

Abstract
A new series of Proteolysis Targeting Chimeras (PROTACs) targeting Bruton’s Tyrosine Kinase (BTK) was synthesized, with the goal of improving the pharmacokinetic properties of our previously reported PROTAC, MT802. We recently described the ability of MT802 to induce degradation of both wild-type and C481S mutant BTK in immortalized cells and patient-derived B-lymphocytes. However, the pharmacokinetic properties of MT802 were not suitable for further in vivo development. Therefore, we undertook a systematic medicinal chemistry campaign to overcome this issue and made a series of PROTACs with structural modifications to the linker and E3-recruiting ligand; more specifically, the new PROTACs were synthesized with different von Hippel-Lindau (VHL) and cereblon (CRBN) ligands while keeping the BTK ligand and linker length constant. This approach resulted in an equally potent PROTAC, SJF620, with a significantly better pharmacokinetic profile than MT802. This compound may hold promise for further in vivo exploration of BTK degradation.
Keywords: PROTACs, Pharmacokinetics, Target protein degradation, BTK, CRBN, VHL
Graphical Abstract
1. Introduction
Bruton’s Tyrosine Kinase (BTK) plays a critical role in all aspects of B cell development, including cell growth, maturation, migration, and apoptosis1, 2. Dysregulation of any of these pathways is associated with conditions such cancer, autoimmunity and inflammation3. Therefore, development of reversible and irreversible inhibitors, has drawn the attention of many researchers and pharmaceutical companies in the last two decades3, 4. As a result of these development efforts, two irreversible BTK inhibitors, ibrutinib and acalabrutinib, have garnered FDA approval (Fig. 1A) and many others are in clinical development3, 4. These inhibitors exploit a cysteine (Cys481) within the active site of BTK to bind covalently to the kinase and inhibit its catalytic activity (Fig. 1B). Since this cysteine is conserved among several other kinases, such as EGFR and ITK, ibrutinib is a relatively promiscuous inhibitor with many side-effects. This realization prompted the development of acalabrutinib, a more potent and BTK selective inhibitor than ibrutinib5. However, in chronic lymphocytic leukemia (CLL) patients subjected to prolonged BTK inhibition, drug resistance often arises via mutation of the active site cysteine to serine (C481S)6. This renders covalent inhibitors capable of only reversibly binding to BTK. As a result, ibrutinib’s IC50 for BTK inhibition shifts from 2.2 nM to 1 μM7. Most recently, several promising reversible inhibitors of BTK, most notably GDC-0853, have been applied to CLL patients with varying degrees of success (Fig. 1B)7, 8. Despite such efforts, no effective second-line treatment options exist for relapsed CLL patients and outcome is poor. As a result, there is still a need for new approaches to address drug resistance in CLL.
Figure 1.

(A) Structures of currently FDA-approved BTK inhibitors. (B) Aligned structures of ibrutinib (Magenta, PDBID: 5P9J) and GDC-0853 (Blue, PDBID: 5VFI) bound to BTK.
Our group has pioneered the development of PROteolysis TArgeting Chimeraes (PROTACs), a technology for the targeted degradation of disease-relevant proteins9. Conceptually, PROTACs are hetero-bifunctional small molecules that simultaneously engage an E3 ubiquitin ligase and a target protein to be degraded. Bringing these two proteins into proximity results in a de-novo complex suitable for interfacing with the protein machinery required for ubiquitination. Once ubiquitinated, this ‘neo-substrate’ is then recognized by the proteasome and degraded (Fig. 2).
Figure 2.

General schematic for how PROTACs work.
The PROTAC mechanism can be summarized as follows: (1) PROTACs are composed of three elements: an E3 ligand, a linker, and a ligand for the target protein. (2) An effective PROTAC induces a complex between the target (e.g. BTK) and E3 ligase (e.g. CRBN or VHL) forming a ternary complex. (3) An E2 enzyme catalyzes transfer of ubiquitin onto the target protein, forming ubiquitin chains, which are eventually recognized by the proteasome. (4) Dissociation of the target from the E3 ligase allows recognition by the proteasome for degradation. (5) PROTACs can function catalytically whereby they can induce multiple rounds of target protein ubiquitination before clearance or metabolism. (6) The amino acids of the degraded protein and ubiquitin are recycled by the cell (Fig. 2). Because of their selectivity10, catalytic nature11 and potency12, PROTACs offer benefits not possible with regular inhibitors13–18.
Our group recently reported the discovery of MT80219, a PROTAC that degrades BTK in cells at low nanomolar concentrations. PROTAC MT802 has a ligand specific for BTK, and a ligand specific to a E3 ligase cereblon (CRBN) connected by polyethylene glycol (PEG) linker (Fig. 3). Since a transient interaction with substrate is enough for ubiquitination, we reasoned that MT802 would be able to induce degradation of both wild-type and C481S BTK, despite the reduced affinity of the non-covalent BTK ligand.
Figure 3.

Chemical structure of MT802
Furthermore, because the BTK-targeting warhead in MT802 does not have the Michael acceptor scaffold does not bind BTK covalently (Fig. 4). As a result, MT802 should be comparatively more selective; and indeed, KinomeScan binding studies show MT802 binds fewer off-target kinases than ibrutinib19. Collectively, these findings make MT-802 and the PROTAC approach a promising avenue for overcoming ibrutinib resistance in CLL.
Figure 4.

Ligand interactions diagrams; a) Crystal structure of BTK bound to ibrutinib (PDB, 5PNI). b) MT802 warhead dock into BTK.
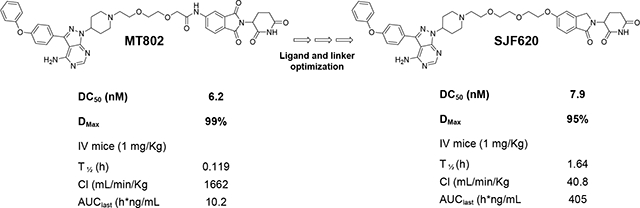
Because of its outstanding degradation profile, we selected MT802 as our initial lead to further advance into preclinical studies. However, the pharmacokinetic data on MT802 in mice showed the compound was not suitable for in vivo studies, Specifically, its clearance was too high (1662 mL/min/kg) and half-life was too short (0.119 h). To address these issues, we undertook a medicinal chemistry campaign to improve the pharmacokinetic properties of an MT802-derived PROTAC series. Herein we present two strategies to improve the pharmacokinetic profile on these PROTACs. Initially, we prepared three PROTACs where the CRBN ligand was replaced with a ligand targeting the von Hippel-Lindau E3 ligase (VHL). However, the VHL series suffered from poor potency and we did not pursue these compounds further. Thereafter, we explored structural modifications on the CRBN ligand of MT802. These modifications resulted in the discovery of SJF620, a new PROTAC that retains potent degradation of BTK in cellular assays and has a superior pharmacokinetic profile to MT802 in mice. Therefore, SJF620 is a promising compound for further exploration of BTK degradation in vivo.
2. Results and discussion
Based on our previous knowledge, we proposed that the replacement of the CRBN Ligand A warhead with a VHL ligand warhead could result in better pharmacokinetic properties. Therefore, our first efforts focused on the syntheses of analogs of MT802, containing VHL Ligands B20 and C21 (Fig. 5a) as different E3-recruiting elements. These two ligands are structurally similar and bind at the same site on VHL, but they have different linker-attaching points (Fig. 5b). Given our recent work10,22 and other studies23, 24 about characterization of PROTAC-induced ternary complexes, we explored both VHL ligands, which likely induce different de-novo interactions between the surfaces of BTK and VHL and therefore different orientations for the E3 ligase (Fig. 5b). Since we were recruiting a new E3 ligase, we could not rely on the optimized linker length identified in MT802. Thus, we screened many PROTACs with varying linker lengths (Table 1).
Figure 5.

a) CRBN and VHL ligands. b) Crystal structures of VHL in complex with VHL ligand (PDB code 4W9H)20.
Table 1. -.
Degradation profile, and pharmacokinetic data after IV compound administration in mice.
| PROTAC | In NAMALWA cells line | IV mice (1 mg/Kg) | |||||
|---|---|---|---|---|---|---|---|
| Compound | E3 ligase recruited | DC50 (nM) | Dmax | Clearance (Cl) mL/min/kg | Half Life (t1/2) h | Cmax**(ug/mL) | Exposure (AUClast) min*ng/mL |
| MT802 | CRBN | 6.2 | 99% | 1662 | 0.119 | 0.073 | 10.2 |
| SJF638 | VHL | 374 | 49% | --- | --- | --- | --- |
| SJF678 | VHL | 162 | 50% | --- | --- | --- | --- |
| SJF690 | VHL | >1000 | --- | --- | --- | --- | --- |
| SJF608 | CRBN | 8.3 | 91% | 102 | 1.62 | 0.831 | 166 |
| SJF620 | CRBN | 7.9 | 95% | 40.8 | 1.64 | 2.1 | 405 |
Tmax (h) = 0.033
The synthesis of the first PROTAC containing VHL Ligand B as a warhead was achieved starting from commercially available 3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine, which was converted under Mitsunobu conditions to piperidine compound 1 following the procedure reported by J. Liu, et.al.25. Alkylation of compound 1 with the ω-bromo t-butyl ester 2 provided the corresponding condensation product 3. Cleavage of the t-butyl ester on 3, followed by amide bond formation with amino VHL Ligand B afforded the final PROTAC SJF638 (Scheme 1).
Scheme 1.

Synthesis of PROTAC SJF638. Reagents and conditions: (a) Et3N, DMF; (b) TFA, DCM; (c) VHL Ligand, PyBOP, Et3N, DMF.
Based on prior experience21, 22, we also prepared PROTACs containing the VHL ligand C, which will induce a protein complex with a different E3 orientation relative to the target, BTK. We initiated the synthesis of one of these PROTACs starting from commercially available 1-bromo-4-chloro-butane, which was condensed with butane-1,4-diol to provide compound 5 (Scheme 2). This chloro-alkyl alcohol 5 was transformed to the corresponding mesylate, followed by in-situ conversion to the chloro-iodo alkyl linker 6. Alkylation of VHL Ligand C with the iodo-linker 6 gave the chloro-compound 7, which was converted under Finkelstein conditions to the iodo-compound 8. Finally, condensation of 8 with the warhead 1 provided the PROTAC SJF678.
Scheme 2.

Synthesis of PROTAC SJF678. Reagents and conditions: (a) 1,4-butanodiol, KOH, DMSO; (b) MsCl, Et3N, DCM; (c) NaI, MeCN; (d) VHL Ligand C, Cs2CO3, DMF; (e), NaI, acetone; (f) 1, Et3N, DMF.
Following the approach shown on Scheme 3, we prepared a second PROTAC containing VHL Ligand C. Michael addition of 3-chloropropanol to t-butyl acrylate provided the chloro-alkyl linker 9, which was transformed to the iodo-compound 10 under Finkelstein conditions. Condensation of VHL-Ligand C with the linker 10 gave t-butyl ester 11. Cleavage of t-butyl ester 11 to the corresponding carboxylic acid, followed amide-bond formation with warhead 1 afforded the expected PROTAC SJF690 in good yields (Scheme 3).
Scheme 3.

Synthesis of PROTAC SJF690. Reagents and conditions: (a) Triton B, MeCN; (b) NaI, acetone; (c) VHL ligand C, Cs2CO3, DMF; (d) TFA, DCM; (e) PyBOP, Et3N, DMF.
Simultaneously, we explored a different strategy also aimed to improve on the pharmacokinetic properties of MT802. In this approach we made structural modifications on the linker and CRBN ligand. We hypothesized that replacing the amide connectivity with an ether moiety between the linker and CRBN would provide a more robust and metabolically stable compound than MT802 (Fig. 6). We initiated the synthesis of this PROTAC by preparing the N-protected-5-hydroxythalidomide 12 following a new methodology that we developed in-house26. Alkylation of 12 with commercially available bis-iodo linker 13 provided the corresponding iodo-compound 14 in good yields. Alkylation of 14 with warhead 1 gave the protected PROTAC 15. SEM deprotection of compounds 15 afforded the final PROTAC SJF608 in very high yields (Scheme 4).
Figure 6.

Structural modifications on linker and CRBN ligand in MT802
Scheme 4.

Synthesis of PROTAC SJF608. Reagents and conditions: (a) Cs2CO3, DMF; (b) Et3N, DMF; (c) TFA, DCM.
We then decided to further modify the CRBN ligand in an effort to improve pharmacokinetic properties. Thus, we eliminated one of the carbonyls on the phthalimide moiety (lenalidomide analog) on MT802 (Fig. 6). This compound was synthesized starting from 5-hydroxy-iso-indolinone 1615, following the procedure described in the literature. Alkylation of compound 16 with commercially available 1,2-bis(2-iodoethoxy)ethane 17 provided the iodo-compound 18, further condensation of this iodo-compound 18 with the warhead 1 gave the uncycled intermediate 19. Finally, acidic catalyzed cyclization of 19 afforded to the final PROTAC SJF620 in overall good yields (Scheme 5).
Scheme 5.

Synthesis of PROTAC SJF620. Reagents and conditions: (a) Cs2CO3, DMF; (b) Et3N, DMF; (c) BSA, MeCN, reflux temperature.
With these synthetic routes fully developed, we evaluated their BTK degradation profile in NAMALWA cells a Burkitt’s lymphoma cell line with robust BTK expression. Unfortunately, PROTACs harboring VHL Ligands B and C as the E3-targeting warhead showed significant reduction of target degradation (Dmax ~ 50%) and less potency (Table 1). However, PROTAC SJF608, which contained the same CRBN ligand as MT802 but without the amide group in its linker, showed a similarly potent degradation profile as MT802 (Table 1 and Fig. 6). We observed similar conservation of potency and efficacy with SJF620, which contained a lenalidomide analog for recruiting CRBN (Table 1 and Fig. 7). Encouragingly, both PROTACs SJF608 and SJF620 proved to have superior pharmacokinetic profiles relative to MT-802, with SJF620 displaying a more robust clearance and exposure profile, and we believe that this compound can be progressed to further in vivo studies.
Figure 7.

BTK degradation profile in NAMALWA cells for PROTACs SJF608 and SJF620. (A) Structures of PROTAC derivatives screened for cellular degradation of BTK. (B) Representative immunoblots showing degradation of BTK relative to GAPDH loading control. All compounds were screened at indicated concentrations in biological duplicate in NAMALWA. (C) Quantitation of DC50 and DMax values of these compounds by non-linear regression.
Experimental section
1. Chemistry
General comments. Unless otherwise indicated, common reagents or materials were obtained from commercial source and used without further purification. Tetrahydrofuran (THF), dimethylformamide (DMF), and dichloromethane (DCM) were dried by a PureSolv™ solvent drying system. Flash column chromatography was performed using silica gel 60 (230–400 mesh). Analytical thin layer chromatography (TLC) was carried out on Merck silica gel plates with QF-254 indicator and visualized by Uv or KMnO4. 1H and 13C NMR spectra were recorded on an Agilent DD2 500 (500 MHz 1H; 125 MHz 13C) or Agilent DD2 600 (600 MHz 1H; 150 MHz 13C) or Agilent DD2 400 (400 MHz 1H; 100 MHz 13C) spectrometer at room temperature. Chemical shifts were reported in ppm relative to the residual CDCl3 (δ 7.26 ppm 1H; δ 77.00 ppm 13C), CD3OD (δ 3.31 ppm 1H; δ 49.00 ppm 13C), or d6-DMSO (δ 2.50 ppm 1H; δ 39.52 ppm 13C). NMR chemical shifts were expressed in ppm relative to internal solvent peaks, and coupling constants were measured in Hz. (bs = broad signal). Only peaks of the major rotamer are reported. Mass spectra were obtained using electrospray ionization (ESI) on a time of flight (TOF) mass spectrometer.
Preparation of 3-(4-phenoxyphenyl)-1-(piperidin-4-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine hydrochloride (Compound 1)
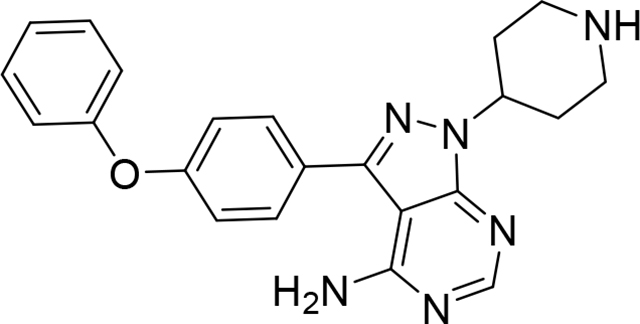
To a solution of triphenylphosphine (864.72 mg, 3.3 mmol) in THF (300 mL) was added DIAD (0.65 mL, 3.3 mmol) at 0 °C. The reaction mixture was stirred at this temperature for 0.5 h under argon, then tert-butyl 4-hydroxypiperidine-1-carboxylate (663.53 mg, 3.3 mmol) was added. The reaction mixture was stirred at 0 °C for 0.5 h. After that, 3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (500 mg, 1.65 mmol) was added. The reaction mixture was allowed to warm to room temperature with stirring for 4 h. The resulting mixture was then concentrated to afford the crude product, which was purified by flash silica gel column chromatography (SiO2-80g, Hex:EtOAc, gradient 3:7 to 100% in 15 min ) to provide the desired Boc-protected compound as an off-white foam (504 mg). 1H NMR (400 MHz, DMSO-d6) δ 8.22 (s, 1H), 7.63 (d, J = 8.7 Hz, 2H), 7.49 – 7.33 (m, 2H), 7.27 – 6.98 (m, 5H), 4.97 – 4.80 (m, 1H), 4.08 (bs, 2H), 2.97 (bs, 2H), 2.12 – 1.75 (m, 4H), 1.40 (s, 9H). Then, this foam was dissolved in EtOAc (10 mL) and 4 N HCl in Dioxane (5 mL) was added. The reaction mixture was stirred at room temperature for 12 h (overnight). After complete conversion of the starting material, the solid was collected by filtration and washed with (EtOAc, 2mLx2) to give an off-white solid (1) (560 mg, 80% yield). 1H NMR (500 MHz, DMSO-d6) δ 9.35 (bs, 1H), 9.02 (bs, 1H), 8.54 (s, 1H), 7.66 (d, J = 8.2 Hz, 2H), 7.45 (t, J = 7.5 Hz, 2H), 7.17 (td, J = 19.2, 17.4, 7.6 Hz, 5H), 5.12 (t, J = 11.1 Hz, 1H), 3.43 (d, J = 12.5 Hz, 2H), 3.19 (q, J = 11.5 Hz, 2H), 2.39 (q, J = 10.9 Hz, 2H), 2.15 (d, J = 12.6 Hz, 2H). LC/MS: [M+H]+ for C22H23N6O calculated. 387.1904. Found 387.1933.
tert-butyl 2-(2-(2-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)ethoxy)ethoxy)acetate (Compound 3)
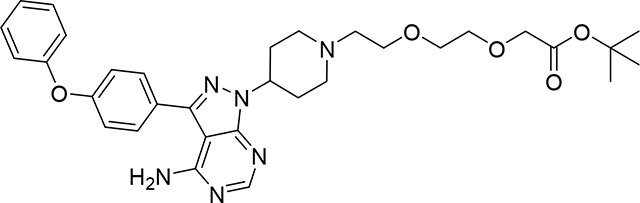
To a solution of 3-(4-phenoxyphenyl)-1-(4-piperidyl)pyrazolo[3,4-d] -pyrimidin-4-amine (1) (7.51 mg, 0.02 mmol) and TEA (0.01 mL, 0.58 mmol) in DMF (1 mL) was added tert-butyl 2-(2-(2-bromoethoxy)ethoxy)acetate (2) (5.5 mg, 0.02 mmol) and the resulting solution stirred for 36 h at room temperature. The reaction mixture was diluted with EtOAc (5 mL) and washed with water/brine (3×5 mL), organic phase was dried (Na2SO4, and evaporated under vacuum. Crude product was purified by PTLC (DCM:MeOH:NH4OH, 91:8:1) to give 3 mg of product (3) (26%). 1H NMR (500 MHz, DMSO-d6) δ 8.24 (s, 1H), 7.66 (d, J = 8.3 Hz, 2H), 7.43 (t, J = 7.7 Hz, 2H), 7.19 (t, J = 7.3 Hz, 1H), 7.14 (dd, J = 12.3, 8.5 Hz, 5H), 4.72 (bs, 1H), 4.00 (s, 2H), 3.74 – 3.44 (m, 6H), 3.14 (bs, 2H), 2.85 – 2.55 (m, 3H), 2.38 – 2.14 (m, 3H), 2.02 – 1.80 (m, 2H), 1.40 (s, 9H). LC/MS (ESI); m/z [M+H]+: Calcd. for C32H41N6O5, 589.3138. Found 589.3347.
(2S,4R)-1-((S)-2-(2-(2-(2-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)ethoxy)ethoxy)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (PROTAC SJF638)
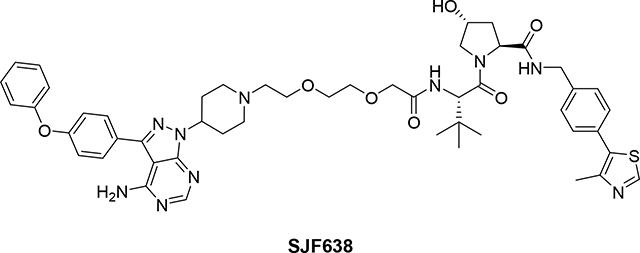
A solution of tert-butyl 2-(2-(2-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4- d]15pyrimidin-1-yl)piperidin-1-yl)ethoxy)ethoxy)acetate (3) (3 mg, 0.01 mmol) in a mixture of TFA (1 mL, 13.46 mmol) and dichloromethane (3 mL) was stirred for 2 h. Then the solvent was removed under vacuum and crude product was dried under high vacuum for 2 h. Crude product was used in the next step without any further purification (2.7 mg, quantitative yield). LC/MS (ESI); m/z: [M+H]+ Calcd. for C28H33N6O5, 533.2512. Found 533.2579. To a solution of 2-(2-(2-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d] -pyrimidin-1-yl)piperidin-1-yl)ethoxy)ethoxy)acetic acid (crude product from previous step) (2.7 mg, 0.01 mmol) and (2S,4R)-1-[(2S)-2-amino-3,3-dimethyl-butanoyl]-4-hydroxy-N-[[4-(4-methylthiazol-5-yl)-phenyl]methyl]pyrrolidine-2-carboxamide; hydro-chloride (VHL ligand B)20 (2.6 mg, 0.006 mmol) in DMF (1 mL) was added TEA (0.1 mL, 0.72 mmol) and PyBOP (2.77 mg, 0.01 mmol) at room temperature. The reaction mixture was stirred for 4 h at the same temperature. TLC (DCM:MeOH:NH4OH, 91:8:1) shows no starting materials. The DMF was removed under high vacuum. Crude product was filtered over a silica-carbonate cartridge using a mixture of DCM:MeOH:NH4OH (91:8:1) as a eluent. Filtrate was evaporated under vacuum and crude product was purified by PTLC (DCM:MeOH:NH4OH, 91:8:1) to give 2.5 mg of product SJF638 (52% yield). 1H NMR (600 MHz, DMSO-d6) δ 8.96 (s, 1H), 8.60 (t, J = 6.0 Hz, 1H), 8.23 (s, 1H), 7.65 (d, J = 8.4 Hz, 2H), 7.48 – 7.34 (m, 7H), 7.18 (t, J = 7.4 Hz, 1H), 7.16 – 7.07 (m, 4H), 5.16 (d, 1H), 4.63 (bs, 1H), 4.57 (d, J = 9.6 Hz, 1H), 4.44 (t, J = 8.2 Hz, 1H), 4.40 – 4.32 (m, 2H), 4.23 (dd, J = 15.8, 5.6 Hz, 1H), 3.98 (s, 2H), 3.72 – 3.50 (m, 8H), 3.01 (bs, 2H), 2.56 (bs, 2H), 2.43 (s, 3H), 2.26 – 2.11 (m, 4H), 2.10 – 1.98 (m, 1H), 1.94 – 1.78 (m, 3H), 0.95 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 171.76, 169.10, 168.57, 158.15, 157.03, 156.29, 155.42, 153.60, 151.42, 147.72, 142.77, 139.42, 131.13, 130.13, 130.01, 129.67, 128.68, 128.11, 127.43, 123.79, 118.99, 118.94, 97.44, 70.51, 69.59, 69.46, 68.88, 58.74, 56.99, 56.58, 55.67, 54.93, 52.72, 52.66, 41.67, 37.90, 35.75, 30.99, 26.19, 15.93. LC/MS (ESI); m/z [M+H]+: Calcd. for C50H61N10O7S, 945.4445. Found 945.4641.
4-(4-chlorobutoxy)butan-1-ol (Compound 5)
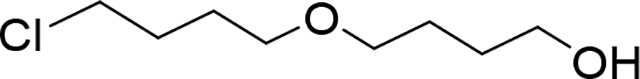
A solution of butane-1,4-diol (23.14 mL, 260.35 mmol) and KOH (2.43 g, 43.39 mmol) in DMSO (135 mL) and distilled water (15 mL) was stirred 15 min at rt. Then the mixture was cooled down to 0 °C and 1-bromo-4-chloro-butane (5 mL, 43.39 mmol) was added dropwise over 1 h. The resulting solution was stirred 1 h at 0 °C and then 12 h (overnight) at rt. After dilution with a mixture of ether/hexane (1:1,250 mL) and water (250 mL), the organic phase was separated. Water layer was extracted with a mixture ether/hexane (2×150 mL). Organic extracts were combined and washed with water (4×150 mL), dried over Na2SO4, and concentrated under vacuum. Crude product was subjected to flash column chromatography (SiO2-120 g, Gradient, Hexane 100% to hexane / EtOAc, 1/1 in 20 minutes) to give 3.18 g (41% yield) of product (5) as an oil. 1H NMR (400 MHz, Chloroform-d) δ 3.64 (t, J = 5.7 Hz, 2H), 3.57 (t, J = 6.5 Hz, 2H), 3.51 – 3.43 (m, 4H), 2.15 (s, 1H), 1.91 – 1.81 (m, 2H), 1.79 – 1.61 (m, 6H). 13C NMR (151 MHz, Chloroform-d) δ 71.09, 70.24, 62.92, 45.06, 30.46, 29.60, 27.13, 27.02. LC/MS (ESI); m/z: [M+H]+ Calcd. for C8H18ClO2, 181.0995. Found 181.0990
Chloro-4-(4-iodobutoxy)butane (Compound 6)
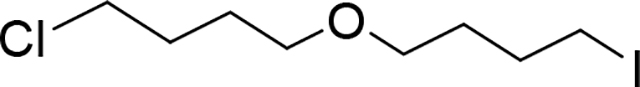
To a solution of 4-(4-chlorobutoxy)butan-1-ol (5)(271 mg, 1.5 mmol) in dichloromethane (5 mL) was added TEA (0.63 mL, 4.5 mmol), then reaction mixture was cooled to 0 °C (water ice/acetone bath) and Mesyl chloride (0.14 mL, 1.8 mmol) was added dropwise. The reaction mixture was stirred for 1 h at the same temperature. By TLC no starting material (Hex:EtOAc, 3:7), and a less polar compound was formed. Reaction mixture was poured into an aqueous solution of NaHCO3 (20 mL) and product extracted with DCM (20mL, 2x), the organic extracts were combined, dried (Na2SO4), and evaporated under vacuum. The crude product (mesylate) was used in the next step without any further purification (>95% pure by NMR): 1H NMR (400 MHz, Chloroform-d) δ 4.26 (t, J = 6.5 Hz, 2H), 3.57 (t, J = 6.6 Hz, 2H), 3.44 (td, J = 6.2, 2.0 Hz, 4H), 3.01 (s, 3H), 1.92 – 1.78 (m, 4H), 1.76 – 1.62 (m, 4H). Crude mixture from previous step was diluted in Acetonitrile (5 mL) and Nal (247.32 mg, 1.65 mmol) was added, the reaction mixture was stirred at room temperature for 72 h (over the weekend). By TLC (Hex:EtOAc, 7:3) and NMR small amount of masylate, the reaction was poured into an aqueous solution of Na2S2O3 (10%, 20 mL) and product was extracted with DCM (2×20 mL). Organic extracts were combined, dried (Na2SO4) and evaporated under vacuum. Crude product was purified by flash chromatography (SiO2-40g, grad. Hex:EtOAc, 2 to 20% in 10 min), to give 310 mg of product (6) as an oil (71% yield): 1H NMR (500 MHz, Chloroform-d) δ 3.57 (t, J = 6.6 Hz, 2H), 3.43 (td, J = 6.3, 2.4 Hz, 4H), 3.22 (t, J = 7.0 Hz, 2H), 2.01 – 1.80 (m, 4H), 1.77 – 1.61 (m, 4H). 13C NMR (151 MHz, Chloroform-d) δ 70.13, 69.77, 45.13, 30.74, 30.56, 29.68, 27.22, 7.02. LC/MS (ESI); m/z: [M+H]+ Calcd. for C8H17ClO, 291.001. Found 291.0060.
(2S,4R)-N-(2-(4-(4-chlorobutoxy)butoxy)-4-(4-methylthiazol-5-yl)benzyl)-4-hydroxy-1-((S)-3-methyl-2-(1-oxoisoindolin-2-yl)butanoyl)pyrrolidine-2-carboxamide (Compound 7)
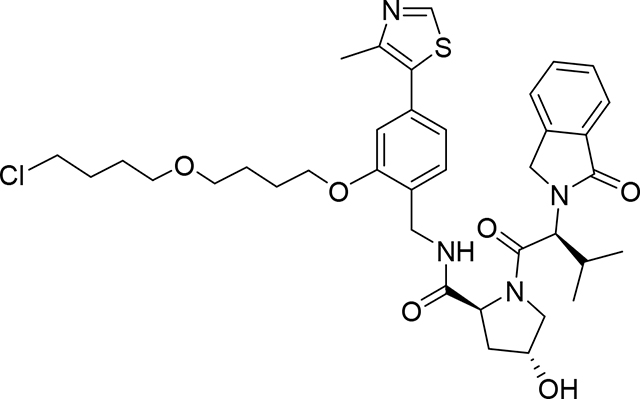
To a mixture of (2S,4R)-4-hydroxy-N-[[2-hydroxy-4-(4-methylthiazol-5-yl)phenyl]methyl]-1-[(2S)-3-methyl-2- (1-oxoisoindolin-2-yl)butanoyl]pyrrolidine-2-carboxamide (VHL ligand C)21 (83 mg, 0.151 mmol) and 1-chloro-4-(4-iodobutoxy)butane (6) (44 mg, 0.151 mmol) in N,N-Dimethylformamide (1 mL) was added Cs2CO3 (49.34 mg, 0.151 mmol). After stirring at room temperature for 4 hrs, the reaction mixture was diluted with EtOAc (10 mL) and washed with water (5×10 mL), organic phase was dried (Na2SO4, and evaporated under vacuum. Crude product was purified by PTLC (DCM:MeOH:NH4OH, 92:7:1) to give 83 mg of product (7) (77% yield). 1H NMR (500 MHz, DMSO-d6) δ 8.99 (s, 1H), 8.36 (t, J = 5.7 Hz, 1H), 7.71 (d, J = 7.5 Hz, 1H), 7.65 – 7.57 (m, 2H), 7.50 (t, J = 7.7 Hz, 1H), 7.33 (d, J = 7.6 Hz, 1H), 7.02 – 6.96 (m, 2H), 5.08 (d, J = 3.9 Hz, 1H), 4.71 (d, J = 10.8 Hz, 1H), 4.59 – 4.18 (m, 6H), 4.07 (t, J = 5.0 Hz, 2H), 3.77 (dd, J = 10.5, 4.2 Hz, 1H), 3.69 (d, J = 10.4 Hz, 1H), 3.63 (t, J = 6.6 Hz, 2H), 3.49 – 3.36 (m, 4H), 2.47 (s, 3H), 2.37 – 2.29 (m, 1H), 2.07 – 2.01 (m, 1H), 1.96 – 1.89 (m, 1H), 1.85 – 1.56 (m, 8H), 0.96 (d, J = 6.4 Hz, 3H), 0.74 (d, J = 6.6 Hz, 3H).13C NMR (151 MHz, DMSO-d6) δ 171.54, 168.09, 167.48, 155.90, 151.48, 147.89, 142.21, 131.60, 131.37, 131.32, 130.98, 127.92, 127.69, 126.96, 123.63, 123.02, 120.77, 111.69, 69.68, 69.14, 68.62, 67.56, 58.70, 57.79, 55.42, 46.82, 45.36, 38.10, 37.6, 29.18, 28.40, 26.63, 25.90, 25.69, 18.89, 18.64, 16.03. LC/MS (ESI); m/z: [M+H]+ Calcd. for C37H48ClN4O6S, 711.2983. Found 711.3224.
(2S,4R)-N-(2-(4-(4-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)butoxy)butoxy)-4-(4-methylthiazol-5-yl)benzyl)-4-hydroxy-1-((S)-3-methyl-2-(1-oxoisoindolin-2-yl)butanoyl)pyrrolidine-2-carboxamide (SJF678)
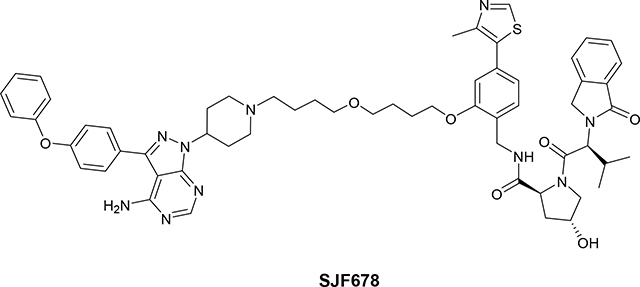
To a solution of (2S,4R)-N-[[2-[4-(4-chlorobutoxy)butoxy]-4-(4-methylthiazol-5-yl)phenyl]methyl]-4-hydroxy-1-[(2S)-3-methyl-2-(1-oxoisoindolin-2-yl)butanoyl]pyrrolidine −2-carboxamide (7) (75 mg, 0.11 mmol) in Acetone (10 mL) was added Nal (158.05 mg, 1.05 mmol). The reaction mixture was stirred at reflux temperature for 48 h, then the solvent was removed under vacuum and crude product was dissolved in EtOAc (15 mL) and an aqueous solution of Na2SO3 (10%, 10 mL), organic layer was separated, washed with water (10 mL), dried (Na2SO4) and evaporated under vacuum. Crude iodo-compound (8) was pure by NMR (quantitative yield) no further purification. 1H (400 MHz, Chloroform-d) δ 8.68 (s, 1H), 7.78 (d, J = 7.6 Hz, 1H), 7.52 (t, J = 7.4 Hz, 1H), 7.47 – 7.37 (m, 2H), 7.34 – 7.23 (m, 2H), 6.96 (d, J = 8.7 Hz, 1H), 6.87 (s, 1H), 4.76 (s, 1H), 4.73 (d, J = 6.7 Hz, 1H), 4.64 (t, J = 7.8 Hz, 1H), 4.57 – 4.36 (m, 5H), 4.05 (t, J = 6.2 Hz, 2H), 3.65 (dd, J = 11.5, 3.5 Hz, 1H), 3.49 (t, J = 6.2 Hz, 2H), 3.44 (t, J = 6.2 Hz, 2H), 3.19 (t, J = 6.9 Hz, 2H), 2.58 – 2.45 (m, 1H), 2.53 (s, 3H), 2.47 – 2.27 (m, 2H), 2.12 – 2.00 (m, 2H), 1.98 – 1.84 (m, 3H), 1.79 (dt, J = 9.4, 6.5 Hz, 2H), 1.71 – 1.58 (m, 2H), 0.89 (dd, J = 6.0 Hz, 6H).13C NMR (126 MHz, Chloroform-d) δ 170.54, 170.47, 169.71, 156.94, 150.41, 148.61, 142.23, 132.35, 131.99, 131.92, 131.73, 129.41, 128.15, 126.47, 123.97, 122.99, 121.65, 112.15, 70.62, 70.13, 69.83, 68.04, 58.81, 58.55, 56.7, 47.59, 39.05, 35.87, 30.75, 30.58, 29.85, 28.89, 26.55, 26.34, 19.23, 16.28, 6.98. LC/MS (ESI); m/z [M+H]+: Calcd. for C37H48IN4O6S, 803.2339. Found 803.2675.
To a solution of 3-(4-phenoxyphenyl)-1-(4-piperidyl)pyrazolo[3,4-d]pyrimidin-4-amine (1) (4.81 mg, 0.01 mmol) and (2S,4R)-4-hydroxy-N-(2-(4-(4-iodobutoxy)butoxy)-4-(4-methylthiazol-5-yl)benzyl)-1-((S)-3-methyl-2-(1-oxoisoindolin-2-yl)butanoyl)pyrrolidine-2-carboxamide (8) (10 mg, 0.01 mmol) in DMF (1 mL) was added TEA (0.01 mL, 0.04 mmol), the resulting solution stirred for 12 h (overnight) at room temperature. The DMF was evaporated under high vacuum and the crude product was filtered over a silica-carbonate cartridge using a mixture of DCM:MeOH:NH4OH (91:8:1) as a eluent. Filtrate was evaporated and crude was purified by PTLC (DCM:MeOH:NH4OH, 91:8:1) to give 4.8 mg of pure product SJF678 (36% yield). 1H NMR (500 MHz, Chloroform-d) δ 8.66 (s, 1H), 8.34 (s, 1H), 7.75 (d, J = 7.6 Hz, 1H), 7.64 (d, J = 8.5 Hz, 2H), 7.50 (t, J = 7.5 Hz, 1H), 7.44 – 7.33 (m, 4H), 7.33 – 7.26 (m, 2H), 7.15 (dd, J = 17.5, 8.0 Hz, 3H), 7.08 (d, J = 8.4 Hz, 2H), 6.95 (d, J = 7.7 Hz, 1H), 6.87 (s, 1H), 5.55 (s, 2H), 4.84 – 4.68 (m, 3H), 4.62 (t, J = 7.8 Hz, 1H), 4.58 – 4.28 (m, 5H), 4.05 (t, J = 6.3 Hz, 2H), 3.67 (dd, J = 11.3, 3.5 Hz, 1H), 3.50 (t, J = 6.3 Hz, 2H), 3.46 – 3.38 (m, 2H), 3.15 – 2.98 (m, 2H), 2.52 (s, 3H), 2.51 – 2.30 (m, 5H), 2.25 – 1.88 (m, 8H), 1.85 – 1.74 (m, 2H), 0.88 (dd, J = 18.6, 6.5 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 170.62, 170.32, 169.48, 158.50, 157.89, 157.00, 156.50, 155.60, 153.97, 150.39, 148.58, 143.44, 142.19, 132.36, 132.36, 131.97, 131.82, 131.75, 130.08, 129.59, 128.16, 128.7, 126.39, 124.12, 123.89, 122.96, 121.60, 119.65, 119.20, 112.09, 98.71, 70.88, 70.54, 69.92, 68.02, 58.72, 58.71, 58.28, 56.07, 52.91,47.53, 39.20, 36.21, 31.37, 29.83, 28.97, 27.85, 26.61, 26.31, 23.99, 19.19, 19.13, 16.27. LC/MS (ESI); m/z [M+H]+: Calcd. for C59H69N10O7S, 1061.5071. Found 1061.5353.
tert-Butyl 3-(3-chloropropoxy)propanoate (Compound 9)
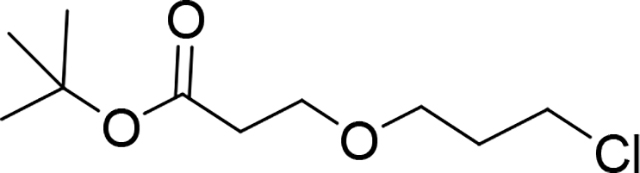
3-chloropropan-1-ol (8.84 mL, 105.78 mmol) in acetonitrile (50 mL) was added tert-butyl prop-2-enoate (15.35 mL, 105.78 mmol) followed by Triton B (1 mL, 2.54 mmol) in. The mixture was stirred at room temperature for 72 hour. The mixture was concentrated in vacuum and crude product was purified by CC (SiO2-250g, gradient Hex:EtOAc,95:5 to 9:1) to give 14.2 g of product (9) as an oil (60% yield). 1H NMR (500 MHz, Chloroform-d) δ 3.66 (t, J = 6.4 Hz, 2H), 3.62 (t, J = 6.5 Hz, 2H), 3.56 (t, J = 5.9 Hz, 2H), 2.51 – 2.42 (m, 2H), 1.99 (p, J = 6.2 Hz, 2H), 1.45 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 171.03, 80.70, 67.32, 66.72, 42.02, 36.46, 32.79, 28.22. LC/MS (ESI); m/z [M+Na]+: Calcd. for C10H19ClO3Na, 245.0920. Found 245.0957.
tert-Butyl 3-(3-iodopropoxy)propanoate (Compound 10)
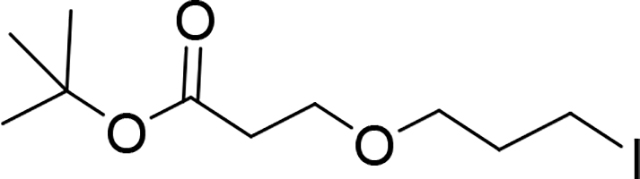
To a solution of tert-butyl 3-(3-chloropropoxy)propanoate (9) (1.36 g, 6.11 mmol) in acetone (100 mL) was added Nal (4.58 g, 30.53 mmol). The reaction mixture was stirred at reflux temperature for 72 h, then the solvent was removed under vacuum and crude product was re-dissolved in EtOAc (100 mL) and washed with water (100 mL), and an aqueous solution of Na2SO3 (10%, 50 mL), organic layer was separated, dried (Na2SO4) and evaporated under vacuum. Crude product (10) was pure by NMR (>98% purity). It was used in the next step without any further purification. 1H NMR (500 MHz, Chloroform-d) δ 3.67 (t, J = 6.4 Hz, 2H), 3.49 (t, J = 5.8 Hz, 2H), 3.25 (t, J = 6.8 Hz, 2H), 2.47 (t, J = 6.4 Hz, 2H), 2.04 (p, J = 6.3 Hz, 2H), 1.45 (s, 9H). 13C NMR (151 MHz, Chloroform-d) δ 171.01,80.72, 70.26, 66.71,36.46, 33.52, 28.25, 3.54. LC/MS (ESI); m/z [M+Na]+: Calcd. for C10H19IO3Na, 337.0276. Found 337.0351.
tert-Butyl 3-(3-(2-(((2S,4R)-4-hydroxy-1-((S)-3-methyl-2-(1-oxoisoindolin-2-yl)butanoyl) - pyrrol-idine-2-carboxamido)methyl)-5-(4-methylthiazol-5-yl)phenoxy)propoxy)propanoate (Compound 11)
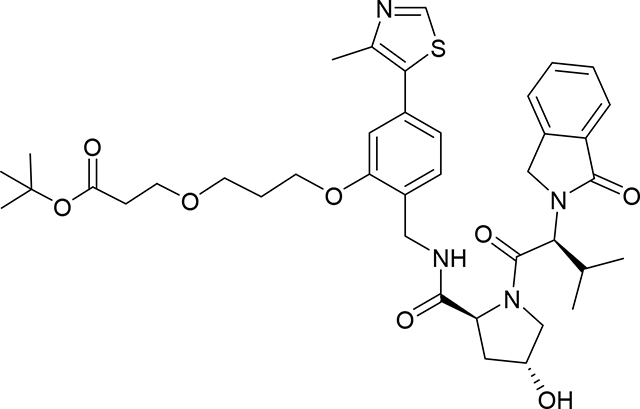
To a solution of (2S,4R)-4-hydroxy-N-[[2-hydroxy-4-(4-methylthiazol-5-yl)phenyl]methyl]-1-[(2S)-3-methyl-2-(1-oxoisoindolin-2-yl)butanoyl]pyrrolidine-2-carboxamide (VHL-Ligand-C)21 (40.0 mg, 0.0729 mmol) and tert-butyl 3-(3-iodopropoxy)propanoate (10) (34.4 mg, 0.109 mmol) in DMF (1 mL) was added Cs2CO3 (71 mg, 0.21 mmol) at room temperature, the reaction mixture was stirred for 12 h (overnight) at the same temperature. Reaction was diluted with EtOAc (10 mL) and washed with water (4×10 mL). Organic extract was dried (Na2SO4), and concentrated under vacuum. Crude product was purified by PTLC (DCM:MeOH:NH4OH, 91:8:1) to give 38 mg of product (11) (71% yield). 1H NMR (500 MHz, DMSO-d6) δ 8.98 (s, 1H), 8.36 (t, J = 5.8 Hz, 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.62 (q, J = 7.5, 6.5 Hz, 2H), 7.50 (t, J = 6.8 Hz, 1H), 7.35 (d, J = 7.7 Hz, 1H), 7.00 (d, J = 7.5 Hz, 2H), 5.09 (d, J = 4.1 Hz, 1H), 4.72 (d, J = 10.8 Hz, 1H), 4.61 – 4.19 (m, 6H), 4.09 (t, J = 6.1 Hz, 2H), 3.78 (dd, J = 10.5, 4.3 Hz, 1H), 3.70 (d, J = 10.6 Hz, 1H), 3.64 – 3.49 (m, 4H), 2.47 (s, 3H), 2.42 (t, J = 6.0 Hz, 2H), 2.39 – 2.27 (m, 1H), 2.12 – 1.87 (m, 4H), 1.34 (s, 9H), 0.97 (d, J = 6.5 Hz, 3H), 0.74 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 171.49, 170.43, 168.09, 167.47, 155.86, 151.42, 147.86, 142.18, 131.56, 131.37, 131.26, 131.00, 127.88, 127.81, 126.99, 123.60, 123.00, 120.83, 111.63, 79.63, 68.61,66.59, 65.95, 64.71, 58.69, 57.78, 55.40, 46.81, 38.08, 37.05, 35.85, 28.98, 28.38, 27.69, 18.88, 18.61, 15.99. LC/MS (ESI); m/z: [M+H]+ Calcd. for C39H51N4O8S, 735.3427. Found 735.3720.
(2S,4R)-N-(2-(3-(3-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)-3-oxopropoxy)propoxy)-4-(4-methylthiazol-5-yl)benzyl)-4-hydroxy-1-((S)-3-methyl-2-(1-oxoisoindolin-2-yl)butanoyl)pyrrolidine-2-carboxamide (SJF690)
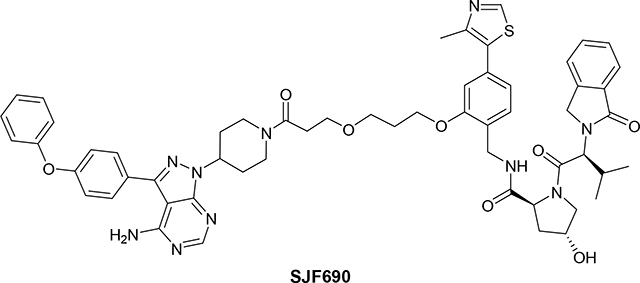
A solution of tert-butyl 3-(3-(2-(((2S,4R)-4-hydroxy-1-((S)-3-methyl-2- (1-oxoisoindolin-2-yl)butanoyl)pyrrolidine-2-carboxamido)methyl)-5-(4-methylthiazol-5-yl)phenoxy)propoxy)-propanoate (19 mg, 0.03 mmol) in a mixture of TFA (0.7 mL, 9.42 mmol) and dichloromethane (2 ml) was stirred for 2 h. Then the solvent was removed under vacuum and crude product was dried under high vacuum for 1 h. Crude carboxylic acid was used in the next step without any further purification (17.5 mg, quantitative yield). LC/MS (ESI); m/z: [M+H]+ Calcd. for C35H43N4O8S, 679.2801. Found 679.3105.
To a solution of 3-(3-(2-(((2S,4R)-4-hydroxy-1-((S)-3-methyl-2-(1-oxoisoindolin-2-yl)butanoyl)-pyrrolidine-2-carboxamido)methyl)-5-(4-methylthiazol-5-yl)phenoxy)propoxy)propanoic acid (from previous step) (17.5 mg, 0.03 mmol) and 3-(4-phenoxyphenyl)-1-(piperidin-4-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (10.96 mg, 0.03 mmol) (1) in DMF(1 mL) was added TEA (0.1 mL, 0.72 mmol) and PyBOP (14.76 mg, 0.03 mmol) at room temperature. The reaction mixture was stirred for 4 h at the same temperature. TLC (DCM:MeOH:NH4OH, 91:8:1) shows no starting materials. The reaction mixture was dissolved in EtOAc (10 mL) and washed with brine/water (3×5mL). Organic extract was concentrated under vacuum and crude product was purified by PTLC (DCM:MeOH:NH4OH, 91:8:1) to give 22 mg of SJF690 (83% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.91 (s, 1H), 8.33 (t, J = 5.5 Hz, 1H), 8.22 (s, 1H), 7.68 (d, J = 7.5 Hz, 1H), 7.63 – 7.52 (m, 4H), 7.50 – 7.43 (m, 1H), 7.43 – 7.34 (m, 2H), 7.31 (d, J = 7.6 Hz, 1H), 7.17 – 7.11 (m, 1H), 7.07 (dd, J = 11.4, 8.4 Hz, 4H), 6.98 – 6.90 (m, 2H), 5.05 (d, J = 3.9 Hz, 1H), 4.98 – 4.87 (m, 1H), 4.68 (d, J = 10.8 Hz, 1H), 4.59 – 4.16 (m, 7H), 4.06 (t, J = 5.6 Hz, 2H), 4.04 – 3.94 (m, 1H), 3.80 – 3.60 (m, 4H), 3.57 (t, J = 6.0 Hz, 2H), 3.20 (t, J = 12.5 Hz, 1H), 2.82 – 2.53 (m, 3H), 2.40 (s, 3H), 2.33 – 2.23 (m, 1H), 2.16 – 1.76 (m, 8H), 0.92 (d, J = 6.3 Hz, 3H), 0.69 (d, J = 6.3 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 171.50, 168.79, 168.08, 167.46, 158.17, 157.01, 156.30, 155.88, 155.52, 153.62, 151.37, 147.85, 143.01, 142.19, 131.56, 131.37, 131.26, 130.99, 130.11, 130.00, 128.02, 127.89, 127.79, 127.00, 123.75, 123.60, 123.00, 120.82, 118.96, 118.91, 111.66, 97.42, 68.62, 66.75, 66.69, 64.81, 58.69, 57.78, 55.42, 53.33, 46.81, 44.11, 38.10, 37.06, 32.68, 31.52, 30.90, 29.05, 28.39, 18.87, 18.62, 15.98. LC/MS (ESI); m/z [M+H]+: Calcd. for C57H63N10O8S, 1047.4551. Found 1047.5190.
2-(2,6-dioxo-1-((2-(trimethylsilyl)ethoxy)methyl)piperidin-3-yl)-5-(2-(2-(2-iodoethoxy) - ethoxy)ethoxy)isoindoline-1,3-dione (Compound 14)
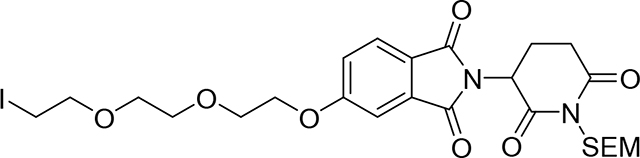
To a mixture of 2-[2,6-dioxo-1-(2-trimethylsilylethoxymethyl)-3-piperidyl]-5-hydroxy -isoindoline-1,3-dione (12)26 (64 mg, 0.16 mmol) and 1,2-bis(2-iodoethoxy)ethane (13) (702.45 mg, 1.9 mmol) in DMF (2 mL) was added Cs2CO3 (309.31 mg, 0.95 mmol). After stirring at room temperature for 2 hrs, the reaction mixture was diluted with EtOAc (10 mL) and washed with water (5×10 mL), organic phase was dried (Na2SO4, and evaporated under vacuum. Crude product was filtered over a short column of SiO2 (DCM 100%, then DCM:MeOH:NH4OH, 92:7:1) to remove the excess of the bis-iodo reactant, then crude product was purified by PTLC (DCM:MeOH:NH4OH, 92:7:1) to give 71 mg of product (14) (69% yield). 1H NMR (500 MHz, DMSO-d6) δ 7.83 (d, J = 8.3 Hz, 1H), 7.45 (d, J = 2.3 Hz, 1H), 7.38 (dd, J = 8.4, 2.3 Hz, 1H), 5.25 (dd, J = 13.1, 5.3 Hz, 1H), 5.08 (s, 2H), 4.41 – 4.23 (m, 2H), 3.87 – 3.76 (m, 2H), 3.66 (t, J = 6.4 Hz, 2H), 3.63 – 3.43 (m, 6H), 3.30 (t, J = 6.5 Hz, 2H), 3.03 (ddd, J = 18.8, 14.3, 5.3 Hz, 1H), 2.84 – 2.74 (m, 1H), 2.57 (qd, J = 13.4, 4.5 Hz, 1H), 2.16 – 2.03 (m, 1H), 0.84 (t, J = 7.7 Hz, 2H), −0.02 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 171.65, 169.90, 166.80, 166.73, 163.95, 133.90, 125.31, 123.04, 120.94, 108.94, 70.99, 69.95, 69.31, 68.68, 68.44, 68.30, 65.96, 49.54, 31.20, 21.07, 17.43, 5.43, −1.32. LC/MS (ESI); m/z: [M+Na]+ Calcd. for C25H35IN2O8SiNa, 669.1105. Found 669.1311.
5-(2-(2-(2-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)ethoxy)ethoxy)ethoxy)-2-(2,6-dioxo-1-((2-(trimethylsilyl)ethoxy)methyl)piperidin-3-yl)isoindoline-1,3-dione (Compound 15)
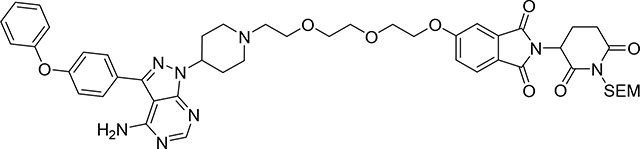
To a solution of 3-(4-phenoxyphenyl)-1-(4-piperidyl)pyrazolo[3,4-d]pyrimidin-4-amine; - hydrochloride (1) (36.63 mg, 0.09 mmol) and TEA (0.1 mL, 0.57 mmol) in DMF (1 mL) was added 2-[2,6-dioxo-1-(2-trimethylsilylethoxymethyl)-3-piperidyl]-5-[2-[2-(2-iodoethoxy) ethoxy]ethoxy] - iso-indoline-1,3-dione (14) (56 mg, 0.09 mmol) and the resulting solution stirred for 36 h at rt. The reaction mixture was evaporated under vacuum. Crude product was purified by PTLC (DCM:MeOH:NH4OH, 92:7:1, and then again DCM:MeOH, 95:5) to give 36.5 mg of pure product (15) (47% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.22 (s, 1H), 7.80 (d, J = 8.3 Hz, 1H), 7.65 (d, J = 8.7 Hz, 2H), 7.50 – 7.33 (m, 4H), 7.15 (dt, J = 22.5, 7.4 Hz, 5H), 5.24 (dd, J = 13.1, 5.4 Hz, 1H), 5.06 (s, 2H), 4.68 – 4.56 (m, 1H), 4.39 – 4.24 (m, 2H), 3.84 – 3.76 (m, 2H), 3.65 – 3.42 (m, 8H), 3.09 – 2.94 (m, 3H), 2.77 (dd, J = 14.1, 3.3 Hz, 1H), 2.64 – 2.50 (m, 3H), 2.26 – 2.01 (m, 5H), 1.92 – 1.80 (m, 2H), 0.82 (t, J = 8.6 Hz, 2H), −0.04 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 171.62, 169.87, 166.78, 166.70, 163.96, 158.13, 157.03, 156.28, 155.41, 153.60, 142.75, 133.87, 130.11, 129.99, 128.11, 125.26, 123.78, 123.00, 120.93, 118.98, 118.92, 108.91, 97.43, 69.95, 69.71,68.65, 68.58, 68.46, 68.29, 65.95, 57.06, 53.89, 52.70, 49.53, 31.19, 31.03, 21.05, 17.41, −1.35. LC/MS (ESI); m/z [M+H]+: Calcd. for C47H57N8O9Si, 905.4017. Found 905.4290.
5-(2-(2-(2-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)ethoxy)ethoxy)ethoxy)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (PROTAC SJF608)
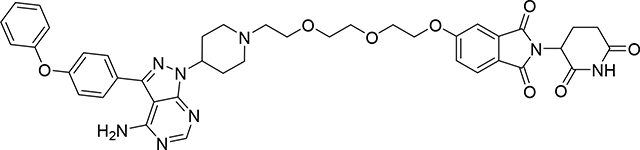
A solution of 5-[2-[2-[2-[4-[4-amino-3-(4-phenoxyphenyl)pyrazolo[3,4-d]pyrimidin −1-yl]-1-piperidyl]ethoxy]ethoxy]ethoxy]-2-[2,6-dioxo-1-(2-trimethylsilylethoxymethyl)-3-piperidyl]isoindoline-1,3-dione (15) (30 mg, 0.03 mmol) in a mixture of TFA (1 mL, 13.46 mmol) and dichloromethane (2 mL) was stirred for 1 h. Then the solvent was removed under vacuum and crude product was dried under high vacuum for 1 h. Crude product was purified by PTLC (DCM:MeOH:NH4OH, 92:7:1) to give 24.2 mg of PROTAC SJF0608 (94% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.11 (bs, 1H), 8.22 (s, 1H), 7.80 (d, J = 8.3 Hz, 1H), 7.65 (d, J = 8.4 Hz, 2H), 7.50 – 7.30 (m, 4H), 7.15 (dt, J = 23.0, 7.7 Hz, 5H), 5.10 (dd, J = 12.9, 5.3 Hz, 1H), 4.72 – 4.62 (m, 1H), 4.39 – 4.23 (m, 2H), 3.86 – 3.74 (m, 2H), 3.70 – 3.45 (m, 6H), 3.14 – 3.00 (m, 1H), 2.93 – 2.81 (m, 1H), 2.73 – 2.50 (m, 4H), 2.37 – 2.14 (m, 4H), 2.07 – 1.98 (m, 1H), 1.96 – 1.82 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 172.76, 169.92, 166.84, 166.77, 163.94, 158.15, 157.05, 156.27, 155.43, 153.61, 142.79, 133.89, 130.12, 130.00, 128.10, 125.24, 123.79, 123.03, 120.90, 119.00, 118.93, 108.87, 97.44, 73.80, 69.94, 69.71, 68.66, 68.48, 68.46, 56.99, 53.79, 52.65, 48.96, 30.96, 22.08. LC/MS (ESI); m/z: [M+H]+ Calcd. for C41H43N8O8, 775.3203. Found 775.3290.
tert-Butyl 5-amino-4-(5-(2-(2-(2-iodoethoxy)ethoxy)ethoxy)-1-oxoisoindolin-2-yl)-5-oxopentanoate (Compound 18)
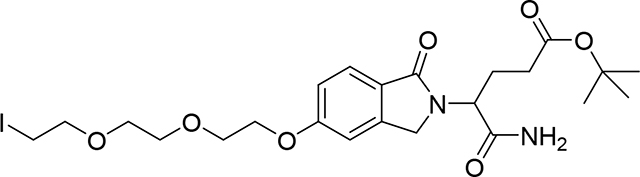
To a mixture of tert-butyl 5-amino-4-(5-hydroxy-1-oxo-isoindolin-2-yl)-5-oxo-pentanoate (16)27 (50 mg, 0.15 mmol) and 1,2-bis(2-iodoethoxy)ethane (663.88 mg, 1.79 mmol) in DMF (2 mL) was added Cs2CO3 (243.61 mg, 0.75 mmol). After stirring at room temperature for 2 hrs, the reaction mixture was diluted with EtOAc (10 mL) and washed with water (5×10 mL), organic phase was dried (Na2SO4), and evaporated under vacuum. Crude product was filtered over a short column of SiO2 (DCM 100%, then DCM:MeOH:NH4OH, 91:8:1) to remove the excess of the bis-iodo reactant. Product was purified again by PTLC (DCM:MeOH:NH4OH, 92:7:1) to give 51 mg of product (18) (59% yield). 1H NMR (500 MHz, Chloroform-d) δ 7.71 (d, J = 8.4 Hz, 1H), 7.00 (dd, J = 8.4, 2.0 Hz, 1H), 6.95 (d, J = 2.1 Hz, 1H), 6.58 (bs, 1H), 5.54 (bs, 1H), 4.86 (dd, J = 8.9, 6.2 Hz, 1H), 4.42 (dd, 2H), 4.18 (t, 2H), 3.89 (t, 2H), 3.81 – 3.61 (m, 6H), 3.25 (t, J = 6.8 Hz, 2H), 2.40 – 2.07 (m, 4H), 1.40 (s, 9H). 13C NMR (126 MHz, Chloroform-d) δ 172.01, 171.93, 169.27, 162.42, 144.08, 125.25, 124.58, 115.72, 108.51, 80.97, 72.12, 70.99, 70.39, 69.75, 68.04, 53.95, 47.16, 32.01, 28.16, 24.31, 2.99. LC/MS (ESI); m/z: [M+H]+ Calcd. for C23H34IN2O7, 577.1410. Found 577.1578.
tert-butyl 5-amino-4-(5-(2-(2-(2-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)ethoxy)ethoxy)ethoxy)-1-oxoisoindolin-2-yl)-5-oxopentanoate (Compound 19)
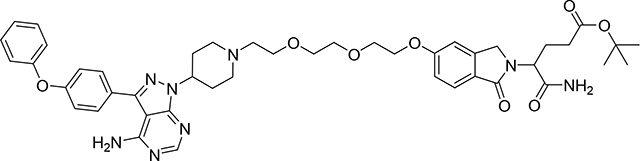
To a solution of 3-(4-phenoxyphenyl)-1-(4-piperidyl)pyrazolo[3,4-d]pyrimidin-4-amine; hydrochloride (36.68 mg, 0.09 mmol) (1) and TEA (0.1 mL, 0.57 mmol) in DMF (1 mL) was added tert-butyl 5-amino-4-[5-[2-[2-(2-iodoethoxy)ethoxy]ethoxy]-1-oxo-isoindolin-2-yl]-5-oxo-pentanoate (18) (50 mg, 0.09 mmol) and the resulting mixture was stirred for 72 h at rt. The reaction mixture was evaporated under vacuum. Crude product was purified by PTLC (DCM:MeOH:NH4OH, 91:8:1) to give 50 mg of pure product (19) (69% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.22 (s, 1H), 7.65 (d, J = 7.6 Hz, 2H), 7.60 – 7.51 (m, 2H), 7.43 (t, J = 7.4 Hz, 2H), 7.25 – 7.07 (m, 7H), 7.02 (d, J = 8.4 Hz, 1H), 4.75 – 4.66 (m, 1H), 4.66 – 4.60 (m, 1H), 4.54 (d, J = 17.5 Hz, 1H), 4.36 (d, J = 17.5 Hz, 1H), 4.24 – 4.12 (m, 2H), 3.83 – 3.74 (m, 2H), 3.69 – 3.46 (m, 6H), 3.05 – 2.96 (m, 2H), 2.57 – 2.43 (m, 2H), 2.29 – 2.04 (m, 7H), 2.02 – 1.80 (m, 3H), 1.31 (s, 9H). 13C NMR (151 MHz, DMSO-d6) δ 171.96, 171.36, 167.69, 161.49, 158.15, 157.04, 156.28, 155.42, 153.61, 144.66, 142.77, 130.12, 130.00, 128.11, 124.41, 124.16, 123.78, 118.99, 118.93, 115.16, 108.58, 97.44, 79.73, 69.93, 69.71,68.79, 68.60, 67.66, 57.06, 53.91, 53.29, 52.71,46.68, 31.77, 31.04, 27.65, 24.88. LC/MS (ESI); m/z [M+H]+: Calcd. for C45H55N8O8.
3-(5-(2-(2-(2-(4-(4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)ethoxy)ethoxy)ethoxy)-1-oxoisoindolin-2-yl)piperidine-2,6-dione (PROTAC SJF620)
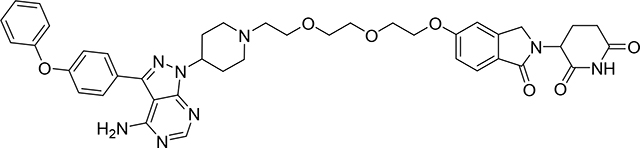
To a solution of tert-butyl 5-amino-4-[5-[2-[2-[2-[4-[4-amino-3-(4-phenoxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]-1-piperidyl]ethoxy]ethoxy]ethoxy]-1-oxo-isoindolin-2-yl]-5-oxo-pentanoate (19) (27.0 mg, 0.0323 mmol) in MeCN (15 mL) was added benzenesulfonic acid (10.2 mg, 0.0647 mmol) and the reaction mixture was heated at reflux temperature for 24 h (Dean-Stark distilling trap, with molecular sieves). The reaction progress was monitored by LC-MS. The reaction mixture was evaporated to dryness under reduced pressure. The crude was purified by PTLC (DCM:MeOH:NH4OH, 91:8:1) to give 13 mg of PROTAC SJF620 (51% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1H), 8.19 (s, 1H), 7.61 (d, J = 8.6 Hz, 2H), 7.56 (d, J = 8.4 Hz, 1H), 7.39 (t, J = 8.0 Hz, 2H), 7.21 – 7.05 (m, 6H), 7.04 – 6.97 (m, 1H), 5.02 (dd, J = 13.3, 4.9 Hz, 1H), 4.69 – 4.52 (m, 1H), 4.40 – 4.07 (m, 4H), 3.84 – 3.68 (m, 2H), 3.63 – 3.43 (m, 6H), 3.06 – 2.92 (m, 2H), 2.92 – 2.77 (m, 1H), 2.67 – 2.46 (m, 3H), 2.39 – 2.23 (m, 1H), 2.25 – 2.04 (m, 4H), 2.00 – 1.74 (m, 3H). 13C NMR (151 MHz, DMSO-d6) δ 172.89, 171.15, 167.89, 161.67, 158.15, 157.05, 156.28, 155.44, 153.62, 144.40, 142.79, 130.13, 130.01, 128.10, 124.31, 124.18, 123.80, 119.00, 118.94, 115.40, 108.63, 97.45, 69.93, 69.72, 68.79, 68.52, 67.72, 57.02, 53.88, 52.67, 51.50, 46.96, 31.25, 30.97, 22.52. LC/MS (ESI); m/z [M+H]+: Calcd. For C41H45N8O7, 761.3411. Found 761.3866
2. Biology
Culturing of Cell Lines. NAMALWA cells line were purchased from ATCC and cultured according to supplier guidelines at 37 °C and 5% CO2 in RPMI 1640 (Gibco) supplemented with 10% fetal bovine serum, 100 pg/mL streptomycin, and 100 units/mL penicillin-G (Gibco).
Cell Treatment and Immunoblotting. For each treatment concentration, 2 × 106 NAMALWA cells were plated and then incubated with PROTAC for 24 hours. The cells were then collected and rinsed once with ice-cold PBS, followed by lysis in buffer containing 20 mM Tris (pH 8.0), 0.25% sodium deoxycholate, and 1% Triton X-100, supplemented with protease inhibitors (Roche) and phosphatase inhibitors (10 mM NaF, 2 mM Na3VO4, 10 mM β-glycerophosphate, and 10 mM sodium pyrophosphate). Lysates were centrifuged at 15000g for 10 min at 4 °C and the supernatant was quantified for total protein content using the BCA protein assay (Thermo Fisher Scientific). Thirty micrograms of protein were loaded onto sodium dodecyl sulfate–polyacrylamide gels (Bio-Rad) for electrophoretic separation, followed by transfer of the resolved proteins onto nitrocellulose membranes, which probed with the specified primary antibodies overnight while being rocked at 4°C in 1×TBS-T (TBS + 0.1% Tween 20) containing 5% nonfat milk. HRP-conjugated secondary antibodies (Pierce) were incubated with the membranes for 1 h at room temperature at 1:10,000 dilutions in 5% nonfat milk in 1× TBS-T. Imaging was performed using the ECL Prime chemiluminescent western blot detection reagents (GE Healthcare) followed by visualization with the Bio-Rad ChemiDoc imaging instrument. All western blots were subsequently processed and quantified using the accompanying Bio-Rad Image Lab software. Anti-BTK (catalog no. 8547) anti-GAPDH (catalog no. 2118) antibodies were purchased from Cell Signaling Technology and used at 1:1000 dilutions in 5% nonfat milk in 1× TBS-T.
Pharmacokinetic Data
Pharmacokinetic properties were estimated externally at Pharmaron Inc.; samples and protocols were prepared following the guidelines of this company.
Acknowledgements
J.H. and C.M.C gratefully acknowledge support from the NIH (R50CA211252 and R35CA197589, respectively). C.M.C. is also supported by an American Cancer Research Professorship.
Footnotes
Disclosures
C.M.C. is consultant and shareholder in Arvinas, Inc., which supports research in his lab.
Declaration of interests
☐ The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
☒The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
C.M.C. is a consultant and shareholder in Arvinas, Inc, which supports research in his laboratory.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Singh SP, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and malignancies. Molecular Cancer. 2018;17(57): 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell RC G; Hawkes E Novel Indications for Bruton’s Tyrosine Kinase Inhibitors, beyond Hematological Malignancies. J.l of Clinical Medicine. 2018;7(4): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Z, Zhang D, Liu Y, et al. Targeting Bruton’s tyrosine kinase for the treatment of B cell associated malignancies and autoimmune diseases: Preclinical and clinical developments of small molecule inhibitors. Arch Pharm (Weinheim). 2018;351(7): e1700369. [DOI] [PubMed] [Google Scholar]
- 4.Liang C, Tian D, Ren X, et al. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: A mini-review. Eur J Med Chem. 2018;151: 315–326. [DOI] [PubMed] [Google Scholar]
- 5.Byrd JC, Harrington B, O’Brien S, et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016;374(4): 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24): 2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reiff SD, Muhowski EM, Guinn D, et al. Noncovalent inhibition of C481S Bruton tyrosine kinase by GDC-0853: a new treatment strategy for ibrutinib-resistant CLL. Blood. 2018;132(10): 1039–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byrd JC, Smith S, Wagner-Johnston N, et al. First-in-human phase 1 study of the BTK inhibitor GdC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget. 2018;9(16): 13023–13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(15): 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bondeson DP, Smith BE, Burslem GM, et al. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem Biol. 2018;25(1): 78–87 e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bondeson DP, Mares A, Smith IE, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nature Chem Biology. 2015;11(8): 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riching KM, Mahan S, Corona CR, et al. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem Biol. 2018;13(9): 2758–2770. [DOI] [PubMed] [Google Scholar]
- 13.Ottis P, Crews CM. Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem Biol. 2017;12(4): 892–898. [DOI] [PubMed] [Google Scholar]
- 14.Churcher I Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones? J Med Chem. 2018;61(2): 444–452. [DOI] [PubMed] [Google Scholar]
- 15.Burslem GM, Smith BE, Lai AC, et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem Biol. 2018;25(1): 67–77 e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lei H, Wang W, Wu Y. Targeting oncoproteins for degradation by small molecules in myeloid leukemia. Leuk Lymphoma. 2017: 1–8. [DOI] [PubMed] [Google Scholar]
- 17.Tan L, Gray NS. When Kinases Meet PROTACs. Chinese J Chem. 2018;36(10): 971–977. [Google Scholar]
- 18.Huang HT, Dobrovolsky D, Paulk J, et al. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader. Cell Chem Biol. 2018;25(1): 88–99 e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buhimschi AD, Armstrong HA, Toure M, et al. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry. 2018;57(26): 3564–3575. [DOI] [PubMed] [Google Scholar]
- 20.Galdeano C, Gadd MS, Soares P, et al. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein Protein Interaction between the von Hippel- Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J of Med Chem. 2014;57(20): 8657–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buckley DL, Raina K, Darricarrere N, et al. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem Biol. 2015;10(8): 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith BE, Wang SL, Jaime-Figueroa S, et al. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat Commun. 2019;10(1): 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gadd MS, Testa A, Lucas X, et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nature Chem Biol. 2017;13(5): 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nowak RP, DeAngelo SL, Buckley D, et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nature Chem Biol. 2018;14(7): 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X, Wang A, Yu K, et al. Discovery of (R)-1-(3-(4-Amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin- 1-yl)-2-(dimethylamino)ethanone (CHMFL-FLT3–122) as a Potent and Orally Available FLT3 Kinase Inhibitor for FLT3-ITD Positive Acute Myeloid Leukemia. J Med Chem. 2015;58(24): 9625–9638. [DOI] [PubMed] [Google Scholar]
- 26.Burslem GM, Ottis P, Jaime-Figueroa S, et al. Efficient Synthesis of Immunomodulatory Drug Analogues Enables Exploration of Structure-Degradation Relationships. ChemMedChem. 2018;13(15): 1508–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qian YC AP; Crews CM; Dong H; Hornberger KR; Wang J Preparation of substituted benzothiophenes as modulators of estrogen receptor proteolysis and associated methods of use. PCT Int. Appl. (2018), WO 2018140809 A1 20180802. [Google Scholar]