

Abstract
Classic galactosemia (CG) is a potentially lethal inborn error of metabolism, if untreated, that results from profound deficiency of galactose‐1‐phosphate uridylyltransferase (GALT), the middle enzyme of the Leloir pathway of galactose metabolism. While newborn screening and rapid dietary restriction of galactose prevent or resolve the potentially lethal acute symptoms of CG, by mid‐childhood, most treated patients experience significant complications. The mechanisms underlying these long‐term deficits remain unclear. Here we introduce a new GALT‐null rat model of CG and demonstrate that these rats display cataracts, cognitive, motor, and growth phenotypes reminiscent of patients outcomes. We further apply the GALT‐null rats to test how well blood biomarkers, typically followed in patients, reflect metabolic perturbations in other, more relevant tissues. Our results document that the relative levels of galactose metabolites seen in GALT deficiency differ widely by tissue and age, and that red blood cell Gal‐1P, the marker most commonly followed in patients, shows no significant association with Gal‐1P in other tissues. The work reported here establishes our outbred GALT‐null rats as an effective model for at least four complications characteristic of CG, and sets the stage for future studies addressing mechanism and testing the efficacy of novel candidate interventions.
Keywords: cognitive, galactosemia, GALT, metabolite, model, rat
1. INTRODUCTION
Classic galactosemia (CG) is a rare autosomal recessive disorder that results from profound deficiency of galactose‐1‐phosphate uridylyltransferase (GALT), the middle enzyme in the Leloir pathway of galactose metabolism (Figure S1).1 Infants with CG may develop bilateral cataracts and experience a rapid and devastating progression of acute symptoms following exposure to breast milk or cow's milk‐based formula,1 both of which contain large quantities of galactose.2 Diagnosed early, infants with CG may be switched to a low galactose formula which prevents or resolves the acute symptoms of disease.3 However, despite early and rigorous lifelong dietary restriction of galactose, a majority of children with CG grow to experience a constellation of long‐term complications including speech, cognitive and behavioural problems, motor difficulties, and mild growth delay, among other challenges.1, 4 Premature ovarian insufficiency is also common among girls and women with CG.5 At present, the mechanisms underlying long‐term outcomes in CG remain unclear, and there is no known intervention that prevents or resolves the long‐term sequelae of disease.3
While a variety of accessible biomarkers have been reported in CG, including differences in glycans, hormones, markers of inflammation,6, 7 and plasma metabolomic profiles,8 perhaps the most easily and commonly followed is the accumulation of galactose metabolites.1 Specifically, galactose, galactose‐1‐phosphate (Gal‐1P), and galactitol (Figure S1) can all be detected at extremely high levels in the blood and tissues of affected infants following exposure to milk, and much lower, but still abnormal, levels following dietary restriction of galactose. However, the link between elevated galactose metabolites and long‐term outcome severity remains unclear,8 and even patients who never consumed milk, never showed marked elevation of galactose metabolites in blood, and never experienced acute symptoms, show the same increased risk for developmental problems as they grow.9
The absence of a clear relationship between elevated galactose metabolites in blood and long‐term outcome severity in CG raises the important question of which, if any, of the galactose metabolites detected in blood accurately reflect their counterparts in other, more relevant, tissues such as brain. This is a fundamental question, with implications for treatment and the definition of biomarkers that accurately reflect the long‐term disease process in CG.
Prior studies have addressed the relationship between galatose metabolites and outcome using a variety of approaches, from cells in culture to GALT‐null microbial, invertebrate, fish, and mouse model systems (reviewed in Coelho et al10). In all of these systems, the expected galactose metabolites were affected under conditions of high galactose exposure, but none of these studies compared galactose metabolites across tissues or developmental times. Furthermore, the only whole‐organism mammalian model—a GALT‐null mouse—was created twice by separate investigators11, 12 and in one instance showed no relevant phenotypes11 and in the other was reported to show some phenotypes, but these were pronounced only following extraordinary dietary galactose intoxication.12, 13
To explore the relationship between metabolites and outcomes, we created and characterised an outbred GALT‐null rat model of CG. As demonstrated here, our GALT‐null rats exhibit a range of phenotypes reminiscent of CG in humans, including cataracts, mild growth delay, and both cognitive impairment and motor deficits in adults. Using this model, we addressed the relationship between metabolite levels in blood and other tissues, and identified striking differences in GALT‐independent galactose metabolism among tissues and across post‐natal time points. These results offer first clues to the tissue‐specific and longitudinal consequences of GALT deficiency in mammals and set the stage for future studies of mechanism and intervention in CG.
2. RESULTS
2.1. Creating and confirming a GALT‐null rat
We produced a GALT‐null Sprague‐Dawley rat as described in Supplemental materials and methods using CRISPR‐Cas9 gene editing with non‐homologous end joining14 to introduce a 2‐base pair insertion mutation into exon 6 of the Galt locus. A single male founder with the mutation was identified, confirmed by Sanger sequencing, and outbred to establish the SD‐Galt M3 strain used here. The Galt M3 mutation (Figure S2) disrupts the his‐pro‐his active site of the GALT enzyme15 and reveals a premature stop codon downstream.
To confirm that the Galt M3 allele is indeed null we performed GALT enzyme assays on samples of liver harvested from newborn wild‐type (+/+), Galt M3 homozygous (−/−), and Galt M3/WT heterozygous (+/−) pups. Galactokinase (GALK) and UDP galactose 4′‐epimerase (GALE) were also tested in all samples. As presented in Table 1, GALT activity was approximately half the normal level in Galt M3 /WT animals (59.07 ± 6.67 vs 128.62 ± 14.21 pmol/μg protein/min) and undetectable in Galt M3 /Galt M3 animals (0.07 ± 0.07 pmol/μg protein/min). These differences were significant as judged by analysis of variance (ANOVA; P = .0287). As expected, both GALK and GALE activities were indistinguishable among all three Galt genotypes (ANOVA; P = .3501 for GALK and P = .6649 for GALE).
Table 1.
Leloir pathway enzyme activities in neonatal rat liver
| Galt genotype (neonatal liver) | GALK enzyme activity (pmol/μg protein/min) | GALT enzyme activity (pmol/μg protein/min) | GALE enzyme activity (pmol/μg protein/min) |
|---|---|---|---|
| WT/WT | 22.33 ± 7.94 | 128.62 ± 14.21 | 50.97 ± 13.83 |
| (n = 6) | (n = 6) | (n = 6) | |
| Galt M3 /WT | 13.13 ± 3.01 | 59.07 ± 6.67 | 53.56 ± 11.55 |
| (n = 6) | (n = 6) | (n = 6) | |
| Galt M3 /Galt M3 | 19.71 ± 2.45 | 0.07 ± 0.07 | 74.58 ± 19.46 |
| (n = 5) | (n = 5) | (n = 5) |
Abbreviations: GALK, galactokinase; GALT, galactose‐1‐phosphate uridylyltransferase.
2.2. Outcomes of GALT‐null rats
The neonatal and ovarian outcomes of our GALT‐null rats are described in Supplemental Results and Discussion (Tables S1 and S2, and Figure S3).
2.2.1. Growth delay
As a group, and as seen in patients,16 GALT‐null pups showed a mild growth delay relative to both heterozygous and wild‐type pups (Figure S4). Specifically, at 1 day after birth (panel A), both male and female GALT‐null pups weighed significantly less than their GALT+ counterparts, and this was true even when the analysis was limited to include only pups from crosses with one heterozygous parent (data not shown). The size disparity continued through weaning (day 21, panel B). By puberty (6 weeks old, panel C), however, GALT‐null male rats were only slightly smaller than their GALT+ counterparts, and GALT‐null females had closed the gap.
2.2.2. Cataracts
GALT‐null pups consuming their mother's milk also displayed striking bilateral cataracts (Figure S5, panel A) that were apparent as soon as the pups opened their eyes (days 13‐14 after birth), and remained visible, albeit slightly diminished in intensity, into adulthood (Figure S5, panel B). No WT/WT or Galt M3 /WT (GALT+) animals showed cataracts at any time in the experiment (Figure S5, panel B).
2.2.3. Motor and cognitive functions
To assess motor and cognitive functions of our GALT‐null rats, we subjected adult males, all 4‐5 months old, to both a rotarod activity17 and a Morris water maze with hidden platform.18 As reported for motor19 and cognitive outcomes in patients,16 we saw a range of performance scores for individual GALT‐null rats on both tests, and there was considerable overlap with the control range. As a group, however, the GALT‐null rats demonstrated significant deficits (P < .05) in both activities (Figure 1, panels A and B).
Figure 1.
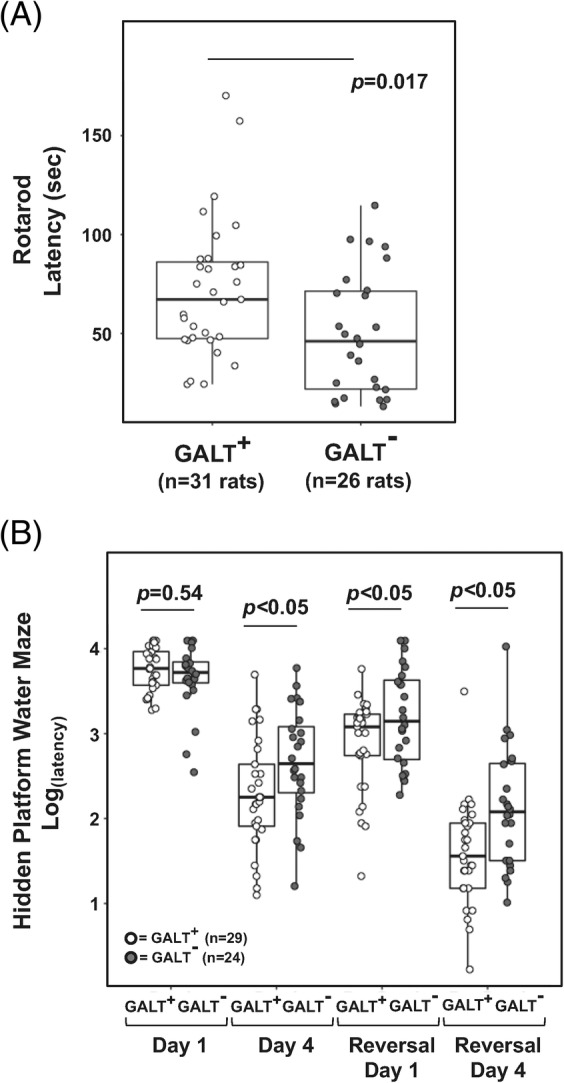
Motor and cognitive deficits in galactose‐1‐phosphate uridylyltransferase (GALT)‐null rats. GALT+ (n = 31) and GALT‐null (n = 26) male rats, all 4‐5 months old, were tested in parallel for evidence of motor and cognitive deficits. A, As a group, the GALT‐null rats exhibited significantly decreased latency to falling off a spinning rotarod device (P = .017), demonstrating a deficit in motor function, balance, and/or coordination. B, As a group, the GALT‐null rats (n = 24) also exhibited significantly increased latency to finding a hidden platform relative to GALT+ rats (n = 29) on day 4 of training, reversal day 1, and reversal day 4 (P < .05), demonstrating a deficit in cognitive function. Of note, there was no deficit (P = .54) observed on day 1, when all rats were new to the activity, and also no difference in swim speed (data not shown), confirming that the deficits observed after training were cognitive in nature
Of note, while GALT‐null rats showed a longer latency in finding the hidden platform in the Morris water maze after 4 days of training (Figure 1, panel B), we saw no difference on the first day of the activity (P = .54), demonstrating that their ability to swim and search was not impaired, only their ability to navigate, learn, and/or remember. As expected, the actual swim speeds of GALT‐null rats were also indistinguishable from those of their GALT+ counterparts (data not shown).
To test whether impaired vision, due to cataracts, might account for the apparent behavioural deficits in our GALT‐null rats, we subjected a subset of both GALT+ and GALT‐null rats to a second water maze activity in which the platform was visible above the water and moved to a new location before each trial. If cataracts present in the GALT‐null rats functionally impaired their ability to see in the water maze, we reasoned this would increase their latency to find the visible platform, but it did not (data not shown).
2.3. Metabolic consequences of GALT‐deficiency in rats
To address the metabolic consequences of GALT‐deficiency in rats, we quantified galactose, galactitol, and Gal‐1P in tissues from GALT+ and GALT‐null animals ranging in age from newborns to older adults. We did not include galactonate in this study because in all tissues sampled, other than testes, galactonate was either near background level or confounded by overlapping peaks whose combined area did not distinguish GALT‐null from GALT+ animals.
2.3.1. Neonates
To quantify metabolites in neonates, we euthanised Galt M3 homozygous, heterozygous, and wild‐type pups within 24‐hours of delivery and collected liver and brain tissues for analysis. Of note, while all pups had nursed prior to euthanasia, as demonstrated by milk spots, the quantity and timing of milk exposure for individual pups was not assessed.
As expected, in both liver and brain from GALT+ pups all three galactose metabolites were detected at very low levels (median values <36 pmol/mg) (Figure S6). In samples from GALT‐null pups, however, the levels were up to orders of magnitude above this baseline. A small number of “outlier” GALT‐null pups demonstrated levels of galactose and galactitol even higher than their counterparts; we presume these might be pups that had consumed more milk, or perhaps consumed milk earlier, or later, than their sibs. P‐values are presented in Table S3.
2.3.2. Nursing and weanling pups
Next, we measured galactose metabolites in pups euthanised at 10 days (Figure S7) and 20 and 23 days (Figure S8) after birth. All of these pups were consuming rat breast milk, which contains about 3% galactose by calorie content, although some of the 20‐23 day old pups (weanlings) may have also been eating some solid food (LabDiet 5001) which was available in their cage and contains about 1.5% calories from galactose.
Tissues studied included plasma, red blood cells (RBC), brain (cerebellum, hippocampus, and frontal cortex), liver, eyes, ovaries, and testes. As expected, we saw extremely low levels of galactose, galactitol, and Gal‐1P in all tissues tested from GALT+ animals, and much higher levels in tissues from GALT‐null animals. We also noted that some metabolite levels were up to nine times higher in GALT‐null samples from 10‐days old pups (Figure S7) compared with 20 to 23‐days old pups (Figure S8). This quantitative difference was seen across tissue types, although the exact ratios varied. Whether this difference reflected altered relative galactose exposure of pups over time, or other factor(s), remains unclear. P‐values are presented in Tables S4 and S5.
2.3.3. Adult rats
By ages 4‐6 months (Figure S9), and 12‐16 months (Figure S10), tissue metabolites measured in GALT‐null animals consuming exclusively LabDiet 5001 had diminished substantially from the levels seen in weanlings. For example, while median galactitol in frontal cortex in weanlings was about 940 pmol/mg, it was about 130 pmol/mg in adults. Similarly, while median liver Gal‐1P in weanlings was 2706 pmol/mg, in adults it was close to 1500 pmol/mg. P‐values are presented in Tables S6 and S7.
2.4. Relative distribution of galactose metabolites differs by tissue and over time in GALT‐null rats
Perhaps our most important observation noted from metabolite studies of GALT‐null rats was that the relative distribution of specific metabolites differed by tissue and over time. To visualise the differences, we graphed the proportion of combined galactose metabolites corresponding to each individual metabolite for each tissue and age cohort (Figure 2). The results demonstrated two clear patterns.
Figure 2.
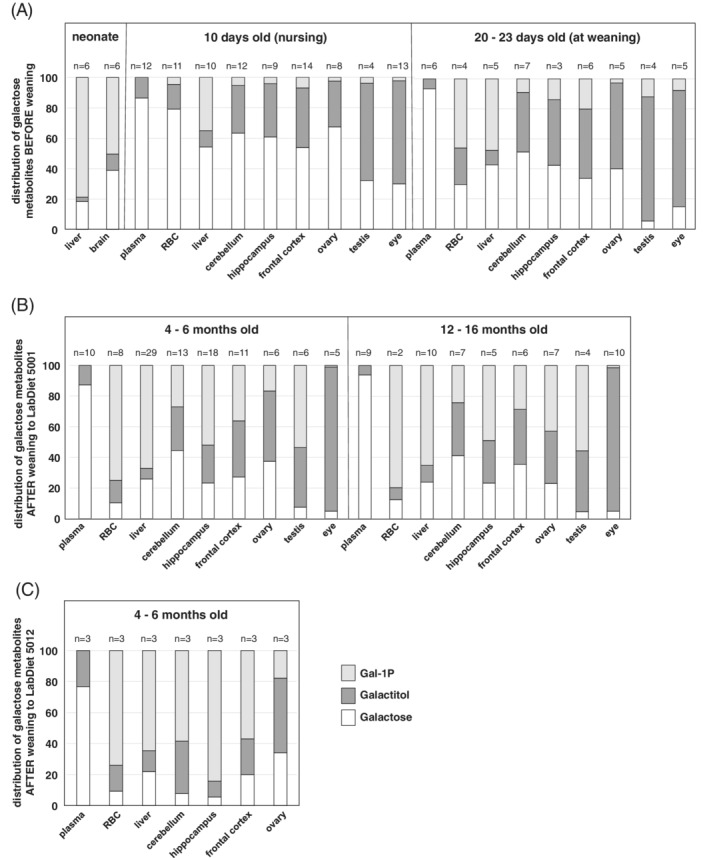
Relative distribution of galactose metabolites in the tissues of galactose‐1‐phosphate uridylyltransferase (GALT)‐null rats. To compare markers of galactose metabolism across tissues (all panels), stages of development (panels A and B), and diet (panel C) in GALT‐null rats, we plotted the relative proportions of total galactose metabolites comprised of galactose, galactitol, and Gal‐1P by tissue and age group for the different cohorts. As presented, the relative distribution of specific metabolites varied markedly among tissues, and as a function of age. LabDiet 5001 contains about 1.5% of calories from galactose; LabDiet 5012 contains about 0.15% calories from galactose. P‐values are presented in Tables S8 and S9
First, which was the predominant metabolite varied by tissue. For example, in weanlings, Gal‐1P predominated in RBC and liver, but free galactose and galactitol predominated in all other tissues tested. Second, which metabolite predominated varied widely by age. For example, the relative abundance of free galactose was greatest in most tissues from 10‐days old pups and diminished as the animals aged. In testis, galactitol predominated in pups but Gal‐1P predominated in adults. In ovary, galactitol predominated in weanlings and 4 to 6 month old adults but was surpassed by Gal‐1P in 12‐16 month old rats. P‐values are presented in Tables S8 and S9.
2.5. RBC Gal‐1P is not an accurate predictor of Gal‐1P in other tissues
As an extension of the observation that galactose metabolites differ among tissues and age groups in GALT‐null rats (Figure 2), we tested how well the Gal‐1P detected in RBCs, and the galactose and galactitol detected in plasma, reflected their counterparts in liver and brain. This is an important question because RBC Gal‐1P is commonly used as a biomarker for diagnosis and clinical follow‐up of patients.1
To address this question, we conducted multiple linear regression testing to assess how well each blood metabolite predicted the corresponding metabolite in liver and brain (frontal cortex, hippocampus, and cerebellum) after adjusting for age, as described in Materials and Methods. Our results (Table 2 and Table S10) confirmed that both plasma galactose and galactitol were significant predictors of the corresponding metabolite levels in brain and liver. However, RBC Gal‐1P was not. To be clear, RBC Gal‐1P showed no significance as a predictor of Gal‐1P level in liver or any of the three brain sections tested.
Table 2.
Relationship between blood and tissue metabolites in rats (all age groups included)
| Predictor variable | Response variable | Significance of predictor on response variablea |
|---|---|---|
| RBC Gal‐1P vs: | ||
| Frontal cortex Gal‐1P | P = .37 | |
| Hippocampus Gal‐1P | P = .37 | |
| Cerebellum Gal‐1P | P = .06 | |
| Liver Gal‐1P | P = .15 | |
| Plasma galactitol vs: | ||
| Frontal cortex galactitol | P = .06 | |
| Hippocampus galactitol | P < .01a | |
| Cerebellum galactitol | P < .01* | |
| Liver galactitol | P < .01* | |
| Plasma galactose vs: | ||
| Frontal cortex galactose | P = .01* | |
| Hippocampus galactose | P < .01* | |
| Cerebellum galactose | P < .01* | |
| Liver galactose | P < .01* | |
Note: Details of the analyses are presented in Table S10.
Calculated using multiple linear regression.
Significance at the α = .05 level.
2.6. Impact of rigorous dietary galactose restriction after weaning on metabolites in GALT‐null rats
Finally, we tested the role of dietary galactose restriction after weaning as a modifier of metabolite accumulation in GALT‐null rats. Specifically, we asked two questions. First, how quickly do elevated galactose metabolite levels drop after weaning, and second, does rigorous dietary galactose restriction fully normalise galactose metabolites in GALT‐null rats?
To address these questions, we weaned a cohort of wild‐type and GALT‐null pups to LabDiet 5012, which is nutritionally similar to our regular chow (LabDiet 5001) but contains 10‐fold less calories from galactose, as a percentage of total (0.15% vs 1.5%). Controls were weaned to LabDiet 5001. We performed monthly tail vein blood draws on animals weaned to each chow and euthanised all animals at 4‐6 months of age for tissue collection.
Galactose metabolites measured in plasma and RBC samples from longitudinal blood draws demonstrated that all three metabolites dropped precipitously, and as a function of diet, after weaning. For example, median plasma galactose in GALT‐null pups at weaning was >1600 pmol/μL, and about 2 weeks later it was about 500 pmol/μL if the pups were consuming LabDiet 5001, and only about 100 pmol/μL if the pups were consuming LabDiet 5012 (Figure S11). For plasma galactitol the drop was similar, from 130 pmol/μL at weaning to 31 pmol/μL about 2 weeks later if the pups were eating LabDiet 5001, and about 12 pmol/μL if the pups were eating LabDiet 5012. The drop in RBC Gal‐1P was also clear, though not quite as striking, perhaps reflecting a slower turnover of intra‐cellular metabolites. After the first post‐wean blood draw, further drops over time were also evident, but these were modest by comparison (Figure S11; P‐values in Table S11).
To test whether rigorous dietary restriction of galactose completely normalised galactose metabolites in GALT‐null rats we compared metabolite levels in the tissues of adult rats weaned either to LabDiet 5012 or 5001 (Figure S12). Two patterns were clear. First, galactose, galactitol, and Gal‐1P all remained higher in GALT‐null rats consuming the diet higher in galactose (LabDiet 5001). Second, even after months of consuming a rigorously galactose‐restricted diet (LabDiet 5012), galactose, galactitol, and Gal‐1P all remained elevated in the tissues of GALT‐null animals compared to their GALT+ counterparts, perhaps reflecting the impact of endogenously produced galactose. The differences seen in brain were smaller than those seen in plasma or liver, but most were nonetheless significant (P‐values listed in Table S12).
3. DISCUSSION
In this paper we introduce and apply a new GALT‐null rat model of CG, documenting phenotypes and defining the relationships among three key galactose metabolites in different tissues and across post‐natal development. Of note, while microbial, tissue culture, invertebrate, fish, and mouse models of GALT‐deficiency have all been reported previously (reviewed in Coelho et al10), the rat model described here is the first GALT‐null mammalian model reported to show learning/memory deficits. Given the prevalence and impact of cognitive disability on CG patient quality of life, this is an important point. That our GALT‐null rats are outbred, rather than inbred like most other models,11, 12 is also key because it minimises concern about potential background effects.
This is also the first study to characterise the relative distribution of galactose, galactitol, and Gal‐1P among tissues and over post‐natal development in a GALT‐null mammal, revealing striking differences and raising the question of whether RBC Gal‐1P is the best biomarker to follow in patients. Of note, while the results presented here have clear implications for clinical practice, they could not have been performed in humans since most tissues relevant to long‐term outcomes in patients are inaccessible.
3.1. Growth
We observed mild growth delay among GALT‐null pups starting the day after birth, which was the first time the pups were weighed. As expected for an autosomal recessive trait, we did not see growth delay among heterozygotes. As is the case for patients,20 the growth delay seen among GALT‐null pups largely resolved following puberty. Whether the growth delay observed reflects a diminished intake of calories, or some other cause, remains unclear. Of note, Tang and colleagues12 also reported growth restriction among their GALT‐null mice.
3.2. Cataracts
The most visually striking phenotype of our GALT‐null rat pups was bilateral cataracts that were plainly evident from the time the pups opened their eyes, and that remained visible through adulthood, even when the rats were weaned to LabDiet 5012 (data not shown) which contains almost no galactose. The formation of cataracts in GALT‐null rats, but not GALK‐null mice unless expressing a human aldose reductase transgene in lens,21 confirms that rats, like humans, naturally express higher levels of aldose reductase than do mice, at least in some tissues.22
3.3. Motor function
To test motor function we subjected adult male rats, ages 4‐5 months, to a traditional rotarod device, assessing performance by how quickly each rat fell off the rotating drum. To be clear, we tested only males because the majority of adult female rats in our colony were used as breeders and therefore not available for behavioural testing. We have no reason to expect a gender bias in the behavioural outcomes we measured, and in the future will test that assumption. That the GALT‐null rats fell off the drum more quickly than their GALT+ counterparts is consistent with reports of motor complications in CG patients.19 Chen and colleagues13 also reported a motor deficit in their GALT‐null mice as revealed using a modified rotarod device.
3.4. Learning and memory
Of particular interest, our GALT‐null rats exhibited a clear deficit in learning and memory as revealed using a Morris water maze with hidden platform. That we saw no increased latency to find the platform on the first day of testing, and also no difference in swim speed between GALT+ and GALT‐null rats confirms that the deficit revealed was cognitive, and not motor, in origin. Retesting a subset of rats using a Morris water maze with visible platform also confirmed that the problem experienced by GALT‐null rats was not caused by a visual deficit.
3.5. Accumulation and distribution of galactose metabolites in the tissues of GALT‐null rats
Perhaps the most clinically relevant result presented here addresses the accumulation and distribution of galactose, galactitol, and Gal‐1P in GALT‐null rats among different tissues and across different points in post‐natal development. In short, we found that different galactose metabolites accumulate to different levels, and in different proportions, in different tissues and at different times in development. Whether this observation reflects differences in the production and/or turnover of the galactose metabolites tested remains unclear.
A related point demonstrated by our metabolic data presented here is that GALT‐null rats, like humans,23, 24, 25 produce endogenous galactose, at least when dietary sources are lacking. Specifically, we found that both extra‐cellular and intra‐cellular galactose metabolites remained slightly elevated in GALT‐null rats relative to controls despite months of rigorous dietary galactose restriction, and this was true in all tissues tested (Figures S11 and S12).
3.6. Conclusions
With regard to mechanism, the results presented here provide a foundation for questioning the relative roles of different galactose metabolites as likely contributors to pathophysiology in CG. With regard to clinical care,3 at minimum, our results raise serious concern about using RBC Gal‐1P as a biomarker for disease, and suggest that plasma galactose or galactitol, and by extension perhaps urinary galactitol, may provide a more meaningful proxy for metabolic status of inaccessible tissues. Finally, the results presented here firmly establish our outbred GALT‐null rat as a powerful new model of CG and set the stage for future experiments applying this model to explore mechanism and test the efficacy of novel candidate interventions for CG.
4. MATERIALS AND METHODS
4.1. Rat handling, breeding, sample collection, and euthanasia
See Supplemental materials and methods.
4.1.1. GALK, GALT, and GALE enzyme assays
Pieces of frozen liver, approximately 10 mg each, were thawed, weighed, and processed essentially as described previously.26 For details see Supplemental materials and methods.
4.2. Quantifying galactose, galactitol, and Gal‐1P in rat blood and tissues
To quantify galactose metabolites we used samples of the following sizes: hippocampus, frontal cortex, and cerebellum, one hemisphere each; eye, one eye; ovary, one ovary; testis, approximately 100 mg; liver, approximately 100 mg; plasma, 100 μL; RBC, 100 to 115 μL. Tissues were stored frozen at −80°C from time of collection until processing.
To extract metabolites, tissue samples were ground, on ice, in 125 μL cold HPLC‐grade water for approximately 30 seconds using a Teflon micropestle and handheld micropestle motor (Kimble Chase Life Science and Research Products LLC). Samples were prepared for metabolite analysis by extraction in chloroform (CHCl3) and methanol following the procedure described previously.27 Further details are provided in Supplemental Materials And Methods.
4.3. Rat behavioural studies
All rat behavioural tests were conducted by the Emory Rodent Behavior Core. Adult male rats between 4 and 6‐months old were tested using both the rotarod17 and Morris Water Maze with hidden platform18 tasks. As a control for effective vision in the presence and absence of cataracts, a subset of animals was also tested using the Morris Water Maze with visible platform.28 This manuscript does not include any human subjects work. All animal work conducted at the Medical College of Wisconsin (MCW) was performed with approval of the MCW Institutional Animal Care and Use Committee (IACUC Protocol AUA2214; PI: Aron Geurts, PhD). All animal procedures, including maintenance and breeding, conducted at Emory University, were performed with approval of the Emory IACUC (Protocol PROTO201700095; PI: JL Fridovich‐Keil, PhD) and oversight of the Emory Division of Animal Resources.
4.3.1. Rotarod
Rats were placed in individual lanes separated by dividers on a drum that was rotating initially at a slow speed (4 rpm). This speed was gradually increased over 5 minutes to a maximum of 40 rpm, and the time until the rat fell off was measured (latency). Each rat was given two practice trials before the first scored run. The maximum fall distance was approximately 12 in.
4.3.2. Morris water maze
Morris water maze with hidden platform testing was conducted using a round (68 in. diameter), water‐filled tub in an environment rich with maze cues and a small platform located in a fixed position 1 cm below the water surface. Water temperature was 25°C at the start of each day of testing, and typically declined to 22°C by the end of the procedure. Non‐toxic white tempera paint was added to the water to make it opaque.
Each daily session was comprised of four consecutive trials in which rats were placed, individually, into the water maze with their paws initially touching the wall from each of four different starting positions (N, S, E, W). The submerged platform was located in the same quadrant on every trial, and once the rat found and mounted the platform, it was left there for 10 seconds before being lifted off, dried, and placed in a dry holding cage with extra paper towels, half on and half off a heating pad, for 10 minutes. Latency to mount the platform (in seconds), swim speed, and total distance travelled before finding the platform were recorded via a video tracking system (MazeScan by Clever Sys Inc.). Following four consecutive days of training with the hidden platform located in the same quadrant, the hidden platform was moved to a different quadrant (reversal day 1) and the training repeated for another four consecutive days.
4.4. Statistical analyses
4.4.1. Rat growth
Separate linear models for each age (1‐day, 21‐days, and 6‐weeks) and gender group (males vs females) were used to compare pup weights. Specifically, for a given model, pup weight measured in grams was the response variable, with Galt genotype and litter size as the predictor variables.
4.4.2. Rat behavioural measures
Mean rotarod latency, measured in seconds, was compared between GALT+ and GALT‐null rats using the non‐parametric Mann‐Whitney U test. The significance of Morris Water Maze latency was calculated using a mixed‐effects model with rat‐specific random intercepts to adjust for correlation among repeated observations. Specifically, log(latency) was modelled as a function of Galt genotype and time, allowing the effects of time to differ by genotype. Time was treated as a categorical variable with four possible values (day 1, day 4, reversal day 1, and reversal day 4).
4.4.3. Success of rat crosses by genotype
The success rate of crosses grouped by maternal genotype (Table S2: WT/WT, Galt M3/WT, and Galt M3/Galt M3) were compared using a Chi‐square test of independence. Crosses were considered successful if the cross yielded at least one pup, regardless of whether that pup survived.
4.4.4. Relationship between blood, brain, and liver metabolite levels in GALT‐null rats
Blood, brain, and liver metabolite levels were compared among rats derived from four distinct age groups that were combined for analysis (Table 2 and Table S10). Because age was determined to be a covariate of metabolite concentration, we adjusted for age in the model. Multiple linear regression analysis was used to evaluate the predictive value of each plasma or RBC galactose metabolite. Blood galactose metabolites were log transformed before further analysis to normalise residuals. Significance of predictor coefficients was determined by fitting separate linear models for each of the following comparisons: Gal‐1P in RBCs against Gal‐1P in the pre‐frontal cortex, hippocampus, cerebellum, and liver; and plasma galactitol and galactose against galactitol and galactose, respectively, in the pre‐frontal cortex, hippocampus, cerebellum, and liver.
4.4.5. Statistics to generate the P‐values presented in Tables S3 to S9, S11 and 1
All P‐values were calculated using the Mann‐Whitney U exact test, which uses a rank‐sum method appropriate for comparing small cohorts where normality of the data cannot be assured. Instead of comparing means, this method orders data from smallest to largest and assigns a rank (1 to n) to each data point. The ranks were summed for both comparison groups, and the sums were compared to the expected rank‐sum, which assumes equality of both groups. P‐values were considered significant if they were less than or equal to α = .05.
CONFLICT OF INTEREST
All of the authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
Shauna Rasmussen helped to design and interprets experiments, performed or oversaw the majority of animal work performed, and participated in writing and editing of the manuscript. Jenna Daenzer helped to design and interpret experiments, performed or oversaw the majority of biochemical and metabolic work performed, and participated in writing and editing of the manuscript. Jessica MacWilliams and Taylor Head helped to design and interpret experiments, performed all of the statistical comparisons included in the manuscript, and participated in writing and editing of the manuscript. Martine Williams helped to interpret experiments, performed some of the animal and metabolic work performed, and participated in editing of the manuscript. Aron Geurts oversaw creation of the GALT‐null rat, helped to design and interpret experiments, and participated in editing of the manuscript. Jason Schroeder and David Weinshenker helped to design and interpret experiments relating to rat behavioural studies and performed or oversaw these tests and participated in editing of the manuscript. Judith Fridovich‐Keil initiated the project, coordinated the activities of all collaborators, helped to design and interpret experiments, collected some of the animal data, and did most of the writing and editing of the manuscript.
Supporting information
Data S1 Supporting information.
ACKNOWLEDGMENTS
We gratefully acknowledge the contributions of Allison Frederick, Marshall Waller, Robert Roundy, Rebecca Anderson, Ronake Desai, Carlos Garcia Gonzalez, Allyson Mateja, Dr Daniel Davis, Dr Sandy Van Calcar, and the many professionals working in the Emory Division of Animal Resources and the Emory IACUC without whom this work could not have been conducted. This work was supported by institutional funds from Emory University School of Medicine (Department of Human Genetics) and by grants from the National Institutes of Health R01DK107900 and R21HD092785 (all to J.L.F.K.).
Rasmussen SA, Daenzer JMI, MacWilliams JA, et al. A galactose‐1‐phosphate uridylyltransferase‐null rat model of classic galactosemia mimics relevant patient outcomes and reveals tissue‐specific and longitudinal differences in galactose metabolism. J Inherit Metab Dis. 2020;43:518–528. 10.1002/jimd.12205
Shauna A. Rasmussen and Jennifer M. I. Daenzer contributed equally to this study.
Communicating Editor: Ina Knerr
Funding information National Institutes of Health, Grant/Award Numbers: R21HD092785, R01DK107900; Emory University School of Medicine (Department of Human Genetics)
REFERENCES
- 1. Berry G. Classic galactosemia and clinical variant galactosemia In: Pagon R, Adam M, Ardinger H, et al., eds. GeneReviews®. Seattle, WA: University of Washington; 2014. [PubMed] [Google Scholar]
- 2. Jenness R. Proceedings: biosynthesis and composition of milk. J Invest Dermatol. 1974;63:109‐118. [DOI] [PubMed] [Google Scholar]
- 3. Welling L, Bernstein LE, Berry GT, et al. International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow‐up. J Inherit Metab Dis. 2017;40:171‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rubio‐Gozalbo ME, Haskovic M, Bosch AM, et al. The natural history of classic galactosemia: lessons from the GalNet registry. Orphanet J Rare Dis. 2019;14:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frederick AB, Zinsli AM, Carlock G, Conneely K, Fridovich‐Keil JL. Presentation, progression, and predictors of ovarian insufficiency in classic galactosemia. J Inherit Metab Dis. 2018;41:785‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Colhoun HO, Rubio Gozalbo EM, Bosch AM, et al. Fertility in classical galactosaemia, a study of N‐glycan, hormonal and inflammatory gene interactions. Orphanet J Rare Dis. 2018a;13:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Colhoun HO, Treacy EP, MacMahon M, et al. Validation of an automated ultraperformance liquid chromatography IgG N‐glycan analytical method applicable to classical galactosaemia. Ann Clin Biochem. 2018b;55:593‐603. [DOI] [PubMed] [Google Scholar]
- 8. Fischer S, Frederick A, Tran V, Li S, Jones D, Fridovich‐Keil J. Metabolic perturbations in classic galactosemia beyond the Leloir pathway: insights from an untargeted metabolomic study. J Inher Metab Dis. 2018;42:254‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hughes J, Ryan S, Lambert D, et al. Outcomes of siblings with classical galactosemia. J Pediatr. 2009;154:721‐726. [DOI] [PubMed] [Google Scholar]
- 10. Coelho AI, Rubio‐Gozalbo ME, Vicente JB, Rivera I. Sweet and sour: an update on classic galactosemia. J Inherit Metab Dis. 2017;40:325‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leslie ND, Yager KL, McNamara PD, Segal S. A mouse model of galactose‐1‐phosphate uridyl transferase deficiency. Biochem Mol Med. 1996;59:7‐12. [DOI] [PubMed] [Google Scholar]
- 12. Tang M, Siddiqi A, Witt B, et al. Subfertility and growth restriction in a new galactose‐1 phosphate uridylyltransferase (GALT)‐deficient mouse model. Eur J Hum Genet. 2014;22:1172‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen W, Caston R, Balakrishnan B, et al. Assessment of ataxia phenotype in a new mouse model of galactose‐1 phosphate uridylyltransferase (GALT) deficiency. J Inherit Metab Dis. 2017;40:131‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wedekind JE, Frey PA, Rayment I. The structure of nucleotidylated histidine‐166 of galactose‐1‐phosphate uridylyltransferase provides insight into phosphoryl group transfer. Biochemistry. 1996;35:11560‐11569. [DOI] [PubMed] [Google Scholar]
- 16. Frederick A, Cutler D, Fridovich‐Keil J. Rigor of non‐dairy galactose restriction in early childhood, measured by retrospective survey, does not associate with severity of five long‐term outcomes quantified in 231 children and adults with classic galactosemia. J Inherit Metab Dis. 2017;40:813‐821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dunham NW, Miya TS. A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc am Pharm Assoc. 1957;46:208‐209. [DOI] [PubMed] [Google Scholar]
- 18. Rorabaugh JM, Chalermpalanupap T, Botz‐Zapp CA, et al. Chemogenetic locus coeruleus activation restores reversal learning in a rat model of Alzheimer's disease. Brain. 2017;140:3023‐3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kuiper A, Grunewald S, Murphy E, et al. Movement disorders and nonmotor neuropsychological symptoms in children and adults with classic galactosemia. J Inher Metab Dis. 2019;42:451‐458. [DOI] [PubMed] [Google Scholar]
- 20. Panis B, Gerver WJ, Rubio‐Gozalbo ME. Growth in treated classical galactosemia patients. Eur J Pediatr. 2007;166:443‐446. [DOI] [PubMed] [Google Scholar]
- 21. Ai Y, Zheng Z, O'Brien‐Jenkins A, et al. A mouse model of galactose‐induced cataracts. Hum Mol Genet. 2000;9:1821‐1827. [DOI] [PubMed] [Google Scholar]
- 22. Wen Y, Bekhor I. Levels of expression of hexokinase, aldose reductase and sorbitol dehydrogenase genes in lens of mouse and rat. Curr Eye Res. 1993;12:323‐332. [DOI] [PubMed] [Google Scholar]
- 23. Berry GT, Moate PJ, Reynolds RA, et al. The rate of de novo galactose synthesis in patients with galactose‐1‐phosphate uridyltransferase deficiency. Mol Genet Metab. 2004;81:22‐30. [DOI] [PubMed] [Google Scholar]
- 24. Berry GT, Nissim I, Lin ZP, Mazur AT, Gibson JB, Segal S. Endogenous synthesis of galactose in normal men and patients with hereditary galactosemia. Lancet. 1995;346:1073‐1074. [DOI] [PubMed] [Google Scholar]
- 25. Schadewaldt P, Kamalanathan L, Hammen HW, Kotzka J, Wendel U. Endogenous galactose formation in galactose‐1‐phosphate uridyltransferase deficiency. Arch Physiol Biochem. 2014;120:228‐239. [DOI] [PubMed] [Google Scholar]
- 26. Sanders RD, Sefton JM, Moberg KH, Fridovich‐Keil JL. UDP‐galactose 4′ epimerase (GALE) is essential for development of Drosophila melanogaster . Dis Model Mech. 2010;3:628‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ross KL, Davis CN, Fridovich‐Keil JL. Differential roles of the Leloir pathway enzymes and metabolites in defining galactose sensitivity in yeast. Mol Genet Metab. 2004;83:103‐116. [DOI] [PubMed] [Google Scholar]
- 28. Packard MG, Vecchioli SF, Schroeder JP, Gasbarri A. Task‐dependent role for dorsal striatum metabotropic glutamate receptors in memory. Learn Mem. 2001;8:96‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Supporting information.