

ABSTRACT
Mutations in the genes for extracellular matrix (ECM) components cause a wide range of genetic connective tissues disorders throughout the body. The elucidation of mutations and their correlation with pathology has been instrumental in understanding the roles of many ECM components. The pathological consequences of ECM protein mutations depend on its tissue distribution, tissue function, and on the nature of the mutation. The prevalent paradigm for the molecular pathology has been that there are two global mechanisms. First, mutations that reduce the production of ECM proteins impair matrix integrity largely due to quantitative ECM defects. Second, mutations altering protein structure may reduce protein secretion but also introduce dominant negative effects in ECM formation, structure and/or stability. Recent studies show that endoplasmic reticulum (ER) stress, caused by mutant misfolded ECM proteins, makes a significant contribution to the pathophysiology. This suggests that targeting ER‐stress may offer a new therapeutic strategy in a range of ECM disorders caused by protein misfolding mutations. Anat Rec, 2019. © 2019 The Authors. The Anatomical Record published by Wiley Periodicals, Inc. on behalf of American Association of Anatomists.
Keywords: genetic disorders, extracellular matrix, ER stress, unfolded protein response
INTRODUCTION
The extracellular matrix (ECM) is a tissue‐specific mixture of interacting proteins, glycoproteins, and proteoglycans which provides the structural networks and scaffolds of tissues. As well as contributing to tissue shape and biomechanics, the ECM mediates important aspects of cellular signaling in growth, development, and connective tissue remodeling and repair (Hynes, 2009; Rozario and DeSimone, 2010; Bonnans et al., 2014; Humphries et al., 2018). To provide a more systematic definition of ECM complexity revealed by the explosion of genomic, transcriptomic, and proteomic data, new bioinformatics tools have been developed to define the “matrisome,” the cohort of genes that encode ECM and ECM‐associated proteins (Naba et al., 2012; Naba et al., 2016). They are defined by the presence of a characteristic set of protein domains, and further curated manually based on structural and functional features. This definition of ECM components provides a useful annotation for identifying ECM signatures in “omics” datasets and is gaining wide application in the field (Naba et al., 2016). The human matrisome (1,027 genes) is composed of 274 “Core Matrisome” genes and 753 “Matrisome‐associated” genes. The core matrisome contains genes encoding ECM glycoproteins (195), proteoglycans (35), and collagens (44). Matrisome‐associated genes included “ECM‐affiliated” (171), “Regulators” (238), and “secreted factors” (344).
The functional importance of the ECM is graphically demonstrated by numerous genetic disorders resulting from mutations in the “Core Matrisome” (Table 1). To date, mutations in 105 core matrisome genes cause human disorders defined in OMIM, and this number should increase rapidly with the wide‐spread clinical application of whole‐exome and whole‐genome sequencing. Of the 195 ECM glycoprotein genes, 67 are implicated in genetic disease or disease susceptibility; 27 of 44 collagen genes, and 11 of 35 proteoglycan genes have disease associations (Table 1).
Table 1.
Core matrisome components associated with genetic disease
| Gene Symbol | Disease | OMIM |
|---|---|---|
| ECM glycoproteins | ||
| AGRN | Myasthenic syndrome, congenital, 8 | 615120 |
| AMBN | Amelogenesis imperfecta, type 1F | 616270 |
| AMELX | Amelogenesis imperfecta, type 1E | 301200 |
| BMPER | Diaphanospondylodysostosis | 608022 |
| CILP | Intervertebral disc disease (susceptibility) | 603932 |
| COCH | Deafness, autosomal dominant, 9 | 601369 |
| COLQ | Myasthenic syndrome, congenital, 5 | 603034 |
| COMP | Pseudoachondroplasia; epiphyseal dysplasia, multiple, 1 | 177170, 132400 |
| CRELD1 | Trioventricular septal defect (susceptibility) | 606217 |
| CTHRC1 | Barrett esophagus | 614266 |
| DMP1 | Hypophosphatemic rickets, autosomal recessive | 241520 |
| DSPP | Deafness, autosomal dominant 39; dentinogenesis imperfecta, shields type III; dentin dysplasia, type II; dentinogenesis imperfecta 1 | 605594, 125500, 125420, 125490 |
| ECM1 | Lipoid proteinosis of Urbach and Wiethe | 247100 |
| EFEMP1 | Doyne honeycomb retinal dystrophy | 126600 |
| EFEMP2 | Cutis laxa, autosomal recessive, type 1B | 614437 |
| ELN | Supravalvular aortic stenosis, cutis laxa, autosomal dominant 1 | 185500, 123700 |
| EYS | Retinitis pigmentosa 25 | 602772 |
| FBLN1 | Synpolydactyly 2 | 608180 |
| FBLN5 | Cutis laxa, autosomal recessive, type IA; cutis laxa, autosomal dominant 2; neuropathy, hereditary, with or without age‐related macular degeneration | 219100, 614434, 608895 |
| FBN1 | Marfan syndrome, ectopia lentis 1, isolated, autosomal dominant; Marfan lipodystrophy syndrome | 154700, 129600, 616914 |
| FBN2 | Arthrogryposis, distal, type 9, macular degeneration, early‐onset | 121050, 616118 |
| FGA | Dysfibrinogenemia, congenital | 616004 |
| FGB | Dysfibrinogenemia, congenital | 616004 |
| FGG | Dysfibrinogenemia, congenital | 616004 |
| FN1 | Spondylometaphyseal dysplasia, corner fracture type; glomerulopathy with fibronectin deposits 2 | 184255, 601894 |
| FRAS1 | Fraser syndrome 1 | 219000 |
| GLDN | Lethal congenital contracture syndrome 11 | 617194 |
| HMCN1 | Macular degeneration, age‐related, 1 | 603075 |
| IGFALS | Acid‐labile subunit deficiency | 615961 |
| IGFBP7 | Retinal arterial macroaneurysm with supravalvular pulmonic stenosis | 614224 |
| KAL1 | Hypogonadotropic hypogonadism 1 with or without anosmia | 308700 |
| LAMA1 | Poretti–Boltshauser syndrome | 615960 |
| LAMA2 | Muscular dystrophy, congenital merosin‐deficient, 1A | 607855 |
| LAMA3 | Epidermolysis bullosa, junctional, herlitz type | 226700 |
| LAMA4 | Cardiomyopathy, dilated, 1 | 615235 |
| LAMB1 | Lissencephaly 5 | 615191 |
| LAMB2 | Pierson syndrome; nephrotic syndrome, type 5, with or without ocular abnormalities | 609049 614199 |
| LAMB3 | Epidermolysis bullosa, junctional, herlitz and non‐herlitz types | 226650 |
| LAMC1 | Epidermolysis bullosa, junctional, non‐herlitz type | 226650 |
| LAMC2 | Epidermolysis bullosa, junctional, non‐herlitz type | 226650 |
| LAMC3 | Cortical malformations, occipital | 614115 |
| LGI1 | Epilepsy, familial temporal lobe, 1 | 600512 |
| LGI4 | Arthrogryposis multiplex congenita, neurogenic, with myelin defect | 617468 |
| LTBP2 | Glaucoma 3, primary congenital, D; Weill–Marchesani syndrome 3 | 613086, 614819 |
| LTBP3 | Dental anomalies and short stature | 601216 |
| LTBP4 | Cutis laxa, autosomal recessive, type IC | 613177 |
| MATN3 | Epiphyseal dysplasia, multiple, 5; spondyloepimetaphyseal dysplasia, matrilin‐3 related | 607078, 608728 |
| MFAP5 | Aortic aneurysm, familial thoracic 9 | 616166 |
| MGP | Keutel syndrome | 245150 |
| OTOG | Deafness, autosomal recessive 18B | 614945 |
| PXDN | Anterior segment dysgenesis 7 | 269400 |
| RELN | Lissencephaly 2; epilepsy, familial temporal lobe, 7 | 257320, 616436 |
| RSPO1 | Palmoplantar hyperkeratosis with squamous cell carcinoma of skin and 46,XX sex reversal | 610644 |
| RSPO2 | Tetraamelia syndrome 2 | 618021 |
| RSPO4 | Nail disorder, nonsyndromic congenital, 4 | 206800 |
| SMOC1 | Microphthalmia with limb anomalies | 206920 |
| SMOC2 | Dentin dysplasia, type I | 125400 |
| SRPX2 | Rolandic epilepsy, mental retardation, and speech dyspraxia, X‐linked | 300643 |
| TECTA | Deafness, autosomal recessive 21; deafness, autosomal dominant 12 | 603629, 601543 |
| TGFBI | Corneal dystrophy—several types—Avellino type; lattice type I; Reis–Bucklers type; Thiel–Behnke type; lattice type IIIA; epithelial basement membrane | 607541, 122200, 608470, 602082, 608471, 121820 |
| THBS2 | Intervertebral disc disease (susceptibility) | 603932 |
| TNXB | Ehlers–Danlos syndrome, classic‐like; vesicoureteral reflux 8 | 606408, 615963 |
| TSPEAR | Deafness, autosomal recessive 98 | 614861 |
| VWA3B | Spinocerebellar ataxia, autosomal recessive 22 | 616948 |
| VWF | Von Willebrand disease, Types 1, 2, and 3 | 193400, 613554, 277480, |
| WISP3 | Arthropathy, progressive pseudorheumatoid, of childhood | 208230 |
| ZP1 | Oocyte maturation defect 1 | 615774 |
| ZP3 | Oocyte maturation defect 3 | 617712 |
| Collagens | ||
| COL1A1 | Osteogenesis imperfecta, types I, II, III, IV (also called OI1, 2, 3, 4); Caffey disease; Ehlers–Danlos syndrome, arthrochalasia type, 1 | 166200, 166210, 259420, 166220, 114000, 130060 |
| COL1A2 | Osteogenesis imperfecta, types I, II, III, IV (also called OI1, 2, 3, 4); Ehlers–Danlos syndrome, arthrochalasia type 2; Ehlers–Danlos syndrome, Cardiac valvular type | 166200, 166210, 259420, 617821, 225320 |
| COL2A1 | Stickler syndrome, type I; Stickler syndrome, type I, nonsyndromic ocular; spondyloepiphyseal dysplasia congenita; kniest dysplasia; avascular necrosis of femoral head, primary, 1; achondrogenesis, type II; Czech dysplasia. osteoarthritis with mild chondrodysplasia; platyspondylic lethal skeletal dysplasia, Torrance type; spondyloepiphyseal dysplasia, Stanescu type; spondyloepimetaphyseal dysplasia, Strudwick type; spondyloperipheral dysplasia; epiphyseal dysplasia, multiple, with myopia and conductive deafness; Legg‐Calve‐Perthes disease | 108300, 609508, 183900, 156550, 608805, 200610, 609162, 604864, 151210, 616583, 184250, 271700, 132450, 150600 |
| COL3A1 | Ehlers–Danlos syndrome, vascular type | 130050 |
| COL4A1 | Brain small vessel disease with or without ocular anomalies; angiopathy, hereditary, with nephropathy, aneurysms, and muscle cramps; porencephaly 1; hemorrhage, intracerebral (susceptibility); schizencephaly | 607595, 175780, 614519, 26160 |
| COL4A2 | Porencephaly 2; hemorrhage, intracerebral (susceptibility) | 614483, 614519 |
| COL4A3 | Alport syndrome, autosomal recessive and autosomal dominant, hematuria, benign familial | 203780, 104200, 141200 |
| COL4A4 | Alport syndrome, autosomal recessive, and autosomal dominant | 203780, 104200 |
| COL4A5 | Alport syndrome, X‐linked | 301050 |
| COL4A6 | Leiomyomatosis, diffuse, with Alport syndrome; deafness, X‐linked 6 | 308940, 300914 |
| COL5A1 | Ehlers–Danlos syndrome, classic type, 1 | 130000 |
| COL5A2 | Ehlers–Danlos syndrome, classic type, 2 | 130010 |
| COL6A1 | Ullrich congenital muscular dystrophy 1; Bethlem myopathy 1 | 254090, 158810 |
| COL6A2 | Ullrich congenital muscular dystrophy 1; Bethlem myopathy 1; myosclerosis, autosomal recessive | 254090, 158810, 255600 |
| COL6A3 | Ullrich congenital muscular dystrophy 1; Bethlem myopathy 1; myosclerosis, autosomal recessive; dystonia 27 | 254090, 158810, 255600, 616411 |
| COL7A1 | Epidermolysis bullosa dystrophica, autosomal recessive and autosomal dominant; nail disorder, nonsyndromic congenital, 8 | 226600, 131750, 607523 |
| COL9A1 | Stickler syndrome, type IV; epiphyseal dysplasia, multiple, 6 | 614134, 614135 |
| COL9A2 | Epiphyseal dysplasia, multiple, 2; Stickler syndrome, type V | 600204, 614284 |
| COL9A3 | Epiphyseal dysplasia, multiple, 3 | 600969 |
| COL10A1 | Metaphyseal chondrodysplasia, Schmid type | 156500 |
| COL11A1 | Stickler syndrome, type II; Marshall syndrome; fibrochondrogenesis 1 | 604841, 154780, 228520 |
| COL11A2 | Otospondylomegaepiphyseal dysplasia, autosomal dominant and autosomal recessive; deafness, autosomal recessive 53; fibrochondrogenesis 2 | 184840, 215150, 609706, 614524 |
| COL12A1 | Bethlem myopathy 2; Ullrich congenital muscular dystrophy 2 | 616471, 616470 |
| COL13A1 | Myasthenic syndrome, congenital, 19 | 616720 |
| COL15A1 | Knobloch syndrome 1 | 267750 |
| COL17A1 | Epidermolysis bullosa, junctional, non‐Herlitz type; epithelial recurrent erosion dystrophy | 226650, 122400 |
| COL18A1 | Knobloch syndrome 1 | 267750 |
| COL25A1 | Fibrosis of extraocular muscles, congenital, 5 | 616219 |
| COL27A1 | Steel syndrome | 615155 |
| Proteoglycans | ||
| ACAN | Short stature and advanced bone age, with or without early‐onset osteoarthritis and/or osteochondritis dissecans; spondyloepiphyseal dysplasia, Kimberley type; spondyloepimetaphyseal dysplasia, aggrecan type | 165800, 608361, 612813 |
| ASPN | Osteoarthritis susceptibility 3 (susceptibility); intervertebral disc disease (susceptibility) | 607850, 603932 |
| BGN | Meester–Loeys syndrome; spondyloepimetaphyseal dysplasia, X‐linked | 300989 |
| DCN | corneal dystrophy, congenital stromal | 610048 |
| HSPG2 | Schwartz–Jampel syndrome, type 1; dyssegmental dysplasia, Silverman‐Handmaker type | 255800, 224410 |
| IMPG1 | Macular dystrophy, vitelliform, 4 | 616151 |
| IMPG2 | Retinitis pigmentosa 56; macular dystrophy, vitelliform, 5 | 613,581, 616152 |
| KERA | Cornea plana 2, autosomal recessive | 217300 |
| NYX | Night blindness, congenital stationary, type 1A | 310500 |
| PRG4 | Camptodactyly‐arthropathy‐coxa vara‐pericarditis syndrome | 208250 |
| VCAN | Wagner vitreoretinopathy | 143200 |
| ACAN | Short stature and advanced bone age, with or without early‐onset osteoarthritis and/or osteochondritis dissecans; spondyloepiphyseal dysplasia, Kimberley type; spondyloepimetaphyseal dysplasia, aggrecan type | 165800, 608361, 612813 |
| ASPN | Osteoarthritis susceptibility 3 (susceptibility); intervertebral disc disease (susceptibility) | 607850, 603932 |
| BGN | Meester–loeys syndrome; spondyloepimetaphyseal dysplasia, X‐linked | 300989 |
| DCN | Corneal dystrophy, congenital stromal | 610048 |
| HSPG2 | Schwartz–Jampel syndrome, type 1; dyssegmental dysplasia, Silverman‐Handmaker type | 255800, 224410 |
| IMPG1 | Macular dystrophy, vitelliform, 4 | 616151 |
| IMPG2 | Retinitis pigmentosa 56; macular dystrophy, vitelliform, 5 | 613581, 616152 |
While the mechanisms of how mutations in many of these core matrisome genes cause disease are poorly understood and will show gene and mutation specificity, mutations in the more intensely studied conditions such osteogenesis imperfecta (OI), Ehlers–Danlos syndrome and numerous chondrodysplasias are offering important insights into molecular pathologies of structural ECM components (Bateman et al., 2009). We will focus our discussion on the core matrisome components involved in these conditions. However, in addition to these, mutations in the broader noncore matrisome genes (Naba et al., 2016), enzymes involved in the ER/Golgi processing machinery, transcription factors and signaling molecules also have the ability to impact on the production and integrity of the ECM.
OSTEOGENESIS IMPERFECTA
The central clinical feature of OI, or “brittle bone disease,” is a decrease in bone mass resulting in bone fragility (Bishop, 2016; Lim et al., 2017; Marini et al., 2017). OI is also often associated with blue sclerae, dental abnormalities (dentinogenesis imperfecta), and progressive hearing loss. As many new OI genes have been discovered, there has been debate around whether clinical classification should be based on the genetic defect or on the clinical phenotype. Both classifications are presented in Table 2. Because of the clinical utility of the classification by phenotype, we will use this as the basis of our discussion. The least severe subtype is OI type 1 with mild to moderate bone fragility and little or no apparent skeletal deformity. Most patients have blue sclerae and hearing loss is common. OI type 2 is lethal in utero or shortly after birth due to severe bone fragility. OI type 3 is progressively deforming with moderate to severe bone deformity, blue sclerae at birth and commonly with hearing loss and abnormal dentition. OI type 4 (common variable OI with normal sclerae) has mild to moderate bone fragility, normal sclerae, and abnormal dentition and hearing loss, albeit less frequently than in OI type 3. The most common mutations causing OI occur in the genes encoding the proα1 or proα2 chain of type I procollagen, COL1A1 and COL1A2. Of the ~2,000 gene mutations now described in OI, more than 85% are heterozygous mutations in these genes. The remainder are predominantly autosomal recessive mutations in components of the collagen biosynthetic and secretion machinery and in key osteogenic transcription factors and signaling pathways (Table 2).
Table 2.
Osteogenesis imperfecta
| Phenotype | Typical features | Clinical type | Genetic type | Inheritance | Gene/protein defect | Protein defect |
|---|---|---|---|---|---|---|
| Non‐deforming form | Mild to moderate bone fragility, normal or near normal stature, most have blue sclerae, normal dentition, hearing loss in ~50%. | OI type 1 | OI type 1 | AD |
COL1A1 COL1A2 |
Collagen I haploinsufficiency |
| Perinatally lethal form | Extreme bone fragility, short stature, long bone bowing | OI type 2 |
OI type 2 OI type 7 OI type 8 OI type 9 |
AD AR |
COL1A1 COL1A2 CRTAP LEPRE1/P3H1 PPIB/CYPB |
Collagen I structural mutations Collagen posttranslational modification and folding machinery |
| Progressively deforming OI |
Moderate to severe bone deformity, blue sclerae at birth, hearing loss and abnormal dentition common. |
OI type 3 |
OI type 3 OI type 7 OI type 8 OI type 9 OI type 10 OI type 11 No type OI type 16
OI Cole‐Carpenter Like
OI type 12
OI type 15 OI type 10
OI type 14
OI type 13 |
AD
AR |
COL1A1 COL1A2 CRTAP LEPRE1/P3H1 PPIB/CYPB FKBP10/FKBP65 SERPINH1/HSP47 PLOD2/ LH2 CREB3L1/OASIS
SEC24D
BMP1
WNT1 SERPINF1/PEDF
TMEM38B/TRIC‐B
SP7/OSX |
Collagen I structural mutations Collagen posttranslational modification and folding machinery
Protein folding/endoplasmic reticulum stress sensor COPII vesicle component—collagen secretion Proteolytic removal of procollagen N‐propeptide Wnt cell signaling pathway Signaling and collagen binding protein, important for mineralization Endoplasmic reticulum cation channel. Transcription factor, bone formation |
| Common Variable OI with normal sclerae | Mild to moderate, bone fragility, normal sclerae, variable dentition, hearing loss in <10%. | OI type 4 |
OI type 4
OI type 15
OI type 7 OI type 11 OI type 9 OI type 6
OI type 17
OI type 13 |
AD
AR |
COL1A1 COL1A2 WNT1
CRTAP FKBP10/FKBP65 PPIB/CYPB SERPINF1/PEDF
SPARC
SP7/OSX |
Collagen I structural mutations
Wnt cell signaling pathway Collagen posttranslational modification and folding machinery Signaling and collagen binding protein, important for mineralization Collagen binding/extracellular matrix assembly Transcription factor, bone formation |
EHLERS–DANLOS SYNDROME
The clinical hallmarks of Ehlers–Danlos syndrome (EDS) are hyperextensible skin and hypermobile joints. EDS classification has been recently revised to identify 13 subtypes (Table 3) (De Paepe and Malfait, 2012; Malfait et al., 2017). Mutations in collagens and collagen processing molecules are a common cause of the condition (Table 3). Classical EDS is most often caused by heterozygous mutations in the genes for collagen V, COL5A1, and COL5A2, and infrequently by COL1A1 mutations. The clinically severe vascular EDS is caused by heterozygous mutations in the gene for type III collagen, COL3A1, which impacts the integrity of tissues where type III collagen is structurally important such as major blood vessels and intestine, resulting in a propensity for catastrophic rupture. Cardiac‐valvular EDS results from homozygous mutations in COL1A2 and arthrochalasia EDS from both COL1A1 and COL1A2 mutations. Dermatosparaxis EDS results from homozygous mutations in ADAMTS2 which encodes the critical proteinase involved in the extracellular removal of the type I collagen N‐propeptide. Retention of this noncollagenous propeptide prevents normal collagen fibril formation which severely impacts the collagen I‐rich tissues such as skin leading to severe skin fragility. Homozygous and heterozygous COL12A1 mutations have been described in Myopathic EDS. In addition to collagen mutations, mutations in the genes for other matrix components, or in their biosynthetic pathways have been described (Table 3).
Table 3.
Ehlers–Danlos syndromea
| Clinical subtype | Abbreviation | Inheritance | Gene defect | Protein defect |
|---|---|---|---|---|
| Classical EDS | cEDS | AD | COL5A1 (major), COL1A1 (rare) | Collagen V, collagen I |
| Classical‐like EDS | clEDS | AR | TNXB | Tenascin XB |
| Cardiac‐valvular EDS | cvEDS | AR | COL1A2 | Collagen I |
| Vascular EDS | vEDS | AD | COL3A1 (major), COL1A1 (rare) | Collagen III, collagen I |
| Hypermobile EDS | hEDS | AD | Unknown | Unknown |
| Arthrochalasia EDS | aEDS | AD | COL1A1, COL1A2 | Collagen I |
| Dermatosparaxis EDS | dEDS | AR | ADAMTS2 | ADAMTS‐2 |
| Kyphoscoliotic EDS | kEDS | AR | PLOD1, FKBP14 | LH1, FKBP22 |
| Brittle cornea syndrome | BCS | AR | ZNF469, PRDM5 | ZNF469, PRDM5 |
| Spondylodysplastic EDS | spEDS | AR | B4GALT7, B3GALT6, SLC39A13 | Β4GalT7, βGalT6, ZIP13 |
| Musculocontractural EDS | mcEDS | AR | CHST14, DSE | D4ST1, DSE |
| Myopathic EDS | mEDS | AR/AD | COL12A1 | Collagen XII |
| Periodontal EDS | pEDS | AD | C1R, C1S | C1r, C1s |
Modified from Malfait et al. (2017).
CHONDRODYSPLASIAS
Of the more than 450 recognized genetic skeletal diseases (Bateman, 2001; Bonafe et al., 2015), at least 200 primarily affect the cartilage with a wide clinical spectrum ranging from lethal conditions in utero to later onset conditions involving the joints and spine. The mutations causing most chondrodysplasias can be broadly grouped into genes involved in local regulation of cartilage growth, genes involved in cartilage metabolic pathways, and genes encoding the cartilage structural proteins (Bateman, 2001; Krakow, 2015). Mutations in growth factors and their receptors, archetypically in fibroblast growth factor receptor 3 (FGFR3), disturb local cartilage growth regulation. FGFR3 signaling is a key negative regulator of growth plate chondrocyte growth and gain‐of‐function FGFR3 mutations result in a common form of human dwarfism, achondroplasia. Cartilage metabolism and homeostasis can also be disturbed by mutations in essential enzymes such the Golgi enzyme arylsulfatase E (ARSE) in chondrodysplasia punctata, the sulfate transporter DTDST in diastrophic dysplasias and the mechano‐sensitive ion channel TRPV4 in metatrophic dysplasia and brachydactyly‐arthropathy.
However, given the role of the cartilage ECM in chondrocyte growth, differentiation, and endochondral bone formation, it is unsurprising that the first characterized and most common skeletal dysplasia mutations are in genes involved in producing the architecturally precise cartilage ECM (Table 4). Heterozygous structural mutations in the main fibril‐forming cartilage collagen, type II (COL2A1), cause disorders including achondrogenesis type II, hypochondrogenesis, spondyloepiphyseal dysplasia congenita, Kneist dysplasia, and early‐onset arthritis. COL2A1 nonsense mutations cause the milder Stickler syndrome (Barat‐Houari et al., 2016; Deng et al., 2016). Mutations in type XI collagen (COL11A1 and COL11A2) which form part of the nucleating core of the collagen II fibrils cause Stickler and Marshall syndrome (Majava et al., 2007). Mutations in collagen IX, which also interacts with and regulates collagen II fibril structure, cause multiple epiphyseal dysplasia (Chapman et al., 2003). Likewise, mutations in other interacting proteins matrilin 3 (MATN3) and cartilage oligomeric matrix protein (COMP) cause multiple epiphyseal dysplasia, and in the case of COMP also pseudoachondroplasia (Chapman et al., 2003). Mutations in the other major structural components of the cartilage pericellular matrix (perlecan; HSPG2) or ECM (aggrecan; ACAN) cause other chondrodysplasias (Gibson and Briggs, 2016). Collagen X (COL10A1), a small network forming collagen which is uniquely expressed in the hypertrophic zone of growth plate cartilage, is defective in metaphyseal chondrodysplasia, type Schmid (Bateman et al., 2005). COL10A1 mutations have been instrumental in understanding the role of endoplasmic stress in the pathophysiology (Tsang et al., 2007; Bateman et al., 2009; Tsang et al., 2010) and is discussed in detail in following sections which explore what we know about how cartilage ECM gene mutations cause pathology.
Table 4.
Chondrodysplasias caused by ECM gene mutations
| Gene | Protein | Group: representative disorders |
|---|---|---|
| Collagens | ||
| COL2A1 | Collagen II | Achondrogenesis Type II, spondyloepiphyseal dysplasia congenita, Kneist dysplasia, Stickler syndrome type 1 |
| COL9A1 | Collagen IX | Multiple epiphyseal dysplasia type 6 |
| COL9A2 | Collagen IX | Multiple epiphyseal dysplasia type 2 |
| COL9A3 | Collagen IX | Multiple epiphyseal dysplasia type 3 |
| COL10A1 | Collagen X | Metaphyseal dysplasia Schmid type |
| COL11A1 | Collagen XI | Stickler syndrome type 2, Marshall syndrome, fibrochondrogenesis type 1 |
| COL11A2 | Collagen XI | Otospondylomegaepiphyseal dysplasia, fibrochondrogenesis type 2 |
| ECM structural proteins | ||
| HGPS2 | Perlecan | Dyssegmental dysplasias, Schwartz–Jampel syndrome |
| ACAN | Aggrecan | Spondyloepiphyseal dysplasia Kimberley type, Spondyloepimetaphyseal dysplasia aggrecan type |
| COMP | Cartilage oligomeric matrix protein | Pseudoachondroplasia, multiple epiphyseal dysplasia type 1 |
| MATN3 | Matrilin 3 | Multiple epiphyseal dysplasia type 5 |
| MGP | Matrix gamma‐carboxyglutamic acid | Keutel syndrome |
| ECM posttranslational modification | ||
| DTDST | SLC6A2 sulfate transporter | Achondrogenesis type 1B, atelogenesis type 2, diastrophic dysplasia |
| PAPSS2 | PAPS‐synthetase 2 | Spondyloepimetaphyseal dysplasia PAPSS2 type, Brachyolmia recessive type |
| CHST3 | Carbohydrate sulfotransferase 3 | Chondrodysplasia with congenital joint dislocations CHST3 type |
| CHST14 | Carbohydrate sulfotransferase 14 | Ehlers–Danlos syndrome CHST14 type |
| ARSE | Arylsulfatase E | Chondrodysplasia punctata group: chondrodysplasia punctate, X‐linked recessive, brachytelephalangic type |
PATHOLOGICAL MECHANISMS OF ECM PROTEIN MUTATIONS
The pathological consequences of ECM protein mutations depend on protein tissue distribution, tissue function, and on the nature of the mutation. The effect of tissue distribution and tissue‐function of the ECM components in pathology is self‐evident, and elucidation of mutations and correlation to molecular pathology has been instrumental in understanding the roles of numerous ECM components. Understanding how each precise mutation affects structure and functions at the molecular level has been more problematic and the focus of countless studies over recent decades. The prevalent paradigm has been that there are two global mechanisms. First, mutations that reduce the production of ECM proteins such as nonsense mutations and frameshift mutations result in reduced matrix integrity due largely to quantitative ECM defects. Second, structural mutations reduce protein secretion and introduce dominant negative effects in ECM formation, structure, and/or stability (Fig. 1). While this “high‐level” model remains broadly true, recent research has added considerable complexity to our understanding of the molecular pathology. In some cases, this has clarified the impact of specific mutations on molecular networks and suggested new therapeutic approaches, and in other cases introducing new complexities that are so far unresolved. The discussion below will address these aspects of ECM disease mechanisms.
Figure 1.
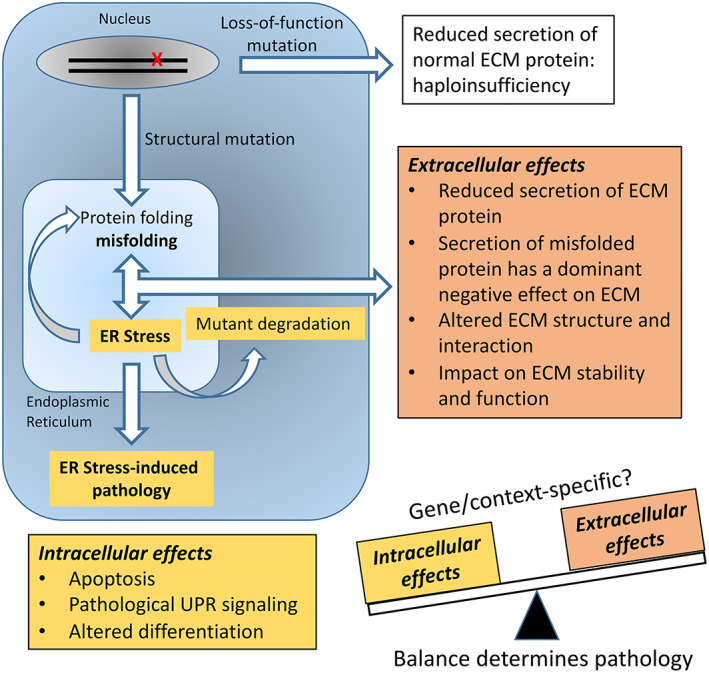
The pathophysiology of ECM mutations. The current model for ECM mutation pathology proposes that the balance between extracellular and intracellular effects determines the pathology and disease severity. Reduced secretion of normal ECM protein as a result of premature termination mutations and nonsense‐mediated mRNA decay or regulatory mutations compromises function and this haploinsufficiency (white box) generally produces relatively mild disease. Structural misfolding mutations have dominant negative consequences leading to reduced secretion, and altered ECM structure and function (orange box). It is now clear that structural misfolding mutations also have intracellular effects that can include mutant degradation, and activation of ER stress pathways and pathological unfolded protein response (UPR) signaling (yellow box). The relative contribution of intracellular and extracellular effects to the pathology is likely to be gene and mutation specific.
Loss‐of‐Function Mutations
Mutations that introduce premature stop codons, either a direct change to a nonsense codon or indels or aberrant splicing which cause a translation frameshift, are a common cause of genetic disease. Premature stop codons commonly trigger an mRNA surveillance process called nonsense‐mediated mRNA decay that degrades the mutant mRNA (Frischmeyer and Dietz, 1999; Fang et al., 2013; Kurosaki and Maquat, 2016). This can result in functional haploinsufficiency in the case of heterozygous mutations, or complete, or near complete, absence of the ECM protein in the case of homozygous mutations (Fig. 1).
Heterozygous loss‐of‐function mutations in COL1A1 and COL1A2 are the predominant cause of mild OI Type 1, and in COL2A1, Stickler, and these decrease the collagenous network. Crucially though, although reduced the ECM contains only structurally normal collagens and a relatively mild phenotype ensues. Heterozygous COL5A1 and COL5A2 premature stop codon mutations in classical EDS do not simply reduce the collagenous matrix but alter its architecture. While type V collagen is a quantitatively minor fibril‐forming collagen, the [α1(V)]2 α2(V)] heterotrimer nucleates type I collagen fibrillogenesis and forms heterotypic type I/V collagen fibrils in tissues such as skin, tendon, and ligaments. Retention of the type V collagen N‐terminal propeptide influences the packing of the Type I monomers into fibrils, thus regulating collagen fibril diameter. The ratio of Type V/I collagen is crucial in determining fibril size and thus haploinsufficiency produces large, irregular, and structurally abnormal Type I collagen fibrils. Type XI collagen, the Type V collagen ortholog in cartilage, plays a comparable role in nucleating collagen II fibrillogenesis and loss‐of‐function mutations in COL11A1 and COL11A2 cause Stickler and Marshall syndromes. Mutations causing aggrecan (ACAN) haploinsuffiency cause spondyloepiphyseal dysplasia, Kimberley type, and idiopathic short stature (Gibson and Briggs, 2016). Aggrecan is a highly sulfated proteoglycan which interacts with hyaluronan to provide an extensive network that endows articular cartilage with its ability to withstand compressive loads.
The major collagen VI isoform (an obligatory α1(VI), α2(VI), α3(VI) hererotrimer) is abundant and found in almost all tissues. Collagen VI is particularly important in muscle and tendon because mutations in the three canonical collagen VI genes, COL6A1, COL6A2, and COL6A3, trigger a spectrum of skeletal muscle disorders with Bethlem myopathy at the mild end and Ullrich congenital muscular dystrophy at the severe end (Lamande and Bateman, 2018). Patients who completely lack collagen VI because of homozygous or compound heterozygous haploinsufficiency mutations in COL6A1, COL6A2, or COL6A3 usually have severe muscular dystrophy. While collagen VI haploinsuffiency mutations in COL6A2 and COL6A3 are well tolerated and produce no apparent pathology, the situation is less clear for COL6A1 haploinsuffiency mutations; sometimes there is no obvious phenotype (Giusti et al., 2005; Foley et al., 2011) and sometimes it causes Bethlem myopathy or mild Bethlem features (Lamande et al., 1998; Baker et al., 2007; Peat et al., 2007). One potential explanation for this conundrum is that half the normal amount of collagen VI is close to the tolerance threshold for pathogenicity and the α1(VI) chain is the limiting chain in collagen VI assembly, meaning that α1(VI) haploinsufficiency reduces total collagen VI synthesis more than either α2(VI) or α3(VI) haploinsufficiency.
In addition to mutations causing haploinsufficiency or null alleles, many of the conditions discussed above are caused by missense mutations. Indeed, these can be the predominant mutation class in many ECMopathies. This is particularly striking with pseudoachondroplasia and multiple epiphyseal dysplasia where structural mutations in COMP, MATN3, COL9A1, COL9A2, and COL9A3 are apparently the only cause of these conditions. Furthermore, the milder clinical phenotypes of heterozygous null mutations in COL1A1 and COL1A2 (OI) and in COL2A1 (Stickler syndrome) further suggest that structural mutations in ECM proteins are commonly more severe, while having less, but structurally normal protein leads to less tissue dysfunction (Fig. 1).
Structural Mutations in ECM Proteins
In contrast to heterozygous null mutations, most of the more severe ECM diseases are caused by heterozygous missense mutations, or less commonly by other mutations such as in‐frame exon‐skipping mutations, that perturb protein structure. Collagen mutations causing OI (COL1A1 and COL1A2) and several chondrodysplasias resulting from COL2A1 and COL10A1 mutations provide the clearest picture of the impact of dominant negative protein structural mutations. Most commonly these mutations result in structurally abnormal collagen pro‐α chains which impair initial assembly into hetero‐ or homo‐trimers or cause abnormal folding of the triple helix. Mutations in the procollagen C‐propeptide can impede the initial trimerization assembly step. Heterotrimerization (procollagen I) and homotrimerization (procollagens II, III, and collagen X) involves the correct registration of the propeptides in the lumen of the ER. This alignment is driven primarily by short discontinuous amino acid sequences presented by the three‐dimensional structures of the individual propeptides. This trimer assembly and subsequent helix formation is also facilitated by the coordinated action of molecular chaperones and posttranslational processing machinery (Ishikawa and Bachinger, 2013). Mutations in the C‐propeptides that perturb the inter‐ and/or intra‐chain disulfide bonding or introduce other structurally deleterious changes can prevent or delay assembly resulting in misfolding and ER retention of the mutant chains. The best studied examples of this are in COL1A1 and COL10A1 and these illustrate pathological processes that can be broadly generalized to other collagen types (Bateman et al., 2009).
Several collagen I C‐propeptide mutations have been mechanistically informative (Bateman et al., 1989; Chessler and Byers, 1993; Lamande et al., 1995; Fitzgerald et al., 1999; Ishida et al., 2009). Mutations that introduced an unpaired cysteine, or a deletion which altered the reading frame and induced abnormal sequence, were shown in OI fibroblasts and transfected cells to impair assembly as evidenced by increased posttranslational modification of the exposed and unassembled chains and reduced secretion. Importantly, the chains bound to BiP (HSPA5; binding immunoglobulin protein) a molecular chaperone located in the lumen of the ER that recognizes and binds to the aberrantly exposed hydrophobic surfaces of misfolded proteins. Furthermore, the intracellularly retained mutant collagens were retro‐translocated to the cytoplasm for degradation by the proteasomal machinery, a key cellular response to unassembled proteins. These are the hallmarks of activation of an ER stress response. A similar story emerged with NC1 (structurally equivalent to C‐propeptide in fibrillar collagens) mutations of COL10A1 in Schmid metaphyseal chondrodysplasia. A range of patient missense mutations prevented collagen X homotrimer assembly and secretion and these led to proteasomal degradation of the unassembled collagen X chains (Chan et al., 1994; Chan et al., 1995; Chan et al., 1996; Chan et al., 1999; Wilson et al., 2002; Wilson et al., 2005). The mutant proteins activated ER stress both in vitro and in vivo (Wilson et al., 2005; Rajpar et al., 2009). The role of ER stress and altered downstream signaling in ECM disease pathology is discussed in the next section.
Mutations that impact initial collagen assembly are relatively rare and the most common mutations in fibril‐forming collagens are glycine substitutions in the triple helical region. Collagen triple helix folding and stability is critically dependent on having a glycine as every third amino acid in the triplet repeat sequence. Replacing glycine with a bulky amino acid has the potential to disrupt helix folding and lead to increased posttranslational lysine hydroxylation and glycosylation, compromised triple helix structural integrity and retention of the mutant trimers in the ER. This has been demonstrated for many COL1A1 and COL1A2 glycine substitutions in OI and for several COL2A1 mutations in chondrodysplasias. The full molecular details of the consequences of these helix‐disrupting mutations is yet to be revealed and the importance of local amino acid sequence context in determining the phenotype needs to be better understood (Marini et al., 2007; Xiao et al., 2015). Despite our incomplete knowledge, the evidence is mounting that mutant collagens can form insoluble aggregates in the ER that are degraded by the autophagosome–endosome system, rather than the proteasome degradation system that is engaged by the C‐propeptide trimerization‐impaired mutants (Ishida et al., 2009). The possible consequences of collagen helix mutations on ER‐stress are discussed below. In addition to Gly substitutions, a number of Arg to Cys substitutions at the X or Y position of the collagen II Gly‐X‐Y repeating amino acid triplet cause a range of mild to severe (nonlethal) chondrodysplasias. While some of these destabilize the collagen II triple helix in in vitro analyses and may also cause ER stress (Hintze et al., 2008; Chung et al., 2009), not all have apparent effects on helix stability, and the pathological mechanism may involve altered extracellular interactions.
It is clear that the cellular quality control mechanisms that bring about ER‐retention and degradation of misfolded collagens is leaky and collagen hetero‐ or homotrimers containing one or several mutant proα‐chains are often secreted. The impact of these on collagen fibril formation and stability and altered interactions with other ECM components needs to be considered. Comprehensive analysis of the types and positions of OI mutations in COL1A1 and COL1A2 revealed some important genotype–phenotype relationships (Marini et al., 2007) including the finding that mutations in the type I collagen major ligand binding domains were lethal. These correlative data strongly suggested that compromised extracellular interactions with ligands such as integrins, MMPs, fibronectin, decorin, and heparin, along with impacts on self‐assembly and intermolecular collagen crosslinking are major contributors to the severe clinical phenotypes. In addition to the likely effect of extracellular mutant‐containing type I collagen molecules on the critical interactions that stabilize the ECM and cell–matrix interactions, mutant collagen deposited into extracellular fibrils can be selectively degraded (Bateman and Golub, 1994), further destabilizing matrix integrity.
Dominant and recessive COL6A1, COL6A2, and COL6A3 structural mutations can cause the full spectrum of collagen VI muscular dystrophies. Collagen VI has a more complex intracellular assembly pathway than the other collagen types, forming first into triple helical monomers, then overlapping antiparallel dimers, then tetramers prior to secretion. Secreted tetramers interact end‐to‐end to form beaded microfibrils in the ECM (Lamande and Bateman, 2018). The phenotype that results from mutations is heavily influenced by the way the mutations affect collagen VI assembly and so understanding genotype/phenotype correlations often requires detailed functional studies. Similar to other collagens, heterozygous glycine substitutions that interrupt the Gly‐X‐Y triplet repeat sequence in the triple helix are the most common disease causing mutations. In collagen VI, the glycine mutations are clustered in Gly‐X‐Y triplets 3–20 at the N‐terminal end of the triple helix (Lamande and Bateman, 2018). This region is critical for tetramer and microfibril formation and so the mutations have a severe dominant negative effect, with intracellular accumulation, reduced secretion, and impaired microfibril assembly (Lamande et al., 1999; Baker et al., 2005; Pace et al., 2008). In general, more C‐terminal glycine mutations prevent incorporation of the chains into triple helical monomers and so are recessive (Lampe et al., 2005; Brinas et al., 2010; Butterfield et al., 2013; Lamande and Bateman, 2018). Similarly, in‐frame deletions toward the N‐terminus of the triple helix (mainly caused by exon skipping mutations) have a dominant negative effect on collagen VI assembly.
Mutations outside the collagen VI triple helix can be dominant or recessive. If the chains cannot be incorporated into monomers they can undergo proteasomal degradation (Zamurs et al., 2015), but if incorporated into monomers, dimers, and tetramers, the mutant chains can prevent microfibril formation (Tooley et al., 2010) or form abundant disorganized microfibrils (Zhang et al., 2010).
Collagen I fibril abnormalities are seen in skin and tendon from collagen VI knock out mice and Ullrich patients (Kirschner et al., 2005; Pan et al., 2013; Sardone et al., 2016) indicating that collagen VI can broadly influence tissue architecture, but how abnormal tissue architecture alters downstream signaling to illicit the cellular consequences which include abnormal mitochondria, increased apoptosis, increased reactive oxygen species, and impaired autophagy (Irwin et al., 2003; Grumati et al., 2010; Menazza et al., 2010; Tagliavini et al., 2013) is not fully resolved.
Collagen IX (COL9A1, COL9A2, and COL9A3) mutations cause multiple epiphyseal dysplasia. These are predominantly exon‐skipping changes and missense mutations concentrated in the N‐terminal COL3 domain (Jackson et al., 2012) which are thought to disturb collagen IX heterotrimer assembly and secretion (Briggs and Chapman, 2002). Collagen IX associates with the collagen II/XI fibril surface and interacts with other ECM networks, thus changes in the amount or structure of the collagen IX have the potential to alter the structure and integrity of the cartilage collagen fibrillar suprastructures.
Cartilage oligomeric protein (COMP) mutations in two chondrodysplasias, pseudoachondroplasia and multiple epiphyseal dysplasia (Briggs and Chapman, 2002; Posey et al., 2008), provide other examples of how structural mutations dominantly affect protein assembly. These mutations, which are predominantly in the calcium binding domains, affect protein folding and assembly leading in many cases to retention of the misfolded protein within the ER (Posey et al., 2018). This can also result in co‐retention of the COMP interacting partners, collagen IX and matrilin‐3 (Holden et al., 2001; Merritt et al., 2007; Posey et al., 2018). Likewise, matrilin‐3 (MATN3) structural mutations also underpin multiple epiphyseal dysplasia and spondyloepimetaphyseal dysplasia (Borochowitz et al., 2004; Jackson et al., 2012). Autosomal‐dominant multiple epiphyseal dysplasia missense mutations map to the MATN3 von Willebrand Factor type A domain and autosomal recessive missense mutation in spondyloepimetaphyseal dysplasia are located in the first EGF‐like protein domain of matrilin‐3. Both these mutation groups impact protein domains important in matrilin‐3 folding and result in reduced secretion and ER stress (Cotterill et al., 2005; Leighton et al., 2007; Suleman et al., 2012; Wang et al., 2015).
IS ER STRESS A DRIVER OF PATHOLOGY WITH ECM PROTEIN STRUCTURAL MUTATIONS?
The ER imposes stringent quality control on protein folding, such that only folded functional proteins leave the ER (van Anken and Braakman, 2005). Unfolded and incorrectly folded proteins that accumulate in the ER bind the molecular chaperone BiP which recognizes exposed hydrophobic protein sequences normally buried within the correctly folded protein structures. This removes BiP from the ER luminal domains of the three canonical ER stress sensors IRE1, ATF6, and PERK, activating them and the cellular response to misfolded proteins, the unfolded protein response (UPR), and thus initiating a cascade of adaptive responses in a coordinated attempt to reduce the load of misfolded proteins (Hetz et al., 2011). IRE1 activation produces a site‐specific RNase that cleaves the mRNA for an inactive transcription factor XBP1, leading to synthesis of an active form, XBP1s. XBP1s then binds to UPR responsive promoter elements to stimulate the synthesis of chaperones in an attempt to cope with the unfolded protein load. ATF6, a member of a bZIP transcription factor family, when activated by BiP removal, transits to the Golgi where it is cleaved to generate an active cytosolic fragment, ATF6p50, which binds to genes promoters containing an ER‐stress responsive element. PERK activation results in downstream phosphorylation of the translational initiation factor eIF2α which prevents formation of the translational initiation complex and downregulates protein translation, thus reducing the protein folding load. While phosphorylated eIF2α inhibits translation of most mRNAs, it paradoxically promotes translation of a subset of stress response genes, such as the transcription factor ATF4. AFT4 upregulates transcription of genes involved with amino acid metabolism and transport, oxidation–reduction reactions, and ER stress‐induced apoptosis such as CHOP (Rutkowski and Kaufman, 2007). In addition, UPR activation stimulates the protein degradation processes, ER‐associated degradation (ERAD) and/or autophagy (Meusser et al., 2005; Yorimitsu and Klionsky, 2007; Ding and Yin, 2008). Initial UPR activation is cytoprotective, offering the cells an opportunity to return to protein folding homeostasis. With increased duration of unfolded protein load, the balance in activities of the three pathways change and a prolonged UPR can have deleterious effects such as apoptosis.
The importance of the UPR in maintaining cellular proteostasis during normal development has been highlighted by situations where components of the UPR machinery are compromised. A striking example is the targeted deletion of BBF2H7 (a member of the ATF6 family of ER stress sensors) in cartilage which results in a severe mouse chondrodysplasia phenotype (Saito et al., 2009). These data showed that UPR activation is necessary during early cartilage development to cope with the massive increase in ECM protein production that occurs during chondrocyte differentiation in the limb bud. UPR activation upregulates Sec23a a key component of the COPII secretory vesicles thus stimulating the machinery necessary for the increased collagen II biosynthesis. Furthermore, disruption to this pathway due to mutations in Site‐1 protease (MBTPS1) which cleaves and activates BBF2H7 also results in a human skeletal dysplasia (Kondo et al., 2018). Likewise mutations in CREB3L1, the gene for OASIS, another ATF6 family member, results in a severe recessive OI by disturbing proteostasis during increased collagen I synthesis by osteoblasts (Symoens et al., 2013).
The importance of the UPR in responding to the challenges of the increased ECM protein folding demands during development raises the question of how UPR activation resulting from intracellular retention of mutant misfolded ECM proteins could relate to disease pathophysiology. It is now clear that UPR activation can contribute to the disease pathology but the precise nature of the UPR pathways activated, and the impact of these intracellular effects relative to the extracellular effects of the mutant abnormal protein is not entirely clear for many ECM protein structural mutations (Fig. 1). The UPR story for COL10A1 mutations in metaphyseal chondrodysplasia, type Schmid is the best understood and is discussed below.
COL10A1 mutations in the C‐terminal NC1 protein domain cause collagen X misfolding and ER retention and induce the canonical cellular UPR pathways, activating all three UPR sensors (Wilson et al., 2005; Tsang et al., 2007; Rajpar et al., 2009; Cameron et al., 2011; Cameron et al., 2015). ER‐chaperones are upregulated and protein degradation by ERAD stimulated. The downstream signaling cascades activated by the UPR block cartilage differentiation and result in growth plate cartilage expansion reduced bone growth (Tsang et al., 2007). Detailed studies in mouse models suggested that the PERK UPR arm was key to the pathology via C/EBP‐β‐mediated chondrocyte differentiation (Cameron et al., 2015). These data raised the important question of whether UPR induction was sufficient to produce the cartilage pathology. This was tested by inducing the UPR in growth plate cartilage, not by misfolded collagen X, but by expressing a misfolding protein not normally made in cartilage, thyroglobulin (Rajpar et al., 2009). The UPR induced by the mutant thyroglobulin resulted in pathology comparable to the authentic collagen X‐induced phenotype, directly demonstrating that in metaphyseal chondrodysplasia, type Schmid, the clinical phenotype was a direct result of the pathological UPR signaling (Rajpar et al., 2009). This suggested that the UPR could be an important therapeutic target in this, and perhaps other ECM disorders.
Although there is increasing evidence that UPR is a common consequence of ECM protein misfolding mutations, the precise pathways of UPR activation and signaling, and the contribution to the pathology are not as clearly understood as they are for COL10A1 mutations. In pseudoachondroplasia and multiple epiphyseal dysplasia, COMP structural mutations result in a characteristic distension of the ER and retention of COMP with mutations in the calcium binding repeats (Hecht et al., 2004; Merritt et al., 2007; Schmitz et al., 2008; Suleman et al., 2012; Posey et al., 2018). With one of these calcium binding domain mutations (D469del) despite ER retention a canonical UPR was apparently not activated although oxidative stress was apparent with consequent cell cycle regulation and apoptosis (Posey et al., 2009; Suleman et al., 2012). Conversely, while a mutation in the C‐terminal globular domain did not result in mutant COMP ER retention, BiP and Chop were upregulated resulting in reduced chondrocyte proliferation and increased and spatially dysregulated apoptosis (Pirog et al., 2014). Apoptosis, the downstream consequence of unresolved ER stress has also been demonstrated in other in vivo and in vitro studies on COMP mutations (Posey et al., 2018). Mutations in matrilin‐3 can also cause the pseudoachondroplasia/multiple epiphyseal dysplasia phenotype, and matrilin misfolding and UPR activation are implicated by reduced chondrocyte proliferation and dysregulated apoptosis (Leighton et al., 2007; Nundlall et al., 2010; Hartley et al., 2013; Jayasuriya et al., 2014; Wang et al., 2015). These data strongly support the hypothesis that a component of the pathophysiology of COMP and matrilin‐3 misfolding mutations is UPR activation although mutation specificity and precise UPR pathways, downstream consequences and the relative contribution of extracellular consequences remain to be fully defined (Fig. 1).
UPR activation by collagen I mutations in OI and collagen II in chondrodysplasias has been reported with strong evidence of misfolding, triple helix destabilization, and intracellular retention of the mutant collagens. However, as with COMP and MATN3, the precise nature of the UPR signaling is not fully resolved and is likely to involve aspects of both mutation and cell specificity. For example, with collagen I mutations in OI clear evidence of UPR activation was demonstrated for a procollagen I C‐propeptide trimerization mutation in the Aga2/+ mouse with upregulation of BiP, Hsp47, and Gadd153 (Chop) resulting in osteoblast apoptosis (Lisse et al., 2008). Recent studies on patient collagen I helix mutations reported upregulation of several components of the UPR, including BiP, PDI, ATF4, phospho‐PERK and XBP1 splicing (Besio et al., 2018). However, the pattern of upregulation of these individual UPR components was different in the five patient mutations studied. For example, BiP was upregulated in only three of five COL1A1 or COL1A2 mutations tested (Besio et al., 2018), highlighting that while a UPR can be activated by these mutations, there is apparent mutation specificity in the strength and nature of the response. In a mouse model of mild to moderate OI due to a Col1a2 missense mutation (α2(I) G610C) which disturbs the collagen triple helix, ER accumulation of the mutant misfolded collagen I results in upregulation of CHOP, eiF2α phosphorylation, and chaperones αβ crystalline and HSP47, and it was concluded this ER‐stress did not involve the canonical UPR pathway (Mirigian et al., 2016). This noncanonical UPR retarded osteoblast differentiation and caused abnormal responses in major signaling pathways.
Mutations in the C‐propeptide of collagen II in mouse models phenocopying platyspondylic skeletal dysplasia, Torrance type, show intracellular retention and upregulation of UPR‐related genes (Furuichi et al., 2011; Kimura et al., 2015) and apoptosis (Kimura et al., 2015). In vitro and in vivo studies have confirmed reduced thermostability, reduced secretion, and UPR hallmarks in cells with mutations toward the C‐terminus of the triple helix (Hintze et al., 2008; Chung et al., 2009; Jensen et al., 2011; Chakkalakal et al., 2018). Interestingly, helix mutations such as R75C, R134C, and R704C located more N‐terminal in the triple helix did not reduce mutant thermal stability compared to wild‐type collagen II. However, the impact of the position in the helix of the more severe glycine substitution mutations has not been studied systematically.
THERAPEUTIC APPROACHES—TARGETING THE UPR?
The cornerstone clinical treatment for ECM disorders caused by structural mutations has relied on approaches to ameliorate the symptoms rather than attempting to intervene in the pathological pathways themselves. For example, in OI clinical management has primarily involved surgical management and bisphosphonate treatment to reduce bone loss (Biggin and Munns, 2017; Bacon and Crowley, 2018). While useful clinically, bisphosphonates do not resolve the important issue of reduced bone quality caused by the collagen structural mutations. Other approaches to improve bone mass under evaluation include anabolic therapies such parathyroid hormone and sclerostin inhibition (Bacon and Crowley, 2018). Since increased TGF‐β signaling has been implicated in OI and inhibiting TGF‐β improved bone mass and strength in mouse models, TGF‐β neutralizing antibody treatment is being evaluated (Bacon and Crowley, 2018). Cell therapy approaches using mesenchymal stem cells and bone‐marrow transplantation are under evaluation for the severe forms of OI (Morello, 2018).
Since structural mutations in collagen I in OI (and other ECM proteins in other ECMopathies) can exert such strong dominant gain‐of‐function effects, a goal of gene targeting approaches is complete, or near‐complete, mutant gene expression knock‐down. Antisense oligonucleotides, ribozymes, and small interfering RNAs have shown experimental efficacy for both COL1A1 (Lindahl et al., 2013; Rousseau et al., 2014) and COMP (Posey et al., 2010) mutations in vitro, and in the case of COMP mutations antisense delivery reduces the severity of the growth plate cartilage pathology in a pseudoachondroplasia mouse model (Posey et al., 2017). However, there are still significant obstacles to clinical delivery, along with uncertainties about possible off‐target effects. The exciting recent developments in CRISPR/Cas9 genome editing (Hess et al., 2017) may find their way into gene correction and cell therapy in OI and other ECM diseases, but the issues of delivery and off‐target effects remain, at least in the short term, with this fast‐moving technology.
Although, as discussed above, there is still much to learn about the precise molecular pathways of the UPR and its contribution to the pathology of ECM disorders, protein misfolding and the UPR does provide a tantalizing therapeutic target. This has been recently comprehensively reviewed in the context of genetic skeletal disease (Briggs et al., 2015). The well‐known therapeutic strategies deployed in many other disorders resulting from aberrant protein folding (Cao and Kaufman, 2013; Rivas et al., 2015) employ approaches that target protein misfolding at three levels (Fig. 2); (1) reducing the mutant protein load by stimulating degradation of the misfolded protein; (2) increasing correct folding of the mutant protein folding; and (3) modifying the pathological consequences of UPR signaling.
Figure 2.
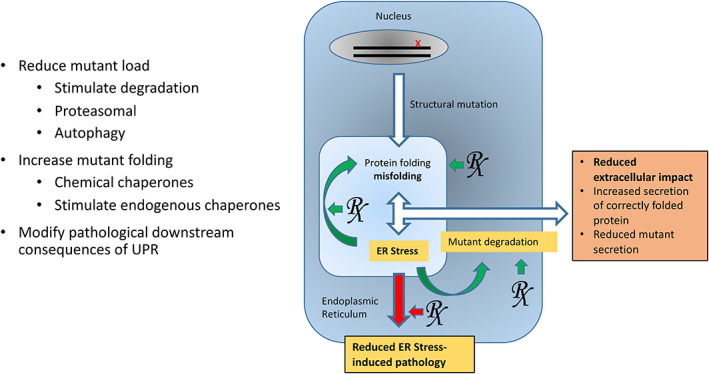
Therapeutically targeting the unfolded protein response. Theoretically the unfolded protein response (UPR) could be modified using three main approaches. First, drugs that can stimulate proteasomal degradation and/or autophagy could reduce the mutant protein load, reduce ER stress, and reduce secretion of mutant protein. A second approach could be to support mutant protein folding by upregulating endogenous chaperones, or treating with chemical chaperones, leading to increased secretion of correctly folded protein. Modifying downstream signaling consequences of the UPR is a third approach that could alleviate the ER stress‐induced pathology.
A promising example of ameliorating the UPR‐induced pathology of an ECM misfolding protein disorder by reducing the mutant protein load was recently reported (Mullan et al., 2017). Treating transfected cells expressing collagen X misfolding mutations with carbamazepine, an autophagy‐stimulating anti‐seizure drug, reduced intracellular accumulation of the mutant collagen X and downregulated UPR markers, BiP, Chop, and Xbp1s. Furthermore, in mice with a knockin metaphyseal chondrodysplasia type Schmid collagen X mutation (Rajpar et al., 2009), carbamazepine reduced ER stress markers in the affected cartilage and reduced the pathological impact of the mutation‐induced UPR in the growth plate cartilage. Interestingly, in in vitro studies on several collagen misfolding mutants, carbamazepine‐stimulated degradation by both autophagy and ERAD via the proteasome. The degradation pathways may be determined by whether the misfolded collagens are retained as predominantly single polypeptide chains which are available for proteasomal degradation, or aggregated forms of collagen which are targeted by autophagy (Ishida et al., 2009). While no other ECM misfolding disorders have been tested in preclinical models yet, this approach to reducing the mutant load by pharmacologically stimulating the proteasomal or autophagy pathways shows clinical promise.
Improving mutant protein folding by upregulating endogenous chaperones or by administering chemical chaperones that can stabilize proteins in their native conformation and rescue mutant protein folding and/or trafficking defects has been a widely used strategy in protein folding disorders (Perlmutter, 2002; Ulloa‐Aguirre et al., 2004; Arakawa et al., 2006; Rivas et al., 2015). The FDA‐approved chemical chaperone sodium 4‐phenylbutyrate (PBA) can reduce ER stress in vitro caused by COL1A1 OI mutations (Besio et al., 2018), COL4A2 (Murray et al., 2014), and COL4A5 (Wang et al., 2017) mutations. In a zebrafish model of OI, PBA administration ameliorated the skeletal pathology (Gioia et al., 2017). While these results offer considerable promise, PBA is also a histone deacetylase inhibitor so that this should be considered in the context of possible off‐target effects. This was highlighted in studies where PBA was used (along with lithium and valproate) to treat a pseudoachondroplasia mouse model caused by a COMP misfolding mutation (Posey et al., 2014). While these drugs reduced the chondrocyte pathology, there were other significant negative effects on mouse growth and development. Other chemical chaperones, such as trimethylamine N‐oxide (TMAO) and the bile acid tauroursodeoxycholic acid have also shown some effectiveness in improving mutant folding and reducing stress (Rivas et al., 2015). In the context of collagen misfolding the potential utility of this approach was demonstrated using TMAO to increase the stability of thermolabile collagen II and improve collagen II secretion and cell survival in vitro (Gawron et al., 2010). Overexpression of the endogenous chaperone BiP can reduce ER‐stress (Reddy et al., 2003), and a small chemical (BIX) that upregulates BiP and protects against ER stress in neurons has been identified (Kudo et al., 2008). However, with these approaches it, is important to consider that the increased folding and secretion of mutant ECM proteins may also lead to dominant negative extracellular effects.
Other therapeutic approaches target UPR sensors (IRE1, PERK, and ATF6) to modify downstream pathological signaling pathways (Maly and Papa, 2014; Jiang et al., 2015; Rivas et al., 2015). The FDA‐approved drugs guanabenz and salubrinal selectively inhibit eIF2α dephosphorylation, which lies downstream of PERK, and can protect cells from ER stress (Boyce et al., 2005). Recent studies have shown that targeting the PERK pathway in a metaphyseal chondrodysplasia mouse model caused by a pathological UPR resulting from collagen X misfolding (Wang et al., 2018) can be beneficial. The small molecule integrated stress response inhibitor which does not prevent PERK activation or eIF2α phosphorylation, but blocks downstream ATF4 expression prevented the PERK pathway‐mediated effects on chondrocyte differentiation and ameliorated the skeletal deformities. Small molecules that modulate the IRE1–XBP1 UPR pathways are exciting possibilities and several are in cancer clinical trials (Rivas et al., 2015). Manipulation of the downstream consequences of the UPR, such as apoptosis, also offers therapeutic potential in the treatment of heritable ECM disorders.
While there are still many questions to be answered about how ECM misfolding mutations interact with cellular quality control mechanisms and the UPR system, the many recent studies investigating the therapeutic potential of re‐purposed clinically approved drugs for treating ECM disorders offer hope that effective therapies are a realistic possibility. Indeed, based on preclinical data in mice (Mullan et al., 2017), carbamazepine is being evaluated as a therapy for metaphyseal chondrodysplasia, type Schmid in a worldwide clinical trial (https://mcds-therapy.eu).
LITERATURE CITED
- Arakawa T, Ejima D, Kita Y, Tsumoto K. 2006. Small molecule pharmacological chaperones: from thermodynamic stabilization to pharmaceutical drugs. Biochim Biophys Acta 1764:1677–1687. [DOI] [PubMed] [Google Scholar]
- Bacon S, Crowley R. 2018. Developments in rare bone diseases and mineral disorders. Ther Adv Chronic Dis 9:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NL, Morgelin M, Pace RA, Peat RA, Adams NE, Gardner RJ, Rowland LP, Miller G, De Jonghe P, Ceulemans B, et al. 2007. Molecular consequences of dominant Bethlem myopathy collagen VI mutations. Ann Neurol 62:390–405. [DOI] [PubMed] [Google Scholar]
- Baker NL, Morgelin M, Peat R, Goemans N, North KN, Bateman JF, Lamande SR. 2005. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet 14:279–293. [DOI] [PubMed] [Google Scholar]
- Barat‐Houari M, Sarrabay G, Gatinois V, Fabre A, Dumont B, Genevieve D, Touitou I. 2016. Mutation Update for COL2A1 Gene Variants Associated with Type II Collagenopathies. Hum Mutat 37:7–15. [DOI] [PubMed] [Google Scholar]
- Bateman JF. 2001. The molecular genetics of inherited cartilage disease. Osteoarthritis Cartilage 9:S141–S149. [PubMed] [Google Scholar]
- Bateman JF, Boot‐Handford RP, Lamande SR. 2009. Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat Rev Genet 10:173–183. [DOI] [PubMed] [Google Scholar]
- Bateman JF, Golub SB. 1994. Deposition and selective degradation of structurally‐abnormal type I collagen in a collagen matrix produced by osteogenesis imperfecta fibroblasts in vitro. Matrix Biol 14:251–262. [DOI] [PubMed] [Google Scholar]
- Bateman JF, Lamande SR, Dahl HH, Chan D, Mascara T, Cole WG. 1989. A frameshift mutation results in a truncated nonfunctional carboxyl‐terminal pro alpha 1(I) propeptide of type I collagen in osteogenesis imperfecta. J Biol Chem 264:10960–10964. [PubMed] [Google Scholar]
- Bateman JF, Wilson R, Freddi S, Lamande SR, Savarirayan R. 2005. Mutations of COL10A1 in Schmid metaphyseal chondrodysplasia. Hum Mutat 25:525–534. [DOI] [PubMed] [Google Scholar]
- Besio R, Iula G, Garibaldi N, Cipolla L, Sabbioneda S, Biggiogera M, Marini JC, Rossi A, Forlino A. 2018. 4‐PBA ameliorates cellular homeostasis in fibroblasts from osteogenesis imperfecta patients by enhancing autophagy and stimulating protein secretion. Biochim Biophys Acta Mol Basis Dis 1864:1642–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggin A, Munns CF. 2017. Long‐Term Bisphosphonate Therapy in Osteogenesis Imperfecta. Curr Osteoporos Rep 15:412–418. [DOI] [PubMed] [Google Scholar]
- Bishop N. 2016. Bone Material Properties in Osteogenesis Imperfecta. J Bone Miner Res 31:699–708. [DOI] [PubMed] [Google Scholar]
- Bonafe L, Cormier‐Daire V, Hall C, Lachman R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R, Sillence D, et al. 2015. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet 167A:2869–2892. [DOI] [PubMed] [Google Scholar]
- Bonnans C, Chou J, Werb Z. 2014. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 15:786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borochowitz ZU, Scheffer D, Adir V, Dagoneau N, Munnich A, Cormier‐Daire V. 2004. Spondylo‐epi‐metaphyseal dysplasia (SEMD) matrilin 3 type: homozygote matrilin 3 mutation in a novel form of SEMD. J Med Genet 41:366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, et al. 2005. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307:935–939. [DOI] [PubMed] [Google Scholar]
- Briggs MD, Bell PA, Wright MJ, Pirog KA. 2015. New therapeutic targets in rare genetic skeletal diseases. Expert Opin Orphan Drugs 3:1137–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs MD, Chapman KL. 2002. Pseudoachondroplasia and multiple epiphyseal dysplasia: mutation review, molecular interactions, and genotype to phenotype correlations. Hum Mutat 19:465–478. [DOI] [PubMed] [Google Scholar]
- Brinas L, Richard P, Quijano‐Roy S, Gartioux C, Ledeuil C, Lacene E, Makri S, Ferreiro A, Maugenre S, Topaloglu H, et al. 2010. Early onset collagen VI myopathies: genetic and clinical correlations. Ann Neurol 68:511–520. [DOI] [PubMed] [Google Scholar]
- Butterfield RJ, Foley AR, Dastgir J, Asman S, Dunn DM, Zou Y, Hu Y, Donkervoort S, Flanigan KM, Swoboda KJ, et al. 2013. Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies. Hum Mutat 34:1558–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron TL, Bell KM, Gresshoff IL, Sampurno L, Mullan L, Ermann J, Glimcher LH, Boot‐Handford RP, Bateman JF. 2015. XBP1‐independent UPR pathways suppress C/EBP‐beta mediated chondrocyte differentiation in ER‐stress related skeletal disease. PLoS Genet 11:e1005505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron TL, Bell KM, Tatarczuch L, Mackie EJ, Rajpar MH, McDermott BT, Boot‐Handford RP, Bateman JF. 2011. Transcriptional profiling of chondrodysplasia growth plate cartilage reveals adaptive ER‐stress networks that allow survival but disrupt hypertrophy. PLoS One 6:e24600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao SS, Kaufman RJ. 2013. Targeting endoplasmic reticulum stress in metabolic disease. Expert Opin Ther Targets 17:437–448. [DOI] [PubMed] [Google Scholar]
- Chakkalakal SA, Heilig J, Baumann U, Paulsson M, Zaucke F. 2018. Impact of Arginine to Cysteine Mutations in Collagen II on Protein Secretion and Cell Survival. Int J Mol Sci 19:541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D, Cole WG, Rogers J, Bateman JF. 1994. A mutation in the conserved NC1 domain of type X collagen prevents in vitro multimer assembly resulting in a Schmid‐type metaphyseal chondrodysplasia. Matrix Biol 14:396–397. [DOI] [PubMed] [Google Scholar]
- Chan D, Cole WG, Rogers JG, Bateman JF. 1995. Type X collagen multimer assembly in vitro is prevented by a Gly618 to Val mutation in the alpha 1(X) NC1 domain resulting in Schmid metaphyseal chondrodysplasia. J Biol Chem 270:4558–4562. [DOI] [PubMed] [Google Scholar]
- Chan D, Freddi S, Weng YM, Bateman JF. 1999. Interaction of collagen alpha1(X) containing engineered NC1 mutations with normal alpha1(X) in vitro. Implications for the molecular basis of schmid metaphyseal chondrodysplasia. J Biol Chem 274:13091–13097. [DOI] [PubMed] [Google Scholar]
- Chan D, Weng YM, Golub S, Bateman JF. 1996. Type X collagen NC1 mutations produced by site‐directed mutagenesis prevent in vitro assembly. Ann N Y Acad Sci 785:231–233. [DOI] [PubMed] [Google Scholar]
- Chapman KL, Briggs MD, Mortier GR. 2003. Review: clinical variability and genetic heterogeneity in multiple epiphyseal dysplasia. Pediatr Pathol Mol Med 22:53–75. [DOI] [PubMed] [Google Scholar]
- Chessler SD, Byers PH. 1993. BiP binds type I procollagen pro alpha chains with mutations in the carboxyl‐terminal propeptide synthesized by cells from patients with osteogenesis imperfecta. J Biol Chem 268:18226–18233. [PubMed] [Google Scholar]
- Chung HJ, Jensen DA, Gawron K, Steplewski A, Fertala A. 2009. R992C (p.R1192C) Substitution in collagen II alters the structure of mutant molecules and induces the unfolded protein response. J Mol Biol 390:306–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotterill SL, Jackson GC, Leighton MP, Wagener R, Makitie O, Cole WG, Briggs MD. 2005. Multiple epiphyseal dysplasia mutations in MATN3 cause misfolding of the A‐domain and prevent secretion of mutant matrilin‐3. Hum Mutat 26:557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paepe A, Malfait F. 2012. The Ehlers‐Danlos syndrome, a disorder with many faces. Clin Genet 82:1–11. [DOI] [PubMed] [Google Scholar]
- Deng H, Huang X, Yuan L. 2016. Molecular genetics of the COL2A1‐related disorders. Mutat Res Rev Mutat Res 768:1–13. [DOI] [PubMed] [Google Scholar]
- Ding WX, Yin XM. 2008. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 4:141–150. [DOI] [PubMed] [Google Scholar]
- Fang Y, Bateman JF, Mercer JF, Lamande SR. 2013. Nonsense‐mediated mRNA decay of collagen ‐emerging complexity in RNA surveillance mechanisms. J Cell Sci 126:2551–2560. [DOI] [PubMed] [Google Scholar]
- Fitzgerald J, Lamande SR, Bateman JF. 1999. Proteasomal degradation of unassembled mutant type I collagen pro‐alpha1(I) chains. J Biol Chem 274:27392–27398. [DOI] [PubMed] [Google Scholar]
- Foley AR, Hu Y, Zou Y, Yang M, Medne L, Leach M, Conlin LK, Spinner N, Shaikh TH, Falk M, et al. 2011. Large genomic deletions: a novel cause of Ullrich congenital muscular dystrophy. Ann Neurol 69:206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischmeyer PA, Dietz HC. 1999. Nonsense‐mediated mRNA decay in health and disease. Hum Mol Genet 8:1893–1900. [DOI] [PubMed] [Google Scholar]
- Furuichi T, Masuya H, Murakami T, Nishida K, Nishimura G, Suzuki T, Imaizumi K, Kudo T, Ohkawa K, Wakana S, et al. 2011. ENU‐induced missense mutation in the C‐propeptide coding region of Col2a1 creates a mouse model of platyspondylic lethal skeletal dysplasia, Torrance type. Mamm Genome 22:318–328. [DOI] [PubMed] [Google Scholar]
- Gawron K, Jensen DA, Steplewski A, Fertala A. 2010. Reducing the effects of intracellular accumulation of thermolabile collagen II mutants by increasing their thermostability in cell culture conditions. Biochem Biophys Res Commun 396:213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BG, Briggs MD. 2016. The aggrecanopathies; an evolving phenotypic spectrum of human genetic skeletal diseases. Orphanet J Rare Dis 11:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioia R, Tonelli F, Ceppi I, Biggiogera M, Leikin S, Fisher S, Tenedini E, Yorgan TA, Schinke T, Tian K, et al. 2017. The chaperone activity of 4PBA ameliorates the skeletal phenotype of Chihuahua, a zebrafish model for dominant osteogenesis imperfecta. Hum Mol Genet 26:2897–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giusti B, Lucarini L, Pietroni V, Lucioli S, Bandinelli B, Sabatelli P, Squarzoni S, Petrini S, Gartioux C, Talim B, et al. 2005. Dominant and recessive COL6A1 mutations in Ullrich scleroatonic muscular dystrophy. Ann Neurol 58:400–410. [DOI] [PubMed] [Google Scholar]
- Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L, et al. 2010. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 16:1313–1320. [DOI] [PubMed] [Google Scholar]
- Hartley CL, Edwards S, Mullan L, Bell PA, Fresquet M, Boot‐Handford RP, Briggs MD. 2013. Armet/Manf and Creld2 are components of a specialized ER stress response provoked by inappropriate formation of disulphide bonds: implications for genetic skeletal diseases. Hum Mol Genet 22:5262–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht JT, Makitie O, Hayes E, Haynes R, Susic M, Montufar‐Solis D, Duke PJ, Cole WG. 2004. Chondrocyte cell death and intracellular distribution of COMP and type IX collagen in the pseudoachondroplasia growth plate. J Orthop Res 22:759–767. [DOI] [PubMed] [Google Scholar]
- Hess GT, Tycko J, Yao D, Bassik MC. 2017. Methods and Applications of CRISPR‐Mediated Base Editing in Eukaryotic Genomes. Mol Cell 68:26–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Martinon F, Rodriguez D, Glimcher LH. 2011. The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol Rev 91:1219–1243. [DOI] [PubMed] [Google Scholar]
- Hintze V, Steplewski A, Ito H, Jensen DA, Rodeck U, Fertala A. 2008. Cells expressing partially unfolded R789C/p.R989C type II procollagen mutant associated with spondyloepiphyseal dysplasia undergo apoptosis. Hum Mutat 29:841–851. [DOI] [PubMed] [Google Scholar]
- Holden P, Meadows RS, Chapman KL, Grant ME, Kadler KE, Briggs MD. 2001. Cartilage oligomeric matrix protein interacts with type IX collagen, and disruptions to these interactions identify a pathogenetic mechanism in a bone dysplasia family. J Biol Chem 276:6046–6055. [DOI] [PubMed] [Google Scholar]
- Humphries JD, Chastney MR, Askari JA, Humphries MJ. 2018. Signal transduction via integrin adhesion complexes. Curr Opin Cell Biol 56:14–21. [DOI] [PubMed] [Google Scholar]
- Hynes RO. 2009. The extracellular matrix: not just pretty fibrils. Science 326:1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GM, et al. 2003. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet 35:367–371. [DOI] [PubMed] [Google Scholar]
- Ishida Y, Yamamoto A, Kitamura A, Lamande SR, Yoshimori T, Bateman JF, Kubota H, Nagata K. 2009. Autophagic elimination of misfolded procollagen aggregates in the endoplasmic reticulum as a means of cell protection. Mol Biol Cell 20:2744–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa Y, Bachinger HP. 2013. A molecular ensemble in the rER for procollagen maturation. Biochim Biophys Acta 1833:2479–2491. [DOI] [PubMed] [Google Scholar]
- Jackson GC, Mittaz‐Crettol L, Taylor JA, Mortier GR, Spranger J, Zabel B, Le Merrer M, Cormier‐Daire V, Hall CM, Offiah A, et al. 2012. Pseudoachondroplasia and multiple epiphyseal dysplasia: a 7‐year comprehensive analysis of the known disease genes identify novel and recurrent mutations and provides an accurate assessment of their relative contribution. Hum Mutat 33:144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasuriya CT, Zhou FH, Pei M, Wang Z, Lemme NJ, Haines P, Chen Q. 2014. Matrilin‐3 chondrodysplasia mutations cause attenuated chondrogenesis, premature hypertrophy and aberrant response to TGF‐beta in chondroprogenitor cells. Int J Mol Sci 15:14555–14573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen DA, Steplewski A, Gawron K, Fertala A. 2011. Persistence of intracellular and extracellular changes after incompletely suppressing expression of the R789C (p.R989C) and R992C (p.R1192C) collagen II mutants. Hum Mutat 32:794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Niwa M, Koong AC. 2015. Targeting the IRE1alpha‐XBP1 branch of the unfolded protein response in human diseases. Semin Cancer Biol 33:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Ichimura S, Sasaki K, Masuya H, Suzuki T, Wakana S, Ikegawa S, Furuichi T. 2015. Endoplasmic reticulum stress‐mediated apoptosis contributes to a skeletal dysplasia resembling platyspondylic lethal skeletal dysplasia, Torrance type, in a novel Col2a1 mutant mouse line. Biochem Biophys Res Commun 468:86–91. [DOI] [PubMed] [Google Scholar]
- Kirschner J, Hausser I, Zou Y, Schreiber G, Christen HJ, Brown SC, Anton‐Lamprecht I, Muntoni F, Hanefeld F, Bonnemann CG. 2005. Ullrich congenital muscular dystrophy: connective tissue abnormalities in the skin support overlap with Ehlers‐Danlos syndromes. Am J Med Genet 132A:296–301. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Fu J, Wang H, Hoover C, McDaniel JM, Steet R, Patra D, Song J, Pollard L, Cathey S, et al. 2018. Site‐1 protease deficiency causes human skeletal dysplasia due to defective inter‐organelle protein trafficking. JCI Insight 3:e121596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krakow D. 2015. Skeletal dysplasias. Clin Perinatol 42:301–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo T, Kanemoto S, Hara H, Morimoto N, Morihara T, Kimura R, Tabira T, Imaizumi K, Takeda M. 2008. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ 15:364–375. [DOI] [PubMed] [Google Scholar]
- Kurosaki T, Maquat LE. 2016. Nonsense‐mediated mRNA decay in humans at a glance. J Cell Sci 129:461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamande SR, Bateman JF. 2018. Collagen VI disorders: insights on form and function in the extracellular matrix and beyond. Matrix Biol 71–72:348–367. [DOI] [PubMed] [Google Scholar]
- Lamande SR, Bateman JF, Hutchison W, McKinlay Gardner RJ, Bower SP, Byrne E, Dahl HH. 1998. Reduced collagen VI causes Bethlem myopathy: a heterozygous COL6A1 nonsense mutation results in mRNA decay and functional haploinsufficiency. Hum Mol Genet 7:981–989. [DOI] [PubMed] [Google Scholar]
- Lamande SR, Chessler SD, Golub SB, Byers PH, Chan D, Cole WG, Sillence DO, Bateman JF. 1995. Endoplasmic reticulum‐mediated quality control of type I collagen production by cells from osteogenesis imperfecta patients with mutations in the pro alpha 1 (I) chain carboxyl‐terminal propeptide which impair subunit assembly. J Biol Chem 270:8642–8649. [DOI] [PubMed] [Google Scholar]
- Lamande SR, Shields KA, Kornberg AJ, Shield LK, Bateman JF. 1999. Bethlem myopathy and engineered collagen VI triple helical deletions prevent intracellular multimer assembly and protein secretion. J BiolChem 274:21817–21822. [DOI] [PubMed] [Google Scholar]
- Lampe AK, Dunn DM, von Niederhausern AC, Hamil C, Aoyagi A, Laval SH, Marie SK, Chu ML, Swoboda K, Muntoni F, et al. 2005. Automated genomic sequence analysis of the three collagen VI genes: applications to Ullrich congenital muscular dystrophy and Bethlem myopathy. J Med Genet 42:108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leighton MP, Nundlall S, Starborg T, Meadows RS, Suleman F, Knowles L, Wagener R, Thornton DJ, Kadler KE, Boot‐Handford RP, et al. 2007. Decreased chondrocyte proliferation and dysregulated apoptosis in the cartilage growth plate are key features of a murine model of epiphyseal dysplasia caused by a matn3 mutation. Hum Mol Genet 16:1728–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Grafe I, Alexander S, Lee B. 2017. Genetic causes and mechanisms of Osteogenesis Imperfecta. Bone 102:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl K, Kindmark A, Laxman N, Astrom E, Rubin CJ, Ljunggren O. 2013. Allele dependent silencing of collagen type I using small interfering RNAs targeting 3'UTR Indels ‐ a novel therapeutic approach in osteogenesis imperfecta. Int J Med Sci 10:1333–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisse TS, Thiele F, Fuchs H, Hans W, Przemeck GK, Abe K, Rathkolb B, Quintanilla‐Martinez L, Hoelzlwimmer G, Helfrich M, et al. 2008. ER Stress‐Mediated Apoptosis in a New Mouse Model of Osteogenesis imperfecta. PLoS Genet 4:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majava M, Hoornaert KP, Bartholdi D, Bouma MC, Bouman K, Carrera M, Devriendt K, Hurst J, Kitsos G, Niedrist D, et al. 2007. A report on 10 new patients with heterozygous mutations in the COL11A1 gene and a review of genotype‐phenotype correlations in type XI collagenopathies. Am J Med Genet 143A:258–264. [DOI] [PubMed] [Google Scholar]
- Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, et al. 2017. The 2017 international classification of the Ehlers‐Danlos syndromes. Am J Med Genet C Semin Med Genet 175:8–26. [DOI] [PubMed] [Google Scholar]
- Maly DJ, Papa FR. 2014. Druggable sensors of the unfolded protein response. Nat Chem Biol 10:892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini JC, Forlino A, Bachinger HP, Bishop NJ, Byers PH, Paepe A, Fassier F, Fratzl‐Zelman N, Kozloff KM, Krakow D, et al. 2017. Osteogenesis imperfecta. Nat Rev Dis Primers 3:17052. [DOI] [PubMed] [Google Scholar]
- Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, Hyland JC, Korkko J, Prockop DJ, De Paepe A, et al. 2007. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat 28:209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menazza S, Blaauw B, Tiepolo T, Toniolo L, Braghetta P, Spolaore B, Reggiani C, Di Lisa F, Bonaldo P, Canton M. 2010. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum Mol Genet 19:4207–4215. [DOI] [PubMed] [Google Scholar]
- Merritt TM, Bick R, Poindexter BJ, Alcorn JL, Hecht JT. 2007. Unique matrix structure in the rough endoplasmic reticulum cisternae of pseudoachondroplasia chondrocytes. Am J Pathol 170:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meusser B, Hirsch C, Jarosch E, Sommer T. 2005. ERAD: the long road to destruction. Nat Cell Biol 7:766–772. [DOI] [PubMed] [Google Scholar]
- Mirigian LS, Makareeva E, Mertz EL, Omari S, Roberts‐Pilgrim AM, Oestreich AK, Phillips CL, Leikin S. 2016. Osteoblast malfunction caused by cell stress response to procollagen misfolding in alpha2(I)‐G610C mouse model of osteogenesis imperfecta. J Bone Miner Res 31:1608–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello R. 2018. Osteogenesis imperfecta and therapeutics. Matrix Biol 71–72:294–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullan LA, Mularczyk EJ, Kung LH, Forouhan M, Wragg JM, Goodacre R, Bateman JF, Swanton E, Briggs MD, Boot‐Handford RP. 2017. Increased intracellular proteolysis reduces disease severity in an ER stress‐associated dwarfism. J Clin Invest 127:3861–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray LS, Lu Y, Taggart A, Van Regemorter N, Vilain C, Abramowicz M, Kadler KE, Van Agtmael T. 2014. Chemical chaperone treatment reduces intracellular accumulation of mutant collagen IV and ameliorates the cellular phenotype of a COL4A2 mutation that causes haemorrhagic stroke. Hum Mol Genet 23:283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO. 2016. The extracellular matrix: Tools and insights for the "omics" era. Matrix Biol 49:10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. 2012. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics 11:014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nundlall S, Rajpar MH, Bell PA, Clowes C, Zeeff LA, Gardner B, Thornton DJ, Boot‐Handford RP, Briggs MD. 2010. An unfolded protein response is the initial cellular response to the expression of mutant matrilin‐3 in a mouse model of multiple epiphyseal dysplasia. Cell Stress Chaperones 15:835–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace RA, Peat RA, Baker NL, Zamurs L, Morgelin M, Irving M, Adams NE, Bateman JF, Mowat D, Smith NJ, et al. 2008. Collagen VI glycine mutations: perturbed assembly and a spectrum of clinical severity. Ann Neurol 64:294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan TC, Zhang RZ, Markova D, Arita M, Zhang Y, Bogdanovich S, Khurana TS, Bonnemann CG, Birk DE, Chu ML. 2013. COL6A3 protein deficiency in mice leads to muscle and tendon defects similar to human collagen VI congenital muscular dystrophy. J Biol Chem 288:14320–14331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peat RA, Baker NL, Jones KJ, North KN, Lamande SR. 2007. Variable penetrance of COL6A1 null mutations: implications for prenatal diagnosis and genetic counselling in Ullrich congenital muscular dystrophy families. Neuromuscul Disord 17:547–557. [DOI] [PubMed] [Google Scholar]
- Perlmutter DH. 2002. Chemical chaperones: a pharmacological strategy for disorders of protein folding and trafficking. Pediatr Res 52:832–836. [DOI] [PubMed] [Google Scholar]
- Pirog KA, Irman A, Young S, Halai P, Bell PA, Boot‐Handford RP, Briggs MD. 2014. Abnormal chondrocyte apoptosis in the cartilage growth plate is influenced by genetic background and deletion of CHOP in a targeted mouse model of pseudoachondroplasia. PLoS One 9:e85145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey KL, Coustry F, Hecht JT. 2018. Cartilage oligomeric matrix protein: COMPopathies and beyond. Matrix Biol 71–72:161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey KL, Coustry F, Veerisetty AC, Hossain M, Gattis D, Booten S, Alcorn JL, Seth PP, Hecht JT. 2017. Antisense Reduction of Mutant COMP Reduces Growth Plate Chondrocyte Pathology. Mol Ther 25:705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey KL, Coustry F, Veerisetty AC, Liu P, Alcorn JL, Hecht JT. 2014. Chondrocyte‐specific pathology during skeletal growth and therapeutics in a murine model of pseudoachondroplasia. J Bone Miner Res 29:1258–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey KL, Liu P, Wang HR, Veerisetty AC, Alcorn JL, Hecht JT. 2010. RNAi reduces expression and intracellular retention of mutant cartilage oligomeric matrix protein. PLoS One 5:e10302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey KL, Veerisetty AC, Liu P, Wang HR, Poindexter BJ, Bick R, Alcorn JL, Hecht JT. 2009. An inducible cartilage oligomeric matrix protein mouse model recapitulates human pseudoachondroplasia phenotype. Am J Pathol 175:1555–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey KL, Yang Y, Veerisetty AC, Sharan SK, Hecht JT. 2008. Model systems for studying skeletal dysplasias caused by TSP‐5/COMP mutations. Cell Mol Life Sci 65:687–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpar MH, McDermott B, Kung L, Eardley R, Knowles L, Heeran M, Thornton DJ, Wilson R, Bateman JF, Poulsom R, et al. 2009. Targeted induction of endoplasmic reticulum stress induces cartilage pathology. PLoS Genet 5:e1000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. 2003. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase‐7 activation. J Biol Chem 278:20915–20924. [DOI] [PubMed] [Google Scholar]
- Rivas A, Vidal RL, Hetz C. 2015. Targeting the unfolded protein response for disease intervention. Expert Opin Ther Targets 19:1203–1218. [DOI] [PubMed] [Google Scholar]
- Rousseau J, Gioia R, Layrolle P, Lieubeau B, Heymann D, Rossi A, Marini JC, Trichet V, Forlino A. 2014. Allele‐specific Col1a1 silencing reduces mutant collagen in fibroblasts from Brtl mouse, a model for classical osteogenesis imperfecta. Eur J Hum Genet 22:667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozario T, DeSimone DW. 2010. The extracellular matrix in development and morphogenesis: a dynamic view. Dev Biol 341:126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Kaufman RJ. 2007. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci 32:469–476. [DOI] [PubMed] [Google Scholar]
- Saito A, Hino S, Murakami T, Kanemoto S, Kondo S, Saitoh M, Nishimura R, Yoneda T, Furuichi T, Ikegawa S, et al. 2009. Regulation of endoplasmic reticulum stress response by a BBF2H7‐mediated Sec23a pathway is essential for chondrogenesis. Nat Cell Biol 11:1197–1204. [DOI] [PubMed] [Google Scholar]
- Sardone F, Traina F, Bondi A, Merlini L, Santi S, Maraldi NM, Faldini C, Sabatelli P. 2016. Tendon extracellular matrix alterations in Ullrich congenital muscular dystrophy. Front Aging Neurosci 8:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz M, Niehoff A, Miosge N, Smyth N, Paulsson M, Zaucke F. 2008. Transgenic mice expressing D469Delta mutated cartilage oligomeric matrix protein (COMP) show growth plate abnormalities and sternal malformations. Matrix Biol 27:67–85. [DOI] [PubMed] [Google Scholar]
- Suleman F, Gualeni B, Gregson HJ, Leighton MP, Pirog KA, Edwards S, Holden P, Boot‐Handford RP, Briggs MD. 2012. A novel form of chondrocyte stress is triggered by a COMP mutation causing pseudoachondroplasia. Hum Mutat 33:218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symoens S, Malfait F, D'Hondt S, Callewaert B, Dheedene A, Steyaert W, Bachinger HP, De Paepe A, Kayserili H, Coucke PJ. 2013. Deficiency for the ER‐stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J Rare Dis 8:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliavini F, Sardone F, Squarzoni S, Maraldi NM, Merlini L, Faldini C, Sabatelli P. 2013. Ultrastructural changes in muscle cells of patients with collagen VI‐related myopathies. Muscles Ligaments Tendons J 3:281–286. [PMC free article] [PubMed] [Google Scholar]
- Tooley LD, Zamurs LK, Beecher N, Baker NL, Peat RA, Adams NE, Bateman JF, North KN, Baldock C, Lamande SR. 2010. Collagen VI microfibril formation is abolished by an {alpha}2(VI) von Willebrand factor type A domain mutation in a patient with Ullrich congenital muscular dystrophy. J Biol Chem 285:33567–33576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang KY, Chan D, Bateman JF, Cheah KS. 2010. In vivo cellular adaptation to ER stress: survival strategies with double‐edged consequences. J Cell Sci 123:2145–2154. [DOI] [PubMed] [Google Scholar]
- Tsang KY, Chan D, Cheslett D, Chan WC, So CL, Melhado IG, Chan TW, Kwan KM, Hunziker EB, Yamada Y, et al. 2007. Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLoS Biol 5:e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa‐Aguirre A, Janovick JA, Brothers SP, Conn PM. 2004. Pharmacologic rescue of conformationally‐defective proteins: implications for the treatment of human disease. Traffic 5:821–837. [DOI] [PubMed] [Google Scholar]
- van Anken E, Braakman I. 2005. Versatility of the endoplasmic reticulum protein folding factory. Crit Rev Biochem Mol Biol 40:191–228. [DOI] [PubMed] [Google Scholar]
- Wang C, Tan Z, Niu B, Tsang KY, Tai A, Chan WCW, Lo RLK, Leung KKH, Dung NWF, Itoh N, et al. 2018. Inhibiting the integrated stress response pathway prevents aberrant chondrocyte differentiation thereby alleviating chondrodysplasia. Elife 7:e37673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Mohammad M, Wang Y, Tan R, Murray LS, Ricardo S, Dagher H, van Agtmael T, Savige J. 2017. The chemical chaperone, PBA, Reduces ER Stress and autophagy and Increases Collagen IV alpha5 Expression in Cultured Fibroblasts From Men With X‐Linked Alport Syndrome and Missense Mutations. Kidney Int Rep 2:739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YC, Liu JS, Chen JY, Wu SQ, Wang GR, Nie J, Zhang SK, Guo QL, Luo JM. 2015. Multiple functions of the first EGF domain in matrilin‐3: secretion and endoplasmic reticulum stress. Int J Mol Med 36:1648–1656. [DOI] [PubMed] [Google Scholar]
- Wilson R, Freddi S, Bateman JF. 2002. Collagen X chains harboring Schmid metaphyseal chondrodysplasia NC1 domain mutations are selectively retained and degraded in stably transfected cells. J Biol Chem 277:12516–12524. [DOI] [PubMed] [Google Scholar]
- Wilson R, Freddi S, Chan D, Cheah KS, Bateman JF. 2005. Misfolding of collagen X chains harboring Schmid metaphyseal chondrodysplasia mutations results in aberrant disulfide bond formation, intracellular retention, and activation of the unfolded protein response. J Biol Chem 280:15544–15552. [DOI] [PubMed] [Google Scholar]
- Xiao J, Yang Z, Sun X, Addabbo R, Baum J. 2015. Local amino acid sequence patterns dominate the heterogeneous phenotype for the collagen connective tissue disease Osteogenesis Imperfecta resulting from Gly mutations. J Struct Biol 192:127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu T, Klionsky DJ. 2007. Endoplasmic reticulum stress: a new pathway to induce autophagy. Autophagy 3:160–162. [DOI] [PubMed] [Google Scholar]
- Zamurs LK, Idoate MA, Hanssen E, Gomez‐Ibanez A, Pastor P, Lamande SR. 2015. Aberrant mitochondria in a Bethlem myopathy patient with a homozygous amino acid substitution that destabilizes the collagen VI alpha2(VI) chain. J Biol Chem 290:4272–4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RZ, Zou Y, Pan TC, Markova D, Fertala A, Hu Y, Squarzoni S, Reed UC, Marie SK, Bonnemann CG, et al. 2010. Recessive COL6A2 C‐globular missense mutations in Ullrich congenital muscular dystrophy: role of the C2a splice variant. J Biol Chem 285:10005–10015. [DOI] [PMC free article] [PubMed] [Google Scholar]