

Summary
Background
High inter‐individual variability in therapeutic response to drugs used in the management of Inflammatory Bowel Disease (IBD) leads to high morbidity and high costs. Genetic variants predictive of thiopurine‐induced myelosuppression, thiopurine‐induced pancreatitis and immunogenicity of Tumour Necrosis Factor alpha (TNFα) antagonists have been identified, but uptake of pre‐treatment pharmacogenetic testing into clinical guidelines has been slow.
Aim
To explore the efficacy of a pharmacogenetic passport for IBD that includes multiple pharmacogenetic predictors of response.
Methods
Patients with IBD exposed to thiopurines and/or TNFα antagonists were retrospectively evaluated for the presence of thiopurine toxicity and/or immunogenicity of TNFα antagonists. All patients were genotyped using both whole‐exome sequencing and the Illumina Global Screening Array. An in‐house‐developed computational pipeline translated genetic data into an IBD pharmacogenetic passport that predicted risks for thiopurine toxicity and immunogenicity of TNFα antagonists per patient. Using pharmacogenetic‐guided treatment guidelines, we calculated clinical efficacy estimates for pharmacogenetic testing for IBD.
Results
Among 710 patients with IBD exposed to thiopurines and/or TNFα antagonists, 150 adverse drug responses occurred and our pharmacogenetic passport would have predicted 54 (36%) of these. Using a pharmacogenetic passport for IBD that includes genetic variants predictive of thiopurine‐induced myelosuppression, thiopurine‐induced pancreatitis, and immunogenicity of TNFα antagonists, 24 patients need to be genotyped to prevent one of these adverse drug responses.
Conclusions
This study highlights the clinical efficacy of a pharmacogenetic passport for IBD. Implementation of such a pharmacogenetic passport into clinical management of IBD may contribute to a reduction in adverse drug responses.
1. INTRODUCTION
Inflammatory Bowel Disease (IBD) is a chronic relapsing inflammatory disease of the gastrointestinal tract primarily consisting of Crohn's disease (CD) and ulcerative colitis (UC). Medical management of IBD aims for lasting disease remission to prevent complications and disease progression. 1 , 2 While both conventional immunosuppressive agents and novel biological agents are effective for IBD treatment, inter‐individual variability in therapy response is high, both with respect to efficacy and toxicity. 3 This inter‐individual variability contributes to high rates of therapeutic failure in IBD, and better patient stratification is therefore needed to maximise patient benefit and minimise the harm caused by adverse events.
Thiopurines (mercaptopurine and its prodrug azathioprine) are conventional immunomodulators commonly used to maintain disease remission in patients with IBD. However, the use of thiopurines is limited by frequently occurring adverse drug reactions, including thiopurine‐induced myelosuppression and thiopurine‐induced pancreatitis, from hereon referred to as myelosuppression and pancreatitis respectively. Myelosuppression is a dose‐dependent adverse reaction with a cumulative incidence of 7%. Most patients with myelosuppression are asymptomatic, but serious opportunistic infections require hospitalisation in 30% of the patients, with an estimated mortality of 1%. 4 Genetic variants in the thiopurine S‐methyltransferase (TPMT) and nudix hydrolase 15 (NUDT15) genes, which both encode enzymes involved in thiopurine metabolism, are associated with an increased risk of myelosuppression. 5 , 6 Pancreatitis occurs in about 4% of thiopurine‐exposed patients, 7 and the pathogenesis of this potentially life‐threatening, idiosyncratic adverse reaction remains poorly understood. The HLA‐DQA1‐HLA‐DRB1 haplotype has been identified as a genetic determinant for pancreatitis. 8 , 9
Tumour Necrosis Factor alpha (TNFα) antagonists, mainly infliximab and adalimumab, are the most commonly prescribed biologicals in the management of IBD. 10 Biological therapy has transformed the management of IBD and has become the largest contribution to IBD healthcare expenditure. 11 Up to 65% of patients treated with infliximab and 38% of patients treated with adalimumab will lose response due to formation of anti‐drug antibodies, a process referred to as immunogenicity. 12 Concomitant use of conventional immunomodulators can reduce immunogenicity. Recently, the HLA‐DQA1*05 haplotype was identified as a genetic determinant of immunogenicity of TNFα antagonists. 13
Given the increasing number of available therapeutic options, and the costs associated with them, there is a clear need for biomarkers predicting individual response to therapy in order to make personalised medicine decisions. We explore pre‐treatment pharmacogenetic testing, which has the potential to maximise patient benefit by optimising drug selection and dose, and minimise the harm caused by drug toxicity. In addition, avoiding (expensive) drugs that are either ineffective or harmful by optimising the use of relatively cheap conventional drugs and achieving optimal dosing as early as possible, will lead to a significant reduction in costs.
Despite this compelling rationale for pre‐treatment pharmacogenetic testing in the context of IBD management, and the falling costs of genetic tests, the implementation of pharmacogenetic‐based guidelines in the clinic has been challenging. In this study, we show that a pharmacogenetic profile or ‘passport’ devised from genetic data, would provide IBD patients with personalised therapeutic recommendations based on their genotypes, leading to a potentially significant reduction in costs and therapeutic failure rates in the management of IBD.
2. METHODS
All patients with IBD treated at the University Medical Center Groningen, a large IBD‐specialised tertiary hospital, are prospectively followed using an electronic IBD‐specific electronic health record. Patients with IBD treated with thiopurines (azathioprine and/or mercaptopurine) and/or TNFα antagonists (infliximab and/or adalimumab) between January 1981 and December 2018 were included in this study. Patients were genotyped and their clinical data were collected. Each patient had been diagnosed with IBD by their gastroenterologist using endoscopic data, histological data, radiological data or a combination of these. During the study period, pharmacogenetic testing was not included as routine clinical practice. All patients provided informed written consent, and the study was approved by the medical ethical board of the University Medical Center Groningen (PSI‐UMCG [IRB no 08/279]).
2.1. Adjudication of patients
Each patient exposed to thiopurine therapy was adjudicated as either an affected case or an unaffected control for myelosuppression and pancreatitis. Case and control adjudication criteria are identical to criteria used in previous IBD pharmacogenetic studies 5 , 8 and are available in the Supplementary Data. All cases were adjudicated by IBD experts using a modified version of the validated Liverpool Adverse Drug Reaction Causality Assessment Tool and assigned a causality category. 14 Probable myelosuppression cases needed to have demonstrated a fall in white cell count to ≤2.5 × 109/L and/or a reduction in neutrophil count to ≤1.0 × 109/L with a clear temporal relationship with thiopurine exposure and without confounding risk factors. In addition, definite myelosuppression cases also developed a second episode of myelosuppression upon thiopurine re‐challenge. For possible myelosuppression cases, confounding causes for the myelosuppression and/or missing data were allowed if the expert opinion still implicated thiopurines as the most likely cause. Probable pancreatitis cases needed to have developed an episode of severe abdominal pain with serum pancreatic enzymes (amylase/lipase) over two times the upper limit of normal within 3 months of starting thiopurine therapy and without confounding causes for pancreatitis. In addition, definite pancreatitis cases also had to have developed a second episode of pancreatitis upon thiopurine re‐challenge. For possible pancreatitis cases, confounding causes for the pancreatitis and/or missing data were allowed if the expert opinion still implicated thiopurines as the most likely cause. In addition to previously used pancreatitis criteria, a fourth pancreatitis causality category was added: suspected pancreatitis cases who developed severe abdominal pain matching pain associated with pancreatitis within 3 months of starting thiopurine therapy, with two independent physicians suspecting pancreatitis, but without available blood amylase or lipase measurements.
Thiopurine‐tolerant controls for both myelosuppression and pancreatitis needed to have been exposed to thiopurines for at least 8 weeks without the development of myelosuppression or pancreatitis. All thiopurine‐tolerant controls, definite and probable myelosuppression cases and all pancreatitis cases were included in subsequent analyses.
Each patient exposed to TNFα antagonist therapy was adjudicated as either an immunogenicity case or unaffected control. Patients with an anti‐drug antibody titres >12 arbitrary units per millilitre (AU/mL), demanding drug withdrawal or start of concomitant immunomodulator therapy, were classified as immunogenicity cases. All other patients exposed to TNFα antagonists for at least 52 weeks with no signs of treatment failure to latest follow‐up or to time of drug withdrawal, or patients with an anti‐drug antibody concentration <12 AU/mL, were adjudicated as controls.
Infliximab and adalimumab antibody titres were obtained using drug‐tolerant assays developed by Sanquin Research (Sanquin, Amsterdam, the Netherlands). ELISA was used to detect drug levels, while a radio‐immuno assay was used to detect anti‐drug antibodies. Radio‐immuno assay test results were converted into arbitrary units per millilitre by comparison with dilutions of a reference serum. The cut‐off level for a positive signal was set at 12 AU/mL (mean + 3 standard deviations) of the pre‐treatment values. Details of these assays, and comparisons to other assays have been published before. 15 , 16 , 17
We reason that all patients exposed to either thiopurine or TNFα antagonist therapy in our cohort are representative of future candidates for these therapies, and thus for pre‐treatment pharmacogenetic screening.
2.2. Genotype data
All patients were genotyped using both whole‐exome sequencing data (Illumina Hiseq 2500, Illumina) and a genome‐wide genotyping array (Infinium Global Screening Array, Illumina). After quality control, 18 alignment to the b37 human reference genome was performed. In total, 86.06 million high‐quality reads were generated per sample. On average, 98.85% of these reads aligned to the human genome (hg19) per sample, resulting in a >30X read depth for 81% of the whole exome. Genetic array data were combined with whole‐exome sequencing data using PLINK v1.9. 19 To account for population stratification, principal component analyses were performed using Eigenstrat v7.2.1 with 1000 Genomes Project data as reference data. 20 , 21 Only data from patients clustering with individuals of non‐Finnish European descent were included in subsequent pharmacogenetic analyses. After extensive quality control, genotype data were phased with the Eagle2 algorithm v2.4. 22
Based on whole‐exome sequencing data, six TPMT star alleles were identified, which together account for > 90% of the variation in TPMT enzyme function in Europeans: TPMT*2, TPMT*3A, TPMT*3B, TPMT*3C, TPMT*9 and TPMT*12. NUDT15 genetic variants are rare in European populations. We could identify NUDT15*3, NUDT15*6 and NUDT15*9, which are the most common NUDT15 variant haplotypes in Europeans. 23 Genetic variation at HLA‐DQA1*02:01‐HLA‐DRB1*07:01 (rs2647087) and HLA‐DQA1*05 (rs2097432) was defined using directly genotyped array data.
2.3. Pharmacogenetic passport
For each patient, genetic variation at TPMT, NUDT15, HLA‐DQA1*02:01‐HLA‐DRB1*07:01 and HLA‐DQA1*05 were translated into a pharmacogenetic passport using an in‐house–developed computational pipeline (Figure S2).
2.3.1. Step 1
Combinations of genetic variants in TPMT or NUDT15, respectively, are referred to as haplotypes. The combinations of two haplotypes (one from each chromosome) were translated into star (*) allele diplotypes provided by the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the Pharmacogenomics Knowledgebase. TMPT*1 and NUDT15*1 were defined as reference. 23 , 24
2.3.2. Step 2
Star allele diplotype assignments were then used to link genotypes with phenotypic function (ie enzyme metabolising activity) according to the CPIC. Patients carrying one deleterious haplotype (ie other than the reference haplotype) in TPMT or NUDT15 are intermediate thiopurine metabolisers. Patient carrying two deleterious haplotypes (eg TPMT*2/TPMT*3A) are poor thiopurine metabolisers.
2.3.3. Step 3
In this step, phenotypic function of TPMT and NUDT15 and HLA genetic variation were translated into a genetic risk of drug‐related effects.
Step 3a. Enzyme metabolising activity for TPMT and NUDT15 was translated into a predicted risk of myelosuppression. Intermediate metabolisers are at intermediate risk of myelosuppression, while poor metabolisers are at high risk of myelosuppression.
Step 3b. Patients homozygous at HLA‐DQA1*02:01‐HLA‐DRB1*07:01 were referred to as patients at high risk of pancreatitis. In this study, patients heterozygous at HLA‐DQA1*02:01‐HLA‐DRB1*07:01 and homozygous reference haplotype carriers were referred to as having a normal risk of pancreatitis.
Step 3c. Patients carrying at least one HLA‐DQA1*05 risk allele (ie heterozygous or homozygous) were referred to as patients at high risk of immunogenicity. In this study, homozygous reference haplotype carriers were referred to as having a normal risk of immunogenicity.
2.3.4. Step 4
In this step, genetic risks were translated into therapeutic recommendations. Therapeutic recommendations regarding TPMT and NUDT15 genetic variation are in line with CPIC guidelines. 23 It remains poorly understood how genetic variation in HLA‐DQA1*02:01‐HLA‐DRB1*07:01 and the HLA‐DQA1*05 risk allele predisposes to pancreatitis or immunogenicity, but our recommendations are in line with previously proposed strategies. 8 , 9 , 13
Step 4a. Patients at intermediate risk of myelosuppression should receive a reduced starting dose (30%‐80% of target dose). For patients at high risk of myelosuppression, alternative treatment should be considered. For patients at normal risk of myelosuppression, starting dose does not need to be altered.
Step 4b. For patients at high risk of pancreatitis, an alternative treatment should be considered. We do not propose drug avoidance in patients heterozygous at HLA‐DQA1*02:01‐HLA‐DRB1*07:01 (2.5‐fold increased risk of pancreatitis) because the high variant carrier frequency would mean drug avoidance in over one third of patients tested. For patients at normal risk of pancreatitis, starting doses do not need to be altered.
Step 4c. Concomitant immunomodulator therapy to prevent antibody development is proposed for patients at high genetic risk of immunogenicity. Patients at normal risk of immunogenicity, intolerant to immunomodulators or patients who are at high risk of opportunistic infections might be treated with TNFα antagonist monotherapy.
2.3.5. Step 5
In this step, our computational pipeline combined all of the above data into one ‘pharmacogenetic passport’ per patient. This pharmacogenetic passport may be used to aid therapeutic decisions.
2.4. Case‐control analyses
Clinical data were compared between cases and exposed controls for myelosuppression, pancreatitis and immunogenicity respectively. Continuous data were described using medians and interquartile ranges, and compared using the Mann‐Whitney test. Categorical data were described as number and percentage, and compared using the Chi‐square test or Fisher exact test. Weight‐adjusted thiopurine dose (milligrams per kilograms) was calculated using the following formulas for mercaptopurine (mercaptopurine dose in milligrams × 2.08/weight in kilograms) and azathioprine (azathioprine dose in milligrams/weight in kilograms). The association of TPMT and/or NUDT15 genetic variation and weight‐adjusted thiopurine dose with myelosuppression and the association of HLA‐DQA1*05 and use of combination therapy with immunogenicity were assessed using multivariable regression analyses. No genome‐wide association study was performed because identifying novel pharmacogenetic interactions was not an aim of this study.
2.5. Clinical efficacy estimates
Predicted clinical efficacy estimates (sensitivity, specificity, negative predictive value and positive predictive value) and numbers needed to genotype, numbers needed to treat and numbers needed to harm were calculated using previously published methods. 5 , 25 , 26 These estimates assumed drug‐response incidence estimates from literature, 4 , 7 the above‐mentioned pharmacogenetic‐based treatment strategies, genotype frequencies from our nation‐wide Dutch IBD cohort and the odds ratio (OR) of the genotype‐phenotype association from our present study (Table S1A‐H).
We first calculated estimates for each pharmacogenetic predictor separately. These were then incorporated into clinical efficacy estimates for a combined IBD pharmacogenetic passport. The number needed to genotype, number needed to treat and number needed to harm to prevent one case (ie of either thiopurine toxicity or immunogenicity of TNFα antagonists) were calculated for a scenario in which an IBD pharmacogenetic genetic passport was established prior to start of either thiopurine or TNFα antagonist therapy. Using the retrospective treatment‐response data from our entire cohort of patients with IBD, we then show the potential consequences of our proposed pharmacogenetic‐based treatment strategies. All statistical calculations were performed using R 3.5.1 (R Foundations for Statistical Computing, Vienna, Austria).
3. RESULTS
We identified 710 patients with IBD who were exposed to thiopurines and/or TNFα antagonists. Of these patients, 695 (98%) were exposed to thiopurines (azathioprine 586 [84%]; mercaptopurine 109 [16%]) and 376 (53%) were exposed to TNFα antagonist therapy (infliximab 284 [76%]; adalimumab 92 [24%]) (Table 1).
TABLE 1.
Patient characteristics of IBD patients exposed to thiopurines. P‐values refer to comparisons between cases and controls
| Characteristic | Patients exposed to thiopurines (n = 695) | Thiopurine‐induced myelosuppression cases (n = 29) | Thiopurine‐induced myelosuppression controls (n = 470) | P‐values | Thiopurine‐induced pancreatitis cases (n = 38) | Thiopurine‐induced pancreatitis controls (n = 555) | P‐values |
|---|---|---|---|---|---|---|---|
| Gender, No. (%) | |||||||
| Female | 409 (59%) | 19 (66%) | 272 (58%) | 0.54 | 27 (71%) | 326 (59%) | 0.19 |
| Male | 286 (41%) | 10 (34%) | 198 (42%) | 11 (29%) | 229 (41%) | ||
| Age, median (IQR), y | 47 (23) | 44 (31) | 47 (24) | 0.72 | 43 (23) | 46 (24) | 0.31 |
| Type of IBD diagnosis, No. (%) | |||||||
| Crohn's disease | 404 (58%) | 19 (66%) | 273 (58%) | 0.32 | 29 (76%) | 320 (58%) | 0.07 |
| Ulcerative colitis | 267 (38%) | 8 (28%) | 182 (39%) | 8 (21%) | 218 (39%) | ||
| IBD unclassified | 24 (3%) | 2 (7%) | 15 (3%) | 1 (3%) | 17 (3%) | ||
| Type of thiopurine, No. (%) | |||||||
| Azathioprine | 586 (84%) | 22 (76%) | 408 (87%) | 0.17 | 32 (84%) | 472 (85%) | 1 |
| Mercaptopurine | 109 (16%) | 7 (24%) | 62 (13%) | 6 (16%) | 83 (15%) | ||
| Weight‐adjusted thiopurine dose, mean (SD), mg/kg | 1.88 (0.62) | 2.17 (0.60) | 1.85 (0.62) | 0.01 | 1.88 (0.53) | 1.83 (0.65) | 0.81 |
| NUDT15 haplotype | |||||||
| Reference/reference | 680 (98%) | 24 (83%) | 466 (99%) | 7.88E‐05 a | 38 (100%) | 544 (98%) | 0.80 |
| Reference/variant | 13 (2%) | 4 (14%) | 4 (1%) | 0 (0%) | 11 (2%) | ||
| Variant/variant | 2 (0.3%) | 1 (3%) | 0 (0%) | 0 (0%) | 0 (0%) | ||
| TPMT haplotype | |||||||
| Reference/reference | 628 (90%) | 23 (79%) | 434 (92%) | 0.065 a | 34 (89%) | 509 (92%) | 0.86 |
| Reference/variant | 67 (10%) | 6 (21%) | 36 (8%) | 4 (11%) | 46 (8%) | ||
| Thiopurine metabolism | |||||||
| Normal metaboliser | 617 (89%) | 18 (65%) | 432 (92%) | 4.55E‐05 b | 34 (89%) | 500 (90%) | 1 |
| Intermediate metaboliser | 76 (11%) | 10 (32%) | 38 (8%) | 4 (11%) | 55 (10%) | ||
| Poor metaboliser | 2 (0.3%) | 1 (3%) | 0 (0%) | 0 (0%) | 0 (0%) | ||
| HLA‐DQA1‐HLA‐DRB1 haplotype | |||||||
| Reference/reference | 416 (60%) | 21 (71%) | 276 (59%) | 0.42 | 17 (45%) | 334 (60%) | 0.14 |
| Reference/variant | 284 (41%) | 8 (29%) | 172 (37%) | 19 (50%) | 199 (36%) | ||
| Variant/variant | 31 (4%) | 0 (0%) | 22 (5%) | 2 (5%) | 22 (5%) | ||
Abbreviations: HLA, human leukocyte antigen; IBD, inflammatory bowel disease; IQR, inter‐quartile range; No., number; NUDT15, nudix hydrolase 15; SD, standard deviation; TPMT, thiopurine S‐methyltransferase.
P‐values from multivariate logistic regression analyses including TPMT variant status, NUDT15 variant status and weight‐adjusted thiopurine dose.
P‐values from multivariate logistic regression analyses including thiopurine metabolism and weight‐adjusted thiopurine dose. NUDT15 heterozygotes are pooled with NUDT15 homozygotes in multivariate logistic regression analyses.
3.1. Case‐control analyses
We first performed case‐control analyses for each pharmacogenetic predictor separately (ie for myelosuppression, pancreatitis or immunogenicity). ORs from these pharmacogenetic case‐control analyses were used as input for clinical efficacy calculations.
3.1.1. Thiopurine‐induced myelosuppression
Of the 695 patients exposed to thiopurines, we identified 29 (4.2%) cases and 470 (68%) controls for myelosuppression. The remaining 196 (28%) patients did not meet our stringent case/control adjudication criteria and were excluded. Myelosuppression cases received a higher weight‐adjusted thiopurine dose compared with controls (2.17 mg/kg vs 1.85 mg/kg; P = 0.01). There were no differences between cases and controls when comparing gender, age, IBD diagnosis or type of thiopurine used. Univariate analysis revealed that carriage of any variant in TPMT (OR 3.13 [95% CI 1.11‐7.80]; P = 0.02) or NUDT15 (OR 24.3 [95% CI 6.06‐104]; P = 5.67E‐6) was associated with myelosuppression. Multivariable regression analyses revealed that carriage of any variant in NUDT15 (OR 20.2 [95% CI 4.4‐94.4]; P = 7.88E‐05) and weight‐adjusted thiopurine dose (OR 2.69 [95% CI 1.39‐5.26]; P = 3.41E‐03) were independently associated with myelosuppression (Table 1). The association of TPMT with myelosuppression failed to reach independent statistical significance using multivariable regression (OR 2.86 [95% CI 0.84‐8.17]; P = 0.065).
3.1.2. Thiopurine‐induced pancreatitis
Of 695 patients exposed to thiopurines, we identified 38 (5.5%) cases and 555 (80%) controls for pancreatitis. The remaining 102 (15%) patients did not meet stringent case/control adjudication criteria and were excluded. There were no differences between cases and controls in gender, age, IBD diagnosis, type of thiopurine used or weight‐adjusted dose. Carriage of the HLA‐DQA1‐HLA‐DRB1 risk haplotype was not significantly associated with the risk of pancreatitis when using a dominant genetic model (OR 1.87 [95% CI 0.97‐3.66]; P = 0.064) or when using an additive genetic model (P = 0.14) (Table 1).
3.1.3. Immunogenicity of TNFα antagonists
Of 376 patients exposed to TNFα antagonist therapy, we identified 85 (23%) cases and 194 (52%) controls for immunogenicity. The remaining 97 (26%) patients did not meet our stringent case/control adjudication criteria and were excluded. There were no differences between cases and controls when comparing gender and type of drug. Immunogenicity cases less frequently received concomitant immunomodulator therapy (32% vs 54%; P = 1.2E‐4), used TNFα antagonist therapy for a shorter period of time (57 weeks vs 244 weeks; P = 2.44E‐14), were of higher age (46 years old vs 42 years old, P = 0.03) and were less frequently diagnosed with CD (69% vs 85%, P = 0.01), compared with controls. Multivariable regression analysis revealed that the use of combination therapy (OR = 0.34 [95% CI 0.19‐0.60], P = 2.33E‐4) and CD (OR = 0.38 [95% CI 0.20‐0.72], P = 3.26E‐3) was significantly associated with a decreased risk of immunogenicity, while carriage of the HLA‐DQA1*05 haplotype increased the risk. However, this genetic association failed to reach statistical significance in our multivariate model (OR = 1.65 [95% CI 0.95‐2.85], P = 0.075) (Table 2).
TABLE 2.
Patient characteristics of patients with IBD exposed to TNFα antagonists. P‐values refer to comparisons between cases and controls
| Characteristic | Patients exposed to TNFα antagonists (n = 376) | Immunogenicity of TNFα antagonists cases (n = 85) | Immunogenicity of TNFα antagonists controls (n = 194) | P‐values |
|---|---|---|---|---|
| Gender, No. (%) | ||||
| Female | 246 (65%) | 59 (69%) | 117 (60%) | 0.19 |
| Male | 130 (35%) | 26 (31%) | 77 (40%) | |
| Age, median (IQR), y | 47 (21) | 46 (27) | 42 (21) | 0.03 |
| Type of IBD diagnosis, No. (%) | ||||
| Crohn's disease | 278 (74%) | 59 (69%) | 164 (85%) | 0.01 |
| Ulcerative colitis | 85 (23%) | 23 (27%) | 25 (13%) | |
| IBD‐unclassified | 13 (3%) | 3 (4%) | 5 (3%) | |
| Type of TNFα antagonist, No (%) | ||||
| Infliximab | 284 (76%) | 63 (74%) | 140 (72%) | 0.10 |
| Adalimumab | 92 (24%) | 22 (26%) | 54 (28%) | |
| Concomitant immunomodulator, No. (%) | ||||
| No | 198 (53%) | 59 (69%) | 88 (45%) | 3.5E‐4 |
| Yes | 178 (47%) | 26 (31%) | 106 (55%) | |
| Duration of use, median (IQR), w | 206 (314) | 18 (161) | 200 (348) | 6.84E‐7 |
| HLA‐DQA1*05 haplotype | ||||
| Reference/reference | 227 (60%) | 45 (53%) | 123 (63%) | 0.075 a |
| Reference/variant | 125 (33%) | 33 (39%) | 60 (31%) | |
| Variant/variant | 24 (6%) | 7 (8%) | 11 (6%) | |
Abbreviations: HLA, human leukocyte antigen; IBD, inflammatory bowel disease; IQR, inter‐quartile range; No., number.
P‐value from multivariate logistic regression analysis including type of IBD diagnosis, age, concomitant immunomodulator use and HLA‐DQA1*05 variant status, using a dominant genetic model.
3.2. Clinical efficacy estimates
We next assessed the clinical efficacy of pharmacogenetic testing for three applications: (a) to predict thiopurine toxicity (ie myelosuppression and pancreatitis) prior to starting thiopurine treatment, (b) to predict immunogenicity prior to starting TNFα antagonist therapy and (c) to predict both thiopurine toxicity and immunogenicity of TNFα antagonists prior to starting either thiopurine or TNFα antagonist therapy (ie an IBD pharmacogenetic passport).
3.2.1. Thiopurine toxicity
Of 695 patients exposed to thiopurines, 590 (85%) were at low genetic risk of thiopurine toxicity, 72 (10%) were at intermediate risk of thiopurine toxicity (ie intermediate thiopurine metabolisers), and 33 (5%) were at high genetic risk of thiopurine toxicity (2 [0.3%] poor thiopurine metaboliser and 31 [5%] HLA‐DQA1‐HLA‐DRB1 homozygotes). Four patients (0.6%) were both homozygous at the HLA‐DQA1‐HLA‐DRB1 haplotype and intermediate thiopurine metabolisers and they were thus classified as being at high genetic risk of thiopurine toxicity. Among patients at intermediate risk of thiopurine toxicity for whom dose reduction would be recommended, we identified 11 (15%) patients who developed myelosuppression and 34 (47%) patients with uncomplicated sustained thiopurine use. Among patients at high genetic risk of thiopurine toxicity for whom drug avoidance would be recommended, we identified 3 (9%) patients who developed either myelosuppression or pancreatitis and 21 (64%) patients with uncomplicated sustained thiopurine use (Figure 1). Again, a number of patients did not meet case/control criteria.
FIGURE 1.
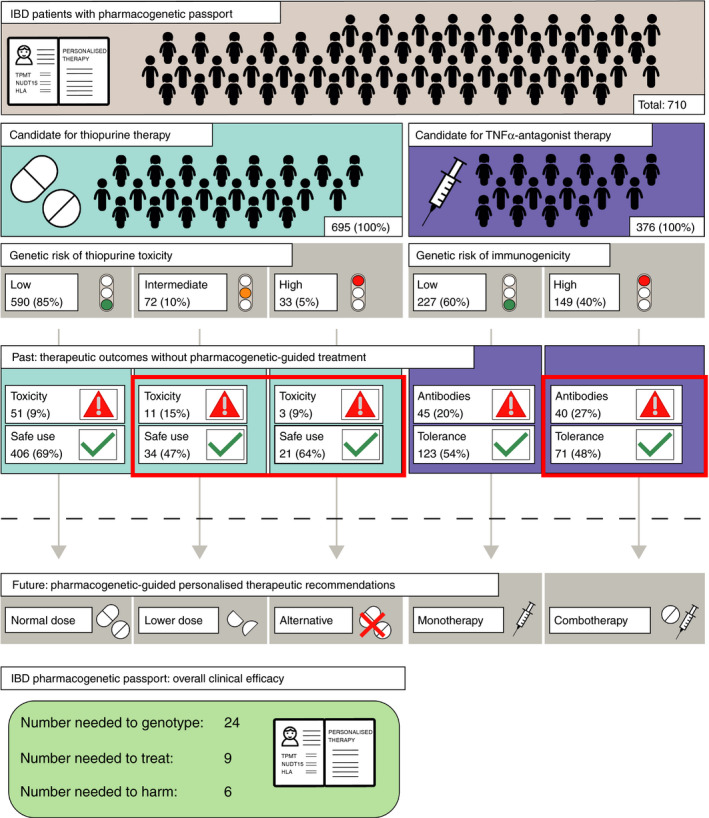
Flowchart representing current therapeutic outcomes (past) and proposed pharmacogenetic‐guided treatment strategies (future). Patients defined as candidates for therapy in this figure were exposed to either thiopurines or TNFα antagonists and are hypothesised to be representative of future candidates. Toxicity refers to either thiopurine‐induced myelosuppression or thiopurine‐induced pancreatitis. Antibodies refers to the formation of anti‐drug antibodies to TNFα antagonists, a process referred to as immunogenicity. Patients in red boxes would receive alternative treatment strategies based on pharmacogenetic testing. IBD: inflammatory bowel disease; TNFα: Tumour Necrosis Factor alpha
We calculated a number needed to genotype of 39, a number needed to treat of 7 and a number needed to harm of 5 to prevent one case of thiopurine toxicity. This means that for every 39 patients genotyped prior to starting thiopurine therapy, 7 will test positive for a predictive variant of thiopurine toxicity and 2 of them would have developed thiopurine toxicity without pharmacogenetic‐guided therapy. However, if these seven patients had received dose reduction or drug avoidance, depending on their predicted genetic risk, one case of thiopurine toxicity would have been prevented. This assumes an overall risk of 7% for myelosuppression 4 and 4% for pancreatitis, 7 an incidence of poor thiopurine metabolisers of 0.27% (nation‐wide Dutch IBD cohort) and an incidence of 6.7% for HLA‐DQA1‐HLA‐DRB1 homozygotes (nation‐wide Dutch IBD cohort) (Table S1A‐E).
3.2.2. Immunogenicity of TNFα antagonists
Among 376 patients exposed to TNFα antagonist therapy, 149 (40%) patients were genetically at risk of immunogenicity. Among these 149 patients, 40 (27%) developed immunogenicity, while 71 (48%) did not (Figure 1). Fifteen of 40 (38%) of these patients received concomitant immunomodulator therapy, which was taken into account in subsequent clinical efficacy calculations. Again, a number of patients did not meet case/control criteria.
We calculated a number needed to genotype of 32, a number needed to treat of 13 and a number needed to harm of 8 to prevent one case of immunogenicity. This means that for every 32 patients genotyped prior to starting TNFα antagonist therapy, 13 will test positive for the HLA‐DQA1*05 haplotype and 5 would have developed immunogenicity without pharmacogenetic‐guided combination therapy. However, if these patients had received concomitant immunomodulator therapy, one case of immunogenicity would have been prevented. This assumes an overall risk of immunogenicity of 30% (this cohort), an HLA‐DQA1*05 variant carrier frequency of 39% (nation‐wide Dutch IBD cohort) and subsequent prescription of a concomitant immunomodulator therapy for patients genetically at risk (Table S1E‐H).
3.2.3. IBD pharmacogenetic passport
To explore the potential benefits of an IBD pharmacogenetic passport, we combined clinical efficacy estimates for preventing thiopurine toxicity and immunogenicity of TNFα antagonists into an overall predicted efficacy. In our total cohort of 710 IBD patients, 150 adverse drug responses occurred (ie either thiopurine toxicity or immunogenicity of TNFα antagonists). An IBD pharmacogenetic passport would have predicted 54 (36%) of these adverse drug responses, while 96 (64%) cases would not have been predicted (Figure 1).
We calculated a number needed to genotype of 24, a number needed to treat of 9 and a number needed to harm of 6 to prevent one adverse drug response (ie myelosuppression, pancreatitis or immunogenicity). This means that, for every 24 candidates for either thiopurine or TNFα antagonist therapy who were genotyped prior to starting treatment, nine patients would test positive for a predictive variant and three of them would have developed an adverse drug response without pharmacogenetic‐guided alternative therapy. However, if a pharmacogenetic‐guided alternative treatment strategy had been applied, one adverse drug response would have been prevented. This calculation makes the same assumptions for separate clinical efficacy estimates that we used above, and a frequency of thiopurine use of 98% relative to a frequency of TNFα antagonist use of 53% based on this cohort (Supplementary Data: IBD pharmacogenetic passport).
4. DISCUSSION
This real‐world IBD cohort provides clinical efficacy estimates for the implementation of an IBD pharmacogenetic passport including genetic variants predictive of both thiopurine toxicity and immunogenicity of TNFα antagonists. If such an IBD pharmacogenetic passport was incorporated into clinical guidelines, better patient stratification would lead to a significant reduction in potentially life‐threatening thiopurine toxicity and higher TNFα antagonist therapy response rates.
Both TPMT and NUDT15 encode enzymes involved in thiopurine metabolism and genetic variation in these genes can lead to deficient thiopurine metabolism. Hence, ‘normal’ starting doses are generally ‘high’ for patients with defective TPMT or NUDT15 alleles. 23 , 27 Indeed, patients carrying defective TPMT or NUDT15 alleles are at increased risk of thiopurine‐induced myelosuppression. We could not replicate the association between TPMT and myelosuppression using a multivariate model, probably due to the relatively small sample size. However, we believe there is enough evidence to support this well‐established association. 6 , 23 , 28 Pharmacogenetic‐guided dose reduction decreases the risk of myelosuppression in these patients without compromising therapeutic efficacy. 29 , 30 Our data show the predicted efficacy of pre‐treatment pharmacogenetic testing including both TPMT and NUDT15.
Genetic variation in HLA‐DQA1‐HLA‐DRB1 is a determinant of thiopurine‐induced pancreatitis, although the mechanism for this remains elusive. We used less stringent criteria for pancreatitis, compared to previous studies, by including suspected cases for whom no biochemical or radiological evidence was obtained. Nevertheless, by doing so, the incidence of pancreatitis in our cohort was similar to what is reported in literature. 7 , 31 The relatively small sample size might explain why we did not replicate the association. Our data show that, if patients were screened for genetic variants in TPMT, NUDT15 and HLA‐DQA1‐HLA‐DRB1, 39 patients would need to be genotyped to prevent one case of thiopurine toxicity.
It is only recently that HLA‐DQA1*05 has been associated with an increased risk of immunogenicity of TNFα antagonists in which the formation of antibodies targeting the TNFα antagonists leads to treatment failure or adverse drug reactions. Although concomitant use of immunomodulators can reduce the risk of immunogenicity, there are concerns about the safety of this combination therapy. Pre‐treatment HLA‐DQA1*05 genetic testing can identify patients at increased risk of immunogenicity and may aid targeted use of combination therapy. Our data show that 32 patients would need to be genotyped prior to starting TNFα antagonist treatment to prevent one patient from developing immunogenicity.
Currently, American Gastroenterology Association and British Society of Gastroenterology guidelines recommend TPMT testing prior to thiopurine therapy in addition to regular hematologic monitoring, while the European Crohn´s and Colitis Organisation guidelines do not recommend any pre‐treatment testing. 32 , 33 , 34 Pharmacogenetic testing prior to TNFα antagonist therapy has not been implemented in routine clinical practice. Insufficient evidence to support clinical efficacy and lack of specialised training for interpretation of complex pharmacogenetic data have so far prevented pharmacogenetic testing from being widely implemented into clinical care. To our knowledge, this study is the first to combine multiple IBD gene‐drug interactions into clinical efficacy estimates. Combining clinical efficacy estimates of thiopurine toxicity and immunogenicity of TNFα antagonists into an overall predicted efficacy, we show that 24 patients need to be genotyped to prevent one of these adverse drug responses.
Our proposed thiopurine dosing strategies based on TPMT and NUDT15 genotypes are in line with guidelines proposed by the Clinical Pharmacogenetics Implementation Consortium. 23 Prospective studies are needed to explore the optimal treatment strategy based on both HLA genetic variants, which will be a trade‐off between the acceptable risk of adverse reactions and a more timely and systematic progression to other (expensive) IBD therapies. Our proposed algorithm excludes patients homozygous at HLA‐DQA1‐HLA‐DRB1 from using thiopurines, while allowing patients heterozygous at this locus to be exposed to thiopurines. This proposed algorithm is based on the high risk (~17%) of pancreatitis in homozygotes, 8 the wide availability of alternative drugs and the relatively low frequency of homozygotes (7%). In contrast, drug avoidance in patients heterozygous at HLA‐DQA1‐HLA‐DRB1 (~9% risk of pancreatitis) would mean that more than one third of patients would be excluded from thiopurine use.
Pre‐treatment pharmacogenetic testing can maximise patient benefit and minimise harm. Moreover, pharmacogenetic testing has the potential to significantly reduce costs associated with IBD treatment, mainly by reducing costs associated with TNFα antagonist treatment failure and on hospitalisation for adverse drug reactions. 11 Approximately 30% of the patients experiencing thiopurine‐induced myelosuppression ultimately require hospitalisation. 4 This hospitalisation is associated with an estimated cost of $5,800 per patient. 4 , 11 Hospitalisation is necessary for 57% of the patients with thiopurine‐induced pancreatitis, with an estimate cost of $33,740 per hospitalisation per patient. 35 , 36 We estimate the costs of a genome‐wide genotyping array, designed to specifically tag pharmacogenetic predictors, to be approximately $50 per individual. Based on an overall “number needed to genotype” of 24, the costs to prevent one adverse drug reaction will be $1,200. Given these numbers, an IBD pharmacogenetic passport should be considered as cost effective to optimise IBD treatment.
We used a combination of whole‐exome sequencing and a genome‐wide genotyping array to reliably identify genetic variation in TPMT, NUDT15 and HLA. With falling costs and wider availability of genotyping, future studies should explore which is the most cost‐effective genotyping platform that is also easy to implement in routine medical care. Furthermore, clinical efficacy estimates presented in this study are calculated using retrospective case‐control analyses, while randomised controlled trials are still considered the gold standard for clinical efficacy studies. However, randomised controlled trials often fail to produce actionable results, since outcomes are averaged across the entire population per study arm, whereas in personalised medicine, treatment is based on the individual patient. Retrospective studies like this present study should be used as a starting point for prospective studies evaluating clinically implemented pharmacogenetic testing.
In conclusion, this is the first study to describe the consequences of pre‐treatment pharmacogenetic testing for multiple gene‐drug interactions using a real‐world IBD cohort. We illustrate that pre‐treatment pharmacogenetic testing can optimise patient benefit and minimise harm. These findings show the predicted efficacy of pharmacogenetic testing prior to start of thiopurine or TNFα antagonist therapy in patients with IBD.
AUTHORSHIP
Guarantors of the article: Amber Bangma and Michiel D. Voskuil.
Author contributions: AB and MDV had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. EAMF and RKW jointly supervised this work. Concept and design: AB, MDV, EAMF, RKW Acquisition, analysis, or interpretation of data: AB, MDV, WTCUV, HB, SH, PL, EAMF, RKW Drafting of the manuscript: AB, MDV, RKW Critical revision of the manuscript: WTCUV, HB, PL, LF, GD, EAMF. All authors approved the final version of this manuscript.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Kate Mc Intyre, Scientific Editor in the Department of Genetics, University Medical Center Groningen, for editing and formatting of this manuscript.
Declaration of personal interests: Rinse K. Weersma received unrestricted research grants from Takeda, Tramedico and Ferring. Eleonora A.M. Festen received an unrestricted research grant from Takeda. The remaining authors disclose no conflicts.
Bangma A, Voskuil MD, Uniken Venema WTC, et al. Predicted efficacy of a pharmacogenetic passport for inflammatory bowel disease. Aliment Pharmacol Ther. 2020;51:1105–1115. 10.1111/apt.15762
The Handling Editor for this article was Professor Roy Pounder, and it was accepted for publication after full peer‐review.
Amber Bangma and Michiel D. Voskuil share first authorship.
Eleonora A.M. Festen and Rinse K. Weersma share last authorship.
Funding information
R.K. Weersma is supported by a Diagnostics Grant from the Dutch Digestive Foundation (D16‐14). E.A.M. Festen is supported by a MLDS Career Development grant (CDG 14‐04).
REFERENCES
- 1. Torres J, Mehandru S, Colombel J‐F, Peyrin‐Biroulet L. Crohn’s disease. Lancet (London, England). 2017;389:1741‐1755. [DOI] [PubMed] [Google Scholar]
- 2. Ungaro R, Mehandru S, Allen PB, Peyrin‐Biroulet L, Colombel J‐F. Ulcerative colitis. Lancet (London, England). 2017;389:1756‐1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Flamant M, Roblin X. Inflammatory bowel disease: towards a personalized medicine. Therap Adv Gastroenterol. 2018;11:1756283X17745029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gisbert JP, Gomollon F. Thiopurine‐induced myelotoxicity in patients with inflammatory bowel disease: a review. Am J Gastroenterol. 2008;103:1783‐1800. [DOI] [PubMed] [Google Scholar]
- 5. Walker GJ, Harrison JW, Heap GA, et al. Association of genetic variants in NUDT15 with thiopurine‐induced myelosuppression in patients with inflammatory bowel disease. JAMA [Internet]. 2019;321:773‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lennard L, Van Loon JA, Weinshilboum RM. Pharmacogenetics of acute azathioprine toxicity: relationship to thiopurine methyltransferase genetic polymorphism. Clin Pharmacol Ther. 1989;46:149‐154. [DOI] [PubMed] [Google Scholar]
- 7. Chaparro M, Ordas I, Cabre E, et al. Safety of thiopurine therapy in inflammatory bowel disease: long‐term follow‐up study of 3931 patients. Inflamm Bowel Dis. 2013;19:1404‐1410. [DOI] [PubMed] [Google Scholar]
- 8. Heap GA, Weedon MN, Bewshea CM, et al. HLA‐DQA1‐HLA‐DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat Genet. 2014;46:1131‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilson A, Jansen LE, Rose RV, et al. HLA‐DQA1‐HLA‐DRB1 polymorphism is a major predictor of azathioprine‐induced pancreatitis in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2018;47:615‐620. [DOI] [PubMed] [Google Scholar]
- 10. Mogilevski T, Sparrow MP. Infliximab Versus Adalimumab in Patients with Biologic‐Naive Crohn’s Disease: Is the Difference Real? Vol. 63, Digestive Diseases and Sciences. United States: 2018;1094‐1096. [DOI] [PubMed] [Google Scholar]
- 11. van der Valk ME, Mangen M‐JJ, Severs M, et al. Evolution of costs of inflammatory bowel disease over two years of follow‐up. PLoS ONE. 2016;11(4):e0142481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vermeire S, Gils A, Accossato P, Lula S, Marren A. Immunogenicity of biologics in inflammatory bowel disease. Therap Adv Gastroenterol. 2018;11:1756283X17750355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sazonovs A, Kennedy NA, Moutsianas L, et al. HLA‐DQA1*05 carriage associated with development of anti‐drug antibodies to infliximab and adalimumab in patients with Crohn’s disease. Gastroenterology. 2020;158:189‐199. [DOI] [PubMed] [Google Scholar]
- 14. Gallagher RM, Kirkham JJ, Mason JR, et al. Development and inter‐rater reliability of the Liverpool adverse drug reaction causality assessment tool. PLoS ONE. 2011;6:e28096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Schouwenburg PA, Bartelds GM, Hart MH, Aarden L, Wolbink GJ, Wouters D. A novel method for the detection of antibodies to adalimumab in the presence of drug reveals “hidden” immunogenicity in rheumatoid arthritis patients. J Immunol Methods. 2010;362:82‐88. [DOI] [PubMed] [Google Scholar]
- 16. Vande Casteele N, Buurman DJ, Sturkenboom MGG, et al. Detection of infliximab levels and anti‐infliximab antibodies: a comparison of three different assays. Aliment Pharmacol Ther. 2012;36:765‐771. [DOI] [PubMed] [Google Scholar]
- 17. Wolbink GJ, Vis M, Lems W, et al. Development of antiinfliximab antibodies and relationship to clinical response in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54(3):711‐715. [DOI] [PubMed] [Google Scholar]
- 18. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20:1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Purchel S, Chank C. PLINK [Internet]. 2019 [cited 2019 Oct 3]. www.cog‐genomics.org/plink/1.9/
- 20. Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature. 2015;526:68‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome‐wide association studies. Nat Genet. 2006;38:904‐909. [DOI] [PubMed] [Google Scholar]
- 22. Loh P‐R, Danecek P, Palamara PF, et al. Reference‐based phasing using the Haplotype Reference Consortium panel. Nat Genet. 2016;48:1443‐1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Relling MV, Schwab M, Whirl‐Carrillo M, et al. Clinical pharmacogenetics implementation consortium guideline for thiopurine dosing based on TPMT and NUDT15 genotypes: 2018 update. Clin Pharmacol Ther. 2018; 105:1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Whirl‐Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92:414‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tonk ECM, Gurwitz D, Maitland‐van der Zee A‐H, Janssens ACJW. Assessment of pharmacogenetic tests: presenting measures of clinical validity and potential population impact in association studies. Pharmacogenomics J. 2017;17:386‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Graaff LCG, van Schaik RHN, van Gelder T. A clinical approach to pharmacogenetics. Neth J Med. 2013;71:145‐152. [PubMed] [Google Scholar]
- 27. Moriyama T, Nishii R, Perez‐Andreu V, et al. NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet. 2016;48:367‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moon W, Loftus EVJ. Review article: recent advances in pharmacogenetics and pharmacokinetics for safe and effective thiopurine therapy in inflammatory bowel disease. Aliment Pharmacol Ther. 2016;43:863‐883. [DOI] [PubMed] [Google Scholar]
- 29. Relling MV, Pui C‐H, Cheng C, Evans WE. Thiopurine Methyltransferase in Acute Lymphoblastic Leukemia. Vol. 107. Blood. United States: 2006;843‐844. [DOI] [PubMed] [Google Scholar]
- 30. Coenen MJH, de Jong DJ, van Marrewijk CJ, et al. Identification of patients with variants in TPMT and dose reduction reduces hematologic events during thiopurine treatment of inflammatory bowel disease. Gastroenterology. 2015;149:907‐917.e7. [DOI] [PubMed] [Google Scholar]
- 31. Gearry RB, Barclay ML, Burt MJ, Collett JA, Chapman BA. Thiopurine drug adverse effects in a population of New Zealand patients with inflammatory bowel disease. Pharmacoepidemiol Drug Saf. 2004;13:563‐567. [DOI] [PubMed] [Google Scholar]
- 32. Feuerstein JD, Nguyen GC, Kupfer SS, Falck‐Ytter Y, Singh S. American gastroenterological association institute guideline on therapeutic drug monitoring in inflammatory bowel disease. Gastroenterology. 2017;153:827‐834. [DOI] [PubMed] [Google Scholar]
- 33. Mowat C, Cole A, Windsor A, et al. Guidelines for the management of inflammatory bowel disease in adults. Gut. 2011;60:571‐607. [DOI] [PubMed] [Google Scholar]
- 34. Gomollon F, Dignass A, Annese V, et al. 3rd European evidence‐based consensus on the diagnosis and management of Crohn’s disease 2016: part 1: diagnosis and medical management. J Crohns Colitis. 2017;11:3‐25. [DOI] [PubMed] [Google Scholar]
- 35. Ledder O, Lemberg DA, Day AS. Thiopurine‐induced pancreatitis in inflammatory bowel diseases. Expert Rev Gastroenterol Hepatol. 2015;9:399‐403. [DOI] [PubMed] [Google Scholar]
- 36. Wadhwa V, Patwardhan S, Garg SK, Jobanputra Y, Lopez R, Sanaka MR. Health care utilization and costs associated with acute pancreatitis. Pancreas. 2017;46:410‐415. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material