

Abstract
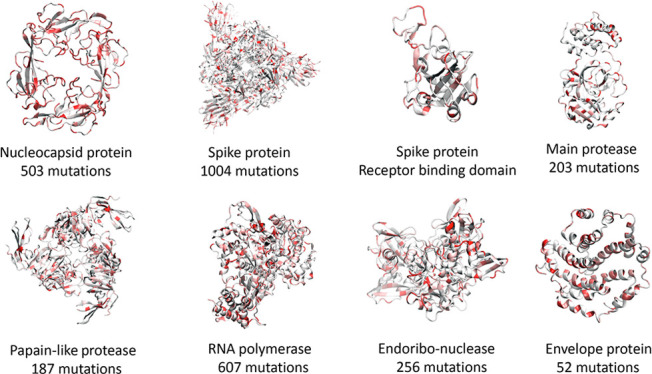
Tremendous effort has been given to the development of diagnostic tests, preventive vaccines, and therapeutic medicines for coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Much of this development has been based on the reference genome collected on January 5, 2020. Based on the genotyping of 15 140 genome samples collected up to June 1, 2020, we report that SARS-CoV-2 has undergone 8309 single mutations which can be clustered into six subtypes. We introduce mutation ratio and mutation h-index to characterize the protein conservativeness and unveil that SARS-CoV-2 envelope protein, main protease, and endoribonuclease protein are relatively conservative, while SARS-CoV-2 nucleocapsid protein, spike protein, and papain-like protease are relatively nonconservative. In particular, we have identified mutations on 40% of nucleotides in the nucleocapsid gene in the population level, signaling potential impacts on the ongoing development of COVID-19 diagnosis, vaccines, and antibody and small-molecular drugs.
1. Introduction
The ongoing pandemic of coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has posed crucial threats to public health and the world economy since it was detected in Wuhan, China, in December 2019.15 As of June 1, 2020, 6 057 853 cases of COVID-19 have been reported in more than 200 countries and territories, resulting in more than 371 166 deaths.27 However, there have been no signs of slowing down nor relief at this monument partially due to the fact there are no specific anti-SARS-CoV-2 drugs and effective vaccines.
SARS-CoV-2 is a positive-strand RNA virus that belongs to the beta coronavirus genus. The genomic information underpins the development of antiviral medical interventions, prophylactic vaccines, and viral diagnostic tests. The first SARS-CoV-2 genome was reported on January 5, 2020.28 It has a genome size of 29.99 kb, which encodes for multiple nonstructural and structural proteins. The leader sequence and ORF1ab encode nonstructural proteins for RNA replication and transcription. Among various nonstructural proteins, viral papain-like (PL) proteinase, main protease (or 3CL protease), RNA polymerase, and endoribonuclease are the common targets in antiviral drug discovery. Yet, it typically takes more than ten years to put an average drug to the market. The downstream regions of the genome encode structural proteins, including spike (S) protein, envelope (E) protein, membrane (M) protein, and nucleocapsid (N) protein. Notably, S-protein uses one of its two subunits to bind directly to the host receptor angiotensin-converting enzyme 2 (ACE2), enabling virus entry into host cells.29 The N protein, one of the most abundant viral proteins, can bind to the RNA genome and is involved in replication, assembly, and host cellular response during viral infection.13 As a virulence factor, the E protein is a small integral membrane protein that regulates cell stress response and apoptosis and promotes inflammation.4 The structural protein, especially, the S protein, is the candidate antigen for vaccine and antibody drug development. Developing safe and effective vaccines is urgently needed to prevent the spread of SARS-CoV-2. However, it typically takes over one year to design and test a new vaccine. Furthermore, the replication in RNA viruses, such as Influenza A, is subject to errors,14 except nidoviruses. Coronaviruses, a kind of nidoviruses, have the ability to proofread their genomes during their genetic replication and recombination.6 Therefore, SARS-CoV-2 might not mutate as fast as Influenza A viruses do, but still has heterogeneous and dynamic populations. The SARS-CoV-2 genome undergoes rapid mutations that are partially stimulated as a response to the challenging immunological environments arising from its transmission to the COVID-19 patients of different races, ages, and medical conditions.
The vaccine developed at one time may not be effective for mitigating the infection by new mutated virus isolates. An alarming fact is that many of these mutations may devastate the ongoing effort in the development of effective medicines, preventive vaccines, and diagnostic tests. Accurate identification of the antigens and their mutations represents the most important roadblock in developing effective vaccines against COVID-19. For example, different vaccines are needed for various geographic locations due to predominant mutations in the corresponding regions. In COVID-19 diagnosis, the diagnostic kits are designed using two major methods: serological tests and molecular tests. Serological tests are to detect specific neutralizing antibodies from COVID-19 infections. Molecular diagnoses look for specific COVID-19 pathogenic genes, which usually rely on the polymerase chain reaction (PCR). Because of the fast mutations of the SARS-CoV-2 genome, genotyping analysis of SARS-CoV-2 may optimize the PCR primer design to detect SARS-CoV safely and to reduce the risk of false-negatives caused by genome sequence variations. In addition, the genotyping analysis may also reveal those highly conserved regions with very few mutations, which can be selected as a target sequence for clinical diagnosis and effective drug therapy.
The evolution pattern through the highly frequent mutations of SARS-CoV-2 can be observable on short time scales. In the early infection period (i.e., February 2020), the SARS-CoV-2 variants were clustered as S and L types.23 Recent genotyping analysis reveals a large number of mutations in various essential genes encoding the S protein, the N protein, and the RNA polymerase in the SARS-CoV-2 population.30 Monitoring the evolutionary patterns and spread dynamics of SARS-CoV-2 is of great importance for COVID-19 control and prevention.
Mutations occur in many different ways. Some mutations occur randomly. Other mutations are enforced by the host immune system surveillance, which induces viral responses. The most preserved mutations and viral evolution can be regarded as the result of the dynamic equilibrium between the random perturbation, host cell defense, and viral fitness. Therefore, the faster and wider the SARS-CoV-2 spread, the more frequent and diverse the mutations will be. The tracking and analysis of COVID-19 dynamics, transmission, and spread are of paramount importance for winning the ongoing battle against COVID-19. Genetic identification and characterization of the geographic distribution, intercontinental evolution, and global trends of SARS-CoV-2 are the most effective approaches for studying COVID-19 genomic epidemiology and offer the molecular foundation for region-specific SARS-CoV-2 vaccine design, drug discovery, and diagnostic development.16 For example, different vaccines for the shell can be designed according to predominant mutations.
This work provides the most comprehensive genotyping to reveal the transmission trajectory and spread dynamics of COVID-19 to date. Based on genotyping 15 140 SARS-CoV-2 genomes from the world as of June 1, 2020, we trace the COVID-19 transmission pathways and analyze the distribution of the subtypes of SARS-CoV-2 across the world. We use K-means methods to cluster SARS-CoV-2 mutations, which provides updated molecular information for the region-specific design of vaccines, drugs, and diagnoses. Our clustering results show that, globally, there are at least six distinct subtypes of SARS-CoV-2 genomes. While, in the U.S., there are four significant SARS-CoV-2 genotypes. We introduce mutation h-index and mutation ratio to characterize conservative and nonconservative proteins and genes. We unveil the unexpected nonconservative genes and proteins, rendering a warning for the current development of diagnostic tests, preventive vaccines, and therapeutic medicines.
2. Results and Discussion
2.1. COVID-19 Evolution and Clustering
Tracking the SARS-CoV-2 transmission pathways and analyzing the spread dynamics are critical to the study of genomic epidemiology. Temporospatially clustering the genotypes of SARS-CoV-2 in the transmission provides insights into diagnostic testing and vaccine development in disease control. In this work, we retrieve and genotype 15 140 SARS-CoV-2 isolates from the world as of June 1, 2020. There are 8309 single mutations in 15 140 SARS-CoV-2 isolates. Based on these mutations, we classify and track the geographical distributions of 15 140 genotype isolates by K-means clustering. The SARS-CoV-2 genotypes, represented as single nucleotide polymorphism (SNP) variants, are clustered as six groups in the world, including the U.S.. In particular, the genotypes in the U.S. are further clustered into four groups. Table 1 lists the co-mutations with the highest number of descendants in different clusters in the world. Optimal clustering groups are established using the Elbow method in the K-means clustering algorithm (Supporting Information).
Table 1. Co-mutations with the Highest Number of Descendants in Six Distinct Clusters in the World.
| cluster | mutation sites | number of descendants |
|---|---|---|
| I | [3037C>T, 14408C>T] | 10875 |
| II | [3037C>T, 14408C>T, 23403A>G] | 10830 |
| III | [14408C>T] | 10923 |
| IV | [3037C>T, 14408C>T, 23403A>G, 28881G>A, 28882G>A, 28883G>C] | 3043 |
| V | [3037C>T, 14408C>T, 23403A>G, 25563G>T] | 4632 |
| VI | [8782C>T, 28144T>C] | 1722 |
The detailed distribution of the SNP variants from the world for each cluster is provided in the Supporting Information. The SNP variant clusters from 76 countries that have a high number of the COVID-19 cases are listed in Table 2. The listed countries are the United States (US), Canada (CA), Australia (AU), United Kingdom (UK), Germany (DE), France (FR), Italy (IT), Russia (RU), China (CN), Japan (JP), Korean (KR), India (IN), Spain (ES), Saudi Arabia (SA), and Turkey (TR). The pie chart plot on the world map is described in Figure 1 which was created by Highcharts (https://www.highcharts.com/maps/demo). The light blue, dark blue, green, red, purple, and yellow represent the Cluster I, II, III, IV, V, and VI, respectively. The color of the dominated cluster decides the base color of each country. The geographic distribution of the SNP variant clusters reflects the approximate transmission pathways and spread dynamics across the world. Several findings can be made from Table 2:
-
1.
Subtypes from clusters III and IV are causing the epidemic in the Asian countries, including those in CN, JP, and KR.
-
2.
The subtypes of SARS-CoV-2 in cluster VI are not spreading in the European countries (UK, DE, FR, IT, RU).
-
3.
All of the subtypes of SARS-CoV-2 in six different clusters can be found in CN, US, CA, AU, and ES. Among them, China initially had samples only in clusters III and VI, and its sample distributions reached to other clusters after the middle of March 2020.
-
4.
The dominant subtypes of SARS-CoV-2 in the COVID-19 pandemic of the United States belong to all of the six clusters.
Table 2. Cluster Distributions of Samples from 15 Countries.
| country | cluster I | cluster II | cluster III | cluster IV | cluster V | cluster VI |
|---|---|---|---|---|---|---|
| US | 844 | 311 | 488 | 156 | 1813 | 975 |
| CA | 12 | 29 | 17 | 16 | 19 | 41 |
| AU | 163 | 149 | 410 | 135 | 146 | 77 |
| UK | 539 | 875 | 908 | 1532 | 119 | 3 |
| DE | 10 | 20 | 21 | 38 | 42 | 0 |
| FR | 41 | 85 | 14 | 12 | 82 | 0 |
| IT | 26 | 24 | 9 | 17 | 0 | 0 |
| RU | 10 | 27 | 1 | 109 | 3 | 0 |
| CN | 8 | 3 | 215 | 1 | 1 | 25 |
| JP | 0 | 3 | 68 | 20 | 3 | 0 |
| KR | 0 | 0 | 28 | 0 | 0 | 0 |
| IN | 93 | 69 | 141 | 10 | 3 | 0 |
| ES | 27 | 100 | 74 | 25 | 3 | 2 |
| SA | 14 | 31 | 9 | 1 | 2 | 0 |
| TR | 25 | 3 | 24 | 9 | 0 | 0 |
Figure 1.

Pie chart plot of six distinct clusters in the world. The light blue, dark blue, green, red, purple, and yellow represent clusters I, II, III, IV, V, and VI, respectively. The base color of each country is decided by the color of the dominant cluster.
The cluster analysis reveals that the Asian countries have three dominant subtype clusters, cluster III [14408C>T], cluster IV [3037C>T, 14408C>T, 23403A>G, 28881G>A, 28882G>A, 28883G>C], and cluster VI [8782C>T, 28144T>C]. Cluster III was detected in the early period of COVID-19 infection in China and other Asian countries. The subtype of SNP mutation in S protein, 23403A>G, is prevalent in the clusters II, IV, and V of European countries. This subtype of S protein mutation may have resulted in the wide spread of SARS-CoV-2 in European countries.
Furthermore, we analyze the statistics of SNP variants located in the United States. In Table 3, we list the number of cases in four different clusters with respect to the west coast states (Washington (WA), California (CA), Alaska (AK), and Oregon (OR)), the east coast cities and states (New York (NY), Washington, D.C. (DC), Pennsylvania (PA), Florida (FL), Massachusetts (MA), Maryland (MD), Virginia (VA)), Wisconsin (WI), Minnesota (MN), Michigan (MI), Georgia (GA), Utah (UT), Connecticut (CT), Arizona (AZ), Idaho (ID), and Illinois (IL). Table 4 lists the co-mutations with the highest number of descendants in different clusters in the United States. Notably, several findings on the genotypes of clusters in the US are as follows:
-
1.
The subtypes of SARS-CoV-2 in all of the clusters are spreading out among the west coast states. Especially, the state of Washington is dominated by cluster B.
-
2.
East coast states are dominated by subtypes from clusters A and C, especially in New York.
-
3.
The subtypes of SARS-CoV-2 in cluster A are spread throughout the United States.
Table 3. Cluster Distributions of Samples from 20 States and Cities in the United States.
| state | cluster A | cluster B | cluster C | cluster D |
|---|---|---|---|---|
| WA | 304 | 805 | 40 | 355 |
| CA | 88 | 53 | 112 | 82 |
| AK | 15 | 0 | 3 | 11 |
| OR | 7 | 4 | 4 | 0 |
| NY | 324 | 15 | 66 | 807 |
| DC | 2 | 1 | 1 | 7 |
| PA | 3 | 0 | 1 | 6 |
| FL | 8 | 2 | 3 | 4 |
| MA | 8 | 0 | 2 | 9 |
| MD | 4 | 0 | 3 | 5 |
| VA | 67 | 11 | 9 | 65 |
| WI | 150 | 8 | 151 | 57 |
| MN | 29 | 28 | 19 | 57 |
| MI | 11 | 2 | 5 | 72 |
| GA | 5 | 2 | 11 | 2 |
| UT | 13 | 9 | 3 | 23 |
| CT | 39 | 1 | 1 | 14 |
| AZ | 31 | 8 | 4 | 35 |
| ID | 31 | 0 | 1 | 8 |
| IL | 23 | 8 | 19 | 20 |
| others | 146 | 13 | 39 | 173 |
| total | 1308 | 970 | 497 | 1812 |
Table 4. Mutation Sites with Highest Frequency in Each Cluster in the United States.
| mutation sites | number of descendants | |
|---|---|---|
| cluster A | [3037C>T, 14408C>T] | 10875 |
| cluster B | [8782C > T, 18060C>T, 28144T>C] | 1127 |
| cluster C | [11083G>T] | 1646 |
| cluster D | [241C>T, 3037C>T, 14408C>T, 23403A>G, 25563G>T] | 4494 |
Figure 2 is the pie chart plot of the four distinct clusters in the US, which was also created by Highcharts. The colors, blue, red, yellow, and green represent clusters A, B, C, and D, respectively. The base color of each state corresponds to its dominant cluster. We note that cluster D in the U.S. is derived from cluster V in the world, with an additional mutation at the leader sequence 241. The high spread in New York is consistent with the high transmission of SARS-CoV-2 in European countries, where the subtype in cluster V is predominant.
Figure 2.
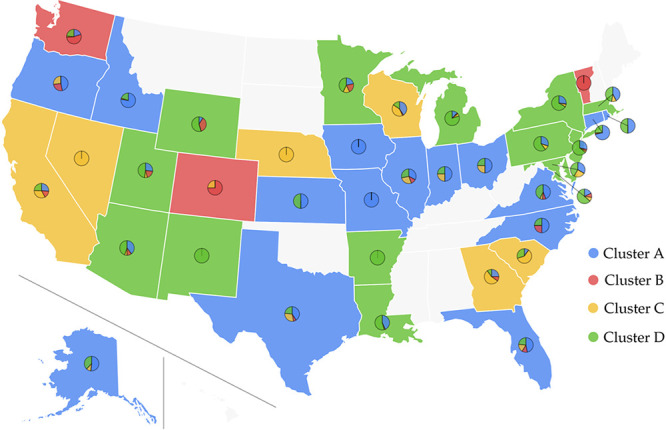
Pie chart plot of four distinct clusters in the US. The blue, red, yellow, and green colors represent clusters A, B, C, and D. The base color of each state corresponds to its dominant cluster.
2.2. Ramifications for COVID-19 Diagnosis, Vaccine, and Medicine
2.2.1. Protein-Specific Mutation Analysis
Figures 3 and 4 depict the distribution and frequencies of SNP mutations of SARS-CoV-2 isolates from 15 140 genome samples in the world with respect to the reference genome of January 5, 2020. The statistics of single mutations on various SARS-CoV-2 proteins that occurred in the recorded genomes between January 5, 2020, and June 1, 2020, are listed in Table 5. The spike protein has the highest number of mutations on gene of 1004, while the envelope protein has the lowest number of mutations of 52. Since the sizes of proteins vary dramatically from 1273 for the spike protein to 75 for the envelope protein, it is useful to consider the mutation ratio, i.e., the number of mutations per residue. In this category, the RNA-dependent RNA polymerase has the lowest score of 0.217, whereas the nucleocapsid protein has the highest score of 0.400, i.e, 503 mutations on its 1257 nucleotides (419 residues). Note that main protease has the second-lowest mutation ratio of 0.221, indicating its conservative nature. Another relatively conservative protein judged by the mutations ratio in terms of gene is the envelope protein, the MRGene = 0.231.
Figure 3.

Distribution of SNP mutations of SARS-CoV-2 isolates from 15 140 genome samples in the world with respect to the reference genome of January 5, 2020 (GenBank access number: NC_045512.2).
Figure 4.

Frequencies of the single SNP mutations of SARS-CoV-2 on the genome samples in the world with respect to the reference genome of January 5, 2020 (GenBank access number: NC_045512.2).
Table 5. Protein-Specific and Gene-Specific Statistics of SARS-CoV-2 Single Mutationsa.
| protein | gene length | protein length | NMGene | NMPro | MRGene | MRPro | mutation h-index |
|---|---|---|---|---|---|---|---|
| spike protein | 3819 | 1273 | 1004 | 391 | 0.263 | 0.307 | 26 |
| main protease | 918 | 306 | 203 | 78 | 0.221 | 0.255 | 16 |
| papain-like protease | 945 | 315 | 187 | 105 | 0.255 | 0.333 | 10 |
| RNA polymerase | 2796 | 932 | 607 | 228 | 0.217 | 0.245 | 21 |
| endoribo-nuclease | 1038 | 346 | 256 | 110 | 0.247 | 0.318 | 12 |
| envelope (E) protein | 225 | 75 | 52 | 23 | 0.231 | 0.307 | 9 |
| membrane protein | 666 | 222 | 165 | 60 | 0.248 | 0.270 | 14 |
| nucleocapsid (N) protein | 1257 | 419 | 503 | 205 | 0.400 | 0.489 | 33 |
NMGene and NMPro are the number of mutations in terms of gene and protein, respectively. MRGene is the mutation ratio of gene, and MRPro is the ratio of the non-degenerated mutations of a protein. Mutation h-index focus on the gene-specific h-index.
Counting the number of single mutations and mutation ratio does not reflect the fact that some mutations occur numerous times over genome samples while other mutations may happen only on a few genome samples. To account for the frequency effect of mutations, we introduce a mutation h-index to measure both the number of mutations and the frequency of mutations of a given protein or genetic section. It is defined as the maximum value of h such that the given protein genetic section has h single mutations that have each occurred at least h times. It is very interesting to note from Table 5 that the mutation h-index correlates very well with the number of mutations on gene; the Pearson correlation coefficient is 0.711. Specifically, N protein has both the highest MRGene of 0.400 and the highest h-index of 33, suggesting that it is the most nonconservative protein in SARS-CoV-2 genomes. In contrast, the envelope protein has the third-lowest number of mutations per residues of 0.231 and the lowest h-index of 9, indicating its relatively conservative nature. By combining the number of mutations per residue and the mutation h-index, we report that the most conservative SARS-CoV-2 proteins is the envelope. It is found that the most nonconservative SARS-CoV-2 proteins are (1) the nucleocapsid protein, (2) the spike protein, and (3) the papain-like protease.
The number of mutations in terms of gene (NMGene) and the number of mutations in terms of protein (NMPro) we reported are accumulated numbers that from all of the 15 140 genome isolates. If we focus on the single genome isolate, the maximum number of mutations on the whole genome sequence is 24.
2.2.2. Diagnosis
Real-time RT-PCR (rRT-PCR) is routinely used in the qualitative detection of nucleic acid from SARS-CoV-2 for diagnostic testing COVID-19.3,24 The primers used in the rRT-PCR are critical for the precise diagnosis of COVID-19 and the discovery of new strains. The primer sequences are specially designed for amplifying the conserved regions across the different existing strains for high specificity and sensitivity and also are subject to genotype changes as the SARS-CoV-2 coronavirus evolves. In diagnostic testing COVID-19, many rRT-PCR primers are designed to detect for three perceived conservative SARS-CoV-2 regions: (1) RNA-dependent RNA polymerase (RdRP) gene in ORF1ab region, (2) the E protein gene, and (3) the N protein gene.3 Our genotyping statistics given in Table 5 indicate that the nucleocapsid protein is the worst choice.
Among the four structural proteins of SARS-CoV-2, the spike surface glycoprotein (S) of 1273 amino acid residues, nucleocapsid protein (N) of 419 amino acid residues, membrane protein (M) of 222 amino acid residues, and envelope protein (E protein) of 75 amino acid residues, the S protein is the most divergent with 1004 unique mutations among the 15 140 SARS-CoV-2 genomes. The N protein has 503 unique mutations, and the envelope (E) protein has 52 mutations. Considering the lengths of the proteins, all the four structural proteins undergo many mutations. The RdRP gene, which is often used in diagnostic testing COVID-19, also has 607 mutations.
Therefore, all three regions in the routine rRT-PCR target, namely RdRP, the N protein gene, and the E protein genes, have significant mutations. Precise and robust diagnosis tools must be re-established according to the conserved regions and predominated mutations in the SARS-CoV-2 genomes detailed in the Supporting Information.
2.2.3. Vaccine Development
Vaccines are mostly associated with the S protein. Compared to SARS-CoV, SARS-CoV-2 has a unique furin cleavage site, where four amino acid residues (PRRA) are inserted into the S1–S2 junction region 681–684 of the S protein.25 The furin cleavage site is crucial for zoonotic transmission of SARS-CoV-2.7 This study reveals crucial mutations near the S1–S2 junction region in the S protein, including 23403A>G-(D614G), 23422C>T-(V620V), 23575C>T-(C671C), 23586A>G-(Q675R), 23611G>A-(R683R), 23707C>T-(P715P), 23731C>T-(T723T), 23849T>C-(L763L), and 23929C>T-(Y789Y). Moreover, these mutations of the S protein SARS-CoV-2 are located at the epitope region, corresponding to the regions 469–882 and 599–620 in SARS-CoV.19
Additionally, many mutated amino acids are on the receptor-binding domain (RBD) of the S protein, as shown in Figure 5. Unfortunately, the S protein is the second most nonconservative protein in the genome based on the number of mutations per residue and mutation h-index. In fact, about half of the receptor-binding domain residues of the S proteins have had mutations in the past few months as shown in Figure 6. Because the surface accessibility of epitope is also important for the interaction of antibody and antigen, these mutations are critical for the antigenicity of the S protein.
Figure 5.

Illustration of SARS-CoV-2 spike protein mutations using 6VXX as a template.
Figure 6.
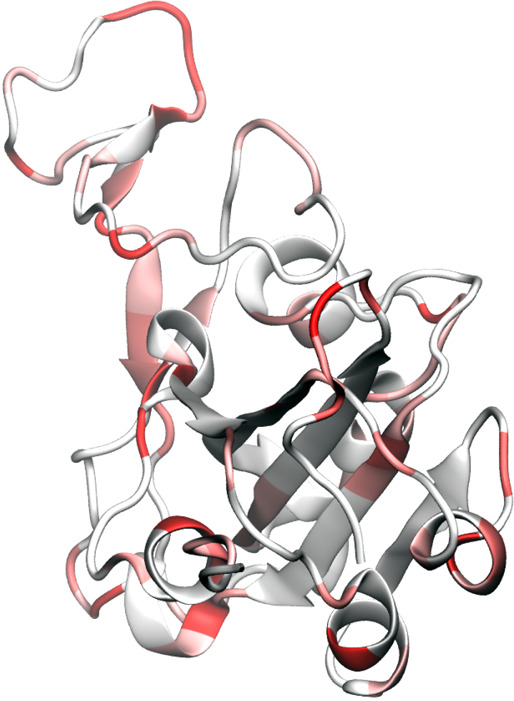
Illustration of SARS-CoV-2 spike-protein receptor binding domain (RBD) mutation using 6M0J as a template. It is noted that nearly half of the residues in RBD have undergone mutations in the few months.
Convalescent COVID-19 patients show a neutralizing antibody response after infection, which is directed mostly against the S protein.18 The neutralizing antibody responses against SARS-CoV-2 could give some defense against SARS-CoV-2 infection, thus having implications for preventing SARS-CoV-2 outbreaks. The divergence of S proteins and the nonconserved regions of the S proteins might contribute to the antigenicity. The highly frequent mutations identified in the S protein may reduce the durability of the SARS-CoV-2 vaccine’s immunity or undermine the current development of vaccines. The existing mutations must be considered when designing a new vaccine. Additionally, a cocktail of multiple vaccines has a better chance of preventing COVID-19 infections.
2.2.4. Drug Discovery
Unfortunately, there is no specific effective drug for SARS-CoV-2 at this point. Potential drugs include small-molecular drugs and antibody drugs. Much of the effort in small-molecular drug discovery focuses on SARS-CoV-2 nonstructural proteins. Among the major nonstructural proteins of SARS-CoV-2, the main protease of 306 amino acids has 78 mutations with 0.255 mutations per residue and the mutation h-index of 16, RNA polymerase of 932 amino acids has 228 mutations with 0.245 mutations per residue and the mutation h-index of 21, and papain-like protease of 945 amino acids has 105 mutations with 0.333 mutations per residue and the mutation h-index of 10. In fact, the main protease is the most popular drug target because there are no similar known genes in the human genome, which implies that SARS-CoV-2 main protease inhibitors will likely be less toxic.10 The present study suggests that the main protease is the second most conservative protein. Therefore, it remains the most attractive target for drug discovery.
Therapeutic antibodies got started from cancer treatments and now applies to infectious diseases by targeting pathogens.1 Antibody drugs are highly specificity and versatile in the treatment of infectious diseases. Their working principle involves the host immune system. The time used to develop antibody therapeutics are usually considerably shorter than that used to develop a vaccine. Many SARS-CoV-2 antibody drugs are isolated from patient blood and target the S proteins. Although there many binding sites on the S protein that antibodies can target, the ones that are most effective in neutralizing SARS-CoV-2 block the receptor-binding domain (RBD) of the host cell angiotensin-converting enzyme 2 (ACE2) receptor. The RBD is a dongle-shape protein at the end of the virus’s spikes. As mentioned above, there are many mutations on the S proteins. The RBD is also prone to mutations. Some mutations that break hydrogen bonds and/or salt bridges in antibody–antigen interactions will have a large impact. However, silent mutations, such as those that replace hydrophobic residues with other hydrophobic residues, will typically have little effect. To avoid the failure of one specific antibody, the cocktail treatments that include several different antibodies might be required to treat SARS-CoV-2 that undergoes antigenic mutations.
2.2.5. Protein-Specific Discussion
2.2.5.1. Spike Glycoprotein
The SARS-CoV-2 spike glycoprotein, or S protein, comprised of two subunits, S1 and S2, of very different properties;25 see Figure 5. Among them, the S1 subunit, as shown in Figure 5, contains the receptor-binding domain (RBD) responsible for binding to the host cell receptor angiotensin-converting enzyme 2 (ACE2). The RBD is also the common binding domain for antibodies. The S2 subunit offers the structural support of the S protein and mediates fusion between the viral and host cell membranes. After the fusion, the virus releases the viral genome into the host cell.
The S1 RBD protein plays key parts in the induction of neutralizing-antibody and T-cell responses, as well as protective immunity. However, S2 and extracellular domain (ECD) of spike protein and their combination are commonly used in recombinant proteins in SARS-CoV-2 antibody development.
As shown in Table 5, the S protein is the most heterogeneous structural protein with a significant number of mutations as shown in Figures 5 and 6 and Table 6. The divergence of the spike protein, the nonconserved regions of the spike protein might contribute to the antigenicity difference in SARS-CoV-2 isolates. We found that most of the high frequent mutations of the S protein are located in the S1 subunit. Figure 6 indicates that near half of the amino acid residues have had mutations since January 5, 2020. One of the important mutations at S1 is 23010T>C (V483A) within the RBD for ACE2 binding, and the total frequency of 23010T>C (V483A) is 23. The structural study revealed that the amino acids 442–487 in the S1 subunit may impact viral binding to human ACE2.9,26 The mutations identified in this study imply the change in ACE2 binding affinity and the transmissibility of SARS-CoV-2 as well as negative impacts in preventive vaccine and diagnostic test development.
Table 6. Top 10 High Frequency Single SNP Genotypes in the Spike Surface Glycoprotein of SARS-CoV-2.
| rank | SNP | protein mutation | total frequency | cluster I | cluster II | cluster III | cluster IV | cluster V | cluster VI |
|---|---|---|---|---|---|---|---|---|---|
| top1 | 23403A>G | D614G | 10969 | 2333 | 2609 | 70 | 2965 | 2991 | 1 |
| top2 | 23731C>T | T723T | 228 | 24 | 0 | 1 | 203 | 0 | 0 |
| top3 | 23929C>T | Y789Y | 228 | 2 | 0 | 225 | 1 | 0 | 0 |
| top4 | 24368G>T | D936Y | 110 | 37 | 0 | 1 | 2 | 70 | 0 |
| top5 | 21575C>T | L5F | 98 | 22 | 9 | 28 | 15 | 14 | 10 |
| top6 | 24862A>G | T1100T | 90 | 14 | 58 | 0 | 18 | 0 | 0 |
| top7 | 24390G>C | S943T | 56 | 20 | 7 | 28 | 1 | 0 | 0 |
| top8 | 24389A>C | S943R | 56 | 20 | 7 | 28 | 1 | 0 | 0 |
| top9 | 24933G>T | G1124V | 47 | 15 | 0 | 21 | 7 | 1 | 3 |
| top10 | 23707C>T | P715P | 44 | 1 | 0 | 39 | 0 | 4 | 0 |
2.2.5.2. Main Protease
SARS-CoV-2 main protease, or 3CL protease, is essential for cleaving the polyproteins that are translated from the viral RNA.10 It operates at multiple cleavage sites on the large polyprotein through the proteolytic processing of replicase polyproteins and plays a pivotal role in viral gene expression and replication. SARS-CoV-2 main protease is one of the most attractive targets for anti-CoV drug design because its inhibition would block viral replication and it is unlikely to be toxic due to no known similar human proteases. Another reason for the focused drug discovery efforts in developing SARS-CoV-2 main protease inhibitors is that this protein is relatively conservative as shown in Table 5.
Figure 7 illustrates the main protease mutation patterns. Figure 8 further highlights the inhibitor binding domain (BD). Indeed, the main protease is relatively conservative compared to the spike protein. Table 7 lists top 10 mutations and their frequency in our data set. It is interesting to see that many mutations, such as D176D, R298R, N151N, are degenerate ones. One possible explanation is that nondegenerates may be nonsilent and likely cause unsurvivable disruption to the virus. Note that mutation G15S mostly occurs in cluster IV. Mutation R298R is restricted to cluster IV. Some other mutations, such as D248E, A266V, N151N, and T45I are specific to certain clusters. Nonetheless, some mutations at the BD shown in Figure 8 are worth noting. They can undermine the ongoing drug discovery effort.
Figure 7.
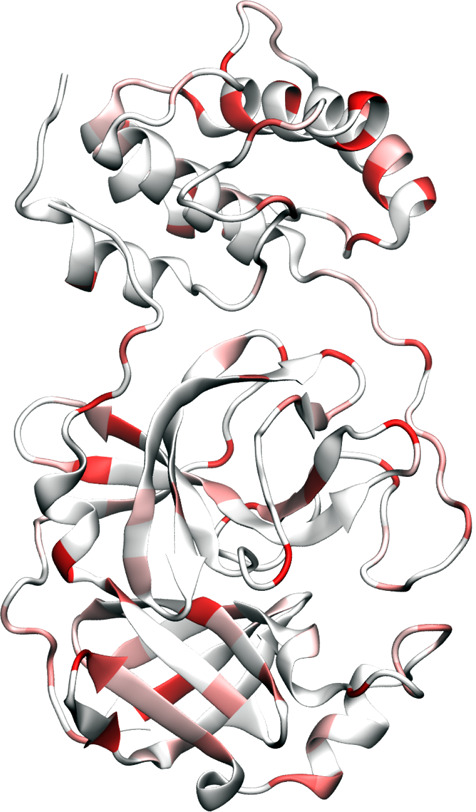
Illustration of SARS-CoV-2 main protease mutations using 6LU7 as a template.10
Figure 8.
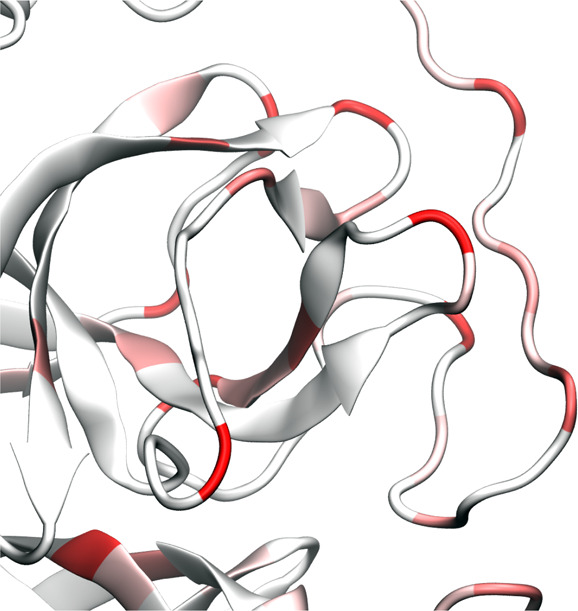
Illustration of SARS-CoV-2 main protease binding domain (BD) mutations of 6LU7.
Table 7. Top 10 High Frequency Single SNP Genotypes in the Main Protease of SARS-CoV-2.
| rank | SNP | protein mutation | total frequency | cluster I | cluster II | cluster III | cluster IV | cluster V | cluster VI |
|---|---|---|---|---|---|---|---|---|---|
| top1 | 10097G>A | G15S | 224 | 23 | 0 | 1 | 200 | 0 | 0 |
| top2 | 10323A>G | K90R | 95 | 8 | 71 | 13 | 1 | 1 | 1 |
| top3 | 10798C>A | D248E | 88 | 44 | 44 | 0 | 0 | 0 | 0 |
| top4 | 10851C>T | A266V | 86 | 25 | 0 | 0 | 0 | 61 | 0 |
| top5 | 10582C>T | D176D | 53 | 20 | 1 | 1 | 0 | 31 | 0 |
| top6 | 10319C>T | L89F | 50 | 28 | 1 | 4 | 0 | 17 | 0 |
| top7 | 10948A>G | R298R | 33 | 0 | 0 | 0 | 33 | 0 | 0 |
| top8 | 10507C>T | N151N | 32 | 3 | 12 | 17 | 0 | 0 | 0 |
| top9 | 10265G>A | G71S | 31 | 3 | 0 | 0 | 28 | 0 | 0 |
| top10 | 10188C>T | T45I | 27 | 23 | 0 | 1 | 0 | 3 | 0 |
2.2.5.3. Papain-like Protease
SARS-CoV-2 papain-like protease (PLPro) is a cysteine cleavage protein located within the nonstructural protein 3 (NSP3) section of the viral genome.17 Like the main protease, PLPro activity is required to cleave the viral polyprotein into functional, mature subunits and, thereby, contributes to the biogenesis of the virus replication. Additionally, PLPro possesses a deubiquitinating activity. The SARS PLPro is also a major therapeutic and diagnostic target.
As shown in Table 5, the SARS PLPro is prone to mutations. Figure 9 shows that mutations are all over the places in PLPro. Table 8 lists the top 10 mutations in PLPro. Three of these mutations are degenerate ones. Note that only two of the top mutations occurred in cluster II. In contrast, cluster I has many different mutations.
Figure 9.
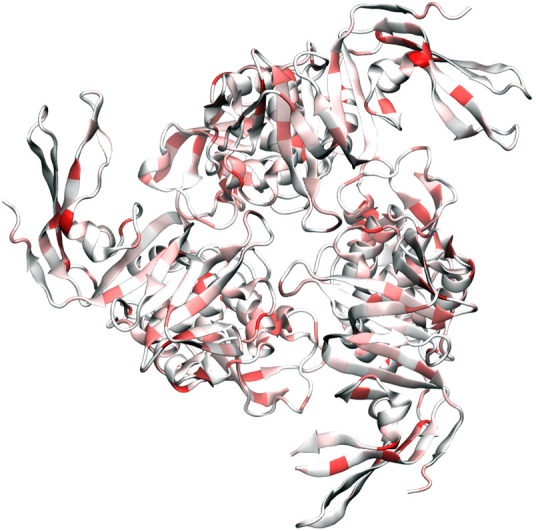
Illustration of SARS-CoV-2 papain-like protease mutations using 6W9C as a template.
Table 8. Top 10 High Frequency Single SNP Genotypes in the Papain-Like Protease of SARS-CoV-2.
| rank | SNP | protein mutation | total frequency | cluster I | cluster II | cluster III | cluster IV | cluster V | cluster VI |
|---|---|---|---|---|---|---|---|---|---|
| top1 | 5142C>T | T808I | 41 | 0 | 0 | 41 | 0 | 0 | 0 |
| top2 | 5730C>T | T1004I | 22 | 3 | 0 | 4 | 9 | 2 | 4 |
| top3 | 5784C>T | T1022I | 19 | 0 | 0 | 0 | 2 | 0 | 17 |
| top4 | 5062G>T | L781F | 15 | 1 | 0 | 14 | 0 | 0 | 0 |
| top5 | 5467C>T | Y916Y | 15 | 10 | 0 | 5 | 0 | 0 | 0 |
| top6 | 5183C>T | P822S | 15 | 2 | 1 | 3 | 2 | 7 | 0 |
| top7 | 5230G>T | K837N | 12 | 7 | 5 | 0 | 0 | 0 | 0 |
| top8 | 5572G>T | M951I | 11 | 0 | 0 | 9 | 0 | 0 | 2 |
| top9 | 5812C>T | D1031D | 10 | 1 | 0 | 5 | 3 | 1 | 0 |
| top10 | 5284C>T | N855N | 10 | 8 | 0 | 1 | 1 | 0 | 0 |
2.2.5.4. RNA Polymerase
SARS RNA-dependent RNA polymerase (RdRP) is an enzyme that catalyzes the synthesis of the SARS RNA strand complementarily to the SARS-CoV-2 RNA template and is thus essential to the replication of SARS-CoV-2 RNA.8 As one of the nonstructural proteins, RdRPs are located in the early part of ORF1b section. Like most other RNA viruses, SARS-CoV-2 RdRPs are considered to be highly conserved to maintain viral functions and thus targeted in antiviral drug development as well as diagnostic tests. On the other hand, the SARS-CoV-2 RNA polymerase lacks proofreading capability and thus its mutations are deemed to happen as shown in Table 5.
Figure 10 illustrates the SARS-CoV-2 RdRP mutations since January 5, 2020. Surprisingly, there are many mutations in SARS-CoV-2 RdRP. Table 9 describes the top 10 mutations. As in other cases, five of these mutations are degenerate ones.
Figure 10.
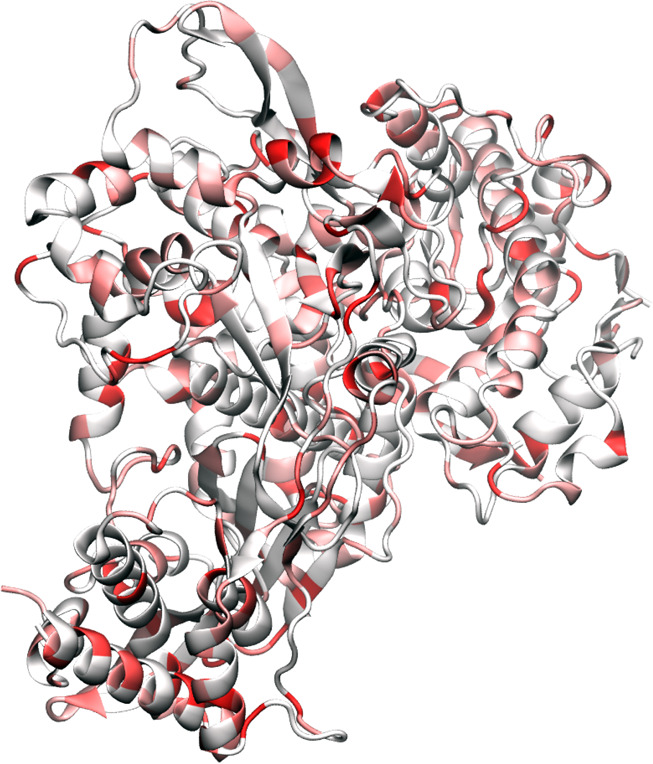
Illustration of SARS-CoV-2 RNA-polymerase mutations using 6M71 as a template.
Table 9. Top 10 High Frequency Single SNP Genotypes in the RNA Dependent Polymerase of SARS-CoV-2.
| rank | SNP | protein mutation | total frequency | cluster I | cluster II | cluster III | cluster IV | cluster V | cluster VI |
|---|---|---|---|---|---|---|---|---|---|
| top1 | 14408C>T | P323L | 10925 | 2309 | 2602 | 68 | 2955 | 2991 | 0 |
| top2 | 14805C>T | Y455Y | 1242 | 9 | 0 | 1202 | 30 | 0 | 1 |
| top3 | 15324C>T | N628N | 405 | 128 | 253 | 18 | 5 | 1 | 0 |
| top4 | 13730C>T | A97V | 263 | 11 | 20 | 232 | 0 | 0 | 0 |
| top5 | 13536C>T | Y32Y | 121 | 23 | 0 | 1 | 92 | 5 | 0 |
| top6 | 13862C>T | T141I | 118 | 61 | 53 | 2 | 0 | 2 | 0 |
| top7 | 14786C>T | A449V | 98 | 53 | 14 | 3 | 22 | 6 | 0 |
| top8 | 15540C>T | V700V | 39 | 1 | 0 | 37 | 1 | 0 | 0 |
| top9 | 13627G>T | D63Y | 36 | 0 | 1 | 35 | 0 | 0 | 0 |
| top10 | 14877C>T | Y479Y | 34 | 2 | 0 | 2 | 1 | 0 | 29 |
2.2.5.5. Endoribo-nuclease
Endoribo-nuclease (NendoU) protein is a nidoviral RNAuridylate-specific enzyme that cleaves RNA.11 It contains a C-terminal catalytic domain belonging to the NendoU family RNA processing. The NendoU protein is presented among coronaviruses, arteriviruses, and toroviruses. The many aspects of the detailed function and activity of SARS-CoV-2 NendoU protein are yet to be revealed.
Figure 11 depicts SARS-CoV-2 NendoU protein mutations. As in most other SARS-CoV-2 proteins, mutations have occurred over different parts. Table 5 shows that NendoU is relatively conservative. Table 10 lists the top 10 high-frequency mutations of the SARS-CoV-2 NendoU protein that occurred in the past few months. Four of these mutations are degenerate ones. The frequencies of these mutations range from 153 to 15. Note that Cluster VI only has one of these mutations.
Figure 11.
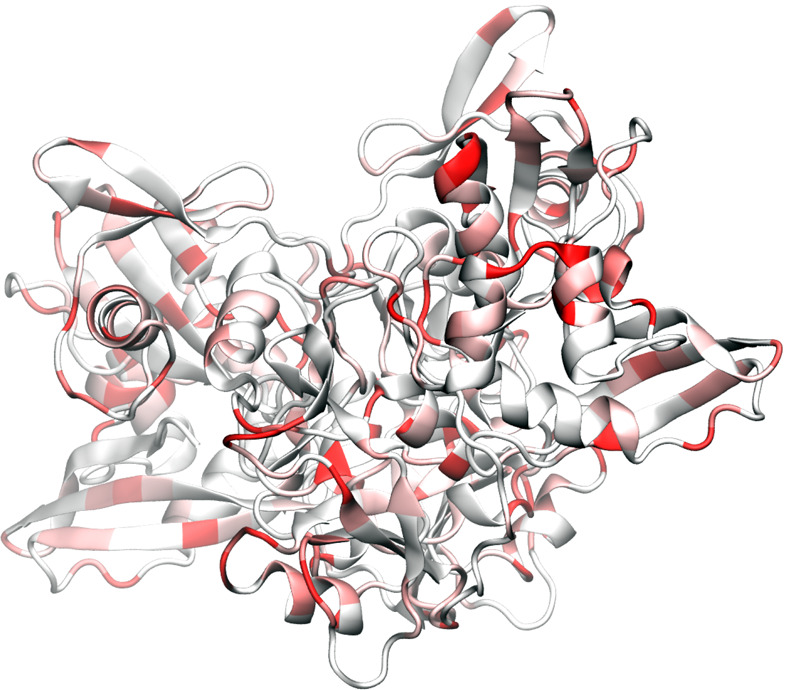
Illustration of SARS-CoV-2 Endoribo-nuclease protein mutations using 6VWW as a template.
Table 10. Top 10 High Frequency Single SNP Genotypes in the Endoribo-nuclease of SARS-CoV-2.
| rank | SNP | protein mutation | total frequency | cluster I | cluster II | cluster III | cluster IV | cluster V | cluster VI |
|---|---|---|---|---|---|---|---|---|---|
| top1 | 19839T>C | N73N | 153 | 7 | 0 | 0 | 146 | 0 | 0 |
| top2 | 19684G>T | V22L | 63 | 2 | 0 | 57 | 4 | 0 | 0 |
| top3 | 20578G>T | V320L | 59 | 42 | 16 | 1 | 0 | 0 | 0 |
| top4 | 20134G>T | V172L | 39 | 1 | 0 | 25 | 10 | 3 | 0 |
| top5 | 20148C>T | F176F | 31 | 3 | 1 | 20 | 5 | 0 | 2 |
| top6 | 19999G>T | V127F | 30 | 14 | 0 | 0 | 1 | 15 | 0 |
| top7 | 20316C>T | F232F | 25 | 0 | 0 | 25 | 0 | 0 | 0 |
| top8 | 20270C>T | A217V | 22 | 3 | 0 | 19 | 0 | 0 | 0 |
| top9 | 20275G>A | D219N | 20 | 1 | 17 | 1 | 0 | 1 | 0 |
| top10 | 20031C>A | A137A | 15 | 1 | 0 | 0 | 15 | 0 | 0 |
2.2.5.6. Envelope Protein
The SARS-CoV-2 envelope (E) protein is one of SARS-CoV’s four structural proteins. As a transmembrane protein, it involves in ion channel activity and thus facilitates viral assembly, budding, envelope formation, pathogenesis, and release of the virus.22 The E protein may not be essential for viral replication, but it is for pathogenesis.
Figure 12 illustrates E protein as a very small pentamer with a few mutations. Table 11 shows its top 10 mutations. Note that the first four mutations are degenerate ones. All other mutations have relatively low frequencies. As shown in Table 5, the SARS-CoV-2 E protein is very conservative.
Figure 12.
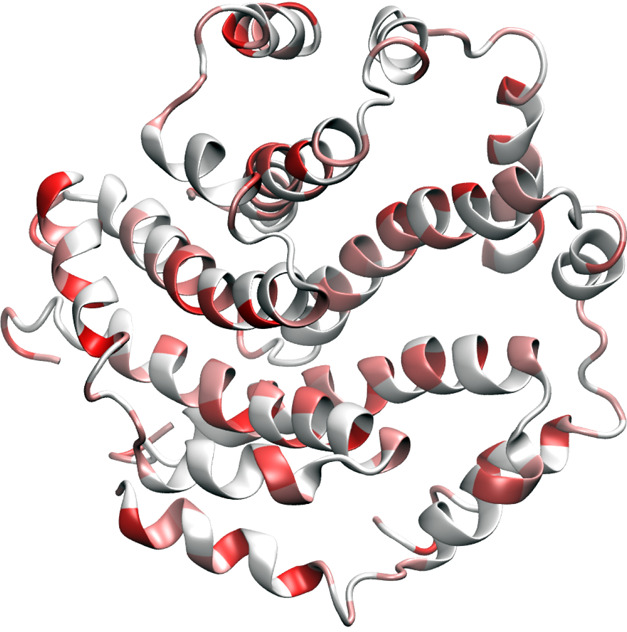
Illustration of SARS-CoV-2 envelope protein mutations using 5X29 as a template.
Table 11. Top 10 High Frequency Single SNP Genotypes in the Envelope (E) Protein of SARS-CoV-2.
| rank | SNP | protein mutation | total frequency | cluster I | cluster II | cluster III | cluster IV | cluster V | cluster VI |
|---|---|---|---|---|---|---|---|---|---|
| top1 | 26340C>T | A32A | 16 | 0 | 2 | 14 | 0 | 0 | 0 |
| top2 | 26256C>T | F4F | 12 | 2 | 6 | 0 | 4 | 0 | 0 |
| top3 | 26319A>T | V25V | 10 | 1 | 0 | 8 | 0 | 0 | 1 |
| top4 | 26319A>G | V25V | 8 | 1 | 0 | 7 | 0 | 0 | 0 |
| top5 | 26270C>T | T9I | 7 | 4 | 1 | 0 | 2 | 0 | 0 |
| top6 | 26416G>T | V58F | 5 | 1 | 0 | 1 | 3 | 0 | 0 |
| top7 | 26326C>T | L28L | 5 | 0 | 0 | 5 | 0 | 0 | 0 |
| top8 | 26314G>A | V24M | 4 | 0 | 0 | 0 | 4 | 0 | 0 |
| top9 | 26262G>A | S6S | 4 | 1 | 0 | 1 | 0 | 2 | 0 |
| top10 | 26370C>T | Y42Y | 4 | 1 | 0 | 3 | 0 | 0 | 0 |
2.2.5.7. Nucleocapsid Protein
SARS-CoV-2 nucleocapsid (N) protein2 is another structural protein. Its primary function is to encapsidate the viral genome. To do so, it is heavily phosphorylated (or charged) and, thereby, can bind with RNA. Additionally, SARS-CoV-2 N protein confirms the viral genome to replicase-transcriptase complex (RTC) and plays a crucial role in viral genome encapsulation. Therefore, it may function completely differently at different stages of the viral life cycle. SARS-CoV-2 N protein is considered to be one of the most conservative SARS-CoV-2 proteins in the literature and is a popular target for diagnosis of vaccine development.3 The present works shown in Table 5 indicate that the SARS-CoV-2 N protein is the worst target of any drug, vaccine, and diagnostic development. Figure 13 is the illustration of SARS-CoV-2 nucleocapsid phosphoprotein mutations using 6VYO as a template.
Figure 13.
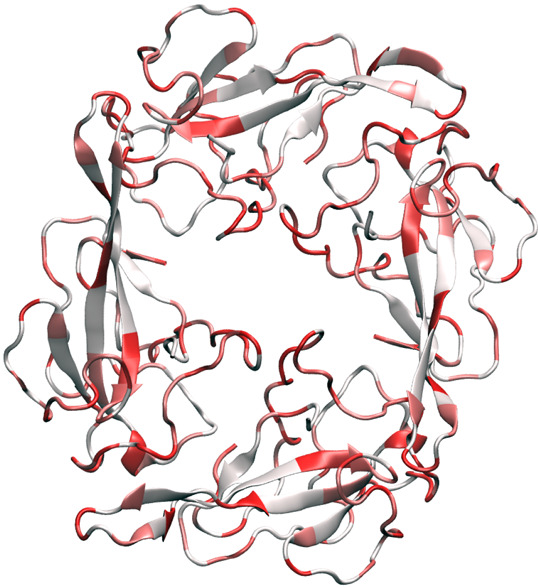
Illustration of SARS-CoV-2 nucleocapsid phosphoprotein mutations using 6VYO as a template.
Table 12 presents the top 10 mutations of the SARS-CoV-2 N protein since January 5, 2020. Note that only 2 out of the top 10 mutations are degenerate ones, which is a significantly lower ratio than that of other proteins. The frequency of 10th mutation is 78, which suggests there are many mutations associated with these mediate-sized proteins. Most top mutations occurred to clusters I, III, and IV. Clusters V and VI have almost none of the top 10 mutations.
Table 12. Top 10 High Frequency Single SNP Genotypes in the Nucleocapsid Phosphoprotein of SARS-CoV-2.
| rank | SNP | protein mutation | total frequency | cluster I | cluster II | cluster III | cluster IV | cluster V | cluster VI |
|---|---|---|---|---|---|---|---|---|---|
| top1 | 28881G>A | R203K | 3083 | 100 | 1 | 17 | 2963 | 1 | 1 |
| top2 | 28882G>A | R203K | 3076 | 96 | 0 | 14 | 2966 | 0 | 0 |
| top3 | 28883G>C | G204R | 3077 | 96 | 1 | 14 | 2966 | 0 | 0 |
| top4 | 28311C>T | P13L | 323 | 1 | 3 | 317 | 1 | 0 | 1 |
| top5 | 28657C>T | D128D | 191 | 1 | 2 | 183 | 3 | 0 | 2 |
| top6 | 28688T>C | L139L | 163 | 1 | 1 | 161 | 0 | 0 | 0 |
| top7 | 28836C>T | S188L | 120 | 64 | 53 | 1 | 0 | 2 | 0 |
| top8 | 28878G>A | S202N | 91 | 0 | 0 | 91 | 0 | 0 | 0 |
| top9 | 28580G>T | D103Y | 79 | 1 | 1 | 3 | 74 | 0 | 0 |
| top10 | 29148T>C | I292T | 78 | 3 | 1 | 1 | 73 | 0 | 0 |
2.2.5.8. Membrane Protein
SARS-CoV-2 membrane (M) protein is another structural protein and plays a central role in viral assembly and viral particle formation. It exists as a dimer in the virion and has certain geometric shapes to enable certain membrane curvature and binding to nucleocapsid proteins. Similar to other SARS-CoV proteins, M protein is also a popular target for viral diagnosis and vaccines.
Table 5 gives SARS-CoV-2 M protein the middle ranking for its conservation. Table 13 details the top 10 mutations in SARS-CoV-2 M protein that occurred in the past few months. Eight of these mutations are degenerate. Clusters I and V have relatively a few of these mutations.
Table 13. Top 10 High Frequency Single SNP Genotypes in the Membrane Glycoprotein of SARS-CoV-2.
| rank | SNP | protein mutation | total frequency | cluster I | cluster II | cluster III | cluster IV | cluster V | cluster VI |
|---|---|---|---|---|---|---|---|---|---|
| top1 | 27046C>T | T175M | 306 | 14 | 1 | 2 | 289 | 0 | 0 |
| top2 | 26530A>G | D3G | 153 | 41 | 110 | 1 | 0 | 0 | 1 |
| top3 | 26729T>C | A69A | 119 | 0 | 0 | 119 | 0 | 0 | 0 |
| top4 | 26951G>A | V143V | 64 | 21 | 1 | 1 | 2 | 39 | 0 |
| top5 | 26750C>T | I76I | 49 | 0 | 1 | 0 | 46 | 1 | 1 |
| top6 | 26681C>T | F53F | 26 | 7 | 1 | 10 | 7 | 1 | 0 |
| top7 | 26864A>G | P114P | 21 | 10 | 4 | 7 | 0 | 0 | 0 |
| top8 | 26936C>T | L138L | 17 | 0 | 2 | 1 | 0 | 1 | 13 |
| top9 | 26873C>T | N117N | 17 | 4 | 2 | 3 | 4 | 4 | 0 |
| top10 | 26625C>T | L35L | 17 | 8 | 0 | 0 | 1 | 8 | 0 |
3. Material and Methods
3.1. Data Collection and Preprocessing
On January 5, 2020, the complete genome sequence of SARS-CoV-2 was first released on GenBank (access number: NC_045512.2) by Zhang’s group at Fudan University.28 Since then, there has been a rapid accumulation of SARS-CoV-2 genome sequences. In this work, 15 140 complete genome sequences with high coverage of SARS-CoV-2 strains from the infected individuals in the world have been downloaded from the GISAID database20 (https://www.gisaid.org/) as of June 1, 2020. All the records in GISAID without the exact submission date were not taken into considerations. To rearrange the 15 140 complete genome sequences according to the reference SARS-CoV-2 genome, multiple sequence alignment (MSA) was carried out by using Clustal Omega21 with default parameters.
3.2. SNP Genotyping
SNP genotyping measures the genetic variations between different members of a species. Establishing the SNP genotyping method for the investigation of the genotype changes during the transmission and evolution of SARS-CoV-2 is of great importance. By analyzing the rearranged genome sequences, SNP profiles which record all of the SNP positions in teams of the nucleotide changes and its corresponding positions can be constructed. The SNP profiles of a given genome of a COVID-19 patient capture all the differences from a complete reference genome sequence and can be considered as the genotype of the individual SARS-CoV-2.
3.3. Distance of SNP Variants
The Jaccard distance measures dissimilarity between sample sets. The Jaccard distance of SNP variants is widely employed in the phylogenetic analysis of human or bacterial genomes.30 In this work, we utilize the Jaccard distance to compare the difference between the SNP variant profiles of SARS-CoV-2 genomes.
The Jaccard similarity coefficient, also known as the Jaccard index, is defined as the intersection size divided by the union of two sets A, B:12
| 1 |
The Jaccard distance of two sets A, B is scored as the difference between one and the Jaccard similarity coefficient and is a metric on the collection of all finite sets:
| 2 |
Therefore, the genetic distance of two genomes corresponds to the Jaccard distance of their SNP variants. If A ∩ B ≠ ⌀, A ⊂ B, and B ⊂ A, then we say these two SNP variants are relatives. If A ⊂ B, then A is the ancestor of B and B is the descendant of A.
In principle, the Jaccard distance measure of SNP variants takes account of the ordering of SNP positions, i.e., transmission trajectory, when an appropriate reference sample is selected. However, one may fail to identify the infection pathways from the mutual Jaccard distances of multiple samples. In this case, the dates of the sample collections offer useful information. Additionally, clustering techniques, such as k-means described below, enable us to characterize the spread of COVID-19 onto the communities.
3.4. K-Means Clustering
K-means clustering
is one of the fundamental unsupervised
algorithms in machine learning which aims at partitioning a given
data set X = {x1, x2, ..., xn, ..., xN}, xn ∈ 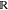 d into k clusters {C1, C2, ..., Ck}, k ≤ N such that the specific
clustering criteria are optimized. More specifically, the standard K-means clustering algorithm starts to pick k points as cluster centers randomly and then allocates each data
to its nearest cluster. The cluster centers will be updated iteratively
by minimizing the within-cluster sum of squares (WCSS) which is defined
by
d into k clusters {C1, C2, ..., Ck}, k ≤ N such that the specific
clustering criteria are optimized. More specifically, the standard K-means clustering algorithm starts to pick k points as cluster centers randomly and then allocates each data
to its nearest cluster. The cluster centers will be updated iteratively
by minimizing the within-cluster sum of squares (WCSS) which is defined
by
| 3 |
where μk is the mean of points located in the kth cluster Ck and nk is the number of points in Ck. Here, ∥•∥2 denotes the L2 distance.
The algorithm above only provides a way to obtain the optimal partition for a fixed number of clusters. However, we are interested in finding the best number of clusters for the SNP variants. Therefore, the Elbow method is applied. By varying the number of clusters k, a set of WCSS can be calculated in the K-means clustering process, and then the plot of WCSS according to the number of clusters k can be carried out. The location of the elbow in this plot will be considered as the optimal number of clusters. To be noticed, the WCSS measures the variability of the points within each cluster which is influenced by the number of points N. Therefore, as the number of total points of N increases, the value of WCSS becomes larger. Additionally, the performance of k-means clustering depends on the selection of the specific distance.
In this work, we propose to implement K-means clustering with the Elbow method for analyzing the optimal number of the subtypes of SARS-CoV-2 SNP variants. The Jaccard distance-based and location-based representations are considered as the input features for the K-means clustering method.
3.4.1. Jaccard Distance-Based Representation
Suppose we have a total of N SNP variants concerning a reference genome in a SARS-CoV-2 sample. The location of the mutation sites for each SNP variant will be saved in the set Si, i = 1, 2, ..., N. The Jaccard distance between two different sets (or samples) Si, Sj is denoted as dJ(Si, Sj). Therefore, the N × N Jaccard distance-based representation will be
| 4 |
3.4.2. Location-Based Representation
Suppose we have N SNP variants with respect to a reference genome in a SARS-CoV-2 sample. Among them, M different mutation sites can be counted. For the ith SNP variant, Vi = [v1i, v2i, ..., vMi], i = 1, 2, ..., N is a 1 × M vector which satisfies the following:
| 5 |
Therefore, an N × M location-based representation will be
| 6 |
3.4.3. Principal Component Analysis (PCA)
Hundreds of complete genome sequences are deposited to GISAID every day, which results in an ever-growing massive quantity of high dimensional data representations for the K-means clustering. For example, if the data set of an organism involves 10 000 SNPs, the initial representation will be a 10 000-dimensional vector for each sample, which can be computationally difficult for a simple K-means clustering algorithm. Therefore, a dimensionality reduction method is used to preprocess the data. The essential idea of PCA-based K-means clustering is to invoke the PCA to obtain a reduced-dimensional representation of each sample before performing the K-means clustering. In practice, one can select a few lowest dimensional principal components as the K-means input for each sample. In ref (5), the authors proved that the principal components are the continuous solution of the cluster indicators in the K-means clustering method, which provides us a rigorous mathematical tool to embed our high-dimensional data into a low-dimensional PCA subspace.
4. Conclusion
The rapid global transmission of coronavirus disease 2019 (COVID-19) has offered some of the most heterogeneous, diverse, and challenging mutagenic environments to stimulate dramatic genetic evolution and response from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). This work provides the most comprehensive genotyping of SARS-CoV-2 transmission and evolution up to date based on 15 140 genome samples and reveals six clusters of the COVID-19 genomes and associated mutations on eight different SARS-CoV-2 proteins. We introduce mutation h-index and mutation ratio to qualify individual protein’s degree of nonconservativeness. We unveil that SARS-CoV-2 envelope protein, main protease, and endoribonuclease protein are relatively the most conservative, whereas SARS-CoV-2 nucleocapsid protein, spike protein, and papain-like protease are relatively the most nonconservative. We report that all of the SARS-CoV-2 proteins have undergone intensive mutations since January 5, 2020, and some of these mutations might seriously undermine ongoing efforts on COVID-19 diagnostic testing, vaccine development, antibody therapeutics, and small-molecular drug discovery.
5. Data Availability
The nucleotide sequences of the SARS-CoV-2 genomes used in this analysis are available, upon free registration, from the GISAID database (https://www.gisaid.org/). Eighteen tables are provided in the Supporting Information for SNP variants of 15 140 SARS-CoV-2 samples across the world, SNP variants of 4587 SARS-CoV-2 samples in the US, SNP variants in six global clusters, SNP variants in four US clusters, and mutation records for eight SARS-CoV-2 proteins. The acknowledgments of the SARS-COV-2 genomes are also given in the Supporting Information.
Acknowledgments
This work was supported in part by NIH grant GM126189, NSF Grants DMS-1721024, DMS-1761320, and IIS1900473, Michigan Economic Development Corporation, Bristol-Myers Squibb, and Pfizer. The authors thank The IBM TJ Watson Research Center, The COVID-19 High Performance Computing Consortium, NVIDIA, and MSU HPCC for computational assistance.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.0c00501.
The authors declare no competing financial interest.
This article is made available via the ACS COVID-19 subset for unrestricted RESEARCH re-use and analyses in any form or by any means with acknowledgement of the original source. These permissions are granted for the duration of the World Health Organization (WHO) declaration of COVID-19 as a global pandemic.
Supplementary Material
References
- Cao Y.; Su B.; Guo X.; Sun W.; Deng Y.; Bao L.; Zhu Q.; Zhang X.; Zheng Y.; Geng C.; Chai X.; He R.; Li X.; Lv Q.; Zhu H.; Deng W.; Xu Y.; Wang Y.; Xie X. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients b cells. Cell 2020, 10.1016/j.cell.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.; Michalska K.; Jedrzejczak R.; Maltseva N.; Endres M.; Godzik A.; Kim Y.; Joachimiak A.. Crystal structure of RNA binding domain of nucleocapsid phosphoprotein from SARS coronavirus 2. Wordwide PDB; 2020; 10.2210/pdb6VYO/pdb. [DOI] [Google Scholar]
- Corman V. M.; Landt O.; Kaiser M.; Molenkamp R.; Meijer A.; Chu D. K.; Bleicker T.; Brnink S.; Schneider J.; Schmidt M. L.; Mulders D. G.; Haagmans B. L.; van der Veer B.; van den Brink S.; Wijsman L.; Goderski G.; Romette J.-L.; Ellis J.; Zambon M.; Peiris M.; Goossens H.; Reusken C.; Koopmans M. P.; Drosten C. Detection of 2019 novel coronavirus (2019-ncov) by real-time RT-PCR. Eurosurveillance 2020, 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeDiego M. L.; Nieto-Torres J. L.; Jiménez-Guardeño J. M.; Regla-Nava J. A.; Alvarez E.; Oliveros J. C.; Zhao J.; Fett C.; Perlman S.; Enjuanes L. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog. 2011, 7 (10), e1002315. 10.1371/journal.ppat.1002315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding C.; He X.. K-means clustering via principal component analysis. In Proceedings of the twenty-first international conference on Machine learning, 2004, p 29.
- Ferron F.; Subissi L.; Silveira De Morais A. T.; Le N. T. T.; Sevajol M.; Gluais L.; Decroly E.; Vonrhein C.; Bricogne G.; Canard B.; Imbert I. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (2), E162–E171. 10.1073/pnas.1718806115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follis K. E.; York J.; Nunberg J. H. Furin cleavage of the SARS coronavirus spike glycoprotein enhances cell–cell fusion but does not affect virion entry. Virology 2006, 350 (2), 358–369. 10.1016/j.virol.2006.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Yan L.; Huang Y.; Liu F.; Zhao Y.; Cao L.; Wang T.; Sun Q.; Ming Z.; Zhang L.; Ge J.; Zheng L.; Zhang Y.; Wang H.; Zhu Y.; Zhu C.; Hu T.; Hua T.; Zhang B.; Yang X.; Li J.; Yang H.; Liu Z.; Xu W.; Guddat L. W.; Wang Q.; Lou Z.; Rao Z. Structure of the rna-dependent rna polymerase from COVID-19 virus. Science 2020, 368 (6492), 779–782. 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M.; Kleine-Weber H.; Schroeder S.; Krüger N.; Herrler T.; Erichsen S.; Schiergens T. S.; Herrler G.; Wu N.-H.; Nitsche A.; Müller M. A.; Drosten C.; Pühlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Du X.; Xu Y.; Deng Y.; Liu M.; Zhao Y.; Zhang B.; Li X.; Zhang L.; Peng C. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Jedrzejczak R.; Maltseva N. I.; Wilamowski M.; Endres M.; Godzik A.; Michalska K.; Joachimiak A. Crystal structure of nsp15 endoribonuclease nendou from SARS-CoV-2. Protein Sci. 2020, 10.1002/pro.3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levandowsky M.; Winter D. Distance between sets. Nature 1971, 234 (5323), 34–35. 10.1038/234034a0. [DOI] [Google Scholar]
- McBride R.; Van Zyl M.; Fielding B. C. The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6 (8), 2991–3018. 10.3390/v6082991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya A.; Holmes E. C.; González-Candelas F. The population genetics and evolutionary epidemiology of RNA viruses. Nat. Rev. Microbiol. 2004, 2 (4), 279–288. 10.1038/nrmicro863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The species severe acute respiratory syndrome-related coronavirus: classifying 2019-ncov and naming it SARS-CoV-2. Nature Microbiology 2020, 5 (4), 536. 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojosnegros S.; Beerenwinkel N. Models of RNA virus evolution and their roles in vaccine design. Immunome Res. 2010, 6 (S2), S5. 10.1186/1745-7580-6-S2-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipiuk J.; Jedrzejczak R.; Tesar C.; Endres M.; Stols L.; Babnigg G.; Kim Y.; Michalska K.; Joachimiak A.. The crystal structure of papain-like protease of SARS CoV-2. Wordwide PDB; 2020; 10.2210/pdb6w9c/pdb. [DOI] [Google Scholar]
- Raoult D.; Zumla A.; Locatelli F.; Ippolito G.; Kroemer G. Coronavirus infections: Epidemiological, clinical and immunological features and hypotheses. Cell Stress 2020, 4 (4), 66. 10.15698/cst2020.04.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y.; Zhou Z.; Liu J.; Lin L.; Li S.; Wang H.; Xia J.; Zhao Z.; Wen J.; Zhou C.; Wang J.; Yin J.; Xu N.; Liu S. A strategy for searching antigenic regions in the SARS-CoV spike protein. Genomics, Proteomics Bioinf. 2003, 1 (3), 207–215. 10.1016/S1672-0229(03)01026-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y.; McCauley J. Eurosurveillance 2017, 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [Google Scholar]
- Sievers F.; Higgins D. G. Clustal omega. Current Protocols in Bioinformatics 2014, 48 (1), 3–13. 10.1002/0471250953.bi0313s48. [DOI] [PubMed] [Google Scholar]
- Surya W.; Li Y.; Torres J. Structural model of the SARS coronavirus E channel in LMPG micelles. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1309–1317. 10.1016/j.bbamem.2018.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Wu C.; Li X.; Song Y.; Yao X.; Wu X.; Duan Y.; Zhang H.; Wang Y.; Qian Z.; Cui J.; Lu J. On the origin and continuing evolution of SARS-CoV-2. Nat. Sci. Rev. 2020, 7, 1012. 10.1093/nsr/nwaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udugama B.; Kadhiresan P.; Kozlowski H. N.; Malekjahani A.; Osborne M.; Li V. Y.; Chen H.; Mubareka S.; Gubbay J.; Chan W. C. Diagnosing COVID-19: The disease and tools for detection. ACS Nano 2020, 14, 3822. 10.1021/acsnano.0c02624. [DOI] [PubMed] [Google Scholar]
- Walls A. C.; Park Y.-J.; Tortorici M. A.; Wall A.; McGuire A. T.; Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y.; Shang J.; Graham R.; Baric R. S.; Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 2020, 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . Coronavirus disease 2019 (COVID-19) situation report 133; 2020. [Google Scholar]
- Wu F.; Zhao S.; Yu B.; Chen Y.-M.; Wang W.; Song Z.-G.; Hu Y.; Tao Z.-W.; Tian J.-H.; Pei Y.-Y.; Yuan M.-L.; Zhang Y.-L.; Dai F.-H.; Liu Y.; Wang Q.-M.; Zheng J.-J.; Xu L.; Holmes E. C.; Zhang Y.-Z. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579 (7798), 265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X.; Chakraborti S.; Dimitrov A. S.; Gramatikoff K.; Dimitrov D. S. The SARS-CoV s glycoprotein: expression and functional characterization. Biochem. Biophys. Res. Commun. 2003, 312 (4), 1159–1164. 10.1016/j.bbrc.2003.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin C. Genotyping coronavirus SARS-CoV-2: methods and implications. Genomics 2020, 10.1016/j.ygeno.2020.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The nucleotide sequences of the SARS-CoV-2 genomes used in this analysis are available, upon free registration, from the GISAID database (https://www.gisaid.org/). Eighteen tables are provided in the Supporting Information for SNP variants of 15 140 SARS-CoV-2 samples across the world, SNP variants of 4587 SARS-CoV-2 samples in the US, SNP variants in six global clusters, SNP variants in four US clusters, and mutation records for eight SARS-CoV-2 proteins. The acknowledgments of the SARS-COV-2 genomes are also given in the Supporting Information.