 24 immunoglobulin (Ig) domains divided into a ROD1 and ROD2 subdomain, and a C‐terminal dimerization domain (van der Flier & Sonnenberg, 2001). The dimerization of two identical filamin proteins is necessary for appropriate function and occurs via Ig‐like domain 24 (Himmel, Van Der Ven, Stocklein, & Furst, 2003). In contrast to filamin A and B, filamin C expression is restricted to striated muscles and localizes around the Z‐disc, the sarcolemma, the myotendinous junction, and the intercalated discs (Thompson et al., 2000). Its main role is maintaining the structural integrity of the sarcomere. This is through crosslinking actin filaments and the anchoring of sarcolemmal proteins to the cytoskeleton. The main interactors of FLNC are either part of the Z‐disc (myotilin, myozenin, myopodin, and calsarcins), signaling molecules (Zhang, Liu, Cheng, Deyoung, & Saltiel, 2007) or sarcolemma‐associated proteins (integrin β1, sarcoglycan delta; Anastasi et al., 2004; Furst et al., 2013; Takada et al., 2001). Proteases such as calpain can regulate the interaction between FLNC and the sarcoglycans by cleaving the corresponding binding domains of FLNC (Guyon et al., 2003). In addition, FLNC interacts with the Xin actin‐binding repeat‐containing proteins (XIRP) and aciculin to fulfill a function in muscle maintenance (Fujita et al., 2012; Leber et al., 2016; Molt et al., 2014). This interaction is mediated via a unique insertion in Ig‐like domain 20, which is absent in the other filamin paralogs. The ROD1 domain (Ig 1–15) is more stretched and lacks interdomain interactions in contrast to the ROD2 domain (Ig 16–23), which is more compact and globularly arranged by domain pairs. These organizational differences between the domains explain why certain ligands bind exclusively to ROD1 or ROD2.
The FLNC gene maps to chromosome 7q32–35 and has two main transcripts, NM_001127487.2 and NM_001458.4. It comprises ~29.5 kb of genomic DNA and is composed of 49 coding exons (Chakarova et al., 2000). The difference between the two transcripts is the presence or absence of exon 31 encoding the hinge region between Ig‐likechroIg‐like domains 15 and 16 (Xie, Xu, Davie, & Chung, 1998). The longest transcript, NM_001458.4, encodes a protein with a molecular mass of 291 kDa and 2.725 amino acids, whereas the shorter transcript NM_001127487.2 encodes a slightly shorter protein (287 kDa, 2.692 amino acids) that is assumed to be less flexible. The exact roles of these two isoforms are unknown, but the long FLNC isoform is more abundantly expressed during cardiac stress while almost absent in the normal situation (Kong et al., 2010). This could potentially alter the integrity and function of the key sarcomeric structures to cope with increased cell stress. The short isoform is mainly expressed in the normal situation and is 3.5 times higher expressed in skeletal compared with cardiac muscle.
Variants in FLNC are traditionally associated with myofibrillar myopathy (MFM; MIM# 609524), but subsequently also with isolated cardiomyopathies (MIM# 617047). To date, most available basic research on FLNC has focused on myopathies. Paradoxically, most genetic variants are described in cardiomyopathy patients, as this clinical entity is more prevalent and studies using high‐throughput sequencing of FLNC are more frequent in cardiomyopathy cohorts. This overview highlights known and novel FLNC variants and focuses on specific pathomechanisms important for distinct cardiac or muscular phenotypes.
2. VARIANTS
All FLNC variants are described according to current Human Genome Variation Society mutation nomenclature guidelines based on Genbank accession number NM_001458.4 (longest transcript). Previously reported and novel variants are interpreted and classified using American College of Medical Genetics and Genomics classification recommendations (Richards et al., 2015).
A total of 285 unique variants could be retrieved from the international peer‐reviewed literature, the Human Gene Mutation Database and the Leiden Open Variation Database (LOVD; Figure 1). We added 40 novel unique variants, leading to a total of 325 unique variants. All variants were submitted to the LOVD. A clear description of the phenotype associated with the variant was a requirement for this overview. One‐hundred variants were excluded from the main analysis, as no clinical information was available (Table S1). Most FLNC variants have been reported over the last 5 years, from the time that FLNC was recognized as a disease‐associated gene in the field of cardiomyopathies. Variant interpretation remains challenging, as the available evidence for pathogenicity is still limited for most types of variants. In general, truncating variants were classified as (likely) pathogenic, and missense variants as a variant of unknown significance (VUS), unless additional evidence from segregation and/or functional experiments was available.
Figure 1.

Variant selection of all FLNC variants and the overview of all variants in association with their phenotype. DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; HGMD, Human Gene Mutation Database; LOVD, Leiden Open Variation Database
2.1. Cardiomyopathies
Cardiomyopathies are a heterogeneous group of myocardial diseases associated with mechanical or electrical dysfunction that exhibit inappropriate ventricular hypertrophy or dilatation (Elliott et al., 2008; Maron et al., 2006). In this review, we distinguish between:
-
1.
Dilated cardiomyopathy (DCM): Characterized by the presence of left ventricular dilatation and contractile dysfunction, in the absence of abnormal loading conditions and severe coronary artery disease (Elliott et al., 2008; Maron et al., 2006).
-
2.
Hypertrophic cardiomyopathy (HCM): The presence of increased left ventricular wall thickness that is not solely explained by abnormal loading conditions (Elliott et al., 2008; Maron et al., 2006).
-
3.
Other cardiac diseases, which do not fulfill these criteria for DCM or HCM.
2.1.1. Dilated cardiomyopathy
Variants predicted to result in a premature stop codon are strongly enriched in DCM, and are classified as (likely) pathogenic, as FLNC is highly intolerant for loss‐of‐function variants (pLI‐score = 1; Figures 1 and 2). The prevalence of FLNC variants in patients with DCM ranges from 1% to 4.5% (Ader et al., 2019; Begay et al., 2018; Janin et al., 2017; Ortiz‐Genga et al., 2016; Table 1).
Figure 2.
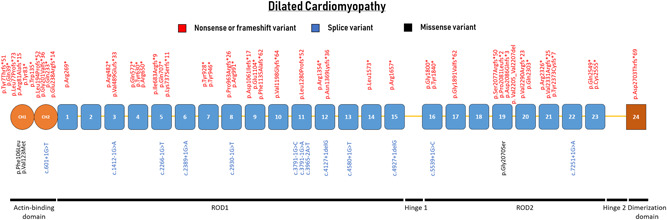
Schematic representation of the FLNC gene with their protein‐coding domains. Numbers inside the boxes refer to the Ig‐like domains of filamin C. Above and below the schematic are all unique variants associated with dilated cardiomyopathy. Variants are annotated at the protein level
Table 1.
FLNC variants found in individuals with dilated cardiomyopathy (DCM) previously reported and from this study
| Exon | c‐Notation | p‐Notation | Variant type | Domain | Location | Reference | Effect |
|---|---|---|---|---|---|---|---|
| 1 | c.19del | p.Tyr7Thrfs*51 | Frameshift | ABD | CH1 | (Ader et al., 2019) | Likely pathogenic |
| 1 | c.115C>T | p.Gln39* | Nonsense | ABD | CH1 | (Janin et al., 2017) | Likely pathogenic |
| 1 | c.230_234delTCAGC | p.Leu77Profs*73 | Frameshift | ABD | CH1 | (Janin et al., 2017) | Likely pathogenic |
| 1 | c.241delC | p.Arg81Alafs*15 | Frameshift | ABD | CH1 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 1 | c.248_265dup | p.Pro88_Arg89insHisArg | In‐frame | ABD | CH1 | Current study | VUS |
| LysPheHisPro | |||||||
| 1 | c.249C>G | p.Tyr83* | Nonsense | ABD | CH1 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 1 | c.318C>G | p.Phe106Leu | Missense | ABD | CH1 | (Reinstein et al., 2016) | Likely pathogenic |
| 1 | c.328G>A | p.Glu110Lys | Missense | ABD | CH1 | Current study | VUS |
| 2 | c.367G>A | p.Val123Met | Missense | ABD | CH1 | Current study | Likely pathogenic |
| 2 | c.404G>A | p.Trp135* | Nonsense | ABD | CH1 | LOVD | Likely pathogenic |
| 2 | c.581_599del19 | p.Leu194Profs*52 | Frameshift | ABD | CH2 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 2 | c.601+1G>T | p.? | Splice | ABD | CH2 | (Janin et al., 2017) | Likely pathogenic |
| 3 | c.602–716_1010delins | p.Gly201Valfs*36 | Frameshift | ABD | CH2 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| TGCCCCGGGAGGGGTGC | |||||||
| CTCAGTCTCCC | |||||||
| TGTCCCTCTG | |||||||
| 3 | c.697C>T | p.Gln233* | Nonsense | ABD | CH2 | Current study | Likely pathogenic |
| 4 | c.711del | p.Glu238Argfs*14 | Frameshift | ABD | CH2 | (Ader et al., 2019) | Likely pathogenic |
| 4 | c.805C>T | p.Arg269* | Nonsense | ROD1 | Ig‐like 1 | (Begay et al., 2018) | Likely pathogenic |
| 6 | c.970‐4A>G | p.? | Splice | ROD1 | Ig‐like 1 | Current study | VUS |
| 9 | c.1412‐1G>A | p.? | Splice | ROD1 | Ig‐like 3 | (Ader et al., 2019) | Likely pathogenic |
| 9 | c.1444C>T | p.Arg482* | Nonsense | ROD1 | Ig‐like 3 | (Tobita et al., 2017) | Likely pathogenic |
| 9 | c.1466_1472del | p.Val489Glyfs*33 | Frameshift | ROD1 | Ig‐like 3 | Current study | Likely pathogenic |
| 9 | c.1466_1473delinsA | p.Val489Glufs*33 | Frameshift | ROD1 | Ig‐like 3 | LOVD | Likely pathogenic |
| 11 | c.1714C>T | p.Gln572* | Nonsense | ROD1 | Ig‐like 4 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 12 | c.1890C>A | p.Tyr630* | Nonsense | ROD1 | Ig‐like 4 | (Janin et al., 2017) | Likely pathogenic |
| 12 | c.1948C>T | p.Arg650* | Nonsense | ROD1 | Ig‐like 4 | LOVD | Likely pathogenic |
| 13 | c.2041_2047dup | p.Ile683Argfs*9 | Frameshift | ROD1 | Ig‐like 5 | (Ader et al., 2019) | Likely pathogenic |
| 13 | c.2119C>T | p.Gln707* | Nonsense | ROD1 | Ig‐like 5 | (Begay et al., 2018) | Likely pathogenic |
| 14 | c.2208delT | p.Lys737Serfs*11 | Frameshift | ROD1 | Ig‐like 5 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 15 | c.2266‐1G>T | p.? | Splice | ROD1 | Ig‐like 5 | (Janin et al., 2017) | Likely pathogenic |
| 15 | c.2389+1G>A | p.? | Splice | ROD1 | Ig‐like 6 | (Nozari et al., 2018) | Likely pathogenic |
| 16 | c.2425G>A | p.Val809Met | Missense | ROD1 | Ig‐like 6 | (Janin et al., 2017) | VUS |
| 18 | c.2784C>G | p.Tyr928* | Nonsense | ROD1 | Ig‐like 7 | (Ader et al., 2019) | Likely pathogenic |
| 19 | c.2838T>A | p.Tyr946* | Nonsense | ROD1 | Ig‐like 7 | LOVD | Likely pathogenic |
| 19 | c.2888delC | p.Pro963Argfs*26 | Frameshift | ROD1 | Ig‐like 8 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 20 | c.2930‐1G>T | p.? | Splice | ROD1 | Ig‐like 8 | (Begay et al., 2018) | Likely pathogenic |
| 20 | c.2971C>T | p.Arg991* | Nonsense | ROD1 | Ig‐like 8 | (Reinstein et al., 2016) | Likely pathogenic |
| 20 | c.3180del | p.Asp1061Ilefs*17 | Frameshift | ROD1 | Ig‐like 9 | Current study | Likely pathogenic |
| 21 | c.3310G>T | p.Glu1104* | Nonsense | ROD1 | Ig‐like 9 | (Janin et al., 2017) | Likely pathogenic |
| 21 | c.3380_3402dup23 | p.Phe1135Alafs*62 | Frameshift | ROD1 | Ig‐like 9 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 21 | c.3592dup | p.Val1198Glyfs*64 | Frameshift | ROD1 | Ig‐like 10 | (Ader et al., 2019) | Likely pathogenic |
| 22 | c.3791‐1G>A | p.? | Splice | ROD1 | Ig‐like 11 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 22 | c.3791‐1G>C | p.? | Splice | ROD1 | Ig‐like 11 | (Deo et al., 2014) | Likely pathogenic |
| 22 | c.3838dup | p.Leu1280Profs*52 | Frameshift | ROD1 | Ig‐like 11 | Current study | Likely pathogenic |
| 23 | c.3965‐2A>T | p.? | Splice | ROD1 | Ig‐like 11 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 23 | c.4060C>T | p.Arg1354* | Nonsense | ROD1 | Ig‐like 12 | (Janin et al., 2017) | Likely pathogenic |
| 23 | c.4106dupA | p.Asn1369Lysfs*36 | Frameshift | ROD1 | Ig‐like 12 | (Cuenca et al., 2016) | Likely pathogenic |
| 23 | c.4127+1delG | p.? | Splice | ROD1 | Ig‐like 12 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 26 | c.4580+1G>T | p.? | Splice | ROD1 | Ig‐like 13 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 27 | c.4700G>A | p.Arg1567Gln | Missense | ROD1 | Ig‐like 14 | (Esslinger et al., 2017) | VUS |
| 27 | c.4718T>A | p.Leu1573* | Nonsense | ROD1 | Ig‐like 14 | (Augusto et al., 2019) | Likely pathogenic |
| 28 | c.4927+1delG | p.? | Splice | ROD1 | Ig‐like 15 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 30 | c.4969C>T | p.Arg1657* | Nonsense | ROD1 | Ig‐like 15 | LOVD | Likely pathogenic |
| 30 | c.5036C>A | p.Thr1679Lys | Missense | ROD1 | Ig‐like 15 | Current study | VUS |
| 32 | c.5398G>T | p.Gly1800* | Nonsense | ROD2 | Ig‐like 16 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 33 | c.5520T>A | p.Tyr1840* | Nonsense | ROD2 | Ig‐like 16 | (Chanavat, Janin, & Millat, 2016) | Likely pathogenic |
| 33 | c.5539+1G>C | p.? | Splice | ROD2 | Ig‐like 16 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 35 | c.5672delG | p.Gly1891Valfs*62 | Frameshift | ROD2 | Ig‐like 17 | (Begay et al., 2018) | Likely pathogenic |
| 35 | c.5791C>T | p.Arg1931Cys | Missense | ROD2 | Ig‐like 17 | Current study | VUS |
| 37 | c.6100G>C | p.Gly2034Arg | Missense | ROD2 | Ig‐like 18 | Current study | VUS |
| 37 | c.6208G>A | p.Gly2070Ser | Missense | ROD2 | Ig‐like 19 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 38 | c.6231delT | p.Ser2077Argfs*50 | Frameshift | ROD2 | Ig‐like 19 | (Cuenca et al., 2016) | Likely pathogenic |
| 38 | c.6240_6259del | p.Pro2081Leufs*2 | Frameshift | ROD2 | Ig‐like 19 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 38 | c.6255_6256del | p.Ile2086Glnfs*3 | Frameshift | ROD2 | Ig‐like 19 | Current study | Likely pathogenic |
| 40 | c.6518G>A | p.Arg2173His | Missense | ROD2 | Intradomain | Current study | VUS |
| 40 | c.6614_6622del | p.Val2205_Val2207del | In‐frame | ROD2 | Intradomain | Current study | Likely pathogenic |
| 41 | c.6790G>A | p.Ala2264Thr | Missense | ROD2 | Ig‐like 20 | Current study | VUS |
| 41 | c.6864_6867dup | p.Val2290Argfs*23 | Frameshift | ROD2 | Ig‐like 20 | Current study | Likely pathogenic |
| 41 | c.6877C>T | p.Arg2293Cys | Missense | ROD2 | Ig‐like 20 | Current study | VUS |
| 41 | c.6907C>T | p.Gln2303* | Nonsense | ROD2 | Ig‐like 20 | Current study | Likely pathogenic |
| 41 | c.6976C>T | p.Arg2326* | Nonsense | ROD2 | Ig‐like 21 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 41 | c.6989dupG | p.Val2331Argfs*25 | Frameshift | ROD2 | Ig‐like 21 | (Janin et al., 2017) | Likely pathogenic |
| 42 | c.7118_7119del | p.Tyr2373Cysfs*7 | Frameshift | ROD2 | Ig‐like 21 | (Ader et al., 2019) | Likely pathogenic |
| 43 | c.7251+1G>A | p.? | Splice | ROD2 | Ig‐like 22 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
| 45 | c.7450G>A | p.Gly2484Ser | Missense | ROD2 | Ig‐like 22 | Current study | VUS |
| 46 | c.7645C>T | p.Gln2549* | Nonsense | ROD2 | Ig‐like 23 | (Ader et al., 2019) | Likely pathogenic |
| 46 | c.7652A>G | p.Asp2551Gly | Missense | ROD2 | Ig‐like 23 | Current study | VUS |
| 46 | c.7665T>A | p.Cys2555* | Nonsense | ROD2 | Ig‐like 23 | (Ader et al., 2019) | Likely pathogenic |
| 48 | c.8107delG | p.Asp2703Thrfs*69 | Frameshift | Dimerization | Ig‐like 24 | (Ortiz‐Genga et al., 2016) | Likely pathogenic |
Abbreviations: ABD, actin‐binding domain; LOVD, Leiden Open Variation Database; VUS, variant of unknown significance.
Three missense variants have been classified as likely pathogenic: p.Phe106Leu, p.Ala123Met, and p.Gly2070Ser. The p.Phe106Leu missense variant occurred on the opposite allele of a nonsense variant (p.Arg991*) in a neonatal patient with DCM (Reinstein et al., 2016). Compound heterozygosity for these FLNC variants led to an early‐onset phenotype. Heterozygous carriers had not developed DCM by age 40 years. Protein levels were decreased for the p.Phe106Leu variant, and the p.Arg991* was not detectable. The p.Val123Met variant reported in the current study is classified as likely pathogenic, due to the well‐investigated p.Val123Ala variant at the same codon in HCM (Valdes‐Mas et al., 2014). The p.Gly2070Ser variant is predicted to alter a canonical splice site, although RNA analysis was not performed (Ortiz‐Genga et al., 2016). All other missense variants are classified as VUS and are not included in Figure 2, but are listed in Table 1.
2.1.2. Hypertrophic cardiomyopathy
Missense variants are mainly associated with HCM with a varying prevalence from 1.3% to 8.7% in HCM cohorts (Ader et al., 2019; Cui et al., 2018; Gomez et al., 2017; Valdes‐Mas et al., 2014; Figure 1; Table 2). Two studies did not detect an excess of rare missense variants between HCM patients and controls, questioning the importance of FLNC missense variants in HCM (Cui et al., 2018; Walsh et al., 2019). Only 13 of the 54 missense variants are supported to be (likely) pathogenic by additional evidence such as functional studies (n = 4) and/or segregation (n = 13; Ader et al., 2019; Cui et al., 2018; Gomez et al., 2017; Valdes‐Mas et al., 2014). Based on current diagnostic classification criteria, all other missense variants would individually be classified as VUS (Table 2). There is a strong clustering of missense variants in the ROD2 domain of the FLNC, which is an important domain for cell signaling (Figure 3). Thus, collectively, missense variants in the ROD2 domain carry an increased likelihood of being pathogenic for HCM.
Table 2.
FLNC variants found in individuals with hypertrophic cardiomyopathy (HCM) previously reported and from this study
| Exon | c‐Notation | p‐Notation | Variant type | Domain | Location | Reference | Effect |
|---|---|---|---|---|---|---|---|
| 1 | c.322G>T | p.Glu108* | Nonsense | ABD | CH1 | (Valdes‐Mas et al., 2014) | Likely pathogenic |
| 2 | c.368T>C | p.Val123Ala | Missense | ABD | CH1 | (Valdes‐Mas et al., 2014) | Likely pathogenic |
| 4 | c.743A>T | p.His248Leu | Missense | ABD | CH2 | Current study | VUS |
| 4 | c.850+4T>G | p.? | Splice | ROD1 | Ig‐like 1 | (Cui et al., 2018) | VUS |
| 5 | c.870C>A | p.Asn290Lys | Missense | ROD1 | Ig‐like 1 | (Valdes‐Mas et al., 2014) | VUS |
| 6 | c.986A>G | p.Asn329Ser | Missense | ROD1 | Ig‐like 1 | (Alejandra Restrepo‐Cordoba et al., 2017) | VUS |
| 7 | c.1076T>C | p.Ile359Thr | Missense | ROD1 | Ig‐like 1 | (Cui et al., 2018) | VUS |
| 7 | c.1132G>T | p.Val378Leu | Missense | ROD1 | Ig‐like 2 | (Cui et al., 2018) | VUS |
| 9 | c.1425C>A | p.Asn475Lys | Missense | ROD1 | Ig‐like 3 | Current study | VUS |
| 12 | c.1882G>A | p.Val628Met | Missense | ROD1 | Ig‐like 4 | (Cui et al., 2018) | VUS |
| 13 | c.2050G>C | p.Val684Leu | Missense | ROD1 | Ig‐like 5 | (Cui et al., 2018) | VUS |
| 13 | c.2084G>A | p.Arg695His | Missense | ROD1 | Ig‐like 5 | Current study | VUS |
| 14 | c.2170G>A | p.Gly724Ser | Missense | ROD1 | Ig‐like 5 | (Cui et al., 2018) | VUS |
| 15 | c.2375G>T | p.Ser792Ile | Missense | ROD1 | Ig‐like 6 | (Jaafar et al., 2016) | VUS |
| 16 | c.2450T>C | p.Ile817Thr | Missense | ROD1 | Ig‐like 6 | (Cirino et al., 2017) | VUS |
| 17 | c.2587C>T | p.Pro863Ser | Missense | ROD1 | Ig‐like 7 | (Cui et al., 2018) | VUS |
| 18 | c.2737G>A | p.Glu913Lys | Missense | ROD1 | Ig‐like 7 | (Cui et al., 2018) | VUS |
| 19 | c.2812‐4A>G | p.? | Splice | ROD1 | Ig‐like 7 | (Cui et al., 2018) | VUS |
| 21 | c.3581C>T | p.Ser1194Leu | Missense | ROD1 | Ig‐like 10 | (Ader et al., 2019) | VUS |
| 21 | c.3623C>T | p.Ala1208Val | Missense | ROD1 | Ig‐like 10 | (Cui et al., 2018) | VUS |
| 24 | c.4271G>T | p.Gly1424Val | Missense | ROD1 | Ig‐like 12 | (Ader et al., 2019) | Likely pathogenic |
| 27 | c.4615G>A | p.Ala1539Thr | Missense | ROD1 | Ig‐like 14 | (Valdes‐Mas et al., 2014) | Likely pathogenic |
| 28 | c.4795A>G | p.Thr1599Ala | Missense | ROD1 | Ig‐like 14 | (Gomez et al., 2017) | VUS |
| 30 | c.5042C>T | p.Thr1681Met | Missense | ROD1 | Ig‐like 15 | (Gomez et al., 2017) | VUS |
| 30 | c.5068C>T | p.Leu1690Phe | Missense | ROD1 | Ig‐like 15 | (Gomez et al., 2017) | VUS |
| 30 | c.5125C>T | p.Pro1709Ser | Missense | ROD1 | Ig‐like 15 | (Cui et al., 2018) | VUS |
| 30 | c.5132C>T | p.Pro1711Leu | Missense | ROD1 | Ig‐like 15 | (Cui et al., 2018) | VUS |
| 36 | c.5888C>T | p.Thr1963Met | Missense | ROD2 | Ig‐like 18 | (Cui et al., 2018) | VUS |
| 36 | c.5954C>T | p.Ser1985Leu | Missense | ROD2 | Ig‐like 18 | Current study | VUS |
| 36 | c.5996G>A | p.Arg1999Gln | Missense | ROD2 | Ig‐like 18 | (Jaafar et al., 2016) | VUS |
| 37 | c.6032G>A | p.Gly2011Glu | Missense | ROD2 | Ig‐like 18 | (Ader et al., 2019) | VUS |
| 37 | c.6053G>A | p.Arg2018His | Missense | ROD2 | Ig‐like 18 | (Chanavat et al., 2016) | VUS |
| 37 | c.6115G>A | p.Gly2039Arg | Missense | ROD2 | Ig‐like 19 | (Ader et al., 2019) | Likely pathogenic |
| 37 | c.6134G>A | p.Arg2045Gln | Missense | ROD2 | Ig‐like 19 | (Chanavat et al., 2016) | VUS |
| 37 | c.6205G>A | p.Ala2069Thr | Missense | ROD2 | Ig‐like 19 | Current study | VUS |
| 39 | c.6398G>A | p.Arg2133His | Missense | ROD2 | Intradomain | (Valdes‐Mas et al., 2014) | Likely pathogenic |
| 39 | c.6397C>T | p.Arg2133Cys | Missense | ROD2 | Intradomain | (Cui et al., 2018) | Likely pathogenic |
| 39 | c.6419G>A | p.Arg2140Gln | Missense | ROD2 | Intradomain | (Gomez et al., 2017) | Likely pathogenic |
| 39 | c.6451G>A | p.Gly2151Ser | Missense | ROD2 | Intradomain | (Valdes‐Mas et al., 2014) | VUS |
| 40 | c.6589C>T | p.Arg2197Trp | Missense | ROD2 | Intradomain | (Alejandra Restrepo‐Cordoba et al., 2017) | VUS |
| 41 | c.6860C>A | p.Thr2287Lys | Missense | ROD2 | Ig‐like 20 | Current study | VUS |
| 41 | c.6892C>T | p.Pro2298Ser | Missense | ROD2 | Ig‐like 20 | (Gomez et al., 2017) | Likely pathogenic |
| 41 | c.6895G>A | p.Gly2299Ser | Missense | ROD2 | Ig‐like 20 | (Ader et al., 2019) | VUS |
| 41 | c.6901C>G | p.Pro2301Ala | Missense | ROD2 | Ig‐like 20 | (Gomez et al., 2017) | Likely pathogenic |
| 41 | c.6943C>A | p.His2315Asn | Missense | ROD2 | Ig‐like 21 | (Valdes‐Mas et al., 2014) | Likely pathogenic |
| 41 | c.6952C>T | p.Arg2318Trp | Missense | ROD2 | Ig‐like 21 | (Gomez et al., 2017) | VUS |
| 42 | c.7030G>A | p.Ala2344Thr | Missense | ROD2 | Ig‐like 21 | (Cui et al., 2018) | VUS |
| 42 | c.7076T>C | p.Ile2359Thr | Missense | ROD2 | Ig‐like 21 | (Ader et al., 2019) | VUS |
| 42 | c.7123G>T | p.Val2375Phe | Missense | ROD2 | Ig‐like 21 | (Gomez et al., 2017) | VUS |
| 42 | c.7123G>C | p.Val2375Leu | Missense | ROD2 | Ig‐like 21 | (Ader et al., 2019) | Likely pathogenic |
| 43 | c.7228C>T | p.Arg2410Cys | Missense | ROD2 | Ig‐like 22 | (Ader et al., 2019) | VUS |
| 43 | c.7250A>C | p.Gln2417Pro | Missense | ROD2 | Ig‐like 22 | (Ader et al., 2019) | VUS |
| 44 | c.7289C>T | p.Ala2430Val | Missense | ROD2 | Ig‐like 22 | (Valdes‐Mas et al., 2014) | Likely pathogenic |
| 45 | c.7484G>A | p.Arg2495His | Missense | ROD2 | Ig‐like 22 | (Ader et al., 2019) | Likely pathogenic |
| 45 | c.7514C>T | p.Pro2505Leu | Missense | ROD2 | Ig‐like 23 | (Cui et al., 2018) | VUS |
| 47 | c.7781G>C | p.Gly2594Ala | Missense | ROD2 | Hinge 2 | (Chanavat et al., 2016) | VUS |
| 47 | c.7853C>T | p.Ser2618Phe | Missense | ROD2 | Hinge 2 | Current study | VUS |
Abbreviations: ABD, actin‐binding domain; VUS, variant of unknown significance.
Figure 3.
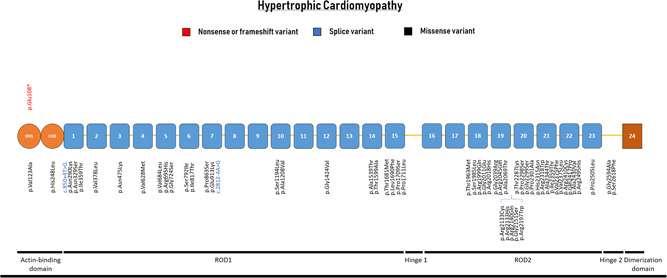
Schematic representation of the FLNC gene with their protein‐coding domains. Numbers inside the boxes refer to the Ig‐like domains of filamin C. Above and below the schematic are all unique variants associated with hypertrophic cardiomyopathy. Variants are annotated at the protein level
2.1.3. Other cardiac phenotypes
FLNC variants have been associated with other cardiac phenotypes such as arrhythmias without detectable structural abnormalities, congenital heart disease, restrictive (RCM), and noncompaction (NCCM) cardiomyopathies (Figure 4; Table 3). The association of FLNC with a broad spectrum of cardiac phenotypes shows an important gap in knowledge. Hence, not all reported variants have proven to be causal. There can also be a large overlap between phenotypes: cardiac noncompaction can be a trait observed in other cardiomyopathies and healthy hearts (Hershberger et al., 2018).
Figure 4.
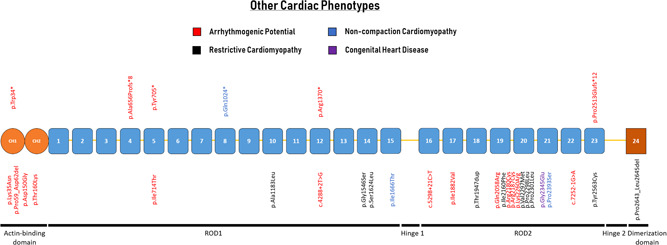
Schematic representation of the FLNC gene with their protein‐coding domains. Numbers inside the boxes refer to the Ig‐like domains of filamin C. Above and below the schematic are all unique variants associated with different cardiac phenotypes. Variants are annotated at the protein level
Table 3.
FLNC variants found in individuals with a variety of cardiac phenotypes as previously reported and from this study
| Exon | c‐Notation | p‐Notation | Variant type | Domain | Location | Phenotype | Reference | Effect |
|---|---|---|---|---|---|---|---|---|
| 1 | c.102G>A | p.Trp34* | Nonsense | ABD | CH1 | ABiMVPS | (Bains et al., 2019) | Likely pathogenic |
| 1 | c.105G>C | p.Lys35Asn | Missense | ABD | CH1 | ACM/SCD | (Hall et al., 2019) | VUS |
| 1 | c.174_185del | p.Pro59_Asp62del | In‐frame | ABD | CH1 | ACM | (Hall et al., 2019) | VUS |
| 2 | c.449A>G | p.Asp150Gly | Missense | ABD | CH1 | ACM | Current study | VUS |
| 2 | c.479C>A | p.Thr160Lys | Missense | ABD | CH2 | ACM/SCD | (Hall et al., 2019) | VUS |
| 12 | c.1965_1966delTG | p.Ala656Profs*8 | Frameshift | ROD1 | Ig‐like 4 | ACM/SCD | (Hall et al., 2019) | Likely pathogenic |
| 13 | c.2115_2120delTGCCCA | p.Tyr705* | Nonsense | ROD1 | Ig‐like 5 | ACM/SCD | (Hall et al., 2019) | Likely pathogenic |
| 14 | c.2141T>C | p.Ile714Thr | Missense | ROD1 | Ig‐like 5 | ACM | (Hall et al., 2019) | VUS |
| 20 | c.3070C>T | p.Gln1024* | Nonsense | ROD1 | Ig‐like 8 | NCCM | (Miszalski‐Jamka et al., 2017) | Likely pathogenic |
| 21 | c.3547_3548delinsCT | p.Ala1183Leu | Missense | ROD1 | Ig‐like 10 | RCM | (Kiselev et al., 2018) | VUS |
| 23 | c.4108C>T | p.Arg1370* | Nonsense | ROD1 | Ig‐like 12 | ACM/SCD | (Hall et al., 2019) | Likely pathogenic |
| 24 | c.4288+2T>G | p.? | Splice | ROD1 | Ig‐like 12 | ACM/SCD | (Hall et al., 2019) | Likely pathogenic |
| 27 | c.4636G>A | p.Gly1546Ser | Missense | ROD1 | Ig‐like 14 | RCM | (Sanoja, Li, Fricker, Kingsmore, & Wallace, 2018) | VUS |
| 28 | c.4871C>T | p.Ser1624Leu | Missense | ROD1 | Ig‐like 14 | RCM | (Brodehl et al., 2016) | Likely pathogenic |
| 30 | c.4997T>C | p.Ile1666Thr | Missense | ROD1 | Ig‐like 15 | NCCM | (Ader et al., 2019) | VUS |
| 31 | c.5298+21C>T | p.? | Splice | ROD2 | Ig‐like 16 | ACM/SCD | (Hall et al., 2019) | VUS |
| 34 | c.5644A>G | p.Ile1882Val | Missense | ROD2 | Ig‐like 17 | ACM | (Hall et al., 2019) | VUS |
| 35 | c.5839_5841dup | p.Thr1947dup | In‐frame | ROD2 | Ig‐like 18 | RCM | (Ader et al., 2019) | VUS |
| 37 | c.6173A>G | p.Gln2058Arg | Missense | ROD2 | Ig‐like 19 | ACM | (Hall et al., 2019) | VUS |
| 39 | c.6478A>T | p.Ile2160Phe | Missense | ROD2 | Intradomain | RCM | (Brodehl et al., 2016) | Likely pathogenic |
| 40 | c.6538C>T | p.Arg2180Cys | Missense | ROD2 | Intradomain | ACM | Current study | VUS |
| 40 | c.6559C>T | p.Arg2187Cys | Missense | ROD2 | Intradomain | ACM | Current study | VUS |
| 41 | c.6779A>G | p.Lys2260Arg | Missense | ROD2 | Ig‐like 20 | ACM | (Hall et al., 2019) | VUS |
| 41 | c.6889G>A | p.Val2297Met | Missense | ROD2 | Ig‐like 20 | RCM | (Tucker et al., 2017) | Likely pathogenic |
| 41 | c.6893C>T | p.Pro2298Leu | Missense | ROD2 | Ig‐like 20 | RCM | (Schubert et al., 2018) | Likely pathogenic |
| 41 | c.6902C>T | p.Pro2301Leu | Missense | ROD2 | Ig‐like 20 | RCM | (Roldan‐Sevilla et al., 2019) | VUS |
| 42 | c.7034G>A | p.Gly2345Glu | Missense | ROD2 | Ig‐like 21 | Congenital heart disease | (Kosmicki et al., 2017) | VUS |
| 43 | c.7177C>T | p.Pro2393Ser | Missense | ROD2 | Ig‐like 21 | NCCM | Current study | Likely pathogenic |
| 44 | c.7252‐1G>A | p.? | Splice | ROD2 | Ig‐like 22 | ACM | (Hall et al., 2019) | Likely pathogenic |
| 45 | c.7536_7548del13 | p.Pro2513Glufs*12 | Frameshift | ROD2 | Ig‐like 23 | Cardiac arrhythmia | (Mangum & Ferns, 2019) | Likely pathogenic |
| 46 | c.7688A>G | p.Tyr2563Cys | Missense | ROD2 | Ig‐like 23 | RCM | (Schubert et al., 2018) | Likely pathogenic |
| 47 | c.7927_7935del | p.Pro2643_Leu2645del | In‐frame | ROD2 | Ig‐like 24 | RCM | (Ader et al., 2019) | VUS |
Abbreviations: ABD, actin‐binding domain; ABiMVPS, arrhythmogenic bileaflet mitral valve prolapse syndrome; ACM, arrhythmogenic cardiomyopathy; NCCM, noncompaction cardiomyopathy; RCM, restrictive cardiomyopathy; SCD, sudden cardiac death; VUS, variant of unknown significance.
One patient with arrhythmogenic bileaflet mitral valve prolapse syndrome (ABiMVPS) was recently reported in association with a truncating variant (p.Trp34*) identified in whole‐exome sequencing data (Bains et al., 2019). It was speculated that FLNC haploinsufficiency was the underlying arrhythmogenic substrate, which was exacerbated by the mitral valve prolapse. Another recent report described familial sudden cardiac death without signs of cardiomyopathy in association with a truncating variant in FLNC (p.Pro2513Glufs*12; Mangum & Ferns, 2019). Both cases highlight the arrhythmogenic potential associated with FLNC truncating variants. However, it remains unknown if alternative diagnostic tools such as global longitudinal strain analysis could detect subtle changes in cardiac function. In addition, arrhythmias can also be accompanied by a cardiomyopathy phenotype with right, left, or biventricular involvement called arrhythmogenic cardiomyopathy (ACM). When there is prominent left ventricular involvement, it is difficult to clinically distinguish it from DCM. A recent cohort study in ACM patients found four truncating variants (3.3%; Table 3; Hall et al., 2019).
FLNC variants in RCM and NCCM are less prevalent compared with DCM and HCM, making it difficult to draw any conclusions on the role of FLNC in these cardiomyopathies. One truncating variant has been described in association with NCCM (p.Gln1024*; Miszalski‐Jamka et al., 2017). Unfortunately, little clinical information was available to assess the arrhythmogenic potential of this individual. In addition, this patient also carried a pathogenic RYR2 variant.
2.2. Myopathies
Classification of myopathies can be based on either clinical presentation, cause or pathology. To enhance clinical utility and prevent miscommunication, we suggest the classification based on clinical presentation, such as distal and/or proximal myopathy. The more specific diagnosis MFM requires finding protein aggregates in muscle.
The first description of a human phenotype related to FLNC was in 2005, when a nonsense variant (p.Trp2710*) was described in a German family with a novel type of autosomal dominant MFM (Vorgerd et al., 2005). The variant was later described in cohorts of varying ethnicity, suggesting codon 2710 to be a mutational hotspot (Kley et al., 2012). FLNC variants are mostly associated with a proximal myopathy, with occasional distal involvement (Figure 5; Table 4; van den Bogaart et al., 2017). There are few associations between variant type or location and the corresponding myopathy phenotype or special features such as cardiac involvement. We observed a cluster of missense variants in Ig‐like domain 10, a domain which is rarely involved in cardiomyopathies. There was no genotype–phenotype association between variant location, and features of MFM, including tissue protein aggregate formation (Figure 5). Missense, in‐frame and nonsense variants are all associated with protein aggregate formation in muscle tissue (Avila‐Smirnow et al., 2010; Luan, Hong, Zhang, Wang, & Yuan, 2010; Vorgerd et al., 2005). Only two truncating variants have been reported to cause myopathy. Both are in Ig‐like 15, and associated with a form of isolated distal myopathy.
Figure 5.
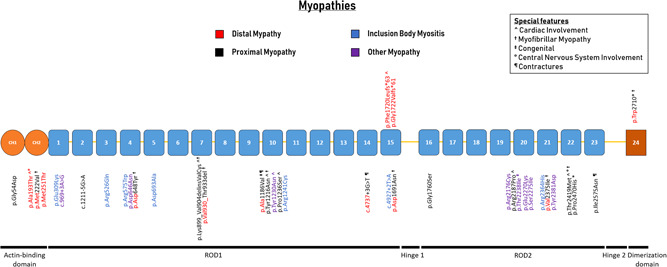
Schematic representation of the FLNC gene with their protein‐coding domains. Numbers inside the boxes refer to the Ig‐like domains of filamin C. Above and below the schematic are all unique variants associated with myopathies. Variants are annotated at the protein level
Table 4.
FLNC variants found in individuals with a muscular phenotype as previously reported and from this study
| Exon | c‐Notation | p‐Notation | Variant type | Domain | Location | Phenotype | Reference | Effect |
|---|---|---|---|---|---|---|---|---|
| 1 | c.161G>A | p.Gly54Asp | Missense | ABD | CH1 | PM | (Fichna et al., 2018) | VUS |
| 2 | c.577G>A | p.Ala193Thr | Missense | ABD | CH2 | DM; PM; Car; CNS | (Duff et al., 2011) | Likely pathogenic |
| 3 | c.664A>G | p.Met222Val | Missense | ABD | CH2 | DM; PM; MFM | (Gemelli et al., 2019) | Likely pathogenic |
| 4 | c.752T>C | p.Met251Thr | Missense | ABD | CH2 | DM; PM | (Duff et al., 2011) | Likely pathogenic |
| 5 | c.925G>A | p.Glu309Lys | Missense | ROD1 | Ig‐like 1 | IBM | (Weihl et al., 2015) | VUS |
| 5 | c.969+3A>G | p.? | Splice | ROD1 | Ig‐like 1 | OM | (Dai et al., 2015) | VUS |
| 8 | c.1211‐5G>A | p.? | Splice | ROD1 | Ig‐like 2 | PM | Current study | VUS |
| 10 | c.1577G>A | p.Arg526Gln | Missense | ROD1 | Ig‐like 3 | IBM | (Weihl et al., 2015) | VUS |
| 11 | c.1723C>T | p.Arg575Trp | Missense | ROD1 | Ig‐like 4 | IBM | (Weihl et al., 2015) | VUS |
| 12 | c.1936G>A | p.Asp646Asn | Missense | ROD1 | Ig‐like 4 | OM | Current study | VUS |
| 12 | p.Asp648Tyr | Missense | ROD1 | Ig‐like 4 | PM; DM; MFM | (Y. T. Zhang et al., 2018) | Likely pathogenic | |
| 13 | c.2078A>C | p.Asp693Ala | Missense | ROD1 | Ig‐like 5 | IBM | (Weihl et al., 2015) | VUS |
| 18 | c.2695_2712del18insGTTTGT | p.Lys899_Val904delinsValCys | In‐frame | ROD1 | Ig‐like 7 | PM; Car; MFM | (Luan et al., 2010) | Likely pathogenic |
| 18 | c.2789_2800del12 | p.Val930_Thr933del | In‐frame | ROD1 | Ig‐like 7 | PM; DM; MFM | (Shatunov et al., 2009) | Likely pathogenic |
| 21 | c.3557C>T | p.Ala1186Val | Missense | ROD1 | Ig‐like 10 | PM; DM; CM; Con | (Ghaoui et al., 2015) | Likely pathogenic |
| 21 | c.3646T>A | p.Tyr1216Asn | Missense | ROD1 | Ig‐like 10 | PM; Car; MFM | (Avila‐Smirnow et al., 2010) | Likely pathogenic |
| 21 | c.3688T>A | p.Tyr1230Asn | Missense | ROD1 | Ig‐like 10 | OM; Car | (Vill et al., 2017) | VUS |
| 21 | c.3706C>T | p.Pro1236Ser | Missense | ROD1 | Ig‐like 10 | PM; Car | (Yu et al., 2017) | VUS |
| 21 | c.3721C>T | p.Arg1241Cys | Missense | ROD1 | Ig‐like 10 | IBM | (Weihl et al., 2015) | VUS |
| 27 | c.4737+3G>T | p.? | Splice | ROD1 | Ig‐like 14 | PM; DM; Con | Current study | VUS |
| 28 | c.4927+2T>A | p.? | Splice | ROD1 | Ig‐like 15 | OM | (Zenagui et al., 2018) | VUS |
| 30 | c.5071G>A | p.Asp1691Asn | Missense | ROD1 | Ig‐like 15 | PM; DM; MFM | (Y. T. Zhang et al., 2018) | Likely pathogenic |
| 30 | c.5160delC | p.Phe1720Leufs*63 | Frameshift | ROD1 | Ig‐like 15 | DM; Car | (Guergueltcheva et al., 2011) | Likely pathogenic |
| 30 | c.5165delG | p.Gly1722Valfs*61 | Frameshift | ROD1 | Ig‐like 15 | DM | (Rossi et al., 2017) | Likely pathogenic |
| 31 | c.5278G>A | p.Gly1760Ser | Missense | ROD2 | Ig‐like 16 | PM | (Yu et al., 2017) | VUS |
| 40 | c.6526C>T | p.Arg2176Cys | Missense | ROD2 | Intradomain | IBM | (Cerino et al., 2017) | VUS |
| 40 | c.6560G>C | p.Arg2187Pro | Missense | ROD2 | Intradomain | PM; Car | Current study | VUS |
| 40 | c.6713C>T | p.Thr2238Ile | Missense | ROD2 | Intradomain | OM; CM | Current study | VUS |
| 41 | c.6808G>A | p.Glu2270Lys | Missense | ROD2 | Ig‐like 20 | OM | Current study | VUS |
| 41 | c.6824G>T | p.Ser2275Ile | Missense | ROD2 | Ig‐like 20 | OM | Current study | VUS |
| 42 | c.7091G>A | p.Arg2364His | Missense | ROD2 | Ig‐like 21 | IBM | (Weihl et al., 2015) | VUS |
| 42 | c.7123G>A | p.Val2375Ile | Missense | ROD2 | Ig‐like 21 | PM; DM; MFM | (Chen et al., 2019) | VUS |
| 43 | c.7141T>G | p.Tyr2381Asp | Missense | ROD2 | Ig‐like 21 | OM | Current study | VUS |
| 44 | c.7256C>T | p.Thr2419Met | Missense | ROD2 | Ig‐like 22 | PM; Car; CNS; MFM | (Tasca et al., 2012) | VUS |
| 45 | c.7409C>A | p.Pro2470His | Missense | ROD2 | Ig‐like 22 | PM; Car | (Reddy et al., 2017) | VUS |
| 46 | c.7724T>A | p.Ile2575Asn | Missense | ROD2 | Ig‐like 23 | PM; Con | Current study | Likely pathogenic |
| 48 | c.8130G>A | p.Trp2710* | Nonsense | Dimerization | Ig‐like 24 | PM; DM; MFM | (Vorgerd et al., 2005) | Likely pathogenic |
Abbreviations: ABD, actin‐binding domain; Car, cardiac involvement; CM, congenital myopathy; CNS, central nervous system involvement; Con, contractures; DM, distal myopathy; IBM, inclusion body myositis; MFM, diagnosis of myofibrillar myopathy using cardiac tissue; OM, other nonspecified myopathy; PM, proximal myopathy; VUS, variant of unknown significance.
2.3. Other nonstriated muscle diseases
Twenty‐two unique FLNC variants have been described in association with noncardiac or muscular phenotypes. These variants are listed in the Supporting Information as they are not the main focus of this overview (Table S2). There is a single large study, which performed high‐throughput sequencing in patients with frontotemporal dementia and found a rare FLNC missense variant in 3.6% of the patients (Janssens et al., 2015).
3. BIOLOGICAL RELEVANCE
FLNC variants are associated with a spectrum of cardiac and muscular phenotypes, suggesting that specific variants fall into three pathomechanisms, as previously suggested (Furst et al., 2013):
-
1.
Variants that are predicted to lead to expression of misfolded proteins, which saturate the proteasome and autophagy pathways.
-
2.
Variants, which give a toxic gain‐of‐function by altering ligand binding properties.
-
3.
Variants causing a premature stop codon and concomitant nonsense‐mediated decay (NMD), resulting in haploinsufficiency.
3.1. Pathomechanisms of FLNC variants in cardiomyopathy phenotypes
A landmark paper regarding FLNC variants in DCM showed a strong association between truncating variants and this disease (Ortiz‐Genga et al., 2016). NMD and subsequent haploinsufficiency were validated for a number of truncating variants as the pathomechanism in FLNC‐associated DCM. Immunohistochemical analysis showed normal FLNC protein in the intercalated discs of patients with DCM. Abnormal FLNC protein aggregates in the cytoplasm were not detectable (Ortiz‐Genga et al., 2016). The absence of aggregates in the cardiac tissue of patients with truncating FLNC variants in the ROD2 domain indicates the lack of an abnormal FLNC protein. In addition, western blot analysis in zebrafish models and rat cardiac myoblasts showed the absence of a truncated protein in the truncating variant models (Begay et al., 2016; Reinstein et al., 2016). Haploinsufficiency affects force transduction of striated muscle, specifically in tissues dependent on high‐force generation, such as the myocardium.
Both HCM and RCM patients have an enrichment of missense variants causing changes in the secondary protein structure resulting in an abnormal protein. Missense variants are strongly clustered in the short ROD2 domain of the protein in HCM (Figure 3). This region of the protein is important for the interaction between FLNC and the Z‐disc. Five missense variants were reported in the intradomain insert (between Ig‐like domain 19 and 20), which mediates the specific targeting to the Z‐disc. Abnormal protein has been observed within aggregates in the tissue of FLNC‐associated HCM and RCM patients in association with marked sarcomeric abnormalities (Kiselev et al., 2018; Valdes‐Mas et al., 2014). The progressive accumulation of protein aggregates in the cardiac muscle eventually leads to sarcomeric disarray. Functional studies including the transfection of missense variants in rat cardiac myoblasts confirmed the formation of insoluble filamin C aggregates (Valdes‐Mas et al., 2014), although there were differences in the size of aggregates and signal strength on western blots per variant. The overall histopathology of FLNC‐associated HCM constituted large nuclei and large fiber diameters, as comparable to established non‐FLNC HCM.
3.2. Pathomechanisms of FLNC variants in myopathy phenotypes with and without protein aggregate formation
The p.Trp2710* variant leads to truncation of Ig‐domain 24, which is needed for the formation of FLNC dimers (Vorgerd et al., 2005). The mutant messenger RNA (mRNA) is stable and not subject to degradation by NMD, probably because the variant is in the last exon of the gene. Instead, the nonsense variant leads to the formation of protein aggregates of mutated filamin fragments, other known MFM‐associated proteins and a number of filamin C binding partners in the skeletal muscle (Kley, Maerkens et al., 2013; Lowe et al., 2007). Interestingly, there is one truncating variant in the last exon described in association with DCM (p.Asp2703Thrfs*69), which is subject to NMD (Ortiz‐Genga et al., 2016). This shows that not only the variant type in the last exon is of importance but also the pathomechanism for the subsequent phenotype.
The autophagy pathways which clear protein aggregates in MFM (mainly the “chaperone‐assisted selective autophagy” [CASA] pathway) are activated but unable to clear the aggregates, preventing recovery of homeostasis (Kley, van der Ven et al., 2013; Ruparelia, Oorschot, Ramm, & Bryson‐Richardson, 2016). More proteins are being associated with the regulation of FLNC proteostasis via autophagy, such as Hspb7 (Mercer, Lin, Cohen‐Gould, & Evans, 2018). Consequently, many of the FLNC binding partners are dispersed, which disturbs the stability between the cytoskeleton and the membrane protein complexes, affecting cell signaling. The formation of protein aggregates in combination with FLNC depletion causes myofiber disintegration and muscle weakness, which is aggravated by muscle activity (Chevessier et al., 2015; Kley et al., 2012).
Two missense variants (p.Ala193Thr and p.Met251Thr) in the ABD are associated with a form of isolated distal myopathy without protein aggregate formation (Duff et al., 2011). In contrast to all other reported missense variants in the ABD, these variants are predicted to change from a hydrophobic to an uncharged amino acid. They are predicted to alter intradomain interactions, thereby increasing the binding affinity of filamin C to actin.
A frameshift variant (p.Phe1720Leufs*63) has also been associated with an isolated distal myopathy without protein aggregates (Guergueltcheva et al., 2011). In contrast to p.Trp2710*, this variant occurs in Ig‐like domain 15 and activates NMD: analysis of RNA and protein in patients' muscle biopsies showed a 50% decrease in FLNC mRNA and protein, implicating haploinsufficiency (Guergueltcheva et al., 2011). When compared with the truncating variants in DCM, this is the only reported frameshift variant in Ig‐like domain 15, which is the domain before hinge 1. This hinge is only present in the long isoform of FLNC which incorporates exon 31 (Xie et al., 1998), and might be important in the isoform switch during cell stress (Kong et al., 2010). There is no comparable frameshift variant encompassing exon 31 in DCM. The different impact of the frameshift on the two isoforms could be a mechanistic explanation for the clinical variation, although this hypothesis needs further testing to accurately determine the importance of the isoform shift.
4. ANIMAL MODELS
Animal studies of FLNC are performed in (zebra) fish, mice, and Drosophila (Figure 6). Shortly after the description of FLNC variants in MFM, the first mouse model was developed by the deletion of the last eight exons of FLNC (Dalkilic, Schienda, Thompson, & Kunkel, 2006). Homozygous mice died shortly after birth due to respiratory failure. Heterozygous mice had less muscle mass and a decreased number of primary muscle fibers. Their muscles also showed excessive fiber size variation, centrally located nuclei and a disorganized muscle structure. This suggests a key role for FLNC in myogenesis as well as in myofiber structure maintenance.
Figure 6.

A timeline representation of animal models generated to study FLNC variants. eGFP indicates enhanced green fluorescent protein
A Medaka fish (Orzyias latipes) model was developed to investigate the cardiac and muscular phenotype of FLNC variants (Fujita et al., 2012). It contained a nonsense variant resulting in truncation at Ig‐15. Despite this variant, these fish had normal myogenesis. However, the myofibrils gradually degenerated and became disorganized, eventually leading to myocardial rupture. This suggests that FLNC is mainly involved in muscle structure maintenance instead of myogenesis, partly by affording protection against mechanical stress related to muscle contraction. Fiber dissolution and protein aggregate formation were not described in this model. These characteristics of MFM were observed in a zebrafish (Danio rerio) model in which the filamin C‐b homolog (flncb) contains a nonsense variant in exon 30 (Ruparelia, Zhao, Currie, & Bryson‐Richardson, 2012). A knockdown of the filamin C‐a homolog (flnca) yielded the same phenotype. However, loss of both homologs leads to a major failure of the muscle fibers. Also here, it was shown that FLNC was mainly involved in fiber protection and maintenance rather than fiber specification and myogenesis. Investigation of the cardiac phenotype in fish with a flncb knockdown showed atrium distention and backflow upon contraction (Deo et al., 2014). Optical mapping showed a decrease in ventricular conduction velocity, suggesting alterations in junctional remodeling, and cell–cell coupling. Later studies also showed sarcomere and Z‐disc disorganization (Begay et al., 2016). A study using Drosophila showed filamin C as an important cohesive element within the Z‐disc, where it acts as a bridge between thin filaments and the elastic scaffold protein titin (Gonzalez‐Morales, Holenka, & Schock, 2017). The Z‐disc requires filamin C to withstand the strong contractile forces acting on the sarcomere.
Other animal models were created to investigate targeted variants by knock‐in experiments in mice or overexpression in zebrafish (Chevessier et al., 2015; Kiselev et al., 2018; Ruparelia et al., 2016). Two models were created to investigate the hotspot variant, p.Trp2710* (Chevessier et al., 2015; Ruparelia et al., 2016). Heterozygous mice developed muscle weakness and myofibrillar instability (Chevessier et al., 2015). In addition to the classical protein aggregates, they also developed filamin C positive lesions between the Z‐discs appearing upon physical exercise. Overexpression of the variant in zebrafish led to the formation of protein aggregates (Ruparelia et al., 2016). In this model, mutant FLNC was localized around the Z‐disc and is able to rescue the disintegration phenotype. This led to the hypothesis that it was mainly the aggregates and the sequestration of FLNC away from the Z‐disc that cause myofibrillar disintegration. The study further showed that the CASA pathway is impaired, making the cell unable to clear the protein aggregates.
5. CLINICAL AND DIAGNOSTIC RELEVANCE
Cardiac involvement is a common clinical manifestation in hereditary muscular dystrophies (Hermans et al., 2010). Conversely, muscular problems in cardiomyopathy patients have also been described (Limongelli et al., 2013), showing the strong molecular link between hereditary muscular dystrophies and cardiomyopathies. In line with this, cardiac involvement is not uncommon in FLNC‐associated myopathy (Figure 5), but muscle involvement has not been described (yet) at the moment of diagnosis in FLNC‐associated cardiomyopathy (Ader et al., 2019). A single patient with DCM developed distal myopathy during follow‐up (Ortiz‐Genga et al., 2016).
5.1. Specific clinical characteristics of FLNC‐associated cardiomyopathies
Compared with the average clinical course of DCM, FLNC‐associated DCM is more malignant, characterized by ventricular arrhythmias, myocardial fibrosis, and a high risk of sudden cardiac death (Ortiz‐Genga et al., 2016). The average age of onset is 39.7 ± 14.5. Considering all available literature at the moment, filaminopathy has a distinct cardiac phenotype. In contrast to arrhythmogenic cardiomyopathies, FLNC‐associated DCM is left‐dominant in the absence of right ventricular involvement (Augusto et al., 2019; Ortiz‐Genga et al., 2016). Diagnostic clues can be inferolateral negative T waves on the electrocardiogram, mild to moderate left ventricular dysfunction and regional dyskinesia. It also has a characteristic ring‐like scar pattern in the left ventricle as detected by cardiovascular magnetic resonance imaging (Augusto et al., 2019). The combination of increased myocardial fibrosis, ventricular arrhythmias, and sudden cardiac death are also found in laminopathies, desminopathies, and desmosomal variants (Hasselberg et al., 2017; Lopez‐Ayala et al., 2014). In contrast to these forms of genetic DCM, cardiac conduction abnormalities such as an atrioventricular block are uncommon in FLNC‐associated DCM while they are common in laminopathies and desminopathies. In addition, desmosomal variants are strongly correlated to isolated or predominant right ventricular involvement. For patients with DCM, the finding of a truncating FLNC is likely causative, and relatives should be screened for this variant (Tayal & Cook, 2016). The finding of a truncating FLNC variant in otherwise healthy subjects outside of a familial context is much less clear at the moment, as there is not enough knowledge regarding penetrance, expression, and clinical correlation. Although the prevalence of truncating FLNC variants is very low in the general population (<0.01% in gnomAD).
The mean age of onset of HCM in FLNC carriers is 35.9 ± 14.8. In a previous cohort of patients with HCM, it was reported that 34% of the FLNC variant carriers had elevated creatine kinase (CK) levels (Valdes‐Mas et al., 2014), also in RCM, there were some patients with mildly elevated CK levels (Brodehl et al., 2016). However, this finding is not consistent across different patient cohorts (Ader et al., 2019).
5.2. Specific clinical characteristics of FLNC‐associated myopathies
The two classic muscular phenotypes are MFM and distal myopathy (with and without protein aggregates respectively; Furst et al., 2013). Other genes have been associated with these phenotypes, such as DES, LDB3, and BAG3 (Hermans et al., 2010). Besides the muscular phenotype, these genes are also associated with isolated cardiomyopathies. Characteristic features of FLNC‐associated MFM is the symmetrical involvement of proximal muscles in the lower extremities, respiratory weakness during the disease course, and a specific set of imaging characteristics for muscle involvement (Kley et al., 2012). About one‐third of the FLNC‐MFM showed cardiac involvement. Distal myopathies due to FLNC variants are characterized by weakness in the hand and calf muscles with an onset in early adulthood (Furst et al., 2013).
6. GENOTYPE/PHENOTYPE CORRELATIONS
Genotype–phenotype correlations are currently incomplete, but some patterns are starting to emerge (Figure 7).
Figure 7.
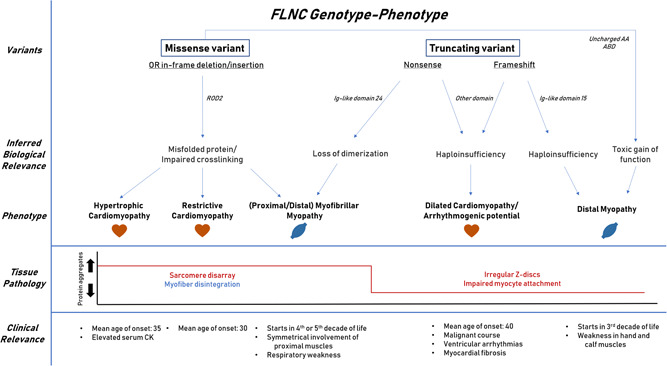
A summary how different variants in FLNC lead to a variety of disease mechanisms eventually giving a spectrum of clinical entities with the corresponding structural histological changes. These FLNC‐associated diseases contain specific clinical characteristics compared with other forms of the corresponding disease
There is no clear clustering of DCM variants in any specific region of the gene, partly because most truncating variants are predicted to result in NMD. Truncating FLNC variants are strongly associated with DCM and arrhythmogenic potential. Just three out of 55 truncating variants are associated with a muscular phenotype. Two are a frameshift in Ig‐like 15 spanning hinge 1 and one is a nonsense variant in Ig‐like 24 of the dimerization domain. The different pathomechanism underlying missense and truncating variants partly explains the structural changes at the histological level and the corresponding clinical phenotype. Missense variants are mainly found in ROD2, which is essential for filamin C dimerization and Z‐disc interaction. These variants interfere with the secondary protein structure, leading to sarcomere disarray and aggregate formation. These histopathological changes are associated with HCM and RCM phenotypes.
-
1.
Truncating variants are found throughout the whole gene, and are expected to invoke haploinsufficiency via NMD. This will lead to Z‐disc disarray and weakened cell–cell adhesion with subsequent impaired mechanotransduction. These structural alterations make the heart prone for developing DCM with arrhythmogenesis and promote fibrogenesis contributing to the arrhythmogenesis.
-
2.
Frameshift variants spanning hinge 1 (exon 31) potentially interfere with flexibility for isoform switching. Both described frameshift variants spanning exon 31 are associated with distal myopathy.
-
3.
Missense variants in the ABD that create an uncharged amino acid give a toxic gain‐of‐function with a stronger actin‐binding activity. These variants have been associated with distal myopathy.
-
4.
A nonsense variant in the dimerization domain interferes with the ability to form FLNC homodimers, although proteins are still translated. These proteins form aggregates in the skeletal muscle leading to MFM.
7. FUTURE PROSPECTS
As FLNC is now included in many genetic screening panels for muscular and cardiac diseases, the number of variants will increase in the coming years. A better understanding of the molecular alterations due to FLNC variants can shed light on potential treatment targets. It can help us in understanding and predicting genotype–phenotype correlations. Appropriate functioning of FLNC depends on multiple interactions with other proteins (correlations Mercer et al., 2018). These interactions could contribute to specific phenotypes. The ROD2 domain encompasses binding sites for the majority of FLNC interactors, and is necessary for mechanosensing and muscle maintenance functions. Chaperones such as HspB1 need to bind to FLNC to ensure these functions (Collier et al., 2019). The inability of HspB1 leads to bind to FLNC can lead to cardiac dysfunction. This is one example how variants in a specific domain could affect protein interactions and contribute to a specific phenotypes. A field that should be further explored. As an example, we formulated the following research questions:
-
1.
Which factors explain the clinical variety among truncating variants in the FLNC gene. For example: Why does p. Phe1720Leufs*63 lead to a distal myopathy while p.Asn1369Lysfs*36 leads to DCM? Does flexibility in isoform switching play a role in this?
-
2.
How does a nonsense variant in Ig‐like 24 (p.Trp2710*) lead to an MFM and a frameshift variant in the same region (p.Asp2703Thrfs*69) lead to arrhythmogenic DCM?
-
3.
What are the exact structural protein changes related to missense variants in the ABD and how do they explain the clinical difference between HCM and distal myopathy?
-
4.
Are truncating variants in certain parts of the gene better tolerated clinically and therefore less penetrant?
-
5.
Which molecular pathways are differentially activated due to FLNC variants and can these pathways serve as potential therapeutic targets?
-
6.
What is the clinical relevance and outcome of (truncating) FLNC variants in the general population?
8. CONCLUSION
Variants in FLNC can lead to myopathies and cardiomyopathies. The difference in phenotypes can be partly explained by the pathomechanism associated with the variant type and location within the gene. As a general rule, interference with the dimerization and folding of the protein leads to aggregate formation, as found in HCM or MFM. Truncating variants with subsequent haploinsufficiency lead to weakened structural adhesion mainly associated with DCM and cardiac arrhythmias.
Supporting information
Supporting information
ACKNOWLEDGMENTS
We would like to thank Margo Eijck‐Vievermans and the Biobank Clinical Genetics Maastricht for their contribution in collecting all FLNC variants.
Verdonschot JAJ, Vanhoutte EK, Claes GRF, et al. A mutation update for the FLNC gene in myopathies and cardiomyopathies. Human Mutation. 2020;41:1091–1111. 10.1002/humu.24004
Job A. J. Verdonschot and Els K. Vanhoutte contributed equally to this work.
REFERENCES
- Ader, F. , De Groote, P. , Reant, P. , Rooryck‐Thambo, C. , Dupin‐Deguine, D. , Rambaud, C. , … Richard, P. (2019). FLNC pathogenic variants in patients with cardiomyopathies: Prevalence and genotype‐phenotype correlations. Clinical Genetics, 96(4), 317–329. 10.1111/cge.13594 [DOI] [PubMed] [Google Scholar]
- Alejandra Restrepo‐Cordoba, M. , Campuzano, O. , Ripoll‐Vera, T. , Cobo‐Marcos, M. , Mademont‐Soler, I. , Gamez, J. M. , … Garcia‐Pavia, P. (2017). Usefulness of genetic testing in hypertrophic cardiomyopathy: An analysis using real‐world data. Journal of Cardiovascular Translational Research, 10(1), 35–46. 10.1007/s12265-017-9730-8 [DOI] [PubMed] [Google Scholar]
- Anastasi, G. , Cutroneo, G. , Trimarchi, F. , Santoro, G. , Bruschetta, D. , Bramanti, P. , … Favaloro, A. (2004). Evaluation of sarcoglycans, vinculin‐talin‐integrin system and filamin2 in alpha‐ and gamma‐sarcoglycanopathy: An immunohistochemical study. International Journal of Molecular Medicine, 14(6), 989–999. [PubMed] [Google Scholar]
- Augusto, J. B. , Eiros, R. , Nakou, E. , Moura‐Ferreira, S. , Treibel, T. A. , Captur, G. , … Lopes, L. R. (2019). Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: A comprehensive genotype‐imaging phenotype study. European Heart Journal Cardiovascular Imaging, 21(3), 326–336. 10.1093/ehjci/jez188 [DOI] [PubMed] [Google Scholar]
- Avila‐Smirnow, D. , Béhin, A. , Gueneau, L. , Claeys, K. , Beuvin, M. , Goudeau, B. , … Mathis, S. J. N. D. (2010). P2. 18 A novel missense FLNC mutation causes arrhythmia and late onset myofibrillar myopathy with particular histopathology features. Neuromuscular Disorders, 20(9), 623–624. [Google Scholar]
- Bains, S. , Tester, D. J. , Asirvatham, S. J. , Noseworthy, P. A. , Ackerman, M. J. , & Giudicessi, J. R. (2019). A novel truncating variant in FLNC‐encoded Filamin C may serve as a proarrhythmic genetic substrate for arrhythmogenic bileaflet mitral valve prolapse syndrome. Mayo Clinic Proceedings, 94(5), 906–913. 10.1016/j.mayocp.2018.11.028 [DOI] [PubMed] [Google Scholar]
- Begay, R. L. , Graw, S. L. , Sinagra, G. , Asimaki, A. , Rowland, T. J. , Slavov, D. B. , … Taylor, M. R. G. (2018). Filamin C truncation mutations are associated with arrhythmogenic dilated cardiomyopathy and changes in the cell‐cell adhesion structures. JACC: Clinical Electrophysiology, 4(4), 504–514. 10.1016/j.jacep.2017.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begay, R. L. , Tharp, C. A. , Martin, A. , Graw, S. L. , Sinagra, G. , Miani, D. , … Taylor, M. R. (2016). FLNC gene splice mutations cause dilated cardiomyopathy. JACC: Basic to Translational Science, 1(5), 344–359. 10.1016/j.jacbts.2016.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart, F. J. , Claeys, K. G. , Kley, R. A. , Kusters, B. , Schrading, S. , Kamsteeg, E. J. , & Voermans, N. C. (2017). Widening the spectrum of filamin‐C myopathy: Predominantly proximal myopathy due to the p.A193T mutation in the actin‐binding domain of FLNC. Neuromuscular Disorders: NMD, 27(1), 73–77. 10.1016/j.nmd.2016.09.017 [DOI] [PubMed] [Google Scholar]
- Brodehl, A. , Ferrier, R. A. , Hamilton, S. J. , Greenway, S. C. , Brundler, M. A. , Yu, W. , … Gerull, B. (2016). Mutations in FLNC are associated with familial restrictive cardiomyopathy. Human Mutation, 37(3), 269–279. 10.1002/humu.22942 [DOI] [PubMed] [Google Scholar]
- Cerino, M. , Gorokhova, S. , Laforet, P. , Ben Yaou, R. , Salort‐Campana, E. , Pouget, J. , … Krahn, M. (2017). Genetic characterization of a French cohort of GNE‐mutation negative inclusion body myopathy patients with exome sequencing. Muscle and Nerve, 56(5), 993–997. 10.1002/mus.25638 [DOI] [PubMed] [Google Scholar]
- Chakarova, C. , Wehnert, M. S. , Uhl, K. , Sakthivel, S. , Vosberg, H. P. , van der Ven, P. F. , & Furst, D. O. (2000). Genomic structure and fine mapping of the two human filamin gene paralogues FLNB and FLNC and comparative analysis of the filamin gene family. Human Genetics, 107(6), 597–611. 10.1007/s004390000414 [DOI] [PubMed] [Google Scholar]
- Chanavat, V. , Janin, A. , & Millat, G. (2016). A fast and cost‐effective molecular diagnostic tool for genetic diseases involved in sudden cardiac death. Clinica Chimica Acta, 453, 80–85. 10.1016/j.cca.2015.12.011 [DOI] [PubMed] [Google Scholar]
- Chen, J. , Wu, J. , Han, C. , Li, Y. , Guo, Y. , & Tong, X. (2019). A mutation in the filamin c gene causes myofibrillar myopathy with lower motor neuron syndrome: A case report. BMC Neurology, 19(1), 198 10.1186/s12883-019-1410-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevessier, F. , Schuld, J. , Orfanos, Z. , Plank, A. C. , Wolf, L. , Maerkens, A. , … Schroder, R. (2015). Myofibrillar instability exacerbated by acute exercise in filaminopathy. Human Molecular Genetics, 24(25), 7207–7220. 10.1093/hmg/ddv421 [DOI] [PubMed] [Google Scholar]
- Cirino, A. L. , Lakdawala, N. K. , McDonough, B. , Conner, L. , Adler, D. , Weinfeld, M. , … Ho, C. Y. (2017). A comparison of whole genome sequencing to multigene panel testing in hypertrophic cardiomyopathy patients. Circulation: Cardiovascular Genetics, 10(5), 1–9. 10.1161/circgenetics.117.001768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier, M. P. , Alderson, T. R. , de Villiers, C. P. , Nicholls, D. , Gastall, H. Y. , Allison, T. M. , … Benesch, J. L. P. (2019). HspB1 phosphorylation regulates its intramolecular dynamics and mechanosensitive molecular chaperone interaction with filamin C. Science Advances, 5(5), eaav8421 10.1126/sciadv.aav8421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenca, S. , Ruiz‐Cano, M. J. , Gimeno‐Blanes, J. R. , Jurado, A. , Salas, C. , Gomez‐Diaz, I. , … Garcia‐Pavia, P. (2016). Genetic basis of familial dilated cardiomyopathy patients undergoing heart transplantation. Journal of Heart and Lung Transplantation, 35(5), 625–635. 10.1016/j.healun.2015.12.014 [DOI] [PubMed] [Google Scholar]
- Cui, H. , Wang, J. , Zhang, C. , Wu, G. , Zhu, C. , Tang, B. , … Wang, S. (2018). Mutation profile of FLNC gene and its prognostic relevance in patients with hypertrophic cardiomyopathy. Molecular Genetics & Genomic Medicine, 6(6), 1104–1113. 10.1002/mgg3.488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, Y. , Wei, X. , Zhao, Y. , Ren, H. , Lan, Z. , Yang, Y. , … Cui, L. (2015). A comprehensive genetic diagnosis of Chinese muscular dystrophy and congenital myopathy patients by targeted next‐generation sequencing. Neuromuscular Disorders: NMD, 25(8), 617–624. 10.1016/j.nmd.2015.03.002 [DOI] [PubMed] [Google Scholar]
- Dalkilic, I. , Schienda, J. , Thompson, T. G. , & Kunkel, L. M. (2006). Loss of FilaminC (FLNc) results in severe defects in myogenesis and myotube structure. Molecular and Cellular Biology, 26(17), 6522–6534. 10.1128/mcb.00243-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo, R. C. , Musso, G. , Tasan, M. , Tang, P. , Poon, A. , Yuan, C. , … Roth, F. P. (2014). Prioritizing causal disease genes using unbiased genomic features. Genome Biology, 15(12), 534 10.1186/s13059-014-0534-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff, R. M. , Tay, V. , Hackman, P. , Ravenscroft, G. , McLean, C. , Kennedy, P. , … Laing, N. G. (2011). Mutations in the N‐terminal actin‐binding domain of filamin C cause a distal myopathy. American Journal of Human Genetics, 88(6), 729–740. 10.1016/j.ajhg.2011.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott, P. , Andersson, B. , Arbustini, E. , Bilinska, Z. , Cecchi, F. , Charron, P. , … Keren, A. (2008). Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. European Heart Journal, 29(2), 270–276. 10.1093/eurheartj/ehm342 [DOI] [PubMed] [Google Scholar]
- Esslinger, U. , Garnier, S. , Korniat, A. , Proust, C. , Kararigas, G. , Muller‐Nurasyid, M. , … Villard, E. (2017). Exome‐wide association study reveals novel susceptibility genes to sporadic dilated cardiomyopathy. PLoS One, 12(3):e0172995 10.1371/journal.pone.0172995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichna, J. P. , Macias, A. , Piechota, M. , Korostynski, M. , Potulska‐Chromik, A. , Redowicz, M. J. , & Zekanowski, C. (2018). Whole‐exome sequencing identifies novel pathogenic mutations and putative phenotype‐influencing variants in Polish limb‐girdle muscular dystrophy patients. Human Genomics, 12(1), 34 10.1186/s40246-018-0167-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier, A. , & Sonnenberg, A. (2001). Structural and functional aspects of filamins. Biochimica et Biophysica Acta/General Subjects, 1538(2‐3), 99–117. 10.1016/s0167-4889(01)00072-6 [DOI] [PubMed] [Google Scholar]
- Fujita, M. , Mitsuhashi, H. , Isogai, S. , Nakata, T. , Kawakami, A. , Nonaka, I. , … Kudo, A. (2012). Filamin C plays an essential role in the maintenance of the structural integrity of cardiac and skeletal muscles, revealed by the medaka mutant zacro. Developmental Biology, 361(1), 79–89. 10.1016/j.ydbio.2011.10.008 [DOI] [PubMed] [Google Scholar]
- Furst, D. O. , Goldfarb, L. G. , Kley, R. A. , Vorgerd, M. , Olive, M. , & van der Ven, P. F. (2013). Filamin C‐related myopathies: Pathology and mechanisms. Acta Neuropathologica, 125(1), 33–46. 10.1007/s00401-012-1054-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemelli, C. , Prada, V. , Fiorillo, C. , Fabbri, S. , Maggi, L. , Geroldi, A. , … Grandis, M. (2019). A novel mutation in the N‐terminal acting‐binding domain of Filamin C protein causing a distal myofibrillar myopathy. Journal of the Neurological Sciences, 398, 75–78. 10.1016/j.jns.2019.01.019 [DOI] [PubMed] [Google Scholar]
- Ghaoui, R. , Cooper, S. T. , Lek, M. , Jones, K. , Corbett, A. , Reddel, S. W. , … Clarke, N. F. (2015). Use of whole‐exome sequencing for diagnosis of limb‐girdle muscular dystrophy: Outcomes and lessons learned. JAMA Neurology, 72(12), 1424–1432. 10.1001/jamaneurol.2015.2274 [DOI] [PubMed] [Google Scholar]
- Gomez, J. , Lorca, R. , Reguero, J. R. , Moris, C. , Martin, M. , Tranche, S. , … Coto, E. (2017). Screening of the Filamin C gene in a large cohort of hypertrophic cardiomyopathy patients. Circulation: Cardiovascular Genetics, 10(2), 1–11. 10.1161/circgenetics.116.001584 [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Morales, N. , Holenka, T. K. , & Schock, F. (2017). Filamin actin‐binding and titin‐binding fulfill distinct functions in Z‐disc cohesion. PLoS Genetics, 13(7):e1006880 10.1371/journal.pgen.1006880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guergueltcheva, V. , Peeters, K. , Baets, J. , Ceuterick‐de Groote, C. , Martin, J. J. , Suls, A. , … Jordanova, A. (2011). Distal myopathy with upper limb predominance caused by filamin C haploinsufficiency. Neurology, 77(24), 2105–2114. 10.1212/WNL.0b013e31823dc51e [DOI] [PubMed] [Google Scholar]
- Guyon, J. R. , Kudryashova, E. , Potts, A. , Dalkilic, I. , Brosius, M. A. , Thompson, T. G. , … Spencer, M. J. (2003). Calpain 3 cleaves filamin C and regulates its ability to interact with gamma‐ and delta‐sarcoglycans. Muscle and Nerve, 28(4), 472–483. 10.1002/mus.10465 [DOI] [PubMed] [Google Scholar]
- Hall, C. L. , Akhtar, M. M. , Sabater‐Molina, M. , Futema, M. , Asimaki, A. , Protonotarios, A. , … McKenna, W. J. (2019). Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. International Journal of Cardiology, 1–8. 10.1016/j.ijcard.2019.09.048 [DOI] [PubMed] [Google Scholar]
- Hasselberg, N. E. , Haland, T. F. , Saberniak, J. , Brekke, P. H. , Berge, K. E. , Leren, T. P. , … Haugaa, K. H. (2017). Lamin A/C cardiomyopathy: Young onset, high penetrance, and frequent need for heart transplantation. European Heart Journal, 39, 853–860. 10.1093/eurheartj/ehx596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans, M. C. , Pinto, Y. M. , Merkies, I. S. , de Die‐Smulders, C. E. , Crijns, H. J. , & Faber, C. G. (2010). Hereditary muscular dystrophies and the heart. Neuromuscular Disorders: NMD, 20(8), 479–492. 10.1016/j.nmd.2010.04.008 [DOI] [PubMed] [Google Scholar]
- Hershberger, R. E. , Givertz, M. M. , Ho, C. Y. , Judge, D. P. , Kantor, P. F. , McBride, K. L. , … Ware, S. M. (2018). Genetic evaluation of cardiomyopathy‐A Heart Failure Society of America Practice Guideline. Journal of Cardiac Failure, 24(5), 281–302. 10.1016/j.cardfail.2018.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmel, M. , Van Der Ven, P. F. , Stocklein, W. , & Furst, D. O. (2003). The limits of promiscuity: Isoform‐specific dimerization of filamins. Biochemistry, 42(2), 430–439. 10.1021/bi026501+ [DOI] [PubMed] [Google Scholar]
- Jaafar, N. , Gomez, J. , Kammoun, I. , Zairi, I. , Amara, W. B. , Kachboura, S. , … Coto, E. (2016). Spectrum of mutations in hypertrophic cardiomyopathy genes among Tunisian patients. Genetic Testing and Molecular Biomarkers, 20(11), 674–679. 10.1089/gtmb.2016.0187 [DOI] [PubMed] [Google Scholar]
- Janin, A. , N'Guyen, K. , Habib, G. , Dauphin, C. , Chanavat, V. , Bouvagnet, P. , … Millat, G. (2017). Truncating mutations on myofibrillar myopathies causing genes as prevalent molecular explanations on patients with dilated cardiomyopathy. Clinical Genetics, 92(6), 616–623. 10.1111/cge.13043 [DOI] [PubMed] [Google Scholar]
- Janssens, J. , Philtjens, S. , Kleinberger, G. , Van Mossevelde, S. , van der Zee, J. , Cacace, R. , … Van Broeckhoven, C. (2015). Investigating the role of filamin C in Belgian patients with frontotemporal dementia linked to GRN deficiency in FTLD‐TDP brains. Acta Neuropathologica Communications, 68(3), 1–18. 10.1186/s40478-015-0246-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselev, A. , Vaz, R. , Knyazeva, A. , Khudiakov, A. , Tarnovskaya, S. , Liu, J. , … Kostareva, A. (2018). De novo mutations in FLNC leading to early‐onset restrictive cardiomyopathy and congenital myopathy. Human Mutation, 39(9), 1161–1172. 10.1002/humu.23559 [DOI] [PubMed] [Google Scholar]
- Kley, R. A. , Maerkens, A. , Leber, Y. , Theis, V. , Schreiner, A. , van der Ven, P. F. , … Marcus, K. (2013). A combined laser microdissection and mass spectrometry approach reveals new disease relevant proteins accumulating in aggregates of filaminopathy patients. Molecular & Cellular Proteomics, 12(1), 215–227. 10.1074/mcp.M112.023176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kley, R. A. , Serdaroglu‐Oflazer, P. , Leber, Y. , Odgerel, Z. , van der Ven, P. F. , Olive, M. , … Furst, D. O. (2012). Pathophysiology of protein aggregation and extended phenotyping in filaminopathy. Brain, 135(Pt 9), 2642–2660. 10.1093/brain/aws200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kley, R. A. , van der Ven, P. F. , Olive, M. , Hohfeld, J. , Goldfarb, L. G. , Furst, D. O. , & Vorgerd, M. (2013). Impairment of protein degradation in myofibrillar myopathy caused by FLNC/filamin C mutations. Autophagy, 9(3), 422–423. 10.4161/auto.22921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, S. W. , Hu, Y. W. , Ho, J. W. , Ikeda, S. , Polster, S. , John, R. , … Pu, W. T. (2010). Heart failure‐associated changes in RNA splicing of sarcomere genes. Circulation: Cardiovascular Genetics, 3(2), 138–146. 10.1161/CIRCGENETICS.109.904698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmicki, J. A. , Samocha, K. E. , Howrigan, D. P. , Sanders, S. J. , Slowikowski, K. , Lek, M. , … Daly, M. J. (2017). Refining the role of de novo protein‐truncating variants in neurodevelopmental disorders by using population reference samples. Nature Genetics, 49(4), 504–510. 10.1038/ng.3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leber, Y. , Ruparelia, A. A. , Kirfel, G. , van der Ven, P. F. , Hoffmann, B. , Merkel, R. , … Furst, D. O. (2016). Filamin C is a highly dynamic protein associated with fast repair of myofibrillar microdamage. Human Molecular Genetics, 25(13), 2776–2788. 10.1093/hmg/ddw135 [DOI] [PubMed] [Google Scholar]
- Limongelli, G. , D'Alessandro, R. , Maddaloni, V. , Rea, A. , Sarkozy, A. , & McKenna, W. J. (2013). Skeletal muscle involvement in cardiomyopathies. Journal of Cardiovascular Medicine (Hagerstown), 14(12), 837–861. 10.2459/JCM.0b013e3283641c69 [DOI] [PubMed] [Google Scholar]
- Lopez‐Ayala, J. M. , Gomez‐Milanes, I. , Sanchez Munoz, J. J. , Ruiz‐Espejo, F. , Ortiz, M. , Gonzalez‐Carrillo, J. , … Gimeno, J. R. (2014). Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: Characterizing a phenotype. Europace: European Pacing, Arrhythmias, and Cardiac Electrophysiology: Journal of the Working Groups on Cardiac Pacing, Arrhythmias, and Cardiac Cellular Electrophysiology of the European Society of Cardiology, 16(12), 1838–1846. 10.1093/europace/euu128 [DOI] [PubMed] [Google Scholar]
- Lowe, T. , Kley, R. A. , van der Ven, P. F. , Himmel, M. , Huebner, A. , Vorgerd, M. , & Furst, D. O. (2007). The pathomechanism of filaminopathy: Altered biochemical properties explain the cellular phenotype of a protein aggregation myopathy. Human Molecular Genetics, 16(11), 1351–1358. 10.1093/hmg/ddm085 [DOI] [PubMed] [Google Scholar]
- Luan, X. , Hong, D. , Zhang, W. , Wang, Z. , & Yuan, Y. (2010). A novel heterozygous deletion‐insertion mutation (2695–2712 del/GTTTGT ins) in exon 18 of the filamin C gene causes filaminopathy in a large Chinese family. Neuromuscular Disorders: NMD, 20(6), 390–396. 10.1016/j.nmd.2010.03.009 [DOI] [PubMed] [Google Scholar]
- Mangum, K. D. , & Ferns, S. J. (2019). A novel familial truncating mutation in the filamin C gene associated with cardiac arrhythmias. European Journal of Medical Genetics, 62(4), 282–285. 10.1016/j.ejmg.2018.08.006 [DOI] [PubMed] [Google Scholar]
- Maron, B. J. , Towbin, J. A. , Thiene, G. , Antzelevitch, C. , Corrado, D. , Arnett, D. , … Young, J. B. (2006). Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation, 113(14), 1807–1816. 10.1161/circulationaha.106.174287 [DOI] [PubMed] [Google Scholar]
- Mercer, E. J. , Lin, Y. F. , Cohen‐Gould, L. , & Evans, T. (2018). Hspb7 is a cardioprotective chaperone facilitating sarcomeric proteostasis. Developmental Biology, 435(1), 41–55. 10.1016/j.ydbio.2018.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miszalski‐Jamka, K. , Jefferies, J. L. , Mazur, W. , Glowacki, J. , Hu, J. , Lazar, M. , … Bainbridge, M. N. (2017). Novel genetic triggers and genotype‐phenotype correlations in patients with left ventricular noncompaction. Circulation: Cardiovascular Genetics, 10(4), e001763 10.1161/circgenetics.117.001763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molt, S. , Bührdel, J. B. , Yakovlev, S. , Schein, P. , Orfanos, Z. , Kirfel, G. , … Fürst, D. O. (2014). Aciculin interacts with filamin C and Xin and is essential for myofibril assembly, remodeling and maintenance. Journal of Cell Science, 127(Pt 16), 3578–3592. 10.1242/jcs.152157 [DOI] [PubMed] [Google Scholar]
- Nozari, A. , Aghaei‐Moghadam, E. , Zeinaloo, A. , Mollazadeh, R. , Majnoon, M. T. , Alavi, A. , … Behjati, F. (2018). A novel splicing variant in FLNC gene responsible for a highly penetrant familial dilated cardiomyopathy in an extended Iranian family. Gene, 659, 160–167. 10.1016/j.gene.2018.03.044 [DOI] [PubMed] [Google Scholar]
- Ortiz‐Genga, M. F. , Cuenca, S. , Dal Ferro, M. , Zorio, E. , Salgado‐Aranda, R. , Climent, V. , … Monserrat, L. (2016). Truncating FLNC mutations are associated with high‐risk dilated and arrhythmogenic cardiomyopathies. Journal of the American College of Cardiology, 68(22), 2440–2451. 10.1016/j.jacc.2016.09.927 [DOI] [PubMed] [Google Scholar]
- Reddy, H. M. , Cho, K. A. , Lek, M. , Estrella, E. , Valkanas, E. , Jones, M. D. , … Kang, P. B. (2017). The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. Journal of Human Genetics, 62(2), 243–252. 10.1038/jhg.2016.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinstein, E. , Gutierrez‐Fernandez, A. , Tzur, S. , Bormans, C. , Marcu, S. , Tayeb‐Fligelman, E. , … Lopez‐Otin, C. (2016). Congenital dilated cardiomyopathy caused by biallelic mutations in Filamin C. European Journal of Human Genetics, 24(12), 1792–1796. 10.1038/ejhg.2016.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Committee, A. L. Q. A. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roldan‐Sevilla, A. , Palomino‐Doza, J. , de Juan, J. , Sanchez, V. , Dominguez‐Gonzalez, C. , Salguero‐Bodes, R. , & Arribas‐Ynsaurriaga, F. (2019). Missense mutations in the FLNC gene causing familial restrictive cardiomyopathy. Circ Genom Precis Med, 12(3):e002388 10.1161/circgen.118.002388 [DOI] [PubMed] [Google Scholar]
- Rossi, D. , Palmio, J. , Evila, A. , Galli, L. , Barone, V. , Caldwell, T. A. , … Sorrentino, V. (2017). A novel FLNC frameshift and an OBSCN variant in a family with distal muscular dystrophy. PLoS One, 12(10):e0186642 10.1371/journal.pone.0186642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruparelia, A. A. , Oorschot, V. , Ramm, G. , & Bryson‐Richardson, R. J. (2016). FLNC myofibrillar myopathy results from impaired autophagy and protein insufficiency. Human Molecular Genetics, 25(11), 2131–2142. 10.1093/hmg/ddw080 [DOI] [PubMed] [Google Scholar]
- Ruparelia, A. A. , Zhao, M. , Currie, P. D. , & Bryson‐Richardson, R. J. (2012). Characterization and investigation of zebrafish models of filamin‐related myofibrillar myopathy. Human Molecular Genetics, 21(18), 4073–4083. 10.1093/hmg/dds231 [DOI] [PubMed] [Google Scholar]
- Sanoja, A. J. , Li, H. , Fricker, F. J. , Kingsmore, S. F. , & Wallace, M. R. (2018). Exome sequencing identifies FLNC and ADD3 variants in a family with cardiomyopathy. Journal of Translational Genetics and Genomics, 2(6), 1–11. [Google Scholar]
- Schubert, J. , Tariq, M. , Geddes, G. , Kindel, S. , Miller, E. M. , & Ware, S. M. (2018). Novel pathogenic variants in filamin C identified in pediatric restrictive cardiomyopathy. Human Mutation, 39(12), 2083–2096. 10.1002/humu.23661 [DOI] [PubMed] [Google Scholar]
- Shatunov, A. , Olive, M. , Odgerel, Z. , Stadelmann‐Nessler, C. , Irlbacher, K. , van Landeghem, F. , … Goldfarb, L. G. (2009). In‐frame deletion in the seventh immunoglobulin‐like repeat of filamin C in a family with myofibrillar myopathy. European Journal of Human Genetics, 17(5), 656–663. 10.1038/ejhg.2008.226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada, F. , Vander Woude, D. L. , Tong, H. Q. , Thompson, T. G. , Watkins, S. C. , Kunkel, L. M. , & Beggs, A. H. (2001). Myozenin: An alpha‐actinin‐ and gamma‐filamin‐binding protein of skeletal muscle Z lines. Proceedings of the National Academy of Sciences of the United States of America, 98(4), 1595–1600. 10.1073/pnas.041609698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasca, G. , Odgerel, Z. , Monforte, M. , Aurino, S. , Clarke, N. F. , Waddell, L. B. , … Goldfarb, L. G. (2012). Novel FLNC mutation in a patient with myofibrillar myopathy in combination with late‐onset cerebellar ataxia. Muscle and Nerve, 46(2), 275–282. 10.1002/mus.23349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayal, U. , & Cook, S. A. (2016). Truncating variants in filamin C: The challenges of genotype‐phenotype correlations in cardiomyopathies. Journal of the American College of Cardiology, 68(22), 2452–2453. 10.1016/j.jacc.2016.05.105 [DOI] [PubMed] [Google Scholar]
- Thompson, T. G. , Chan, Y. M. , Hack, A. A. , Brosius, M. , Rajala, M. , Lidov, H. G. , … Kunkel, L. M. (2000). Filamin 2 (FLN2): A muscle‐specific sarcoglycan interacting protein. Journal of Cell Biology, 148(1), 115–126. 10.1083/jcb.148.1.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobita, T. , Nomura, S. , Morita, H. , Ko, T. , Fujita, T. , Toko, H. , … Komuro, I. (2017). Identification of MYLK3 mutations in familial dilated cardiomyopathy. Scientific Reports, 7(1):17495 10.1038/s41598-017-17769-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker, N. R. , McLellan, M. A. , Hu, D. , Ye, J. , Parsons, V. A. , Mills, R. W. , … Ellinor, P. T. (2017). Novel mutation in FLNC (Filamin C) causes familial restrictive cardiomyopathy. Circulation: Cardiovascular Genetics, 10(6), e001780 10.1161/circgenetics.117.001780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdes‐Mas, R. , Gutierrez‐Fernandez, A. , Gomez, J. , Coto, E. , Astudillo, A. , Puente, D. A. , … Lopez‐Otin, C. (2014). Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nature Communications, 5, 5326 10.1038/ncomms6326 [DOI] [PubMed] [Google Scholar]
- Vill, K. , Blaschek, A. , Glaser, D. , Kuhn, M. , Haack, T. , Alhaddad, B. , … Muller‐Felber, W. (2017). Early‐onset myopathies: Clinical findings, prevalence of subgroups and diagnostic approach in a single neuromuscular referral center in Germany. Journal of Neuromuscular Diseases, 4(4), 315–325. 10.3233/jnd-170231 [DOI] [PubMed] [Google Scholar]
- Vorgerd, M. , van der Ven, P. F. , Bruchertseifer, V. , Lowe, T. , Kley, R. A. , Schroder, R. , … Huebner, A. (2005). A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. American Journal of Human Genetics, 77(2), 297–304. 10.1086/431959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, R. , Mazzarotto, F. , Whiffin, N. , Buchan, R. , Midwinter, W. , Wilk, A. , … Ware, J. S. (2019). Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: The case of hypertrophic cardiomyopathy. Genome Medicine, 11(1), 5 10.1186/s13073-019-0616-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihl, C. C. , Baloh, R. H. , Lee, Y. , Chou, T. F. , Pittman, S. K. , Lopate, G. , … Harms, M. B. (2015). Targeted sequencing and identification of genetic variants in sporadic inclusion body myositis. Neuromuscular Disorders: NMD, 25(4), 289–296. 10.1016/j.nmd.2014.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, Z. , Xu, W. , Davie, E. W. , & Chung, D. W. (1998). Molecular cloning of human ABPL, an actin‐binding protein homologue. Biochemical and Biophysical Research Communications, 251(3), 914–919. 10.1006/bbrc.1998.9506 [DOI] [PubMed] [Google Scholar]
- Yu, M. , Zheng, Y. , Jin, S. , Gang, Q. , Wang, Q. , Yu, P. , … Wang, Z. (2017). Mutational spectrum of Chinese LGMD patients by targeted next‐generation sequencing. PLoS One, 12(4):e0175343 10.1371/journal.pone.0175343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenagui, R. , Lacourt, D. , Pegeot, H. , Yauy, K. , Juntas Morales, R. , Theze, C. , … Cossee, M. (2018). A reliable targeted next‐generation sequencing strategy for diagnosis of myopathies and muscular dystrophies, especially for the giant Titin and Nebulin genes. Journal of Molecular Diagnostics, 20(4), 533–549. 10.1016/j.jmoldx.2018.04.001 [DOI] [PubMed] [Google Scholar]
- Zhang, M. , Liu, J. , Cheng, A. , Deyoung, S. M. , & Saltiel, A. R. (2007). Identification of CAP as a costameric protein that interacts with filamin C. Molecular Biology of the Cell, 18(12), 4731–4740. 10.1091/mbc.e07-06-0628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. T. , Pu, C. Q. , Ban, R. , Liu, H. X. , Shi, Q. , & Lu, X. H. (2018). Clinical, pathological, and genetic features of two Chinese cases with filamin C myopathy. Chinese Medical Journal (England), 131(24), 2986–2988. 10.4103/0366-6999.247208 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information