

Abstract
The Lewis acidities of a series of [n]magnesocenophanes (1 a–d) have been investigated computationally and found to be a function of the tilt of the cyclopentadienyl moieties. Their catalytic abilities in amine borane dehydrogenation/dehydrocoupling reactions have been probed, and C[1]magnesocenophane (1 a) has been shown to effectively catalyze the dehydrogenation/dehydrocoupling of dimethylamine borane (2 a) and diisopropylamine borane (2 b) under ambient conditions. Furthermore, the mechanism of the reaction with 2 a has been investigated experimentally and computationally, and the results imply a ligand‐assisted mechanism involving stepwise proton and hydride transfer, with dimethylaminoborane as the key intermediate.
Keywords: amine boranes, catalysis, dehydrocoupling, magnesium, metallocenophanes
Magnesocenophanes in catalysis: The dehydrocoupling of dimethylamine borane to cyclic tetramethyldiborazane under ambient conditions, catalyzed by a [1]magnesocenophane, is described. This is the first example of a magnesium‐based catalyst for this reaction that operates at room temperature. Based on experimental investigations and DFT calculations, a ligand‐assisted mechanism involving stepwise proton and hydride transfer is proposed.
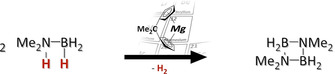
Introduction
Amine boranes have attracted much attention over the last decades, primarily for their possible applications as solid hydrogen storage materials1 or as precursors in the preparation of B−N materials.2 Although hydrogen is eliminated from many amine boranes at high temperatures, these reactions often afford ill‐defined product mixtures or suffer from mediocre yields with respect to the released hydrogen equivalents.1c Since catalytic routes can overcome these shortcomings, various homogeneous and heterogeneous catalysts for dehydrogenation/dehydrocoupling of amine boranes have been reported, most of which are based on d‐block elements, many on precious metals, such as rhodium or iridium.3 Although these operate at room temperature and with low catalyst loadings, main group element and in particular s‐block metal‐based catalysts have been investigated in view of lower element costs and ready availability.3e, 3f, 3g With respect to magnesium, for example, Hill and co‐workers reported the use of dialkyl‐ and 1,3‐diketimine magnesium compounds in the dehydrocoupling of dimethylamine borane (Figure 1; I–III).4 In this pioneering work,4a a relatively high catalyst loading of 10 mol % and an elevated temperature of 333 K were necessary to facilitate the reaction. Recently, Kays and co‐workers succeeded in devising a catalytically much more active magnesium complex for the dehydrocoupling of dimethylamine borane (Figure 1; IV), allowing for shorter reaction times at lower catalyst loadings, although an elevated temperature of 333 K was still required (Figure 1).5 In addition, room temperature dehydrocoupling reactions of cyclic monohydrido‐boranes with amines and of arylamine boranes, catalyzed by magnesium compounds, have been reported.6 Although these systems are less powerful than many transition‐metal‐based catalysts, magnesium‐based catalysts have notable advantages, namely the high abundance of magnesium in the Earth's crust, its non‐toxic nature, and its ready availability at low cost.7
Figure 1.
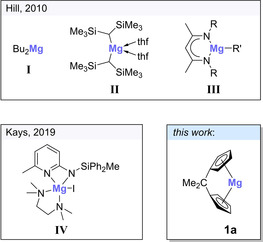
Examples of magnesium (pre)catalysts for dimethylamine borane dehydrogenation/dehydrocoupling.
Inspired by these aspects and in continuation of our group's interest in metallocenophanes of main group elements,8 we were intrigued to investigate the reactivity of [n]magnesocenophanes towards amine boranes. Various silicon‐ and carbon‐bridged [1]‐ and [2]magnesocenophanes (1 a–d) have been reported in the literature over the last decade (Figure 2).8b, 9
Figure 2.
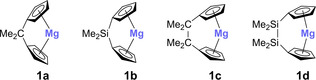
Silicon‐ and carbon‐bridged [1]‐ and [2]magnesocenophanes 1 a–d.
Results and Discussion
Structural and electronic properties of [n]magnesocenophanes
To better understand the different geometric features and electronic properties of magnesocenophanes 1 a–d, a computational study was carried out prior to any experimental investigations. In this context, it is important to note that the reported crystal structures of magnesocenophanes 1 a–d 8b, 9 are of solvated complexes, in which the dihedral angle α of the cyclopentadienide planes is strongly influenced by the coordination of the donor solvent molecule(s) to the magnesium atom.10 Therefore, dihedral angles α of the Cp planes in 1 a–d differ significantly between the geometries obtained from DFT calculations11 (B3LYP‐D3/def2‐TZVP) under quasi‐gas‐phase conditions, without donor solvent coordination to the magnesium atom, and the experimental solid‐state structures of the solvated complexes. This is also evident from the crystal structure of 1 a⋅dme (dme=dimethoxyethane) (Figure 3), which features a dihedral angle of α=80°, whereas a dihedral angle of α=56° is calculated when no solvent molecule is coordinated to the magnesium atom, illustrating the high flexibility of the Mg−Cp bonds due to their high ionic character.
Figure 3.
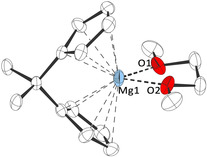
Molecular structure of 1 a⋅dme in the crystal (thermal ellipsoids at 30 % probability level; hydrogen atoms omitted for clarity; selected bond lengths and angle: Mg1−O1/O2: 205.3, Mg1−Cpcentroid: 230.4 pm; α=80.1°).
Moreover, the solvent coordination in 1 a⋅dme was found to be reversible in vacuo at room temperature,12a unlike in the case of 1 a⋅(thf)2. The calculated binding energies of the solvent molecules to the magnesium atom are 139.6 kJ mol−1 for dme in 1 a⋅dme and 197.0 kJ mol−1 for thf in 1 a⋅(thf)2 (100.5 kJ mol−1 for binding of the first thf molecule and 96.5 kJ mol−1 for binding of the second thf molecule).
Regarding electronic properties, DFT calculations predict an increase in fluoride ion affinity (FIA)13 and global electrophilicity index ω (GEI)14 as a function of tilt of the cyclopentadienyl moieties, quantified by the dihedral angle α, in solvent‐free magnesocenophanes 1 a–d (Figure 4). This is primarily a result of a lowering in energy of the LUMO in these systems, which has a large coefficient of high s character at the magnesium atom.
Figure 4.
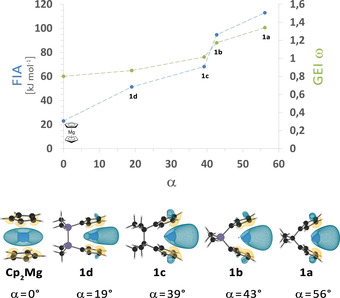
Calculated fluoride ion affinities (FIA) and global electrophilicity indices ω (GEI) as a function of the dihedral angle between the Cp planes (α), and isosurface plots of LUMOs (isovalue=0.035) of magnesocene (Cp2Mg) and magnesocenophanes 1 a–d (calculated at the B3LYP‐D3/def2‐TZVP level of theory11).
Therefore, according to these results, C[1]magnesocenophane 1 a should be the most Lewis acidic compound in this series. This might result in a higher catalytic reactivity, as Lewis acidity can be important for substrate binding, although it has to be taken into consideration that magnesocenophanes 1 a–d will exist as solvent adducts in solutions in donor solvents.
Catalytic studies
Following the computational results, we started our experimental investigations regarding catalytic dehydrogenation/dehydrocoupling of amine boranes by focusing mainly on C[1]magnesocenophane 1 a. Indeed, this compound proved to be a very potent catalyst for dehydrogenation of dimethylamine borane 2 a and diisopropylamine borane 2 b (Scheme 1).
Scheme 1.
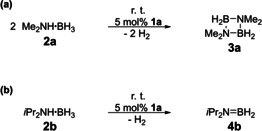
Dehydrogenations of a) Me2NH⋅BH3 (2 a) and b) iPr2NH⋅BH3 (2 b) catalyzed by 1 a.
When a solution of dimethylamine borane 2 a and 5 mol % C[1]magnesocenophane 1 a in dme was stirred at room temperature,12a dehydrogenation occurred to afford cyclic diborazane 3 a, with ≥95 % conversion12b being achieved after 16 h (Figure 5).
Figure 5.
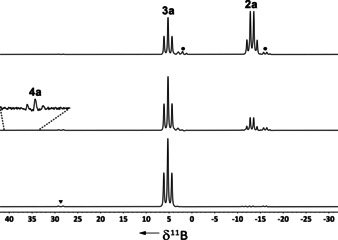
11B NMR spectra of the reaction of dimethylamine borane (2 a) with 5 mol % 1 a in dme at room temperature (from top to bottom: 4, 8, 16 h; ▾=(Me2N)2BH); •=5).
Similarly, diisopropylamine borane 2 b was also dehydrogenated to give diisopropylaminoborane 4 b in the presence of 5 mol % of 1 a, with ≥95 % conversion after 24 h (Figure S15). When the catalyst loading was lowered for the reaction with 2 a, 52 % conversion to 3 a was observed in the presence of 3 mol % of 1 a after 24 h and ≥95 % conversion was achieved after 48 h. As expected, when the catalyst loading was increased, the reaction proceeded at a faster rate, giving ≥95 % conversion to 3 a after 8 h in the presence of 10 mol % of 1 a (Table 1). Interestingly, the catalytic activity of 1 a towards primary amine boranes proved to be lower. When methylamine borane 2 c was treated with 5 mol % of 1 a in dme at room temperature, dehydrocoupling occurred to give different borazane species (Scheme 2), as indicated by 11B NMR spectroscopy and ESI mass spectrometry (Figures S16 and S26).15
Table 1.
Reaction parameters for the catalytic dehydrocoupling of dimethylamine borane 2 a (c0=850 mm) with magnesocenophanes 1 a–d, magnesocene (Cp2Mg), and di‐n‐butylmagnesium (nBu2Mg) under ambient conditions.
|
(Pre)catalyst (loading [mol %]) |
Reaction time [h] |
Solvent |
Conversion [%] |
TON/TOF [h−1] |
|---|---|---|---|---|
|
1 a (5) |
16 |
dme |
≥95 |
20/1.2 |
|
1 a (10) |
8 |
dme |
90 |
9/1.1 |
|
1 a (3) |
24 |
dme |
52 |
17/0.7 |
|
1 a (3) |
48 |
dme |
≥95 |
31/0.6 |
|
1 a (5) |
24 |
thf |
42 |
8/0.3 |
|
1 a (5) |
24 |
Et2O |
28 |
6/0.2 |
|
1 a (5) |
24 |
C6H4F2 |
17 |
4/0.2 |
|
1 a (5) |
24 |
C6D6 |
15 |
3/0.1 |
|
1 b (5) |
16 |
dme |
76 |
15/0.9 |
|
1 b (5) |
24 |
dme |
≥95 |
19/0.8 |
|
1 c (5) |
24 |
dme |
50 |
10/0.4 |
|
1 d (5) |
24 |
dme |
23 |
5/0.2 |
|
Cp2Mg (5) |
16 |
dme |
75 |
14/0.9 |
|
Cp2Mg (5) |
24 |
dme |
≥95 |
16/0.7 |
|
nBu2Mg (5) |
24 |
dme |
≤5 |
– |
Scheme 2.
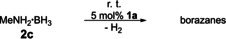
Dehydrocoupling of MeNH2 ⋅BH3 (2 c) catalyzed by magnesocenophane 1 a.
However, only around 20 % conversion was observed for this reaction after 24 h. When the catalyst loading was increased to 10 mol %, around 30 % conversion was observed after 24 h at room temperature. This profound deviation might be due to catalyst degradation in the presence of the more reactive substrate 2 c, but the exact reason remains unclear at this point.
In previously reported lithium‐ and magnesium‐based systems, strong solvent effects were observed, insofar as dehydrocoupling reactions were slower in donor solvents, such as thf, but faster in non‐coordinating solvents, such as aromatic hydrocarbons.5, 16 This can be rationalized in terms of coordination of the solvent to the metal atom of the catalyst, essentially blocking binding of the substrate. In contrast, in the case of magnesocenophane 1 a, the reaction proceeded much more rapidly in dme than in benzene, toluene, or 1,2‐difluorobenzene, which can primarily be attributed to the extremely low solubility of 1 a in non‐coordinating solvents. Comparing coordinating solvents, the reaction is faster in dme than in thf (Table 1), which might be related to the observed reversibility of dme coordination to the magnesium atom, suggesting that thf is more tightly bound than dme (see above). In addition, it should be noted that different solvents can have a significant impact on equilibria between different species, which is believed to be important in the present catalysis (see below).
Remarkably, the present reactions proceed at room temperature, whereas previously reported magnesium (pre)catalysts I–IV operate only at elevated temperatures. Moreover, this represents a rare example of a main group metallocene being employed in homogeneous catalysis. It should be noted, however, that Kays’ catalyst IV fully converts dimethylamine borane 2 a into cyclic diborazane 3 a within 150 min at 333 K at 5 mol % [Mg]. In order to benchmark our system against this, we also conducted catalytic experiments at 333 K. At this temperature, magnesocenophane 1 a catalyzes the reaction at a much faster rate, with ≥95 % conversion of 2 a into 3 a being observed after just 90 min in the presence of 5 mol % 1 a in dme (TON=20; TOF=13.4 h−1),15 making it even faster than Kays’ catalyst IV.
To follow the reaction conveniently and to determine kinetic data, we applied in situ 11B NMR spectroscopy. The reaction of 2 a and 5 mol % 1 a was conducted in an NMR tube at 323 or 333 K within the spectrometer, and spectra were acquired at appropriate intervals (Figure 6; Figures S3–S8 and S13). The results suggested pseudo‐first‐order kinetics for the conversion of 2 a at 323 K, as a linear fit for ln [2 a] vs. t was obtained. For the reaction at 333 K, however, a significant deviation from a linear fit was observed, especially at lower substrate concentration, indicating a complex mechanism with more than one reaction route. Comparable reaction profiles were observed for the catalysis at room temperature, monitored by discontinuous 11B NMR measurements (Figures S1 and S2).15
Figure 6.
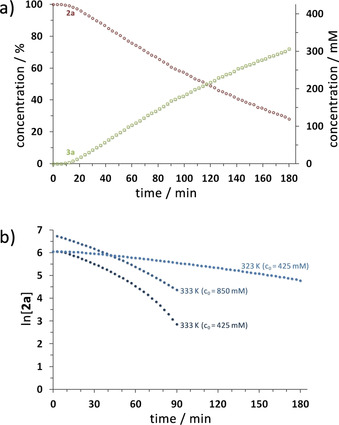
a) Plots of [2 a] and [3 a] versus time for the reaction of Me2NH⋅BH3 (2 a) with 5 mol % 1 a in dme at 323 K. b) Plots of ln [2 a] versus time for different reactions of Me2NH⋅BH3 (2 a) with 5 mol % 1 a in dme at 323 and 333 K (reactions at 323 K were monitored until >70 % conversion; reactions at 333 K were monitored until >90 % conversion).
Having obtained these intriguing results, we also investigated the catalytic activities of magnesocenophanes 1 b–d and magnesocene (Cp2Mg) towards dimethylamine borane 2 a. Interestingly, although these compounds also catalyze the dehydrogenation of 2 a at room temperature, they do so at slower rates (Table 1).
For the reaction of 2 a with 5 mol % Si[1]magnesocenophane 1 b in dme at room temperature, 76 % conversion was observed after 16 h, whereas only 50 % conversion was observed after 24 h in the case of C[2]magnesocenophane 1 c, and just 23 % after 24 h in the case of Si[2]magnesocenophane 1 d. Notably, magnesocene performed similarly to Si[1]magnesocenophane 1 b, giving 75 % conversion after 16 h, but it must not be overlooked in this context that magnesocene exhibits better solubility in many solvents than [1]magnesocenophanes 1 a,b. Overall, catalytic activity is evidently strongly influenced by the ligand system. In particular, the differences between [1]‐ and [2]magnesocenophanes are considerable (Table 1). This indicates that the ansa ligand system plays an important role in the dehydrocoupling catalysis and suggests that magnesocenophanes are not simply catalyst precursors (precatalysts).
We proceeded to investigate the possible coordination of dimethylamine borane 2 a to magnesocenophanes 1 a–d computationally. Based on known coordination modes of hydridoamines to magnesocene (Figure 7, left)10b, 10d and our DFT calculations, we predict a coordination mode whereby the amine borane binds to the magnesium atom through two B−H moieties and forms a hydrogen bond between its N−H group and the π system of one cyclopentadienyl ligand under elongation of the corresponding Mg−Cp bond (Figure 7, center).
Figure 7.
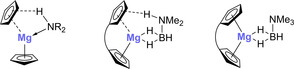
Coordination of hydrido‐amines to magnesocene (left), of hydrido‐amine boranes to magnesocenophanes (center), and of trimethylamine borane to magnesocenophanes (right).
Our computational investigation of the coordination of dimethylamine borane 2 a to magnesocenophanes 1 a–d reveals that the complexation energies roughly correlate with the dihedral angles of the Cp planes in the latter, with the largest binding energy being calculated for C[1]magnesocenophane 1 a, which is predicted to have the largest dihedral angle α, and lower binding energies being calculated for the less bent 1 b–d with smaller dihedral angles α (Table 2).
Table 2.
Calculated complexation energies E and G 298 between dimethylamine borane (2 a) and magnesocenophanes 1 a–d (Figure 7, center).
|
Compound |
E[a] |
[n][b] |
E [kJ mol−1] |
G 298 [kJ mol−1] |
|---|---|---|---|---|
|
1 a |
C |
1 |
−116.5 |
−63.3 |
|
1 b |
Si |
1 |
−104.3 |
−53.2 |
|
1 c |
C |
2 |
−66.2 |
−17.8 |
|
1 d |
Si |
2 |
−61.4 |
−10.8 |
[a] E=bridging atom(s) of ansa bridge; [b] n=number of bridging atom(s) in ansa bridge.
This is in part a result of the influence of the ansa bridge on the Cp⋅⋅⋅H−N hydrogen bonds. Shorter ansa bridges promote stronger hydrogen bonds due to a more bent geometry of the Cp groups, whereas longer ansa bridging motifs result in more parallel oriented Cp ligands, disfavoring the establishment of Cp⋅⋅⋅H−N hydrogen bonds. Moreover, Lewis acidity of the magnesium center, which is predicted to be highest for C[1]magnesocenophane 1 a, and to decrease in 1 b–d as a function of dihedral angle α, favors stronger coordination of the B−H moieties to the magnesium atom. This is evident from the complexation energies for trimethylamine borane (Figure 7, right; Table 3). Stronger Mg−H−B coordination favors B−H bond activation, which might explain the differences in catalytic performance. Assuming that, under the catalytic conditions in solution, substrate complexes (1 a–d)⋅2 a exist in equilibrium with the corresponding dme adducts (1 a–d)⋅dme and solvent adducts of the amine borane complexes (1 a–d)⋅dme⋅2 a, these differences in binding energies will have an important impact on such equilibria and therefore on the substrate activation, and might explain the observed differences in overall catalytic activities. It should be noted, however, that other effects, such as solubilities and ligand stabilities, must also be taken into account, and that binding energies of dme are different for each of the magnesocenophanes 1 a–d.17
Table 3.
Calculated complexation energies E and G 298 between trimethylamine borane and magnesocenophanes 1 a–d (Figure 7, right).
|
Compound |
E[a] |
[n][b] |
E [kJ mol−1] |
G 298 [kJ mol−1] |
|---|---|---|---|---|
|
1 a |
C |
1 |
−81.4 |
−32.7 |
|
1 b |
Si |
1 |
−64.5 |
−16.7 |
|
1 c |
C |
2 |
−51.6 |
−3.0 |
|
1 d |
Si |
2 |
−30.9 |
+15.1 |
[a] E=bridging atom(s) of ansa bridge; [b] n=number of bridging atom(s) in ansa bridge.
Mechanistic investigations
The most intriguing aspect of the dehydrocoupling catalysis presented here is that the conversion of dimethylamine borane 2 a into cyclic diborazane 3 a proceeds at room temperature, unlike with previously reported magnesium‐based systems, which required elevated temperatures.4, 5 We therefore investigated the mechanism in detail, and suggest that the reaction proceeds in a ligand‐assisted cooperative manner, with stepwise proton and hydride transfer from the amine borane molecule to the magnesocenophane and subsequent hydrogen elimination.
In principle, two different intermediates can be envisaged for the dehydrocoupling of dimethylamine borane 2 a to cyclic diborazane 3 a, namely dimethylaminoborane 4 a, which is short‐lived at room temperature and could undergo an off‐metal [2+2] cycloaddition to give 3 a (Scheme 3 a), and linear diborazane 3 b, which could undergo catalyzed ring‐closing dehydrocoupling to give 3 a (Scheme 3 b).
Scheme 3.
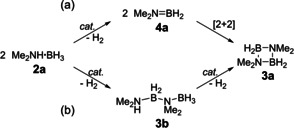
Dehydrocoupling of Me2NH⋅BH3 (2 a) to give [Me2NBH2]2 (3 a) via a) Me2N=BH2 (4 a) or b) Me2NH−BH2−NMe2−BH3 (3 b) as intermediates.
In both pathways, the N−H and B−H bonds of substrate 2 a have to be activated and cleaved. Therefore, selective deuteration of substrate 2 a should provide insight into which of these bonds is broken in the rate‐determining step. Investigations of kinetic isotope effects (KIEs) for starting material conversion with the deuterated substrate derivatives Me2ND⋅BH3 (2 aND), Me2NH⋅BD3 (2 aBD), and Me2ND⋅BD3 (2 aNDBD) gave KIEs of k(2 a)/k(2 aND)=2.24 for deuteration on nitrogen, k(2 a)/k(2 aBD)=1.62 for deuteration on boron, and k(2 a)/k(2 aNDBD)=3.46 for the fully deuterated substrate (rate constants: k(2 a)=1.22(1)×10−4 s−1, k(2 aND)=5.45(1)×10−5 s−1, k(2 aBD)=7.51(9)×10−5 s−1, and k(2 aNDBD)=3.53(2)×10−5 s−1; Figure 8).
Figure 8.
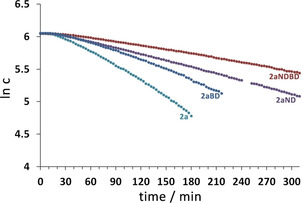
Plots of ln [2 a], ln [2 aND], ln [2 aBD], and ln [2 aNDBD] versus time for the reactions of Me2NH⋅BH3 (2 a), Me2ND⋅BH3 (2 aND), Me2NH⋅BD3 (2 aBD), and Me2ND⋅BD3 (2 aNDBD) with 5 mol % 1 a in dme at 323 K (reactions were monitored until >50 % conversion in all cases).
These KIEs are different to those reported by Kays et al. for their magnesium‐based catalyst IV, but are similar to those observed for an iron diketiminate catalyst.18 This suggests that cleavage of the N−H bond might occur in the rate‐determining step. The results must be interpreted with caution, however, as the mechanism is complex and most likely involves different catalytic cycles (see below).
In their early work, Hill et al. succeeded in structurally characterizing a key intermediate, I, in the dehydrocoupling of dimethylamine borane 2 a with dibutylmagnesium.4a In the borylamido magnesium compound, the magnesium atom is complexed by two deprotonated, and hence anionic, linear diborazane moieties. This compound was shown to form cyclic diborazane 3 a at temperatures above 333 K. We therefore investigated whether a similar intermediate plays a role in the dehydrocoupling reaction of dimethylamine borane 2 a catalyzed by magnesocenophane 1 a. First, we performed a reaction under the conditions normally applied for 1 a (r.t., 16 h, dme), but using 5 mol % of dibutylmagnesium. In this experiment, virtually no formation of 3 a was observed after 16 h by 11B NMR (Table 1), although signals of a magnesium complex, similar to those described by Hill and co‐workers, were observed. We therefore directly synthesized, isolated, and structurally characterized this compound, following Hill's protocol,4a but using dme as solvent (Scheme 4, Figure 9).15
Scheme 4.
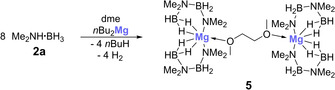
Synthesis of borylamido magnesium complex 5.
Figure 9.
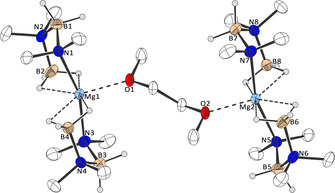
Molecular structure of 5 in the crystal (thermal ellipsoids at 50 % probability level; hydrogen atoms except (B)−H omitted for clarity; selected bond lengths: Mg1/2−O1/2: 218.1, Mg1/2−N1/3/5/7: 215.9/216.1 pm).
Complex 5 exhibits two crystallographically equivalent magnesium centers in a distorted pentagonal‐bipyramidal configuration, in which the equatorial plane is defined by the four hydrogen atoms and the oxygen atom, with the nitrogen atoms in axial positions.15 The Mg−N bond lengths are very similar to those in the thf analogue, but the Mg−O bond lengths are around 10 pm shorter in the thf complex.4a In this context, it is also worth noting that heptacoordinated magnesium complexes are quite rare.19 Compound 5 proved to be stable at room temperature under an inert atmosphere for many weeks and in organic solvents for at least several days. It does not undergo conversion to cyclic diborazane 3 a, even in the presence of additional amounts of dimethylamine borane 2 a, exactly how Hill and co‐workers described for the thf analogue.4a However, heating a solution of 5 to temperatures in excess of 333 K does slowly produce 3 a, again in line with what was reported by Hill and co‐workers.4a We therefore conclude that this compound is not a key intermediate in the room temperature dehydrocoupling of 2 a. However, when a solution of complex 5 in dme or thf was treated with 2,2‐dicyclopentadienylpropane, the neutral protonated ligand of magnesocenophane 1 a at room temperature, some formation of 3 a was evident from the 11B NMR spectrum (Scheme 5, Figure S21).15 Moreover, most significantly, two triplets in the 1H NMR spectrum of the solution in thf at δ 1H=5.60 and 5.70 ppm clearly indicated the formation of magnesocenophane 1 a⋅(thf)2 from the reaction of complex 5 with 2,2‐dicyclopentadienylpropane (Figure S22).15
Scheme 5.
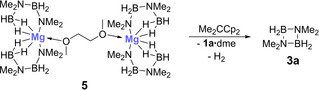
Reaction of magnesium complex 5 with 2,2‐dicyclopentadienyl‐propane to give cyclic diborazane 3 a.
This clearly shows that the ansa ligand plays an important role in the room temperature dehydrocoupling catalysis, and that magnesocenophane 1 a is formed from complex 5 and 2,2‐dicyclopentadienylpropane. Under catalytic conditions, magnesium complex 5 most likely exists in equilibrium with magnesocenophane 1 a (Scheme 6), as both the neutral ligand, 2,2‐dicyclopentadienylpropane, as well as 1 a, are observed by 1H NMR spectroscopy. Accordingly, we believe that 1 a is the relevant precatalyst and that 1 a⋅dme⋅2 a may be the catalytically active species in the dehydrogenation/dehydrocoupling process.
Scheme 6.
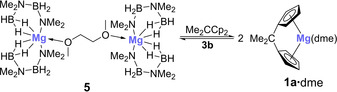
Reaction of magnesium complex 5 with 2,2‐dicyclopentadienyl‐propane to give magnesocenophane 1 a.
Since linear diborazane 3 b must be formed in at least small quantities through this equilibrium, we decided to directly study its reactivity towards magnesocenophane 1 a under catalytic conditions. When 3 b was treated with 5 mol % of 1 a in dme at room temperature, dehydrocoupling to cyclic diborazane 3 a was observed, but at a slower rate, giving only 63 % of 3 a after 16 h (≥95 % conversion was observed after 16 h for the reaction of 2 a with 5 mol % 1 a under these conditions). This implies that linear diborazane 3 b is most likely not the sole key intermediate in the catalysis, and that there must be a second, faster reaction route.
Interestingly, the reaction does not proceed exclusively through simple ring‐closing dehydrocoupling, but also appears to proceed through a disproportionation route, as significant amounts of dimethylamine borane 2 a were observed by 11B NMR spectroscopy (Scheme 7, Figure S17).
Scheme 7.
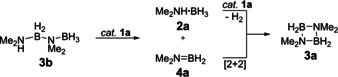
Dehydrocoupling of Me2NH−BH2−NMe2−BH3 (3 b) catalyzed by 1 a.
Close inspection of the 11B NMR spectrum of the reaction mixture obtained from dimethylamine borane 2 a and 5 mol % of magnesocenophane 1 a reveals four small signals, indicative of intermediates and side products formed in the reaction (Figure 5). A set of a quartet and a triplet observed at δ 11B=−15.4 (q, 1 J BH=89 Hz) and 3.7 ppm (t, 1 J BH=102 Hz) corresponds to complex 5. These signals were observed at an early stage of the reaction, did not increase with time, and then disappeared at the end of the reaction (≥95 % conversion), consistent with the assumed equilibrium between complex 5 and magnesocenophane 1 a. A doublet observed at δ 11B=28.7 ppm (1 J BH=132 Hz) corresponds to bis(dimethylamino)borane, a well‐known side product in the dehydrocoupling of 2 a,4 and most likely not an intermediate in the catalytic cycle. Most importantly, a low‐field shifted triplet observed at δ 11B=37.7 ppm (1 J BH=128 Hz) is indicative of the formation of dimethylaminoborane 4 a, which is known to be a key intermediate in the dehydrogenation/dehydrocoupling of 2 a in many catalytic systems.3 Thus, this gives an important indication as to how the reaction might proceed. Because 4 a undergoes rapid, off‐metal [2+2] cycloaddition at room temperature to form cyclic diborazane 3 a, it is short‐lived and only observed in small amounts in the reaction mixture. Therefore, to further verify its formation, the reaction was carried out in the presence of cyclohexene as a hydroboration trapping agent. In this case, some of the formed 4 a reacted with the cyclohexene to form dimethylamino‐dicyclohexylborane, which is persistent at room temperature and could be detected in the 11B NMR spectrum at δ 11B=44.2 ppm (ν1/2=218 Hz) (Figure S14).15 This provides strong evidence for the presence of 4 a in the reaction solution.
Based on these findings, we propose the reaction mechanism outlined in Scheme 8. Magnesium complex 5 exists in equilibrium with magnesocenophane 1 a. Linear diborazane 3 b is formed from the reaction of complex 5 with 2,2‐dicyclopentadienylpropane. Formation of magnesocenophane 1 a from 5 could be observed by 1H NMR spectroscopy (see above), and it is important to point out that complex 5 has been reported previously,4a and has been confirmed to be inactive in the ring‐closing dehydrocoupling of linear diborazane 3 b to cyclic diborazane 3 a under ambient conditions. In a minor catalytic cycle (Scheme 8, left side), magnesocenophane may dehydrocouple 3 b to 3 a. This was found to occur at a slower rate than the conversion of 2 a to 3 a, and to produce 2 a in the process (see above). On the other hand, in what we believe to be the main cycle (Scheme 8, right side), 2 a is directly dehydrogenated by magnesocenophane 1 a to give dimethylaminoborane 4 a, which undergoes an off‐metal [2+2] cycloaddition to form 3 a. This pathway is supported by the observation and trapping of 4 a (see above). Here, the first reaction step is protonation of one of the Cp moieties by the proton of the amine group to give intermediate I1. According to the observed KIEs (Figure 8), this is the rate‐determining step. The resulting intermediate I1 might rearrange to a magnesium amidoborane intermediate I2. Such metal amido complexes have often been implicated as intermediates in amine borane dehydrocoupling reactions.3 In the following step, B−H bond activation and hydrogen elimination form 4 a and regenerate 1 a. Notably, HD is observed by 1H NMR spectroscopy (δ 1H=4.50 ppm, 1 J HD=43 Hz), even when the fully deuterated substrate 2 aNDBD is used, and deuterated Cp carbon atoms are observed by 13C NMR spectroscopy (Figures S24 and S25). This is in agreement with the proposed protonation and deprotonation of a Cp moiety of magnesocenophane 1 a. A similar ligand‐assisted route with consecutive proton and hydride transfer has also been suggested by Kays and co‐workers for their magnesium catalyst IV.5
Scheme 8.
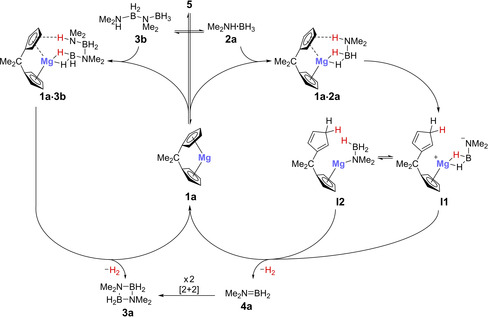
Simplified proposed catalytic cycle (coordinating solvent molecules omitted) for the dehydrogenation of Me2NH⋅BH3 (2 a) and Me2NH−BH2−NMe2−BH3 (3 b) to cyclic diborazane 3 a, catalyzed by magnesocenophane 1 a.
Following the experimental investigations, we turned to computational modeling of the proposed reaction pathway. To ensure that these DFT calculations were as close as possible to the experimental system, solvent coordination was considered and a dme molecule was placed on the magnesium atom in all structures. Indeed, DFT calculations at the B3LYP‐D3/def2‐TZVP level of theory supported the proposed catalytic cycle for dehydrogenation of dimethylamine borane 2 a and formation of dimethylaminoborane 4 a (Scheme 8, right side; Figure 10).11
Figure 10.
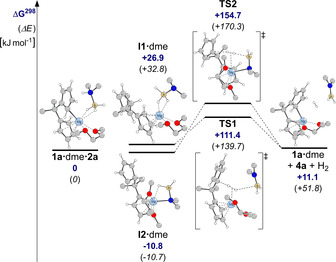
Calculated reaction pathways for the dehydrogenation of dimethylamine borane (2 a) via dimethylaminoborane (4 a), catalyzed by 1 a⋅dme (calculated at the B3LYP‐D3/def2‐TZVP level of theory[11]; hydrogen atoms of CH2 and CH3 groups omitted for clarity).
It is predicted that protonation of the cyclopentadienyl moiety to give intermediate I1⋅dme is endergonic, but that the amido‐magnesium intermediate I2⋅dme is lower in energy (ΔG298(I1⋅dme/I2⋅dme)=37.7 kJ mol−1) and exergonic with respect to the initial complex 1 a⋅dme⋅2 a. Two different transition states, TS1 and TS2, for the metal‐mediated B−H bond activation, hydrogen elimination, and formation of dimethylaminoborane 4 a, starting from intermediates I1⋅dme and I2⋅dme, could be located on the potential energy surface, leading to the same product. Transition state TS2, connected to I2⋅dme, is higher in energy than transition state TS1, connected to I1⋅dme (ΔG298(TS1/TS2)=43.3 kJ mol−1), which suggests that the pathway via intermediate I1/TS1 might be more feasible than that via intermediate I2/TS2, although neither can be ruled out completely.
When solvent coordination to the magnesium center was neglected and no dme molecule was coordinated to the magnesium atom, a similar pathway could be located on the potential energy surface (Figure S29).15
Although the reaction was calculated to be slightly endergonic, the overall driving forces are the elimination of hydrogen and the dimerization of dimethylaminoborane 4 a to give cyclic diborazane 3 a, both of which prevent back‐reactions. In this context, it is noteworthy that the reaction of dimethylamine borane 2 a with 5 mol % magnesocenophane 1 a in thf was found to proceed significantly more slowly when performed in a sealed reaction vessel (42 % conversion after 24 h with 5 mol % 1 a) than in an open reaction vessel (95 % conversion after 24 h with 5 mol % 1 a). Overall, the calculated reaction pathway is in agreement with our experimental observations.
Conclusions
In summary, we have reported the first magnesium‐based catalyst for the dehydrocoupling of dimethylamine borane 2 a to cyclic tetramethyldiborazane 3 a that operates at room temperature. In addition, at elevated temperatures, it outperforms previously reported magnesium‐based catalysts in the field,4, 5 and represents a rare example of a main‐group metallocene being employed as a catalyst. Furthermore, different [1]‐ and [2]magnesocenophanes have been investigated, and significant differences in the catalytic activities of these compounds have been discerned, which we believe to be a function of the tilt of the cyclopentadienyl moieties and related differences in substrate binding.
Experimental and computational investigations of the mechanism suggest a pre‐equilibrium between magnesocenophane 1 a and magnesium complex 5 and clearly indicate that 1 a is the catalyst in the reaction, which is believed to operate through a ligand‐assisted route involving stepwise proton and hydride transfer, with dimethylaminoborane 4 a as the key intermediate.
Overall, magnesocenophane 1 a represents the most active magnesium catalyst for amine borane dehydrogenation/dehydrocoupling reported to date, and future studies will focus on the dehydrocoupling of various other substrates.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Prof. Dr. G. Kickelbick is thanked for his continuous support. Dr. K. Hollemeyer at the Mass Spectrometry Service of Saarland University is thanked for ESI‐MS measurements. S. Harling is thanked for elemental analyses. Funding by the Deutsche Forschungsgemeinschaft (DFG; SCHA 1915b/3‐1) and the Fonds der Chemischen Industrie is gratefully acknowledged.
L. Wirtz, W. Haider, V. Huch, M. Zimmer, A. Schäfer, Chem. Eur. J. 2020, 26, 6176.
References
- 1.
- 1a. Keaton R. J., Blacquiere J. M., Baker R. T., J. Am. Chem. Soc. 2007, 129, 1844–1845; [DOI] [PubMed] [Google Scholar]
- 1b. Peng B., Chen J., Energy Environ. Sci. 2008, 1, 479–483; [Google Scholar]
- 1c. Hamilton C. W., Baker R. T., Staubitz A., Manners I., Chem. Soc. Rev. 2009, 38, 279–293; [DOI] [PubMed] [Google Scholar]
- 1d. Staubitz A., Robertson A. P. M., Manners I., Chem. Rev. 2010, 110, 4079–4124. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Staubitz A., Presa Soto A., Manners I., Angew. Chem. Int. Ed. 2008, 47, 6212–6215; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6308–6311; [Google Scholar]
- 2b. Marinelli D., Fasano F., Najjari B., Demitri N., Bonifazi D., J. Am. Chem. Soc. 2017, 139, 5503–5519; [DOI] [PubMed] [Google Scholar]
- 2c. Ayhan O., Eckert T., Plamper F. A., Helten H., Angew. Chem. Int. Ed. 2016, 55, 13321–13325; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13515–13519; [Google Scholar]
- 2d. Lorenz T., Lik A., Plamper F. A., Helten H., Angew. Chem. Int. Ed. 2016, 55, 7236–7241; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7352–7357; [Google Scholar]
- 2e. Ledoux A., Larini P., Boisson C., Monteil V., Raynaud J., Lacôte E., Angew. Chem. Int. Ed. 2015, 54, 15744–15749; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15970–15975; [Google Scholar]
- 2f. Grosche M., Herdtweck E., Peters F., Wagner M., Organometallics 1999, 18, 4669–4672; [Google Scholar]
- 2g. Resendiz-Lara D. A., Stubbs N. E., Arz M. I., Pridmore N. E., Sparkes H. A., Manners I., Chem. Commun. 2017, 53, 11701–11704. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Leitao E. M., Jurca T., Manners I., Nat. Chem. 2013, 5, 817–829; [DOI] [PubMed] [Google Scholar]
- 3b. Waterman R., Chem. Soc. Rev. 2013, 42, 5629–5641; [DOI] [PubMed] [Google Scholar]
- 3c. Johnson H. C., Hooper T. N., Weller A. S., Top. Organomet. Chem. 2015, 49, 153–220; [Google Scholar]
- 3d. Melen R. L., Chem. Soc. Rev. 2016, 45, 775–788; [DOI] [PubMed] [Google Scholar]
- 3e. Stennett T. E., Harder S., Chem. Soc. Rev. 2016, 45, 1112–1128; [DOI] [PubMed] [Google Scholar]
- 3f. Rossin A., Peruzzini M., Chem. Rev. 2016, 116, 8848–8872; [DOI] [PubMed] [Google Scholar]
- 3g. Boom D. H. A., Jupp A. R., Slootweg J. C., Chem. Eur. J. 2019, 25, 9133–9152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Liptrot D. J., Hill M. S., Mahon M. F., MacDougall D. J., Chem. Eur. J. 2010, 16, 8508–8515; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Hill M. S., Hodgson M., Liptrot D. J., Mahon M. F., Dalton Trans. 2011, 40, 7783–7790; [DOI] [PubMed] [Google Scholar]
- 4c. Bellham P., Anker M. D., Hill M. S., Kociok-Köhn G., Mahon M. F., Dalton Trans. 2016, 45, 13969–13978. [DOI] [PubMed] [Google Scholar]
- 5. Ried A. C. A., Taylor L. J., Geer A. M., Williams H. E. L., Lewis W., Blake A. J., Kays D. L., Chem. Eur. J. 2019, 25, 6840–6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Spielmann J., Bolte M., Harder S., Chem. Commun. 2009, 6934–6936; [DOI] [PubMed] [Google Scholar]
- 6b. Liptrot D. J., Hill M. S., Mahon M. F., Wilson A. S. S., Angew. Chem. Int. Ed. 2015, 54, 13362–13365; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13560–13563. [Google Scholar]
- 7. Anderson D. L., Chemical Composition of the Mantle in Theory of the Earth, Blackwell Scientific Publications, Boston, 1989, Chapter 8, pp. 147–175. [Google Scholar]
- 8.
- 8a. Schäfer A., Rohe K., Grandjean A., Huch V., Eur. J. Inorg. Chem. 2017, 35–38; [Google Scholar]
- 8b. Haider W., Huch V., Schäfer A., Dalton Trans. 2018, 47, 10425–10428; [DOI] [PubMed] [Google Scholar]
- 8c. Stahlich A. S. D., Huch V., Grandjean A., Rohe K., Leszczyńska K. I., Scheschkewitz D., Schäfer A., Chem. Eur. J. 2019, 25, 173–176. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Shapiro P. J., Lee S.-J., Perrotin P., Cantrell T., Blumenfeld A., Twamley B., Polyhedron 2005, 24, 1366–1381; [Google Scholar]
- 9b. Perrotin P., Shapiro P. J., Williams M., Twamley B., Organometallics 2007, 26, 1823–1826; [Google Scholar]
- 9c. Perrotin P., Twamley B., Shapiro P. J., Acta Crystallogr. Sect. E 2007, 63, m1277–m1278. [Google Scholar]
- 10.
- 10a. Olmstead M. M., Grigsby W. J., Chacon D. R., Hascall T., Power P. P., Inorg. Chim. Acta 1996, 251, 273–284; [Google Scholar]
- 10b. Xia A., Knox J. E., Heeg M. J., Schlegel H. B., Winter C. H., Organometallics 2003, 22, 4060–4069; [Google Scholar]
- 10c. Jaenschke A., Paap J., Behrens U., Organometallics 2003, 22, 1167–1169; [Google Scholar]
- 10d. Jaenschke A., Behrens U., Z. Naturforsch. B 2014, 69, 655–664. [Google Scholar]
- 11.All DFT calculations were carried out using the Gaussian 09 Revision D.01 package of programs. See the Supporting Information for further details, optimized geometries, and references.
- 12.
- 12a.Room temperature/ambient conditions correspond to about 298 K;
- 12b.the amounts of starting material and product(s) were determined by integration of the 11B NMR spectra.
- 13.Fluoride ion affinities were calculated according to a literature established method from the isodesmic reactions with Et3B/Et3BF− For more details, see, for example: Großekappenberg H., Reißmann M., Schmidtmann M., Müller T., Organometallics 2015, 34, 4952–4958. [Google Scholar]
- 14.Global electrophilicity indices ω were calculated following the method of Stephan et al. (Method C): Jupp A. R., Johnstone T. C., Stephan D. W., Inorg. Chem. 2018, 57, 14764–14771. [DOI] [PubMed] [Google Scholar]
- 15.See the Supporting Information for further details.
- 16. McLellan R., Kennedy A. R., Orr S. A., Robertson S. D., Mulvey R. E., Angew. Chem. Int. Ed. 2017, 56, 1036–1041; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1056–1061. [Google Scholar]
- 17.Calculated binding energies of dme to magnesocenophanes 1 a–d (B3LYP-D3/def2-TZVP)[11]: 1 a: E=−139.6 kJ mol−1, G 298=−87.1 kJ mol−1; 1 b: E=−127.4 kJ mol−1, G 298=−72.2 kJ mol−1; 1 c: E=−98.8 kJ mol−1, G 298=−44.0 kJ mol−1; 1 d: E=−82.2 kJ mol−1, G 298=−24.1 kJ mol−1.
- 18. Coles N. T., Mahon M. F., Webster R. L., Organometallics 2017, 36, 2262–2268. [Google Scholar]
- 19. Seitz M., Kaiser A., Stempfhuber S., Zabel M., Reiser O., Inorg. Chem. 2005, 44, 4630–4636. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary