

Summary
Background
Non‐alcoholic steatohepatitis (NASH) is a severe form of non‐alcoholic fatty liver disease (NAFLD) characterised by liver fat accumulation, inflammation and progressive fibrosis. Emerging data indicate that genetic susceptibility increases risks of NAFLD, NASH and NASH‐related cirrhosis.
Aims
To review NASH genetics and discuss the potential for precision medicine approaches to treatment.
Method
PubMed search and inclusion of relevant literature.
Results
Single‐nucleotide polymorphisms in PNPLA3, TM6SF2, GCKR, MBOAT7 and HSD17B13 are clearly associated with NASH development or progression. These genetic variants are common and have moderate‐to‐large effect sizes for development of NAFLD, NASH and hepatocellular carcinoma (HCC). The genes play roles in lipid remodelling in lipid droplets, hepatic very low‐density lipoprotein (VLDL) secretion and de novo lipogenesis. The PNPLA3 I148M variant (rs738409) has large effects, with approximately twofold increased odds of NAFLD and threefold increased odds of NASH and HCC per allele. Obesity interacts with PNPLA3 I148M to elevate liver fat content and increase rates of NASH. Although the isoleucine‐to‐methionine substitution at amino acid position 148 of the PNPLA3 enzyme inactivates its lipid remodelling activity, the effect of PNPLA3 I148M results from trans‐repression of another lipase (ATGL/PNPLA2) by sequestration of a shared cofactor (CGI‐58/ABHD5), leading to decreased hepatic lipolysis and VLDL secretion. In homozygous Pnpla3 I148M knock‐in rodent models of NAFLD, targeted PNPLA3 mRNA knockdown reduces hepatic steatosis, inflammation and fibrosis.
Conclusion
The emerging genetic and molecular understanding of NASH paves the way for novel interventions, including precision medicines that can modulate the activity of specific genes associated with NASH.
1. INTRODUCTION
Non‐alcoholic fatty liver disease (NAFLD) affects about 25% of the global population. 1 NAFLD includes non‐alcoholic fatty liver (NAFL) and non‐alcoholic steatohepatitis (NASH). Patients whose NAFL develops into NASH have increased overall and liver‐specific mortality 1 , 2 and increased risks of cirrhosis, liver failure and hepatocellular carcinoma (HCC). 3 , 4 , 5 The burden of NASH is expected to increase in line with the global epidemic of obesity, type 2 diabetes and metabolic syndrome. 6 NASH is fast becoming the leading cause of chronic liver disease, and is set to overtake hepatitis C as the leading cause of liver transplantation in the US. 7 , 8
Hepatic steatosis in people with NAFL is characterised by substantial accumulation of lipid droplets within hepatocytes. The progression to NASH is marked by hepatic inflammation and hepatocellular injury, with or without hepatic fibrosis, in histological examinations of liver biopsies. 3 , 4 , 5 Limited evidence suggests that NAFL progresses to NASH in up to 44% of patients undergoing random or voluntary biopsy, 1 , 9 with higher rates in those referred for biopsy because NASH is suspected. 1 Progressive fibrosis drives poor liver‐related clinical outcomes, and develops in 35%‐41% of patients with NASH, according to meta‐analyses of paired biopsy studies. 1 , 2 About 20% of patients with NASH develop end‐stage cirrhosis or HCC. 10
Advances in human genetics present new opportunities to address the unmet need for NASH therapeutics, based on improved understanding of the multifactorial pathogenesis of NASH and the interaction between genetic and environmental risk factors. These new findings have opened up the possibility of precision medicine for patients with NASH based on inherited genetic variants. In this approach, the identification of people who carry a specific genetic variant predisposing them to NASH allows targeting of the same specific gene or molecular pathway to halt or reverse their hepatic steatosis, inflammation and fibrosis.
Here, we review the associations of five key genetic variants with NASH and discuss the potential for targeted interventions in particular disease pathways (Section 3). We then focus on a variant in the gene patatin‐like phospholipase domain‐containing protein 3 (PNPLA3), a common and strong genetic risk factor for NASH (Section 4). We assess the extensive genetic, molecular and cell biological evidence that therapies able to modulate PNPLA3 expression levels may represent precision medicine approaches in patients with NASH.
2. METHODS
This was not a formal systematic review, but a comprehensive search strategy was used to identify and prioritise published findings for inclusion. A PubMed search was performed without language or date restrictions, aiming to identify publications on known genetic variants associated with NASH including genetic, epidemiological, clinical, pre‐clinical and molecular cell biological studies. Search terms included ‘non alcoholic fatty liver disease’ or ‘non alcoholic steatohepatitis’ and ‘genetic’ or ‘genetics’ or ‘PNPLA3’ or ‘TM6SF2’ or ‘MBOAT7’ or ‘HSD17B13’ or ‘GCKR’ or ‘epidemiology’ or ‘prevalence’ or ‘treatment’ or ‘insulin resistance’ or ‘risk factors’. Hits were refined based on the relevance of the title and abstract to the aims of the review. Following peer‐review, 24 articles were added, of which four were suggested directly by the peer‐reviewers. A total of 163 articles were included in the final review.
3. IDENTIFYING TARGETABLE PATHWAYS FOR PRECISION MEDICINE
3.1. Environmental risk factors
Modern sedentary lifestyles and the overconsumption of food drive a consistent positive energy balance and fuel the obesity epidemic, with subsequent increases in incidence of type 2 diabetes, metabolic syndrome and NAFLD. In the US, the estimated prevalence of NASH rises from 12% in middle‐aged adults to 22% among those with diabetes and 33% among those with obesity. 11 , 12 Worldwide, NASH prevalence increases from 15% to 30% in people with obesity to up to 70% in those with morbid obesity. 10 Among patients with NASH worldwide, a reported 31%‐89% have obesity and 33%‐56% have diabetes. 10 Lifestyle interventions are the cornerstone of NAFLD management, and are discussed in Section 4.3. Not all obese individuals with fatty liver develop NASH, however, and some lean individuals do develop NASH, 1 indicating interaction with heritable risk factors.
3.2. Genetic risk factors
Hepatic steatosis and fibrosis cluster in families, with a heritability value of about 0.5 in a twin study, after adjustment for age, sex and ethnicity. 13 In a familial cohort study, the risk of advanced fibrosis was 12 times higher in first‐degree relatives of people with NAFLD and cirrhosis than in population controls, even after adjustment for other risk factors. 14 Of over 100 loci examined in genome‐wide association studies and candidate gene studies, 15 genetic variations in five genes have emerged as reproducibly and robustly predisposing individuals to development of NASH (PNPLA3, TM6SF2, GCKR, MBOAT7 and HSD17B13), as previously reviewed. 16 Unexplained variance remains despite these discoveries, indicating that future genome‐wide studies may reveal additional associations. 13
Table 1 shows published allele frequencies for the five principal genetic variants associated with NAFLD, NASH and HCC, together with allelic odds ratios for each disease. Odds ratios from different cohorts are not comparable with one another, and those from populations with liver disease may represent overestimates (eg liver biopsy cohorts). Nevertheless, published allelic odds ratios provide a useful indication of the approximate magnitude of the effects of the five principal variants on liver disease.
Table 1.
Published allele frequencies and odds ratios for the principal genetic variants associated with NAFLD, NASH and HCC
| Gene | SNP | Minor allele frequency, 1000G (max) | NAFLD allelic odds ratio a (95% CI) | NASH allelic odds ratio a (95% CI) | HCC allelic odds ratio a (95% CI) |
|---|---|---|---|---|---|
| PNPLA3 |
rs738409 c.444 C > G p.I148M |
0.26 (0.72)33 | 1.91 (1.64, 2.21)24 | 2.54 (2.03, 3.16)24 | |
| TM6SF2 |
rs58542926 c.499 G > A p.E167K |
0.07 (0.16)41 | 1.82 (1.59, 2.08) d , 39 | 1.37 (1.11, 1.72) d , e , 39 | 1.72 (1.27, 2.38) d , f , 39 |
| GCKR |
rs1260326 c.1337 C > T p.P446L |
0.29 (0.59)46 |
1.38 (1.25, 1.53)44 1.49 (1.09, 2.05)45 |
1.55 (1.10, 2.17)45 | 1.84 (1.23, 2.75) f , 44 |
| MBOAT7/TMC4 |
rs641738 g.54173068 C > T/ c.50 G > A p.G17E |
0.37 (0.63)51 | 1.42 (1.07, 1.91) g , 47 | 1.18 (1.00, 1.40) g , 47 | 2.10 (1.33, 3.31) h , 48 |
| HSD17B13 |
rs72613567 i c.704/812 + 2dup (usually referred to as T to TA insertion) j |
0.18 (0.40)58 | 0.84 (0.78, 0.91) k , 52 | 0.86 (0.72, 1.02) k , 52 |
Abbreviations: 1000G, 1000 Genomes Project phase 3; CI, confidence interval; HCC, hepatocellular carcinoma; NAFLD, non‐alcoholic fatty liver disease; NASH, non‐alcoholic steatohepatitis; NR, not reported; SNP, single‐nucleotide polymorphism.
Note that odds ratios are from different cohorts and are not comparable across variants or diseases.
In people with obesity; liver fibrosis not assessed.
In patients with NAFLD (76.2% with NASH and 34.4% with fibrosis stage 3 and 4).
Calculated as reciprocal of published odds ratio for the protective allele. 39
NASH cirrhosis.
Severity of liver fibrosis not assessed.
Liver biopsy cohort.
UK/Italian cohort without advanced fibrosis/cirrhosis.
Other liver disease‐associated SNPs in HSD17B13 are rs72613567 (linkage with rs72613567), rs143404524 and rs62305723.
TA to TAA insertion on the chromosomal forward strand is a TA to TTA duplication in HDS17B13 on the opposite strand, affecting introns of two transcripts.
Data shown from Geisinger Health System cohorts; generally similar results in Dallas Heart Study cohort.
Severity of liver fibrosis not assessed.
Adjusted for age, sex, fibrosis stage and aetiology.
The genetic variants associated with NASH are common, with minor allele frequencies of 7%‐37%, but nevertheless have moderate to large effect sizes for NAFLD, NASH and HCC (Table 1). This observation contrasts with the evolutionary theory that decreased reproductive fitness should select against genetic variants that confer disease risk. 17 Present‐day reproductive fitness may not, however, reflect the pressures that have shaped genetic variation throughout evolution. 17 The five genes known to be associated with NASH (Table 1) are all involved in glucose and fat homeostasis regulatory pathways. The modern obesogenic environment may expose a disease risk associated with genetic variants that are advantageous when food supplies are erratic. Increased liver fat levels in people with the genetic variation in PNPLA3 that predisposes to NASH may represent just such an example.
3.2.1. PNPLA3
The first genetic variant found to be associated with NASH is a nonsynonymous single‐nucleotide polymorphism (SNP) in PNPLA3 known as rs738409 c.444 C > G p.I148M. 18 The C to G substitution at nucleotide position 444 of PNPLA3 encodes an isoleucine to methionine substitution at amino acid position 148 of PNPLA3 protein (also known as adiponutrin). This genetic variant (herein referred to as PNPLA3 I148M) is associated with hepatic steatosis, steatohepatitis, elevated plasma liver enzyme levels, hepatic fibrosis and cirrhosis. 18 , 19 , 20 , 21 , 22 Associations between PNPLA3 I148M and NAFLD have been demonstrated in multiple different geographical regions and ethnicities 22 and in populations of all ages including children and adolescents. 23
PNPLA3 I148M has allelic odds ratios of approximately two to three for risks of NAFLD, NASH and HCC (Table 1). 24 , 25 , 26 Compared with allelic odds ratios, genotypic odds ratios are lower in PNPLA3 148IM heterozygotes and higher in PNPLA3 148MM homozygous risk allele carriers. 25 , 26 , 27 In a meta‐analysis of 13 817 patients, the allelic odds ratio for NASH was 2.54 (2.03, 3.16) and the genotypic odds ratios were 1.75 (1.24, 2.46) for heterozygotes and 4.44 (2.92, 6.76) for homozygotes. 24
Elevated risks of hepatic decompensation and liver‐related death were also associated with PNPLA3 I148M in a recent Italian prospective study. 25 PNPLA3 I148M was strongly associated with liver‐related death in an analysis of the US National Health and Nutrition Examination Survey with a median follow‐up of 23 years. 28 Genetic association studies have established PNPLA3 I148M as a strong genetic determinant of NAFLD in multiple populations worldwide. 29 , 30 , 31 The penetrance of PNPLA3 I148M in 148MM homozygotes in Europeans is similar to that of mutations causing canonical monogenic liver disorders. 32 The PNPLA3 I148M allele is common, with a frequency of 26% in the 1000 Genomes Project phase 3 combined population. 33 The minor allele frequency rises to 50% or higher in Latin American populations, contributing to explanation of the high incidence of NASH in this ethnic group (Table 1). 33
3.2.2. TM6SF2
A SNP in transmembrane 6 superfamily member 2 (TM6SF2) is associated with increased liver fat content, 34 NASH, advanced hepatic fibrosis and cirrhosis. 35 , 36 Rs58542926 c.499 G > A p.E167K is a G to A substitution encoding a glutamate to lysine substitution at amino acid position 167 of TM6SF2 protein (E167K), leading to a loss of its function in the hepatic very low‐density lipoprotein (VLDL) secretion pathway. 34 , 37
TM6SF2 variants have a moderate to large effect on the risk of NAFLD, 36 , 38 with the 167K allele having an allelic odds ratio of 1.82 for steatosis (Table 1). 39 Consistent with these findings, the 167EE homozygous ancestral genotype was associated with a significantly reduced risk of NAFLD in recent meta‐analyses. 39 , 40 The TM6SF2 E167K allele has a frequency of 7% in the 1000 Genomes Project phase 3 combined population (Table 1). 36 , 41
3.2.3. GCKR
Variations in the glucokinase regulator (GCKR) gene are associated with histological NAFLD 30 and have a modest effect on the risk of NAFLD. 38 A SNP in GCKR, rs1260326 c.1337 C > T p.P446L, is a C to T substitution encoding a proline to leucine substitution at amino acid position 446 of GCKR protein (P446L). 42 GCKR P446L is a loss‐of‐function variant that increases de novo lipogenesis by inducing glycolysis. 43 This variant is associated with increased susceptibility to NAFLD, NASH and NASH‐derived HCC (Table 1). 44 , 45 The minor allele frequency of GCKR P446L in the 1000 Genomes Project phase 3 combined population is 29%. 46 GCKR P446L interacts with PNPLA3 I148M in elevating susceptibility to NASH in people with both risk alleles. 45
3.2.4. MBOAT7
A SNP downstream of the gene encoding membrane bound O‐acyltransferase domain‐containing 7 (MBOAT7) has been linked with an increased risk of NAFLD, inflammation and fibrosis, and may predispose to HCC (Table 1). 38 , 47 , 48 The rs641738 g.54173068 C > T variant may be associated with downregulation of MBOAT7 at an mRNA and protein level. 38 The same SNP also affects another gene, TMC4 (rs641738 c.50 G > A p.G17E), with a resulting glycine to glutamate substitution in transmembrane channel‐like protein 4. Unlike MBOAT7, TMC4 lacks any known function related to lipid metabolism. 49 Rs641738 was identified first as a susceptibility locus for cirrhosis in alcohol abusers, 50 then for NAFLD 47 and HCC. 48 The minor allele frequency of rs641738 in the 1000 Genomes Project phase 3 combined population is 37% (Table 1). 51
3.2.5. HSD17B13
Inactivating variants in the HSD17B13 gene, which encodes the hepatic lipid droplet protein hydroxysteroid 17‐β dehydrogenase 13, have recently been linked with a reduced risk of chronic liver disease. 52 , 53 , 54 The rs72613567 T to TA insertion variant adjacent to the donor splice site downstream of exon 6 of HSD17B13 may affect mRNA splicing and lead to the production of a truncated protein. 52 Rs72613567 is in strong linkage disequilibrium with rs6834314, which is associated with decreased steatohepatitis and serum liver enzyme levels. 54 Another variant in HSD17B13 (rs143404524) is a deletion and frameshift that also leads to production of a truncated protein, and is associated with a reduced risk of chronic liver disease. 53 Finally, rs62305723 is a missense variant (c.778 C > T p.P260S) that also confers loss of enzymatic activity and is associated with decreased steatohepatitis. 54
Compared with ancestral allele homozygotes (T/T), HSD17B13 rs72613567 insertion variant allele heterozygotes (T/TA) have an 83% odds ratio and homozygotes (TA/TA) have a 70% odds ratio for developing non‐alcoholic liver disease. 52 In a bariatric surgery cohort, the prevalence of NASH decreased with each TA allele, and there was evidence of reduced progression from simple steatosis to NASH or fibrosis in patients carrying the insertion variant. 52 Associations between the variant and reduced odds of HCC have been reported in three different populations (Table 1). 55 , 56 Rs72613567 TA has also been reported to protect against histological steatohepatitis and fibrosis and to reduce plasma alanine aminotransferase (ALT) levels in people with NAFLD. 54 , 55 , 57 Rs72613567 interacted with PNPLA3 I148M in one cohort studied, such that additional HSD17B13 TA alleles reduced the effect of additional PNPLA3 I148M alleles on serum ALT levels. 52
The minor allele frequency of rs72613567 in the 1000 Genomes Project phase 3 combined population is 18% (Table 1). 58 The rs143404524 frameshift variant in HSD17B13 has a minor allele frequency of 6% in the 1000 Genomes Project phase 3 combined population, rising to 2.9% in Latin America and up to 33% in Africa. 59 Reported allelic odds ratios for chronic liver disease are 0.24 (95% confidence interval [CI]: 0.07, 0.76) in black populations and 0.10 (0.01, 0.79) in Hispanic children in the US. 53 HSD17B13 P260S (rs62305723) has a frequency of 2% in the 1000 Genomes Project phase 3 combined population. 60
3.3. Interaction of genetic and environmental risk factors
3.3.1. Interaction of obesity with genetic risk factors
Obesity exposes the association of PNPLA3 I148M with increased liver fat levels and risk of NASH, as revealed by studies in children, adolescents and adults 61 , 62 , 63 soon after its discovery. 18 PNPLA3 I148M has a more extreme effect on liver injury in people with obesity than in lean individuals, and confers genetic susceptibility from a young age. 61 , 62 , 63 Odds ratios for elevated ALT levels in PNPLA3 I148M homozygous children with obesity increased with abdominal fat levels in an Italian cohort study, from 1.2 (95% CI: 0.7, 2.4) to 4.9 (3.2, 7.8) in subgroups of low and high waist‐to‐height ratio, respectively. 64
Liver fat levels increased dramatically with each additional PNPLA3 I148M allele in people with high visceral abdominal fat levels, but there was no association in those with low visceral abdominal fat levels, in a European‐American cohort study. 65 The effect of PNPLA3 I148M on liver fat significantly increased with body mass index (BMI) in the Dallas Heart Study cohort. 66 In lean individuals (BMI <25 kg/m2), liver fat content was only about 50% higher in PNPLA3 148MM than in 148II homozygotes, but in those with obesity (BMI >35 kg/m2), liver fat content was 300% higher in PNPLA3 148MM than in 148II homozygotes. 66 The effect of high BMI in amplifying the risk of steatosis in carriers of PNPLA3 148M may be mediated by insulin resistance. 67 The prevalence of NASH ranged from 9% in lean 148II homozygotes to 84% in 148MM homozygotes with obesity. 66 Adiposity also amplified the interaction of PNPLA3 I148M with ALT levels and cirrhosis in other cohorts. 66 Interactions of obesity with TM6SF2 E167K and GCKR P446L have also been reported. 66 , 68
Taken together, published evidence indicates that the common PNPLA3 I148M variant is usually benign in lean individuals, perhaps reflecting a selective advantage of increased liver fat storage in our evolutionary past. 69 , 70 In people with lipodystrophy, however, impaired expansion of adipose tissue may lead to lipid accumulation at ectopic sites (including the liver). 71 , 72 A polygenic risk score for lipodystrophy has been associated with increased hepatic steatosis and fibrosis. 73 Whether PNPLA3 I148M plays any pathophysiological role in a subset of lean patients with NAFLD requires further investigation.
In contrast, an obesogenic environment transforms PNPLA3 I148M into a major factor in NAFLD and NASH pathophysiology. Furthermore, evidence presented in Section 4.3.1 suggests that PNPLA3 I148M may modify the response to treatments that can lower body weight and liver fat levels in patients with NAFLD (omega‐3 fatty acids, lifestyle modification and bariatric surgery).
3.3.2. Insulin resistance and NAFLD genetic risk factors
Insulin resistance and type 2 diabetes are both significant risk factors for development of NASH. The identified genetic risk factors for elevated liver fat and NASH do not associate with insulin resistance, except in individuals with severe obesity. 18 , 61 , 74 In liver lipidomic analyses, NAFLD associated with PNPLA3 I148M was characterised by high levels of hepatic polyunsaturated triacylglycerols, 75 , 76 but NAFLD associated with insulin resistance was characterised by high levels of saturated and mono‐unsaturated triacylglycerols, free fatty acids and ceramides. 76 The altered lipid composition in the liver in carriers of PNPLA3 148M is reflected in reduced polyunsaturated triglyceride levels in very low‐density lipoprotein particles. 77 Furthermore, hepatic diacylglycerols are implicated in the development of insulin resistance, and were elevated in PNPLA3 ancestral allele carriers but not I148M carriers in another lipidomic study of people with hepatic steatosis. 78
PNPLA3 I148M was, however, associated with a small increase in the risk of type 2 diabetes in a very large genome‐wide association study of type 2 diabetes (allelic odds ratio 1.04 [95% CI: 1.01, 1.07]). 79 , 80 A phenome‐wide analysis confirmed the association of PNPLA3 I148M with increased risk of type 2 diabetes (odds ratio 1.08) 81 reported in a fine‐mapping meta‐analysis (odds ratio 1.05 [95% CI: 1.03, 1.07]). 82 In a Mendelian randomisation study, the genetic risk score for hepatic fat accumulation showed a causal relationship with insulin resistance, but this relationship disappeared when the model was adjusted for liver fibrosis. 79 This suggests that insulin resistance is not caused by genetically determined high liver fat levels per se, but rather that it develops as subsequent liver disease progresses and may be related to the inflammatory and pro‐fibrotic environment. A limitation of Mendelian randomisation approaches is that they do not provide insights on the underlying mechanisms. 79
The causes and consequences of liver steatosis and inflammation in patients with NASH may differ between PNPLA3 I148M carriers and those lacking the variant. The effect of PNPLA3 I148M on retinol metabolism in hepatic stellate cells (detailed in Section 4.2) may trigger or exacerbate hepatic inflammation in carriers with other risk factors for NASH. Further research into NASH pathophysiology in the presence and absence of specific genetic risk factors is needed to understand the relationships between NAFLD, fibrosis and insulin resistance in different patients.
3.3.3. Interaction of cardiovascular disease and NAFLD
Although advanced forms of NAFLD associate with increased risk of coronary artery disease, there is no evidence proving that accumulation of liver fat causes atherosclerosis, as recently reviewed in depth by one of the authors. 83 Indeed, PNPLA3 I148M may be associated with a very small reduction in the risk of ischaemic heart disease, with odds ratios of 0.98 (95% CI: 0.96, 1.00; P = 0.79) in a recent large meta‐analysis (N = 279 013) 84 and 0.96 (0.94, 0.97; P = 4 × 10−8) in a recent large exome‐wide lipidomic study (N > 300 000). 85 PNPLA3 I148M was associated with liver‐related and all‐cause mortality but not with cardiovascular mortality, in a retrospective US general population survey with median follow‐up of 23 years. 28 Circulating triglyceride and LDL cholesterol levels are reduced or unchanged in PNPLA3 I148M carriers compared with noncarriers in multiple studies. 85 , 86 , 87 , 88 Furthermore, NAFLD itself increases mortality, at least in populations with high rates of obesity. 89 The evidence therefore indicates that the association of NAFLD with coronary artery disease is mainly due to shared underlying risk factors, depending on the pathophysiology of NAFLD.
3.4. Potential pathways for targeted therapeutic manipulation
The genetic variants most robustly associated with development of NAFLD (Table 1) highlight pathophysiological processes that may represent new targets for therapeutic intervention in patients with NASH. These include lipid remodelling in lipid droplets, hepatic VLDL secretion and de novo lipogenesis.
3.4.1. PNPLA3 and lipid droplet remodelling
The molecular mechanisms underpinning the strong association of the common PNPLA3 I148M variant with NAFLD are the most well characterised among the genetic associations identified to date. Section 4 is entirely devoted to PNPLA3 I148M and the rationale for a precision medicine that can modulate PNPLA3 expression in patients with NASH carrying the variant. PNPLA3 148M protein acts as a trans‐repressor of hepatocyte lipid droplet lipase activity by competing for a shared co‐activator, leading to lipid accumulation in hepatocytes. 90 , 91 PNPLA3 I148M also impairs retinol production by hepatic stellate cells. 90 Section 4.2 and Figure 1 present current understanding of how PNPLA3 I148M drives the pathogenesis of NASH in people with overweight or obesity.
Figure 1.
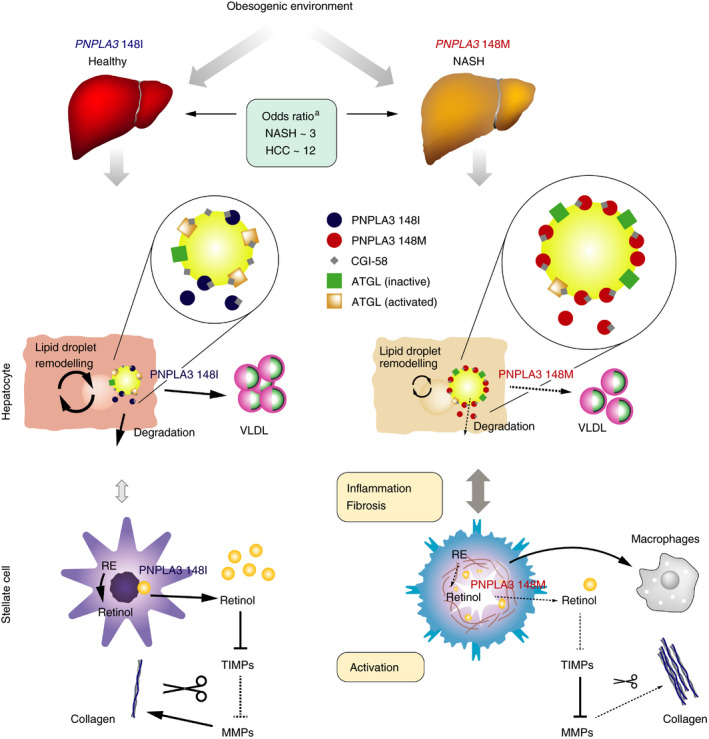
Role of PNPLA3 in the pathophysiology of NASH. Abbreviations: ATGL, adipose triglyceride lipase; HCC, hepatocellular carcinoma; MMP, matrix metalloproteinase; NASH, non‐alcoholic steatohepatitis; RE, retinol esters; TIMP, tissue inhibitor of metalloproteinase; VLDL, very low‐density lipoprotein. aLiu et al 27
Evidence that reduced expression of PNPLA3 attenuates the effect of the I148M variant on liver fat levels is also provided by genetic association studies. Another SNP in PNPLA3 associated with NAFLD (rs2294918 c.1300 G > A p.E434K) encodes a glutamate to lysine substitution in PNPLA3 protein (E434K). 92 In vitro, E434K has no effect on the enzymatic activity of PNPLA3, but it is associated with reduced levels of PNPLA3 mRNA. People who inherit E434K together with I148M variant are partially protected from the NASH‐promoting effect of I148M, as seen in a genetic study of associations with ALT levels. 92
3.4.2. TM6SF2 and VLDL secretion
TM6SF2 plays a role in the pathway for hepatic VLDL secretion. 34 , 38 Selective knockdown of TM6SF2 protein expression in mice led to a threefold increase in liver triglyceride content and a 50% decrease in VLDL secretion, indicating that TM6SF2 normally promotes VLDL secretion. 34 In people with the TM6SF2 E167K variant, loss of function of TM6SF2 may lead to increased hepatic triglyceride content. 34 TM6SF2 E167K protein was associated with increased de novo lipogenesis and reduced secretion of apolipoprotein B particles in a recent study using 3D spheroid cultures of primary human hepatocytes. 37 A precision medicine able to restore deficient TM6SF2 activity in E167K carriers with NASH might also increase hepatic VLDL secretion and reduce triglyceride levels in the liver. This approach might, however, elevate the risk of adverse cardiovascular events, which is reduced in E167K carriers. 35 The modest odds ratios for the association of TM6SF2 E167K with NASH suggest that any potential therapeutic benefit associated with restored function may also be modest. Furthermore, TM6SF2 E167K is rare, so only a small population of patients would be targetable (Table 1).
3.4.3. GCKR and de novo lipogenesis
GCKR is a fructose‐6‐phosphate‐dependent inhibitor of glucokinase involved in regulating de novo lipogenesis. 42 , 93 The GCKR P446L variant disrupts negative regulation of glucokinase by GCKR in response to fructose‐6‐phosphate, leading to constitutive glucokinase activation. 38 , 42 This increases hepatic glucose uptake, glucose metabolism and malonyl CoA production. 38 , 42 Malonyl CoA is a substrate for de novo lipogenesis and blocks fatty acid oxidation (via inhibition of carnitine‐palmitoyltransferase), and thereby favours hepatic fat accumulation. 38 , 42 These findings may explain the association of GCKR P446L with hepatic steatosis and increased susceptibility to NASH. 45 In vitro and animal model data on whether this pathway may be amenable to modulation with a precision medicine are currently lacking.
3.4.4. MBOAT7 and phospholipid remodelling
MBOAT7 is a membrane‐anchored enzyme with six transmembrane domains and is involved in remodelling endomembrane phospholipid acyl chains. 47 , 94 MBOAT7 expression levels are reduced in people with obesity and in rodent models of obesity compared with controls. 95 The rs641738 variant of MBOAT7 may predispose to NAFLD and NASH by changing the acyl remodelling of phospholipids in the liver. 47 Some genetic studies have failed to detect an association of this variant with NAFLD, most likely because they were underpowered to detect the small effect size. 40 The details of the enzymatic activity of MBOAT7, including the preferential acyl donor and phospholipid substrate, are subjects of research. 94 Recent findings in rodent models suggest that acetylation of lysophosphatidylinositol lipids by MBOAT7 may play a protective role against development of liver steatosis in an obesogenic environment. 95
3.4.5. HSD17B13 and lipid droplets
HSD17B13 has been identified as a hepatic lipid droplet‐associated protein with retinol dehydrogenase activity, and the variants that protect against NAFLD confer loss of this enzymatic activity. 52 , 54 The physiological function of HSD17B13 is not well characterised, but other members of the hydroxysteroid 17‐β dehydrogenase family are involved in steroid and fatty acid metabolism. 52 , 96 A role for HSD17B13 in oestradiol metabolism has been proposed, and it has enzymatic activity against bioactive lipid mediators, such as leukotriene B4, which are involved in lipid‐mediated inflammation. 52 Whether loss of HSD17B13 retinol dehydrogenase activity directly affects retinoic acid homeostasis within hepatocytes to modulate retinol levels is the subject of ongoing research. 54 Section 4.2 provides further detail on the role of retinol in modulating hepatic stellate cell fibrogenesis.
Significant upregulation of HSD17B13 protein expression has been observed in the livers of patients with NAFLD, and hepatic overexpression of human HSD17B13 led to a fatty liver phenotype in C57BL/6 mice. 97 The rs72613567 TA loss‐of‐function variant of HSD17B13 was associated with a reduced risk of NASH in human liver samples. 52 It also mitigated liver injury in people genetically predisposed to hepatic steatosis by PNPLA3 I148M and was associated with reduced PNPLA3 mRNA expression. 52 In patients with functional variants of HSD17B13, carriers of PNPLA3 I148M might be a relevant subpopulation for potential therapeutic inhibition of the activity or expression of HSD17B13. 52
4. PNPLA3 PRECISION MEDICINE IN PATIENTS WITH NASH
4.1. Global prevalence of NASH and PNPLA3 I148M
An increasingly large number of people could benefit from precision medicine approaches to treating NASH in carriers of PNPLA3 I148M. NASH prevalence is predicted to rise from 2.4% in China, 3.6%–4.4% in five European countries and 5.3% in the US in 2016 to 3.4% in China, 5.0%–6.3% in five European countries and 7.6% in the US in 2030, 6 with a similar rise predicted in Saudi Arabia (4.2% to 6.8% from 2017 to 2030). 98 Figure 2 combines these data with current PNPLA3 genotype frequencies, based on the 1000 Genomes Project and other published data. 6 , 21 , 33 , 98 , 99 , 100 , 101 , 102 , 103 In China, Japan, Germany, Italy, the UK and the US, the total population of PNPLA3 148MM homozygotes with NASH will increase from about 12.5 million to over 18 million by 2030 (Figure 2).
Figure 2.
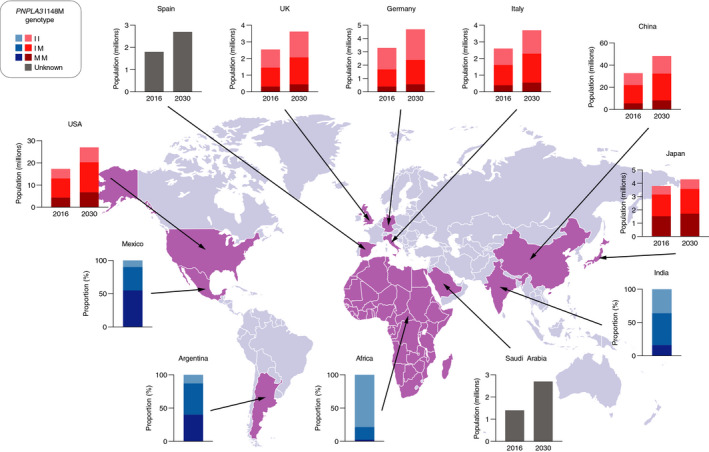
Worldwide prevalence of NASH and PNPLA3 I148M genotypes approximated from their frequencies in patients with NAFLD. Abbreviations: NAFLD, non‐alcoholic fatty liver disease; NASH, non‐alcoholic steatohepatitis; PNPLA3 I148M, rs738409 c.444 C > G p.I148M. Green: only NASH prevalence data available. Blue: only PNPLA3 genotype data available. Red: NASH prevalence and PNPLA3 genotype data available. Data are approximations based on published NASH prevalence estimates and genotype frequencies in patients with NAFLD as follows: Africa, 1000 Genomes Project (general population) 33 ; Argentina, calculated from allele frequency 21 , 99 ; China, data from Peng 6 , 99 ; France 6 ; Germany, data from Kantartzis 99 , 100 ; India, calculated from allele frequency 101 ; Italy, data from Valenti 6 , 99 ; Japan, data from Kitamoto 6 , 99 ; Mexico 102 ; Saudi Arabia, data from 2017 98 ; Spain 6 ; UK 103 ; US, data from Rotman and Speliotes and calculated from allele frequency. 6 , 99 Africa includes Esan in Nigeria (II, 78%; IM, 19%; MM, 3%), Gambia (II, 81%; IM, 18%; MM, 2%), Luhya in Kenya (II, 85%; IM, 13%; MM, 2%), Mende in Sierra Leone (II, 80%; IM, 18%; MM, 2%), Yoruba in Nigeria (II, 78%; IM, 21%; MM, 1%), Afro‐Caribbean in Barbados (II, 76%; IM, 22%; MM, 2%) and African ancestry in south‐west US (II, 72%; IM, 21%; MM, 7%) 33
South America and the Middle East have the highest prevalence of NAFLD globally (>30%). 1 , 104 The frequency of PNPLA3 I148M genotypes is particularly high in Mexico and Latin America (Figure 2), and in populations of Latin American origin in the US (48% MM, 42% IM, 10% II). 99 Further epidemiological and genetic studies are needed to understand the increasing prevalence of NASH and the contribution of genetic variability in the many different heritage groups in South America 105 and elsewhere.
4.2. PNPLA3 structure and function in health and disease
PNPLA3 is a member of a family of patatin‐domain containing lipid hydrolases with specificity for an array of different substrates, including triacylglycerols, phospholipids and retinol esters. 106 PNPLA3 expression levels are highest in hepatocytes and hepatic stellate cells in humans and in adipocytes in mice, followed by the retina and other tissues in both species. 22 High carbohydrate levels upregulate PNPLA3 levels in mice 107 , 108 and in human hepatocytes 109 , 110 by increasing transcription and reducing protein turnover. In contrast, fasting downregulates PNPLA3 levels. 111 Knockdown of wild‐type Pnpla3 expression in rats on a high‐fat diet reduced liver fat content by diminishing fatty acid esterification, 112 consistent with data showing that Pnpla3 overexpression can promote lipogenesis in mammalian cells in vitro. 113
In vitro studies indicate that PNPLA3 localises to the surface of lipid droplets, 114 has triglyceride lipase activity, 115 , 116 , 117 and is involved in lipid remodelling and hepatic retention of polyunsaturated fatty acids (Figure 1). 77 , 118 Recent data indicate that the enzymatic activity of PNPLA3 also mediates the transfer of polyunsaturated fatty acids from triglycerides to phospholipids in hepatocytes, 119 with potentially broad effects on hepatic lipid metabolism. Furthermore, PNPLA3 has retinyl‐palmitate lipase activity in vitro and is involved in retinol release by hepatic stellate cells. 120
Evidence indicates that the NASH‐associated I148M substitution abolishes the enzymatic activity of PNPLA3. This includes both the in vitro triglyceride lipase activity 114 , 117 , 121 and the ex vivo lipid remodelling activity. 119 Molecular dynamics simulations show that the substitution of methionine for isoleucine at position 148 prevents access of fatty acid substrates to the catalytic dyad (serine 47 and aspartate 166). 122
Although lipolysis and VLDL secretion are reduced in PNPLA3 I148M hepatocytes, 110 , 123 hepatic fat accumulation cannot be explained by an absence of PNPLA3 protein. Instead, impairment of hepatic triglyceride mobilisation results from build‐up of PNPLA3 on lipid droplets (Figure 1). Genetic deletion of the mouse Pnpla3 gene from conception does not influence hepatic fat accumulation. 124 , 125 Hepatic steatosis develops in mice overexpressing exogenous human PNPLA3 148M, but not those overexpressing the ancestral 148I protein. 126 Hepatic steatosis also develops in knock‐in mice carrying the I148M variant in endogenous mouse Pnpla3 when they are fed a high‐sucrose diet, 127 and this can be ameliorated by silencing Pnpla3 expression with antisense oligonculeotides. 128 Furthermore, PNPLA3 protein levels on the surface of lipid droplets are higher for 148M than 148I in both the PNPLA3‐overexpressing and knock‐in mouse models (Figure 1). This difference results from decreased ubiquitination and proteasomal degradation of PNPLA3 148M, 129 not from increased mRNA levels. 126 , 127
Increased PNPLA3 levels on lipid droplets appear to reduce hepatic lipolysis via sequestration of a lipase cofactor, CGI‐58 (also known as 1‐acylglycerol‐3‐phosphate O‐acyltransferase, or ABHD5). Enzymatically inactive PNPLA3 148M is still able to bind CGI‐58 and prevent it from activating other lipases present on lipid droplets. 91 Competition for CGI‐58 between PNPLA3 and adipose triglyceride lipase (ATGL, encoded by PNPLA2) has been reported in brown adipocytes. 130 Furthermore, a loss‐of‐function variant in ABHD5 (encoding CGI‐58) is associated with a rare autosomal dominant form of inherited NAFLD. 131 A ubiquitination‐resistant but fully enzymatically active variant of PNPLA3 has recently been shown to accumulate on lipid droplets and increase hepatic triglyceride levels in transgenic mice. 132 Increased de novo lipogenesis does not appear to be involved in the effect of PNPLA3 148M. 133 , 134 Reduced degradation of lipid droplets via autophagy is one potential mechanism for the increase in hepatic triglyceride levels in the presence of PNPLA3 148M. 135
Current understanding is therefore that the association of PNPLA3 I148M with hepatic steatosis may result less from direct loss of PNPLA3 lipase/lipid remodelling activity than from indirect reduction of CGI58‐mediated ATGL activity. PNPLA3 148M acts as a trans‐repressor of lipid droplet lipase activity by competing for a shared co‐activator, and this trans‐repression is what causes hepatic lipid accumulation (Figure 1), rather than the loss of PNPLA3 enzymatic activity itself. 91
PNPLA3 I148M may also disrupt retinol release by hepatic stellate cells, potentially leading to fibrosis (Figure 1). PNPLA3 promotes release of retinol by hepatic stellate cells in response to insulin and transforming growth factor β in vitro. 120 , 136 The I148M variant is associated with a loss of retinyl‐palmitate lipase activity and a resulting impairment in retinol production by hepatic stellate cells. 120 , 136 Impaired retinoid production may lead to reduced secretion of matrix metalloproteinases and tissue inhibitors of metalloproteinase, resulting in extracellular matrix deposition (Figure 1). 136 , 137 Hepatic stellate cells expressing PNPLA3 148M also secrete elevated levels of pro‐inflammatory cytokines, which may potentiate their fibrogenic potential compared with ancestral PNPLA3 148I. 137 In agreement with these in vitro findings, PNPLA3 I148M was associated with reduced circulating retinol and increased intrahepatic retinol levels in individuals with NAFLD or obesity. 138 , 139 Furthermore, reduced levels of retinoic acid metabolites in the liver may promote activation of hepatic stellate cells by macrophages in response to internalisation of apoptotic cells (mediated by c‐mer tyrosine kinase). 140
The effects of PNPLA3 I148M on lipid droplet remodelling in hepatocytes and retinol production by hepatic stellate cells (Figure 1) suggest that a precision medicine able to reduce PNPLA3 levels could provide therapeutic benefits to I148M‐carrying patients with NASH. Silencing of Pnpla3 expression with hepatocyte‐targeted N‐acetylgalactosamine‐conjugated antisense oligonucleotides ameliorated steatohepatitis and liver fibrosis in homozygous Pnpla3 148MM knock‐in mice, but not in wild‐type Pnpla3 148II littermates fed a NASH‐inducing diet. 128 Furthermore, liver steatosis was reduced in Pnpla3 148MM knock‐in mice fed a high‐fructose diet following either knockdown of Pnpla3 expression with short hairpin RNA (expressed from an adeno‐associated virus vector), or lowering of PNPLA3 protein levels with a proteolysis‐targeting chimera (PROTAC). 132 , 141
Modulating levels of PNPLA3 or its interaction with lipase cofactors with these or other approaches could provide routes to therapeutic intervention in PNPLA3 I148M‐carrying patients with NASH, as previously suggested. 142 Reduced levels of PNPLA3 148M protein may, however, have broad effects on lipid metabolism or may be compensated for by multiple mechanisms. Improved understanding of downstream mediators could also provide potential routes to therapeutic intervention in patients with NASH, including those not carrying PNPLA3 I148M.
4.3. Impact of PNPLA3 I148M on treatment of NASH
4.3.1. PNPLA3 I148M and response to treatment
No pharmacological therapies are approved for the treatment of NASH, and liver transplantation is the only available treatment for cirrhosis. 143 , 144 Guidelines recommend reducing body weight through lifestyle interventions, dietary restriction and physical activity, 145 but the effectiveness of lifestyle interventions is often limited and short term. 146 , 147 Bariatric surgery can significantly reduce hepatic steatosis, steatohepatitis and fibrosis in patients with obesity, as detailed in two recent meta‐analyses, 148 , 149 but it carries a significant risk of complications. 145
Very limited evidence suggests that PNPLA3 I148M may modulate the response to treatment in patients with NASH. Lifestyle modification and bariatric surgery have been reported to be more effective in reducing liver fat levels in PNPLA3 I148M carriers than in noncarriers (Table 2). 61 , 150 , 151 , 152 In contrast, omega‐3 fatty acid supplementation may be less effective in decreasing liver fat levels in PNPLA3 I148M carriers than in noncarriers in randomised‐controlled trials (Table 3). 153 , 154 , 155 , 156 An increased effect of high dietary omega‐6 to omega‐3 polyunsaturated fatty acid ratio on liver fat levels has also been reported in homozygotes (Table 3). 157 The modest effect size of omega‐3 fatty acids on liver fat levels in clinical trials and the small number of homozygous participants make the magnitude of the genotypic effect difficult to quantify.
Table 2.
Interaction of weight‐loss interventions with PNPLA3 genotype in NAFLD or obesity
| Reference | Participants | Interventions | Study design | Main liver‐related outcome | PNPLA3 I148M association |
|---|---|---|---|---|---|
| Shen et al150 | Adults with NAFLD (N = 154) | 12‐mo dietician‐led programme or standard care | Parallel‐group | Greater decrease in liver fat a than standard care | Increased reduction in liver fat a in the intervention group |
| Krawczyk et al151 | Adults with suspected NAFLD (N = 143) | 4‐mo dietician‐led programme | Prospective, observational | Significant decrease in liver fat b from baseline | None |
| Krawczyk et al152 | Adults with obesity (N = 84) | Bariatric surgery | Prospective, observational | Decrease from baseline in liver fat c | Increased reduction in liver fat c |
| Palmer et al61 | Adults with obesity (N = 3473) | Bariatric surgery | Parallel‐group | Reduced BMI and serum triglycerides in surgery group only d | Increased reduction in BMI and serum triglycerides |
Abbreviations: BMI, body mass index; NAFLD, non‐alcoholic fatty liver disease.
Magnetic resonance spectroscopy.
Ultrasonography.
Magnetic resonance imaging proton density fat fraction.
Liver fat not assessed.
Table 3.
Interaction of omega‐3 fatty acid intake with PNPLA3 genotype in NAFLD
| Reference | Participants | Interventions | Study design | Main liver‐related outcome | PNPLA3 I148M association |
|---|---|---|---|---|---|
| Oscarsson et al155 | Adults with NAFLD and lipidaemia (N = 78) | ω‐3 carboxylic acids, fenofibrate or placebo | Randomised, double‐blind, parallel‐group | No significant effect on liver fat a versus placebo | No effect on response to either treatment |
| Eriksson et al156 | Adults with NAFLD and T2DM (N = 84) | Dapagliflozin, ω‐3 carboxylic acids, both, or placebo | Randomised, double‐blind, parallel‐group | Decreased liver fat a with all active treatments versus placebo | Trend towards reduced response to treatment with ω‐3 carboxylic acids |
| Scorletti et al154 | Adults with NAFLD (N = 103) | ω‐3 ethyl esters (EPA + DHA) or placebo | Randomised, double‐blind, parallel‐group | Association between DHA enrichment and decreased liver fat b | Reduced response to treatment |
| Nobili et al153 | Children with NAFLD (N = 60) | DHA or placebo | Randomised, double‐blind, parallel‐group | Decreased liver fat c versus placebo | Reduced response to treatment |
| Santoro et al157 | Children and adolescents with obesity (N = 127) | None | Genetic and dietary association | Dietary ω‐6/ω‐3 PUFA ratio associated with liver fat a | Increased effect of high dietary ω‐6/ω‐3 PUFA ratio in homozygotes |
Abbreviations: DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; NAFLD, non‐alcoholic fatty liver disease; PUFA, polyunsaturated fatty acid; ω, omega.
Magnetic resonance imaging proton density fat fraction.
Magnetic resonance spectroscopy.
Ultrasonography.
Associations of PNPLA3 I148M with reduced protective effects of statins on steatosis and NASH in clinical trials have been reported. 156 , 158 PNPLA3 I148M has also been associated with an increased risk of ALT elevation in patients receiving potentially hepatotoxic medications, such as asparaginase for acute lymphoblastic leukaemia, 159 , 160 or diabetes medications that can increase liver fat levels. 161 , 162
Although patients with NASH carrying PNPLA3 I148M may lose more liver fat than noncarriers after a successful intervention, they are also likely to start from a worse pre‐treatment baseline. Patients may benefit most from personalised therapies that act upstream to reduce the liver fat accumulation associated with the variant.
4.3.2. Risk stratification based on PNPLA3 I148M genotype
Guidelines recommend that people with important risk factors such as type 2 diabetes, insulin resistance, obesity and metabolic syndrome should be screened for NASH because of its prognostic implications. 145 While recognising that PNPLA3 I148M may allow risk stratification for tailored HCC surveillance, guidelines do not recommend routine genotyping of any variant. 145 The recent association of PNPLA3 I148M with both liver‐related and all‐cause mortality in a general US population sample highlights the unmet need for effective targeted therapies to prevent disease progression and death in carriers. 28 , 89
Polygenic risk scores adjusted for conventional risk factors may, in the future, have the potential to guide care of patients with NAFLD. Age, sex, BMI, fasting glucose levels and risk variants in PNPLA3, TM6SF2 and MBOAT7 all emerged as independent predictors of liver damage in a logistic regression analysis, with an additive effect of the genetic variants on hepatic triglyceride content. 47 Polygenic risk scores for coronary artery disease, however, are the furthest advanced but have still not yet entered routine clinical practice. 163 Additional studies and evidence are needed before speculating on whether genotyping could be used to guide treatment decisions in patients at risk of NASH.
5. CONCLUSION
The identification of genes associated with development and progression of NASH provides important insights into the pathophysiology that may in time provide novel opportunities for therapeutic intervention. Genetic discoveries provided the impetus for cell and molecular biological studies aiming to elucidate the mechanism responsible for the association between genetic variants and liver disease progression. PNPLA3 148M acts as a trans‐repressor of lipid droplet lipase activity by competing for a shared co‐activator. This indicates that reducing PNPLA3 expression levels could potentially attenuate its negative effect on hepatic lipolysis. Consistent with this possibility, people with a genetic variant that reduces PNPLA3 expression levels are less susceptible to the effect of I148M on liver fat than those without the expression‐reducing variant. Knowledge of the underlying mechanisms remains incomplete, with current research focusing on cofactor recruitment to lipid droplets and lipidomic analyses of alterations to lipid metabolism in hepatocytes and hepatic stellate cells. Despite these uncertainties, an aetiological distinction can be drawn between NASH associated with PNPLA3 I148M and other forms of NASH that are primarily driven by insulin resistance. This possibility presents opportunities for the development of a precision medicine that can modulate the activity of a specific gene (PNPLA3) in a specific organ (the liver) of a specific group of patients (I148M carriers with NASH). Other genes associated with NASH, including HSD17B13, may provide future targets for intervention strategies. All novel therapies require extensive assessment of safety and efficacy in clinical trials. Progress towards proof‐of‐concept studies of a precision medicine for patients with NASH is ultimately driven by the robust human genetic and molecular and cell biological evidence base.
AUTHORSHIP
Guarantors of the article: Stefano Romeo and Rohit Loomba act as guarantors of the article and take responsibility for the integrity of the work.
Author contributions: All authors conceived the review, revised the manuscript critically for important intellectual content and approved the final version including the authorship list.
ACKNOWLEDGEMENTS
Declarations of personal interests: Björn Carlsson, Daniel Lindén, Gabriella Brolén, Mathias Liljeblad and Mikael Bjursell are employees of AstraZeneca and own stock in AstraZeneca. Stefano Romeo has served as a speaker, a consultant or an advisory board member for Akcea, Amgen, AstraZeneca, CAMP4, Foresite Labs, GlaxoSmithKline, MEDACorp, Mercodia, Pfizer and Sanofi; and his institution has received research funding from Amgen, AstraZeneca and Sanofi. Rohit Loomba is co‐founder of Liponexus, Inc and has served as a speaker, a consultant or an advisory board member for Arrowhead Pharmaceuticals, AstraZeneca, Bird Rock Bio, Boehringer Ingelheim, Bristol‐Myers Squibb, Celgene, Cirius, CohBar, Conatus, Eli Lilly and Company, Galmed, Gemphire, Gilead, Glympse Bio, GNI, GRI Bio, Intercept, Ionis, Janssen, Merck, Metacrine, Inc, NGM Biopharmaceuticals, Novartis, Novo Nordisk, Pfizer, Prometheus, Sanofi, Siemens and Viking Therapeutics; and his institution has received research funding from Allergan, Boehringer Ingelheim, Bristol‐Myers Squibb, Cirius, Eli Lilly and Company, Galectin Therapeutics, Galmed, GE, Genfit, Gilead, Grail, Intercept, Janssen, Madrigal Pharmaceuticals, Merck, NGM Biopharmaceuticals, NuSirt, Pfizer, pH Pharma, Prometheus and Siemens.
Carlsson B, Lindén D, Brolén G, et al. Review article: the emerging role of genetics in precision medicine for patients with non‐alcoholic steatohepatitis. Aliment Pharmacol Ther. 2020;51:1305–1320. 10.1111/apt.15738
The Handling Editor for this article was Dr Stephen Ryder, and this uncommissioned review was accepted for publication after full peer‐review.
Funding information
Under the direction of the authors, Dr Matt Cottingham of Oxford PharmaGenesis provided medical writing support funded by AstraZeneca. Although Dr Cottingham was involved in drafting and revising the paper, he does not meet the ICMJE authorship criteria because he did not contribute to the conception or design of the work or the acquisition, analysis, or interpretation of data for the work. AstraZeneca develops and markets treatments for metabolic diseases. AstraZeneca reviewed this publication, without influencing the opinions of the authors, to ensure medical and scientific accuracy, and to protect intellectual property. The corresponding authors had the final responsibility for the decision to submit the manuscript for publication.
Contributor Information
Stefano Romeo, Email: stefano.romeo@wlab.gu.se.
Rohit Loomba, Email: roloomba@ucsd.edu.
REFERENCES
- 1. Younossi ZM, Koenig AB, Abdelatif D, et al. Global epidemiology of nonalcoholic fatty liver disease – meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73‐84. [DOI] [PubMed] [Google Scholar]
- 2. Singh S, Allen AM, Wang Z, et al. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta‐analysis of paired‐biopsy studies. Clin Gastroenterol Hepatol. 2015;13:643‐654.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263‐2273. [DOI] [PubMed] [Google Scholar]
- 4. Milic S, Lulic D, Stimac D. Non‐alcoholic fatty liver disease and obesity: biochemical, metabolic and clinical presentations. World J Gastroenterol. 2014;20:9330‐9337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of non‐alcoholic fatty liver disease: practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55:2005‐2023. [DOI] [PubMed] [Google Scholar]
- 6. Estes C, Anstee QM, Arias‐Loste MT, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J Hepatol. 2018;69:896‐904. [DOI] [PubMed] [Google Scholar]
- 7. Goldberg D, Ditah IC, Saeian K, et al. Changes in the prevalence of hepatitis C virus infection, nonalcoholic steatohepatitis, and alcoholic liver disease among patients with cirrhosis or liver failure on the waitlist for liver transplantation. Gastroenterology. 2017;152:1090‐1099.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cholankeril G, Wong RJ, Hu M, et al. Liver transplantation for nonalcoholic steatohepatitis in the US: temporal trends and outcomes. Dig Dis Sci. 2017;62:2915‐2922. [DOI] [PubMed] [Google Scholar]
- 9. McPherson S, Hardy T, Henderson E, et al. Evidence of NAFLD progression from steatosis to fibrosing‐steatohepatitis using paired biopsies: implications for prognosis and clinical management. J Hepatol. 2015;62:1148‐1155. [DOI] [PubMed] [Google Scholar]
- 10. Povsic M, Wong OY, Perry R, Bottomley J. A structured literature review of the epidemiology and disease burden of non‐alcoholic steatohepatitis (NASH). Adv Ther. 2019;36:1574‐1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non‐alcoholic fatty liver disease and non‐alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274‐285. [DOI] [PubMed] [Google Scholar]
- 12. Williams CD, Stengel J, Asike MI, et al. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle‐aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124‐131. [DOI] [PubMed] [Google Scholar]
- 13. Loomba R, Schork N, Chen C‐H, et al. Heritability of hepatic fibrosis and steatosis based on a prospective twin study. Gastroenterology. 2015;149:1784‐1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Caussy C, Soni M, Cui J, et al. Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J Clin Invest. 2017;127:2697‐2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sookoian S, Pirola CJ. Genetics of nonalcoholic fatty liver disease: from pathogenesis to therapeutics. Semin Liver Dis. 2019;39:124‐140. [DOI] [PubMed] [Google Scholar]
- 16. Valenti LVC, Baselli GA. Genetics of nonalcoholic fatty liver disease: a 2018 update. Curr Pharm Des. 2018;24:4566‐4573. [DOI] [PubMed] [Google Scholar]
- 17. Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Singal AG, Manjunath H, Yopp AC, et al. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: a meta‐analysis. Am J Gastroenterol. 2014;109:325‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dongiovanni P, Donati B, Fares R, et al. PNPLA3 I148M polymorphism and progressive liver disease. World J Gastroenterol. 2013;19:6969‐6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sookoian S, Pirola CJ. Meta‐analysis of the influence of I148M variant of patatin‐like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53:1883‐1894. [DOI] [PubMed] [Google Scholar]
- 22. Bruschi FV, Tardelli M, Claudel T, Trauner M. PNPLA3 expression and its impact on the liver: current perspectives. Hepat Med. 2017;9:55‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goyal NP, Schwimmer JB. The genetics of pediatric nonalcoholic fatty liver disease. Clin Liver Dis. 2018;22:59‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salameh H, Hanayneh MA, Masadeh M, et al. PNPLA3 as a genetic determinant of risk for and severity of non‐alcoholic fatty liver disease spectrum. J Clin Transl Hepatol. 2016;4:175‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grimaudo S, Pipitone RM, Pennisi G, et al. Association between PNPLA3 rs738409 C>G Variant and liver‐related outcomes in patients with non‐alcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2019;18:935‐944.e3. [DOI] [PubMed] [Google Scholar]
- 26. Burza MA, Pirazzi C, Maglio C, et al. PNPLA3 I148M (rs738409) genetic variant is associated with hepatocellular carcinoma in obese individuals. Dig Liver Dis. 2012;44:1037‐1041. [DOI] [PubMed] [Google Scholar]
- 27. Liu YL, Patman GL, Leathart JB, et al. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non‐alcoholic fatty liver disease associated hepatocellular carcinoma. J Hepatol. 2014;61:75‐81. [DOI] [PubMed] [Google Scholar]
- 28. Unalp‐Arida A, Ruhl CE. Patatin‐like phospholipase domain‐containing protein 3 I148M and liver fat and fibrosis scores predict liver disease mortality in the United States population. Hepatology. 2019;71:820‐834. [DOI] [PubMed] [Google Scholar]
- 29. Kitamoto T, Kitamoto A, Yoneda M, et al. Genome‐wide scan revealed that polymorphisms in the PNPLA3, SAMM50, and PARVB genes are associated with development and progression of nonalcoholic fatty liver disease in Japan. Hum Genet. 2013;132:783‐792. [DOI] [PubMed] [Google Scholar]
- 30. Speliotes EK, Yerges‐Armstrong LM, Wu J, et al. Genome‐wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chambers JC, Zhang W, Sehmi J, et al. Genome‐wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. 2011;43:1131‐1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krawczyk M, Stokes CS, Romeo S, Lammert F. HCC and liver disease risks in homozygous PNPLA3 p.I148M carriers approach monogenic inheritance. J Hepatol. 2015;62:980‐981. [DOI] [PubMed] [Google Scholar]
- 33. Ensembl. rs738409 (SNP) ‐ Population genetics ‐ Homo sapiens, 2019. http://www.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=22:43928347‐43929347;v=rs738409;vdb=variation;vf=64400301
- 34. Kozlitina J, Smagris E, Stender S, et al. Exome‐wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46:352‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dongiovanni P, Petta S, Maglio C, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology. 2015;61:506‐514. [DOI] [PubMed] [Google Scholar]
- 36. Liu Y‐L, Reeves HL, Burt AD, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non‐alcoholic fatty liver disease. Nat Commun. 2014;5:4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Prill S, Caddeo A, Baselli G, et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci Rep. 2019;9:11585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: clinical impact. J Hepatol. 2018;68:268‐279. [DOI] [PubMed] [Google Scholar]
- 39. Chen X, Zhou P, De L, Li B, Su S. The roles of transmembrane 6 superfamily member 2 rs58542926 polymorphism in chronic liver disease: a meta‐analysis of 24,147 subjects. Mol Genet Genomic Med. 2019;7:e824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xia Y, Huang CX, Li GY, et al. Meta‐analysis of the association between MBOAT7 rs641738, TM6SF2 rs58542926 and nonalcoholic fatty liver disease susceptibility. Clin Res Hepatol Gastroenterol. 2019;43:533‐541. [DOI] [PubMed] [Google Scholar]
- 41. Ensembl. rs58542926 (SNP) – Population genetics – Homo sapiens, 2019. http://www.ensembl.org/Homo_sapiens/Variation/Explore?db=core;r=19:19268240‐19269240;v=rs58542926;vdb=variation;vf=142541937
- 42. Santoro N, Zhang CK, Zhao H, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology. 2012;55:781‐789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Raimondo A, Rees MG, Gloyn AL. Glucokinase regulatory protein: complexity at the crossroads of triglyceride and glucose metabolism. Curr Opin Lipidol. 2015;26:88‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kawaguchi T, Shima T, Mizuno M, et al. Risk estimation model for nonalcoholic fatty liver disease in the Japanese using multiple genetic markers. PLoS ONE. 2018;13:e0185490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tan H‐L, Zain SM, Mohamed R, et al. Association of glucokinase regulatory gene polymorphisms with risk and severity of non‐alcoholic fatty liver disease: an interaction study with adiponutrin gene. J Gastroenterol. 2014;49:1056‐1064. [DOI] [PubMed] [Google Scholar]
- 46. Ensembl. rs1260326 (SNP) – Population genetics – Homo sapiens, 2019. http://www.ensembl.org/Homo_sapiens/Variation/Explore?db=core;r=2:27507573‐27508573;v=rs1260326;vdb=variation;vf=194662350
- 47. Mancina RM, Dongiovanni P, Petta S, et al. The MBOAT7‐TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology. 2016;150:1219‐1230.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Donati B, Dongiovanni P, Romeo S, et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non‐cirrhotic individuals. Sci Rep. 2017;7:4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sookoian S, Flichman D, Garaycoechea ME, et al. Lack of evidence supporting a role of TMC4‐rs641738 missense variant – MBOAT7‐intergenic downstream variant – in the susceptibility to nonalcoholic fatty liver disease. Sci Rep. 2018;8:5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Buch S, Stickel F, Trépo E, et al. A genome‐wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol‐related cirrhosis. Nat Genet. 2015;47:1443‐1448. [DOI] [PubMed] [Google Scholar]
- 51. Ensembl. rs641738 (SNP) – Population genetics – Homo sapiens, 2019. http://www.ensembl.org/Homo_sapiens/Variation/Explore?db=core;v=rs641738;vdb=variation
- 52. Abul‐Husn NS, Cheng X, Li AH, et al. A protein‐truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med. 2018;378:1096‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kozlitina J, Stender S, Hobbs HH, Cohen JC HSD17B13 and chronic liver disease in blacks and hispanics. N Engl J Med. 2018;379:1876‐1877. [DOI] [PubMed] [Google Scholar]
- 54. Ma Y, Belyaeva OV, Brown PM, et al. 17‐ß‐hydroxysteroid dehydrogenase 13 is a hepatic retinol dehydrogenase associated with histological features of nonalcoholic fatty liver disease. Hepatology. 2019;69:1504‐1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gellert‐Kristensen H, Nordestgaard BG, Tybjaerg‐Hansen A, Stender S. High risk of fatty liver disease amplifies the alanine transaminase‐lowering effect of a HSD17B13 variant. Hepatology. 2019;71:56‐66 [DOI] [PubMed] [Google Scholar]
- 56. Yang J, Trepo E, Nahon P, et al. A 17‐ß‐hydroxysteroid dehydrogenase 13 variant protects from hepatocellular carcinoma development in alcoholic liver disease. Hepatology. 2019;70:231‐240. [DOI] [PubMed] [Google Scholar]
- 57. Pirola CJ, Garaycoechea M, Flichman D, et al. Splice variant rs72613567 prevents worst histologic outcomes in patients with nonalcoholic fatty liver disease. J Lipid Res. 2019;60:176‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ensembl. rs72613567 (SNP) ‐ Population genetics ‐ Homo sapiens, 2019. http://www.ensembl.org/Homo_sapiens/Variation/Explore?db=core;v=rs641738;vdb=variation
- 59. Ensembl. rs80182459 (DELETION) ‐ Population genetics ‐ Homo sapiens, 2019. http://www.ensembl.org/Homo_sapiens/Variation/Population?db=core;r=4:87313445‐87314445;v=rs80182459;vdb=variation;vf=251372883
- 60. Ensembl. rs62305723 (SNP) ‐ Population genetics ‐ Homo sapiens, 2019. http://www.ensembl.org/Homo_sapiens/Variation/Explore?r=4:87309777‐87310777;v=rs62305723;vdb=variation;vf=250939314
- 61. Palmer CN, Maglio C, Pirazzi C, et al. Paradoxical lower serum triglyceride levels and higher type 2 diabetes mellitus susceptibility in obese individuals with the PNPLA3 148M variant. PLoS ONE. 2012;7:e39362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Romeo S, Sentinelli F, Cambuli VM, et al. The 148M allele of the PNPLA3 gene is associated with indices of liver damage early in life. J Hepatol. 2010;53:335‐338. [DOI] [PubMed] [Google Scholar]
- 63. Romeo S, Sentinelli F, Dash S, et al. Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int J Obes (Lond). 2010;34:190‐194. [DOI] [PubMed] [Google Scholar]
- 64. Miraglia del Giudice E, Grandone A, Cirillo G, et al. The association of PNPLA3 variants with liver enzymes in childhood obesity is driven by the interaction with abdominal fat. PLoS ONE. 2011;6:e27933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Graff M, North KE, Franceschini N, et al. PNPLA3 gene‐by‐visceral adipose tissue volume interaction and the pathogenesis of fatty liver disease: the NHLBI family heart study. Int J Obes (Lond). 2013;37:432‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stender S, Kozlitina J, Nordestgaard BG, et al. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet. 2017;49:842‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Barata L, Feitosa MF, Bielak LF, et al. Insulin resistance exacerbates genetic predisposition to nonalcoholic fatty liver disease in individuals without diabetes. Hepatol Commun. 2019;3:894‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Azuma K, Kadowaki T, Cetinel C, et al. Higher liver fat content among Japanese in Japan compared with non‐Hispanic whites in the United States. Metabolism. 2009;58:1200‐1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wattacheril J, Sanyal AJ. Lean NAFLD: an underrecognized uutlier. Curr Hepatol Rep. 2016;15:134‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen F, Esmaili S, Rogers GB, et al. Lean NAFLD: a distinct entity shaped by differential metabolic adaptation. Hepatology. 2019;71:1213‐1227 [DOI] [PubMed] [Google Scholar]
- 71. Mann JP, Savage DB. What lipodystrophies teach us about the metabolic syndrome. J Clin Invest. 2019;130:4009‐4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lotta LA, Gulati P, Day FR, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49:17‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen VL, Wright AP, Halligan B, et al. Body composition and genetic lipodystrophy risk score associate with nonalcoholic fatty liver disease and liver fibrosis. Hepatol Commun. 2019;3:1073‐1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Petäjä EM, Yki‐Jarvinen H. Definitions of normal liver fat and the association of insulin sensitivity with acquired and genetic NAFLD – a systematic review. Int J Mol Sci. 2016;17:633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Peter A, Kovarova M, Nadalin S, et al. PNPLA3 variant I148M is associated with altered hepatic lipid composition in humans. Diabetologia. 2014;57:2103‐2107. [DOI] [PubMed] [Google Scholar]
- 76. Luukkonen PK, Zhou Y, Sädevirta S, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non‐alcoholic fatty liver disease. J Hepatol. 2016;64:1167‐1175. [DOI] [PubMed] [Google Scholar]
- 77. Luukkonen PK, Nick A, Hölttä‐Vuori M, et al. Human PNPLA3 I148M variant increases hepatic retention of polyunsaturated fatty acids. JCI Insight. 2019;4:e127902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Franko A, Merkel D, Kovarova M, et al. Dissociation of fatty liver and insulin resistance in I148M PNPLA3 carriers: differences in diacylglycerol (DAG) FA18:1 lipid species as a possible explanation. Nutrients. 2018;10:1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Dongiovanni P, Stender S, Pietrelli A, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med. 2018;283:356‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fuchsberger C, Flannick J, Teslovich TM, et al. The genetic architecture of type 2 diabetes. Nature. 2016;536:41‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Diogo D, Tian C, Franklin CS, et al. Phenome‐wide association studies across large population cohorts support drug target validation. Nat Commun. 2018;9:4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mahajan A, Wessel J, Willems SM, et al. Refining the accuracy of validated target identification through coding variant fine‐mapping in type 2 diabetes. Nat Genet. 2018;50:559‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Santos RD, Valenti L, Romeo S. Does nonalcoholic fatty liver disease cause cardiovascular disease? Current knowledge and gaps. Atherosclerosis. 2019;282:110‐120. [DOI] [PubMed] [Google Scholar]
- 84. Lauridsen BK, Stender S, Kristensen TS, et al. Liver fat content, non‐alcoholic fatty liver disease, and ischaemic heart disease: Mendelian randomization and meta‐analysis of 279 013 individuals. Eur Heart J. 2018;39:385‐393. [DOI] [PubMed] [Google Scholar]
- 85. Liu DJ, Peloso GM, Yu H, et al. Exome‐wide association study of plasma lipids in >300,000 individuals. Nat Genet. 2017;49:1758‐1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hyysalo J, Gopalacharyulu P, Bian H, et al. Circulating triacylglycerol signatures in nonalcoholic fatty liver disease associated with the I148M variant in PNPLA3 and with obesity. Diabetes. 2014;63:312‐322. [DOI] [PubMed] [Google Scholar]
- 87. Speliotes EK, Butler JL, Palmer CD, et al. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology. 2010;52:904‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sliz E, Sebert S, Würtz P, et al. NAFLD risk alleles in PNPLA3, TM6SF2, GCKR and LYPLAL1 show divergent metabolic effects. Hum Mol Genet. 2018;27:2214‐2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Stender S, Loomba R. PNPLA3 genotype and risk of liver and all‐cause mortality. Hepatology. 2020;71:777‐779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Pingitore P, Romeo S. The role of PNPLA3 in health and disease. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864:900‐906. [DOI] [PubMed] [Google Scholar]
- 91. Wang Y, Kory N, BasuRay S, Cohen JC, Hobbs HH. PNPLA3, CGI‐58, and inhibition of hepatic triglyceride hydrolysis in mice. Hepatology. 2019;69:2427‐2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Donati B, Motta BM, Pingitore P, et al. The rs2294918 E434K variant modulates patatin‐like phospholipase domain‐containing 3 expression and liver damage. Hepatology. 2016;63:787‐798. [DOI] [PubMed] [Google Scholar]
- 93. Irwin DM, Tan H. Evolution of glucose utilization: glucokinase and glucokinase regulator protein. Mol Phylogenet Evol. 2014;70:195‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Caddeo A, Jamialahmadi O, Solinas G, et al. MBOAT7 is anchored to endomembranes by six transmembrane domains. J Struct Biol. 2019;206:349‐360. [DOI] [PubMed] [Google Scholar]
- 95. Helsley RN, Varadharajan V, Brown AL, et al. Obesity‐linked suppression of membrane‐bound O‐acyltransferase 7 (MBOAT7) drives non‐alcoholic fatty liver disease.eLife. 2019;8:e49882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Moeller G, Adamski J. Integrated view on 17beta‐hydroxysteroid dehydrogenases. Mol Cell Endocrinol. 2009;301:7‐19. [DOI] [PubMed] [Google Scholar]
- 97. Su W, Wang Y, Jia X, et al. Comparative proteomic study reveals 17beta‐HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA. 2014;111:11437‐11442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Alswat K, Aljumah AA, Sanai FM, et al. Nonalcoholic fatty liver disease burden – Saudi Arabia and United Arab Emirates, 2017–2030. Saudi J Gastroenterol. 2018;24:211‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Xu R, Tao A, Zhang S, Deng Y, Chen G. Association between patatin‐like phospholipase domain containing 3 gene (PNPLA3) polymorphisms and nonalcoholic fatty liver disease: a HuGE review and meta‐analysis. Sci Rep. 2015;5:9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kantartzis K, Peter A, Machicao F, et al. Dissociation between fatty liver and insulin resistance in humans carrying a variant of the patatin‐like phospholipase 3 gene. Diabetes. 2009;58:2616‐2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Vernekar M, Singhal R, Amarapurkar D. PNPLA3 gene polymorphisms and the associated risk of non‐alcoholic fatty liver disease in Indian subjects. Clin Gastroenterol Hepatol. 2017;15:e39. [Google Scholar]
- 102. Martínez LA, Larrieta E, Kershenobich D, Torre A. The expression of PNPLA3 polymorphism could be the key for severe liver disease in NAFLD in Hispanic population. Ann Hepatol. 2017;16:909‐915. [DOI] [PubMed] [Google Scholar]
- 103. Valenti L, Al‐Serri A, Daly AK, et al. Homozygosity for the patatin‐like phospholipase‐3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1209‐1217. [DOI] [PubMed] [Google Scholar]
- 104. Younossi Z, Anstee QM, Marietti M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15:11‐20. [DOI] [PubMed] [Google Scholar]
- 105. Pinto Marques Souza de Oliveira C, Pinchemel Cotrim H, Arrese M. Nonalcoholic fatty liver disease risk factors in Latin American populations: current scenario and perspectives. Clin Liver Dis (Hoboken). 2019;13:39‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kienesberger PC, Oberer M, Lass A, Zechner R. Mammalian patatin domain containing proteins: a family with diverse lipolytic activities involved in multiple biological functions. J Lipid Res. 2009;50(Suppl):S63‐S68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Baulande S, Lasnier F, Lucas M, Pairault J. Adiponutrin, a transmembrane protein corresponding to a novel dietary‐ and obesity‐linked mRNA specifically expressed in the adipose lineage. J Biol Chem. 2001;276:33336‐33344. [DOI] [PubMed] [Google Scholar]
- 108. Huang Y, He S, Li JZ, et al. A feed‐forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci USA. 2010;107:7892‐7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Dubuquoy C, Robichon C, Lasnier F, et al. Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes. J Hepatol. 2011;55:145‐153. [DOI] [PubMed] [Google Scholar]
- 110. Perttilä J, Huaman‐Samanez C, Caron S, et al. PNPLA3 is regulated by glucose in human hepatocytes, and its I148M mutant slows down triglyceride hydrolysis. Am J Physiol Endocrinol Metab. 2012;302:E1063‐E1069. [DOI] [PubMed] [Google Scholar]
- 111. Hoekstra M, Li Z, Kruijt JK, et al. The expression level of non‐alcoholic fatty liver disease‐related gene PNPLA3 in hepatocytes is highly influenced by hepatic lipid status. J Hepatol. 2010;52:244‐251. [DOI] [PubMed] [Google Scholar]
- 112. Kumashiro N, Yoshimura T, Cantley JL, et al. Role of patatin‐like phospholipase domain‐containing 3 on lipid‐induced hepatic steatosis and insulin resistance in rats. Hepatology. 2013;57:1763‐1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kumari M, Schoiswohl G, Chitraju C, et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012;15:691‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. He S, McPhaul C, Li JZ, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285:6706‐6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Jenkins CM, Mancuso DJ, Yan W, et al. Identification, cloning, expression, and purification of three novel human calcium‐independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem. 2004;279:48968‐48975. [DOI] [PubMed] [Google Scholar]
- 116. Lake AC, Sun Y, Li J‐L, et al. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J Lipid Res. 2005;46:2477‐2487. [DOI] [PubMed] [Google Scholar]
- 117. Huang Y, Cohen JC, Hobbs HH. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J Biol Chem. 2011;286:37085‐37093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ruhanen H, Perttilä J, Hölttä‐Vuori M, et al. PNPLA3 mediates hepatocyte triacylglycerol remodeling. J Lipid Res. 2014;55:739‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Mitsche MA, Hobbs HH, Cohen JC. Patatin‐like phospholipase domain–containing protein 3 promotes transfers of essential fatty acids from triglycerides to phospholipids in hepatic lipid droplets. J Biol Chem. 2018;293:6958‐6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Pirazzi C, Valenti L, Motta BM, et al. PNPLA3 has retinyl‐palmitate lipase activity in human hepatic stellate cells. Hum Mol Genet. 2014;23:4077‐4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Pingitore P, Pirazzi C, Mancina RM, et al. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim Biophys Acta. 2014;1841:574‐580. [DOI] [PubMed] [Google Scholar]
- 122. Xin Y‐N, Zhao Y, Lin Z‐H, et al. Molecular dynamics simulation of PNPLA3 I148M polymorphism reveals reduced substrate access to the catalytic cavity. Proteins. 2013;81:406‐414. [DOI] [PubMed] [Google Scholar]
- 123. Pirazzi C, Adiels M, Burza MA, et al. Patatin‐like phospholipase domain‐containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J Hepatol. 2012;57:1276‐1282. [DOI] [PubMed] [Google Scholar]
- 124. Chen W, Chang B, Li L, Chan L. Patatin‐like phospholipase domain‐containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology. 2010;52:1134‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Basantani MK, Sitnick MT, Cai L, et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J Lipid Res. 2011;52:318‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Li JZ, Huang Y, Karaman R, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest. 2012;122:4130‐4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Smagris E, BasuRay S, Li J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology. 2015;61:108‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lindén D, Ahnmark A, Pingitore P, et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock‐in mice. Mol Metab. 2019;22:49‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. BasuRay S, Smagris E, Cohen JC, Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology. 2017;66:1111‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Yang A, Mottillo EP, Mladenovic‐Lucas L, Zhou L, Granneman JG. Dynamic interactions of ABHD5 with PNPLA3 regulate triacylglycerol metabolism in brown adipocytes. Nat Metabol. 2019;1:560‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Youssefian L, Vahidnezhad H, Saeidian AH, et al. Inherited non‐alcoholic fatty liver disease and dyslipidemia due to monoallelic ABHD5 mutations. J Hepatol. 2019;71:366‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc Natl Acad Sci USA. 2019;116:9521‐9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Mancina RM, Matikainen N, Maglio C, et al. Paradoxical dissociation between hepatic fat content and de novo lipogenesis due to PNPLA3 sequence variant. J Clin Endocrinol Metab. 2015;100:E821‐E825. [DOI] [PubMed] [Google Scholar]
- 134. Sevastianova K, Santos A, Kotronen A, et al. Effect of short‐term carbohydrate overfeeding and long‐term weight loss on liver fat in overweight humans. Am J Clin Nutr. 2012;96:727‐734. [DOI] [PubMed] [Google Scholar]
- 135. Negoita F, Blomdahl J, Wasserstrom S, et al. PNPLA3 variant M148 causes resistance to starvation‐mediated lipid droplet autophagy in human hepatocytes. J Cell Biochem. 2019;120:343‐356. [DOI] [PubMed] [Google Scholar]
- 136. Pingitore P, Dongiovanni P, Motta BM, et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum Mol Genet. 2016;25:5212‐5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Bruschi FV, Claudel T, Tardelli M, et al. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology. 2017;65:1875‐1890. [DOI] [PubMed] [Google Scholar]
- 138. Mondul A, Mancina RM, Merlo A, et al. PNPLA3 I148M variant influences circulating retinol in adults with nonalcoholic fatty liver disease or obesity. J Nutr. 2015;145:1687‐1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Kovarova M, Konigsrainer I, Konigsrainer A, et al. The genetic variant I148M in PNPLA3 is associated with increased hepatic retinyl‐palmitate storage in humans. J Clin Endocrinol Metab. 2015;100:E1568‐E1574. [DOI] [PubMed] [Google Scholar]
- 140. Cai B, Dongiovanni P, Corey KE, et al. Macrophage MerTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab. 2020;31:406‐421.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Chi KR. Drug developers delve into the cell's trash‐disposal machinery. Nat Rev Drug Discov. 2016;15:295‐297. [DOI] [PubMed] [Google Scholar]
- 142. Valenti L, Dongiovanni P. Mutant PNPLA3 I148M protein as pharmacological target for liver disease. Hepatology. 2017;66:1026‐1028. [DOI] [PubMed] [Google Scholar]
- 143. Wattacheril J, Issa D, Sanyal A. Nonalcoholic steatohepatitis (NASH) and hepatic fibrosis: emerging therapies. Annu Rev Pharmacol Toxicol. 2018;58:649‐662. [DOI] [PubMed] [Google Scholar]
- 144. Sanyal AJ, Friedman SL, McCullough AJ, et al. Challenges and opportunities in drug and biomarker development for nonalcoholic steatohepatitis: findings and recommendations from an American Association for the Study of Liver Diseases‐U.S. Food and Drug Administration Joint Workshop. Hepatology. 2015;61:1392‐1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO). EASL‐EASD‐EASO Clinical Practice Guidelines for the management of non‐alcoholic fatty liver disease. J Hepatol. 2016;64:1388‐1402. [DOI] [PubMed] [Google Scholar]
- 146. Baran B, Akyüz F. Non‐alcoholic fatty liver disease: what has changed in the treatment since the beginning? World J Gastroenterol. 2014;20:14219‐14229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Gitto S, Vitale G, Villa E, Andreone P. Treatment of nonalcoholic steatohepatitis in adults: present and future. Gastroenterol Res Pract. 2015;2015:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Laursen TL, Hagemann CA, Wei C, et al. Bariatric surgery in patients with non‐alcoholic fatty liver disease – from pathophysiology to clinical effects. World J Hepatol. 2019;11:138‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lee Y, Doumouras AG, Yu J, et al. Complete resolution of nonalcoholic fatty liver disease after bariatric surgery: a systematic review and meta‐analysis. Clin Gastroenterol Hepatol. 2019;17:1040‐1060.e11. [DOI] [PubMed] [Google Scholar]
- 150. Shen J, Wong GL, Chan HL, et al. PNPLA3 gene polymorphism and response to lifestyle modification in patients with nonalcoholic fatty liver disease. J Gastroenterol Hepatol. 2015;30:139‐146. [DOI] [PubMed] [Google Scholar]
- 151. Krawczyk M, Stachowska E, Milkiewicz P, Lammert F, Milkiewicz M. Reduction of caloric intake might override the prosteatotic effects of the PNPLA3 p.I148M and TM6SF2 p.E167K variants in patients with fatty liver: ultrasound‐based prospective study. Digestion. 2016;93:139‐148. [DOI] [PubMed] [Google Scholar]
- 152. Krawczyk M, Jimenez‐Aguero R, Alustiza JM, et al. PNPLA3 p.I148M variant is associated with greater reduction of liver fat content after bariatric surgery. Surg Obes Relat Dis. 2016;12:1838‐1846. [DOI] [PubMed] [Google Scholar]
- 153. Nobili V, Bedogni G, Donati B, Alisi A, Valenti L. The I148M variant of PNPLA3 reduces the response to docosahexaenoic acid in children with non‐alcoholic fatty liver disease. J Med Food. 2013;16:957‐960. [DOI] [PubMed] [Google Scholar]
- 154. Scorletti E, West AL, Bhatia L, et al. Treating liver fat and serum triglyceride levels in NAFLD, effects of PNPLA3 and TM6SF2 genotypes: results from the WELCOME trial. J Hepatol. 2015;63:1476‐1483. [DOI] [PubMed] [Google Scholar]
- 155. Oscarsson J, Önnerhag K, Risérus U, et al. Effects of free omega‐3 carboxylic acids and fenofibrate on liver fat content in patients with hypertriglyceridemia and non‐alcoholic fatty liver disease: a double‐blind, randomized, placebo‐controlled study. J Clin Lipidol. 2018;12:1390‐1403.e4. [DOI] [PubMed] [Google Scholar]
- 156. Eriksson JW, Lundkvist P, Jansson PA, et al. Effects of dapagliflozin and n‐3 carboxylic acids on non‐alcoholic fatty liver disease in people with type 2 diabetes: a double‐blind randomised placebo‐controlled study. Diabetologia. 2018;61:1923‐1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Santoro N, Savoye M, Kim G, et al. Hepatic fat accumulation is modulated by the interaction between the rs738409 variant in the PNPLA3 gene and the dietary omega6/omega3 PUFA intake. PLoS ONE. 2012;7:e37827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Dongiovanni P, Petta S, Mannisto V, et al. Statin use and non‐alcoholic steatohepatitis in at risk individuals. J Hepatol. 2015;63:705‐712. [DOI] [PubMed] [Google Scholar]
- 159. Liu Y, Fernandez CA, Smith C, et al. Genome‐wide study links PNPLA3 variant with elevated hepatic transaminase after acute lymphoblastic leukemia therapy. Clin Pharmacol Ther. 2017;102:131‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Gutierrez‐Camino A, Martin‐Guerrero I, Garcia‐Orad A. PNPLA3 rs738409 and hepatotoxicity in children with B cell acute lymphoblastic leukemia: a validation study in a Spanish cohort. Clin Pharmacol Ther. 2017;102:906. [DOI] [PubMed] [Google Scholar]
- 161. Guzman CB, Duvvuru S, Akkari A, et al. Coding variants in PNPLA3 and TM6SF2 are risk factors for hepatic steatosis and elevated serum alanine aminotransferases caused by a glucagon receptor antagonist. Hepatol Commun. 2018;2:561‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Pillai S, Duvvuru S, Bhatnagar P, et al. The PNPLA3 I148M variant is associated with transaminase elevations in type 2 diabetes patients treated with basal insulin peglispro. Pharmacogenomics J. 2018;18:487‐493. [DOI] [PubMed] [Google Scholar]
- 163. Rao AS, Knowles JW. Polygenic risk scores in coronary artery disease. Curr Opin Cardiol. 2019;34:435‐440. [DOI] [PubMed] [Google Scholar]