

Abstract
We present herein a novel nitroxide spin label‐containing RNA triphosphate TPT3NO and its application for site‐specific spin‐labeling of RNA through in vitro transcription using an expanded genetic alphabet. Our strategy allows the facile preparation of spin‐labeled RNAs with sizes ranging from short RNA oligonucleotides to large, complex RNA molecules with over 370 nucleotides by standard in vitro transcription. As a proof of concept, inter‐spin distance distributions are measured by pulsed electron paramagnetic resonance (EPR) spectroscopy in short self‐complementary RNA sequences and in a well‐studied 185 nucleotide non‐coding RNA, the B. subtilis glmS ribozyme. The approach is then applied to probe for the first time the folding of the 377 nucleotide A‐region of the long non‐coding RNA Xist, by PELDOR.
Keywords: EPR spectroscopy, PELDOR, RNA, spin labeling, unnatural base pairs
UBPs meet EPR: Unnatural base pairs (UBPs) allow facile site‐specific introduction of nitroxide spin labels into long RNAs by in vitro transcription. Distances are determined in the glmS ribozyme and the 377 nt A‐region of the long non‐coding RNA Xist.
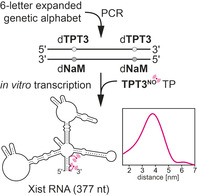
Introduction
An increasing number of regulatory, non‐coding RNA molecules with lengths of several hundred nucleotides has been identified in recent years. In order to study global folding and structural changes of such large tertiary RNA structures by spectroscopic methods, the site‐specific introduction of reporter groups is important. Electron paramagnetic resonance (EPR) spectroscopy provides valuable tools for studying RNA dynamics and folding and requires the site‐specific introduction of unpaired electrons as probes into the RNA of interest.1 Applications of pulsed EPR techniques, for example, pulsed electron‐electron double resonance (PELDOR or DEER)2 have been reported for short duplex RNA1a, 3 but rarely for long (>100 nucleotides) RNA molecules4 due to the difficulties of spin labeling large, complexly folded RNAs.
Site‐specifically spin‐labeled RNAs are usually prepared by solid‐phase synthesis.3a, 3e In this case, the spin label is exposed to the reagents used in nucleic acid synthesis, some of which can partially reduce the nitroxide.3g To circumvent this, a combination with post‐synthetic introduction3b, 3c, 3e, 3f of nitroxide spin labels, for example through click chemistry3c or a photolabile protecting group for nitroxides are used.3d Above all, the efficient chemical synthesis of RNA strands is restricted to sequences with less than a hundred nucleotides and enzymatic ligation strategies4a, 4b, 4c to assemble large functional RNAs are challenging for complex folded structures. Yet, a size of 150–400 nucleotides is common for various naturally occurring RNAs such as ribozymes or riboswitches and studying folding and structure of those regulatory RNAs is of high interest.4c, 5 New ligation and deoxyribozyme‐based methods have recently been developed for the site‐specific introduction of spin labels into RNA, but they are restricted by the accessibility of the modification site.6 Thus, novel approaches for spin labeling such large RNA molecules for structural investigation, in particular for studying conformational changes, are urgently needed.
We provide herein a novel method for the fast and facile preparation of large spin‐labeled RNA molecules by in vitro transcription using an expanded genetic alphabet. Hydrophobic unnatural base pairs,7 such as the dTPT3‐dNaM 8 base pair developed by Romesberg and co‐workers, can be employed to direct the site‐specific introduction of a functionalized unnatural triphosphate into RNA from a DNA template by standard T7 in vitro transcription.9 In this context, our group has recently demonstrated that norbornene‐ and cyclopropene‐modified TPT3 nucleotides can be employed to prepare labeled functional RNAs using copper‐free click chemistry.9h, 9i, 9j We describe the synthesis of a TPT3 triphosphate derivative functionalized with a pyrroline nitroxyl spin label (TPT3NO TP) for the template‐directed introduction of the spin label into RNA (Figure 1 A,B). The general procedure is outlined in Figure 1. A double‐stranded DNA template containing two unnatural dTPT3‐dNaM 8 base pairs at predefined positions is prepared in two ways. Either a six‐letter fusion polymerase chain reaction (PCR) is employed using DNA fragments that contain the non‐natural dNaM nucleotide and are prepared by solid‐phase synthesis (Figure 1 A) or by a six‐letter PCR procedure amplifying from a plasmid template using forward and reverse primers that contain the modified nucleotides (dNaM and d5SICS as dTPT3 analog, respectively, Figure 1 B). The template is transcribed into RNA by T7 RNA polymerase in the presence of the four natural ribonucleoside triphosphates and the additional spin‐labeled non‐natural triphosphate TPT3NO TP (Figure 1), which is complementary to the dNaM units in the DNA template. The result is an in vitro transcribed RNA, which contains two spin labels at predefined positions of its sequence.
Figure 1.
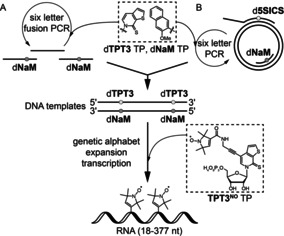
Novel strategy for the enzymatic preparation of site‐specifically spin‐labeled long RNAs using an expanded genetic alphabet. Top: the full‐length double‐stranded DNA template is either generated A) by solid‐phase DNA synthesis of several short oligonucleotides followed by a six‐letter fusion PCR using unnatural nucleoside triphosphates (TPs) dTPT3 TP8 and dNaM TP8 in addition to the canonical nucleoside triphosphates or B) by six letter PCR using dTPT3 TP and dNaM TP amplifying from a plasmid template employing modified forward and reverse primers. Bottom: the novel nitroxyl‐modified nucleotide TPT3NO TP (1) is incorporated into RNA through genetic alphabet expansion transcription.9
Using this approach, two RNA duplexes assembled from self‐complementary oligonucleotides bearing one spin label as well as a functional 185 nucleotide (nt) non‐coding RNA, the B. subtilis glmS ribozyme10 and the 377 nucleotide long A‐region of the M. musculus long non‐coding (lnc) RNA Xist, both possessing two spin labels at predefined positions were prepared. Inter‐spin distance distributions were determined by pulsed EPR techniques (PELDOR).
Results and Discussion
We first set out to assemble a small and rigid variant of the unnatural TPT3 nucleotide containing the TPA1e (7) spin label, TPT3rNO TP (2) (Scheme 1, right path).
Scheme 1.
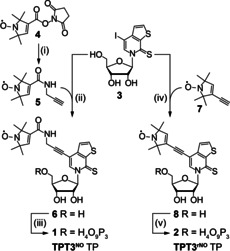
Synthesis of the nitroxyl spin labeled nucleoside triphosphates TPT3NO TP (1) and TPT3rNO TP (2) from TPT3I (3).9i TPT3NO TP (1): (i) propargylamine, CH2Cl2, r.t., 0.5 h, quant. (ii) CuI, NEt3, Pd(PPh3)4, DMF, r.t., o.n., 66 %. (iii) POCl3, Me3PO4, proton sponge, 0 °C, 3 h, then (Bu3NH)2PPi, NBu3, 0 °C, 30 min, 13 %. TPT3rNO TP (2): (iv) CuI, NEt3, Pd(PPh3)4, DMF, r.t., o.n., 96 %. (v) 2‐chloro‐4H‐1,2,3‐dioxaphosphirine‐4‐one, 1‐4‐dioxane, r.t., 40 min, then (Bu3NH)2PPi, NBu3, DMF, r.t., 40 min, then I2, py/H2O, 5 %.
Template‐directed incorporation of TPT3rNO TP (2) into an 18 nt RNA sequence by in vitro transcription was achieved and verified by ESI mass spectrometry and EPR (Figures S5, S6, and S18). However, it resulted in approximately 90 % truncation prior to incorporation of the unnatural triphosphate (see Figure 2 A, lane 1) likely because the T7 RNA polymerase is significantly inhibited incorporating this bulky and rigid unnatural triphosphate. To improve the incorporation efficiency of a spin‐labeled TPT3 derivative during transcription, a more flexible linker was designed. As a reasonable compromise between rigidity desired for EPR versus transcription efficiency, the triphosphate TPT3NO (1) was chosen as the synthetic target in which nitroxide 5 is attached to the TPT3 core through an amide propyl linker: An amide coupling between tempyo NHS ester 4 and propargylamine quantitatively gave alkyne spin label 5, which was attached to the unnatural nucleoside 3 by Sonogashira coupling yielding nucleoside 6 (66 %). The nucleoside was then converted into triphosphate 1 (TPT3NO TP, 13 %).
Figure 2.
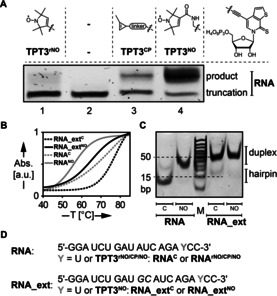
Site‐specific incorporation of spin‐labeled unnatural triphosphates into RNA by in vitro transcription using an expanded genetic alphabet. A) 20 % denaturing PAGE analysis of in vitro transcripts from a DNA template containing the unnatural nucleobase dNaM (DNANaM, for sequence see the Supporting Information) yielding a self‐complementary 18‐nt RNA duplex bearing one nitroxide spin label. The incorporation efficiency of both synthesized TPT3 derivatives TPT3 rNO TP (lane 1) and TPT3NO TP (lane 4) by T7 RNA polymerase was compared with that of cyclopropene‐modified TPT3CP TP9h (lane 3). If no unnatural triphosphate is added to the in vitro transcription reaction, formation of full‐length product is not observed (lane 2). B) UV melting curves (265 nm) of self‐complementary RNA sequences RNANO and RNA_extNO in comparison to unmodified sequences bearing canonical U–A base pairs instead of TPT3NO–A mispairs. C) Native PAGE (20 %) of duplexes RNANO, RNA_extNO and unmodified duplexes RNAC and RNA_extC. D) RNA sequences used in this study.
Subsequently, TPT3NO TP (1) could efficiently be incorporated into 18‐nt and 20‐nt self‐complementary RNA sequences RNANO and RNA_extNO, respectively, during in vitro transcription (Figure 2 A,D, lane 4 and Figures S7–S10). The incorporation efficiency of TPT3NO TP into RNA through T7 in vitro transcription was assessed by the ratio of full‐length to truncated transcript from band intensities and was found to be excellent with 78±2 % for 18 nt RNANO and 72±3 % for 20 nt RNA_extNO (Figure S11).
Both transcribed RNA sequences RNANO and RNA_extNO are self‐complementary and form duplexes with in total two TPT3NO‐A mismatches. The influence of these mismatches on the stability of the RNA duplexes was investigated by UV melting experiments (Figure 2 B and Figure S14), native gel electrophoresis (Figure 2 C) and CD measurements (Figure S13) in comparison to the corresponding unmodified sequences. We observed a reduction in melting temperature of 9.9 °C and 6.2 °C per TPT3NO residue for duplex RNANO and RNA_extNO respectively, which is still comparable to the introduction of other spin‐label modifications on natural nucleobases forming canonical base pairs (4–5 °C per label).3c On a native polyacrylamide gel, the modified sequences formed stable duplex structures as unmodified self‐complementary RNAs (Figure 2 C). Thus, stable duplexes are formed despite the TPT3NO‐A mismatch being present. Most likely the TPT3 nucleobase adopts a stacking interaction within the duplex as shown crystallographically for DNA duplexes.11
The efficiency of the TPT3NO TP (1) incorporation into RNA duplexes RNANO and RNA_extNO was tested by room temperature cw‐EPR spectroscopic analysis (Figure S19), yielding a spin labeling efficiency of 99 % and 60 %, respectively. We further verified that the TPT3NO spin label remains stable in conditions of the in vitro transcription (Figure S20).
We then performed Q‐band PELDOR experiments (at 50 K) for both RNA duplexes (Figure 3 A,B) yielding well‐modulated time traces with modulation depths of 19 % and 7 % for RNANO and RNA_extNO, respectively. The modulation depths are shallower than the 35 % expected for Q‐band PELDOR on nitroxides and are due to the labeling efficiency and the formation of singly labeled hairpins (Figure 2 C).3f Nevertheless, both duplexes provide sharp distance distributions with well‐defined peaks at 5.0 and 5.5 nm for the short and the by‐two‐base‐pairs‐extended RNA duplex (Figure 3 C and Figure S22), respectively. In order to translate the distance distributions into structural information, we performed all‐atom molecular dynamics (MD) simulations in explicit solvent (TIP3P water model) using the GROMACS12 software package in combination with the CHARMM3613 force field. The TPT3NO residue was parametrized and implemented in the CHARMM36 force field (force field parameters of TPT3NO are described in Table S2). Stable RNA duplexes are retained for RNANO and RNA_extNO over 2 μs simulation with an averaged N–N distance (nitroxyl group) of 5.2 nm (RNANO) and 5.6 nm (RNA_extNO) (Figure 3 C and Figure S29 A,B). The experimental distances are in excellent agreement with the MD‐derived distance distributions and fit exactly with the geometry and dimensions of an A‐form RNA duplex. This shows that our approach can be utilized as an accurate measure to determine distances in spin‐labeled RNAs prepared by genetic alphabet expansion transcription.
Figure 3.
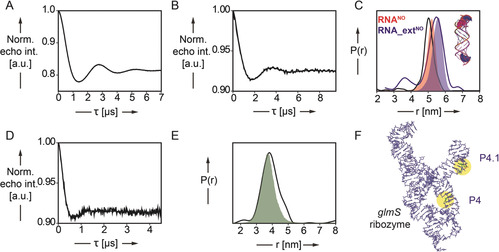
PELDOR‐derived distance distributions in spin‐labeled RNA duplexes RNANO and RNA_extNO and in the 185‐nucleotide‐long glmS ribozyme. A,B) Background corrected PELDOR time traces of RNANO (A, 16.7 μm RNA) and RNA_extNO (B, 15.0 μm) C) Inter‐label distance distributions of RNA duplexes RNANO (red curve) and RNA_extNO (blue curve) overlaid with predicted N‐N (nitroxyl) distance distributions (red and blue shading) by MD simulation. D) Background corrected PELDOR time traces of glmSNO4_4.1 (30 μm). E) Inter‐label distance distributions of glmS ribozyme construct glmSNO4_4.1 (green curve) overlaid with predicted N–N (nitroxyl) distance distributions (green shading) by MD simulation. F) Representative snapshot (cluster analysis) of glmSNO4_4.1. The two‐spin‐labeled TPT3NO residues in helix P4 and P4.1 are colored in green and highlighted in yellow.
We then tested, whether it is possible to introduce two TPT3NO labels into a larger RNA molecule at predefined sequence positions through in vitro transcription to study the folding of a functional non‐coding RNA, in particular a ribozyme. The well‐studied, cofactor‐dependent glmS ribozyme from B. subtilis (for sequence, see the Supporting Information) folds into three parallel helical stacks consisting of three pseudoknots. Binding of its cofactor glucosamine‐6‐phosphate (GlcN6P), which is essential for acid–base catalysis, does not induce conformational changes in the RNA structure.14, 15 We introduced the unnatural triphosphate TPT3NO TP (1) site‐specifically at internal positions within the B. subtilis glmS ribozyme sequence to measure distances in the global fold of the ribozyme. Preparation of the double‐stranded, site‐specifically modified DNA template for the transcription of the glmS ribozyme was achieved by an assembly PCR (Figure 1 A, for sequences see the Supporting Information) in the presence of canonical dNTPs and the two unnatural nucleoside triphosphates of dTPT3 and dNaM. In vitro transcription in the presence of 1 gave construct glmSNO4_4.1 bearing two TPT3NO residues in helix P4 and P4.1, respectively. The spin‐labeled ribozyme construct was able to cleave in vitro (Figures S15 and S16), thus the introduced modifications do not interfere with the fundamental fold and activity of the ribozyme.
In our initial transcription setup, we did observe non‐templated 3′‐extension of the transcript with TPT3NO TP resulting in an overall high spin‐labeling efficiency of the RNA but a low modulation depth in PELDOR experiments. Non‐templated 3′‐extension by canonical nucleotides during in vitro transcription reactions of long DNA templates using T7 RNA polymerase is commonly observed.16 We experimentally verified our hypothesis quantifying the amount of untemplated incorporation of TPT3NO TP at the 3′‐end by an in vitro transcription reaction from an unmodified DNA template in the presence of TPT3NO TP followed by cw‐EPR spectroscopic analysis. For the present glmS ribozyme construct, the untemplated 3′ spin label contributes to the spin‐labeling efficiency with 60 % (Figure S22 A). In contrast, the two duplexes described above did not show any indication of untemplated 3′‐extension, which fits with the previously reported observation that its efficiency depends on the sequence and length of the transcript.16 In order to avoid the unspecific 3′‐extension for the glmS RNA, we constructed the glmS template DNA using two 2′‐OMe modifications in the reverse DNA primer for PCR. This generates the DNA template strand with two 2′‐OMe modified nucleotides at its 5′‐end, resulting in transcription termination. In this way, the unspecific labeling at the 3′‐end of the transcript could be suppressed below the detection limit (see Figure S22 B, cw‐EPR spectra). Under these conditions, the spin‐labeling efficiency for glmSNO4_4.1 is 57 % (Figure S21 A) based on cw‐EPR spin counting. The successful incorporation of both labels is also reflected in the line‐shape of the cw‐EPR spectrum indicating restricted mobility of both spin labels.
From the PELDOR time trace with a modulation depth of 8 %, a distance distribution is obtained with a single peak at 3.8 nm (Figure 3 D,E and Figures S23 C and S24 C). The reduced modulation depth is again related to the labeling efficiency and the presence of mis‐ or unfolded glmS RNA.
To verify the experimentally observed distance distribution, we performed MD simulations on the entire glmS ribozyme (Figure 3 F). The starting model of the glmS ribozyme was constructed based on the crystal structure from Bacillus anthracis (PDB code: 3L3C14) and corresponding nucleobases are mutated according to the B. subtilis glmS ribozyme sequence used in this study (for details see Figure S28). The ribozyme fold is stable over 2 μs simulation (for details see the Supporting Information) and the computationally determined inter‐nitroxide distances overlay well with the main peak at 3.8 nm in the PELDOR‐derived distance distribution (Figure 3 E and Figure S24 C, for MD see Figure S29 C).
Having established a novel method for spin‐labeling during in vitro transcription, we aimed at testing the potential of this approach by examining a structurally underexplored long non‐coding RNA, the A‐region of the lncRNA Xist17 (X inactive specific transcript). Xist regulates X chromosome inactivation in female mammals and acts as a scaffold for protein recruitment.17b The A‐repeat region Xist RNA is highly conserved in mammals including humans and is indispensable for early gene silencing.17b So far, structural information on the Xist A‐region is restricted to structural models proposed by chemical and enzymatic probing.17 By introducing two spin labels in the sequence of the M. musculus Xist A‐region, we aimed to probe one of the multiple structural models proposed in which the 5′ and the 3′‐end of the A‐region fold into a stable duplex.17a This model had been established by Targeted Structure‐Seq, a method combining in vivo DMS chemical probing with next‐generation sequencing.17a
Preparation of the site‐specifically modified DNA template for transcription of the Xist RNA was achieved by six‐letter PCR from a plasmid template containing the M. musculus Xist gene using modified primers9g introducing the UBPs. Two constructs of the Xist A‐region were prepared, one bearing two spin labels at its 3′‐end (XistNO3_3, Figure 4 A and Figure 4 E, right panel) flanking both ends of the potential duplex being formed and one with both spin labels positioned 363 nt apart from each other in its 5′ and 3′‐end (XistNO5_3, Figure 4 A and Figure 4 G, right panel). Spin labeling of both Xist constructs was verified by room temperature cw‐EPR spectroscopic analysis (Figure S21, spin‐labeling efficiency of XistNO3_3: 44 % and of XistNO5_3: 76 %).
Figure 4.
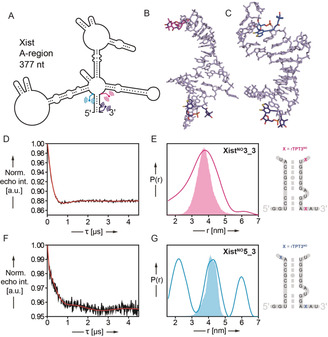
PELDOR‐derived distance distributions in the spin‐labeled A‐region of the long non‐coding RNA Xist (377 nt). A) Schematic of the 377 nt Xist A‐region prepared and site‐specifically modified by TPT3NO in this work based on the folding model of Fang et al.17a The positions of the spin label pairs in two Xist constructs XistNO3_3 and XistNO5_3 are indicated in pink and dark blue (XistNO3_3) or blue and dark blue (XistNO5_3). B,C) Representative snapshots (cluster analysis) of 1 μs MD simulation of the RNA duplexes shown in (E) and (G) (right panel) respectively. D,F) Background corrected PELDOR time traces of 377 nt RNAs XistNO3_3 (D, 7.5 μm RNA) and XistNO5_3 (F, 7.2 μm RNA). E,G) Inter‐label distance distributions of RNAs XistNO3_3 (pink curve, E) and XistNO5 3 (blue curve, G) overlaid with predicted N–N distance distributions (XistNO3_3: pink shading, G and XistNO5_3: blue shading, G) by MD simulation.
From the PELDOR experiment on XistNO3_3, a modulation depth of 12 % is obtained matching the labeling efficiency. The corresponding distance distribution gave a relatively broad distance distribution peaking at 3.8 nm (Figure 4 D,E). Assuming duplex formation between the 5′ and 3′‐ends of the Xist A‐region as proposed earlier17a and depicted schematically in Figure 4 A, a model duplex for MD simulation was constructed as shown in Figure 4 E. The main peak of 1 μs simulation (3.9 nm) overlaps well with the PELDOR derived distance distribution (Figure 4 B,E). With both spin labels positioned at the 3′‐end of the Xist A‐region, similar distances between both labels can be predicted for the diverse alternative folds of the Xist A‐region, which have been proposed previously17 (for an overview of the different structural models see Figure S25) and which may explain the large width of the peak. We reason that the Xist A‐region is not homogenously folded and most likely several of the predicted folds contribute to the broad distance distribution measured by PELDOR.
Only one18 of these proposed models17a, 17c, 17d predicts duplex formation between the 5′ and 3′‐end of the Xist A‐region. With our second construct, XistNO5_3, we aimed testing the validity of this model by PELDOR. The modulation depth in this experiment is low (Figure 4 F), most likely due to conformational heterogeneity in the Xist RNA. In all other folding models, the 5′‐end does not interact with the 3′‐end, which would space both spin labels far apart from each other positioning the 5′‐label in a presumably less structured region of the RNA.17c, 17d In this case, the long distances contribute to the intermolecular background, which is removed during data analysis. In our PELDOR experiment, we observe a distance distribution (Figure 4 G) with three peaks at 2.2, 4.3, and 6.0 nm. The peak at 4.3 nm is in excellent agreement with the MD derived distance distribution of the model duplex (4.3 nm, Figure 4 C,G) and consequently arises from duplex formation between the 5′ and the 3′‐end of the Xist A‐region. As modeling of the entire Xist A‐region is not reasonable at the current status of the structural information available with multiple potential conformations, we cannot yet suggest a model explaining the shorter (2.2 nm) and the longer (6.0 nm) distance. Remarkably, our PELDOR data strongly supports the unique folding model proposed by Fang et al.17a in which 5′‐ and 3′‐end of the Xist A‐region form a stable duplex. Besides this, the Xist RNA shows an explicit conformational heterogeneity in solution as suggested previously.17e
Conclusion
In summary, the described approach is the only known strategy so far for the direct and site‐specific introduction of spin labels into RNA through in vitro transcription, allowing accurate distance measurements on large RNAs. DNA templates for in vitro transcription can be prepared either by assembly PCR from shorter, synthetic DNA oligonucleotides or by direct amplification of plasmid DNA in a six letter PCR. Our method will give access to information on the structure of large non‐coding RNA molecules, which is currently difficult to assess for example, by crystallization. We demonstrated its capability by examining the structure of the A‐region of lncRNA Xist with 377 nt in length. In combination with stable spin labels for in‐cell applications, our approach might in the future even allow spin labeling of functional RNAs in living cells.18
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work has received financial support from the Deutsche Forschungsgemeinschaft (KA 3699/6‐1) and the Boehringer Ingelheim Stiftung. We further thank Jule Mehl for support in chemical synthesis.
C. Domnick, F. Eggert, C. Wuebben, L. Bornewasser, G. Hagelueken, O. Schiemann, S. Kath-Schorr, Angew. Chem. Int. Ed. 2020, 59, 7891.
Contributor Information
Prof. Dr. Olav Schiemann, Email: schiemann@pc.uni-bonn.de.
Dr. Stephanie Kath‐Schorr, Email: skath@uni-bonn.de.
References
- 1.
- 1a. Kamble N. R., Granz M., Prisner T. F., Sigurdsson S. T., Chem. Commun. 2016, 52, 14442–14445; [DOI] [PubMed] [Google Scholar]
- 1b. Endeward B., Marko A., Denysenkov V. P., Sigurdsson S. T., Prisner T. F., Methods Enzymol. 2015, 564, 403–425; [DOI] [PubMed] [Google Scholar]
- 1c. Krstić I., Hänsel R., Romainczyk O., Engels J. W., Dötsch V., Prisner T. F., Angew. Chem. Int. Ed. 2011, 50, 5070–5074; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 5176–5180; [Google Scholar]
- 1d. Schiemann O., Cekan P., Margraf D., Prisner T. F., Sigurdsson S. T., Angew. Chem. Int. Ed. 2009, 48, 3292–3295; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3342–3345; [Google Scholar]
- 1e. Schiemann O., Piton N., Plackmeyer J., Bode B. E., Prisner T. F., Engels J. W., Nat. Protoc. 2007, 2, 904–923; [DOI] [PubMed] [Google Scholar]
- 1f. Piton N., Mu Y., Stock G., Prisner T. F., Schiemann O., Engels J. W., Nucleic Acids Res. 2007, 35, 3128–3143; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1g. Piton N., Schiemann O., Mu Y., Stock G., Prisner T., Engels J. W., Nucleosides Nucleotides Nucleic Acids 2005, 24, 771–775; [DOI] [PubMed] [Google Scholar]
- 1h. Schiemann O., Weber A., Edwards T. E., Prisner T. F., Sigurdsson S. T., J. Am. Chem. Soc. 2003, 125, 3434–3435. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Milov A., Salikhov K., Shirov M., Fiz. Tverd. Tela 1981, 23, 975–982; [Google Scholar]
- 2b. Martin R. E., Pannier M., Diederich F., Gramlich V., Hubrich M., Spiess H. W., Angew. Chem. Int. Ed. 1998, 37, 2833–2837; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 2993–2998. [Google Scholar]
- 3.
- 3a. Sicoli G., Wachowius F., Bennati M., Höbartner C., Angew. Chem. Int. Ed. 2010, 49, 6443–6447; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 6588–6592; [Google Scholar]
- 3b. Babaylova E. S., Ivanov A. V., Malygin A. A., Vorobjeva M. A., Venyaminova A. G., Polienko Y. F., Kirilyuk I. A., Krumkacheva O. A., Fedin M. V., Karpova G. G., Bagryanskaya E. G., Org. Biomol. Chem. 2014, 12, 3129–3136; [DOI] [PubMed] [Google Scholar]
- 3c. Kerzhner M., Abdullin D., Wiecek J., Matsuoka H., Hagelueken G., Schiemann O., Famulok M., Chem. Eur. J. 2016, 22, 12113–12121; [DOI] [PubMed] [Google Scholar]
- 3d. Weinrich T., Jaumann E. A., Scheffer U., Prisner T. F., Göbel M. W., Chem. Eur. J. 2018, 24, 6202–6207; [DOI] [PubMed] [Google Scholar]
- 3e. Gophane D. B., Endeward B., Prisner T. F., Sigurdsson S. T., Org. Biomol. Chem. 2018, 16, 816–824; [DOI] [PubMed] [Google Scholar]
- 3f. Halbmair K., Seikowski J., Tkach I., Höbartner C., Deniz Sezer D., Bennati M., Chem. Sci. 2016, 7, 3172–3180; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3g. Shelke S. A., Sigurdsson S. T., Eur. J. Org. Chem. 2012, 2291–2301. [Google Scholar]
- 4.
- 4a. Wachowius F., Höbartner C., ChemBioChem 2010, 11, 469–480; [DOI] [PubMed] [Google Scholar]
- 4b. Esquiaqui J. M., Sherman E. M., Ye J. D., Fanucci G. E., Methods Enzymol. 2014, 549, 287–311; [DOI] [PubMed] [Google Scholar]
- 4c. Esquiaqui J. M., Sherman E. M., Ye J. D., Fanucci G. E., Biochemistry 2016, 55, 4295–4305; [DOI] [PubMed] [Google Scholar]
- 4d. Duss O., Yulikov M., Jeschke G., Allain F. H., Nat. Commun. 2014, 5, 3669; [DOI] [PubMed] [Google Scholar]
- 4e. Babaylova E. S., Malygin A. A., Lomzov A. A., Pyshnyi D. V., Yulikov M., Jeschke G., Krumkacheva O. A., Fedin M. V., Karpova G. G., Bagryanskaya E. G., Nucleic Acids Res. 2016, 44, 7935–7943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Mattick J. S., Makunin I. V., Hum. Mol. Genet. 2006, 15, R17–R29; [DOI] [PubMed] [Google Scholar]
- 5b. Morris K. V., Mattick J. S., Nat. Rev. Genet. 2014, 15, 423–437; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Hövelmann F., Seitz O., Acc. Chem. Res. 2016, 49, 714–723. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Lebars I., Vileno B., Bourbigot S., Turek P., Wolff P., Kieffer B., Nucleic Acids Res. 2014, 42, e117; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Wawrzyniak-Turek K., Höbartner C., Methods Enzymol. 2014, 549, 85–104. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Malyshev D. A., Romesberg F. E., Angew. Chem. Int. Ed. 2015, 54, 11930–11944; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12098–12113; [Google Scholar]
- 7b. Seo Y. J., Matsuda S., Romesberg F. E., J. Am. Chem. Soc. 2009, 131, 5046–5047; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Malyshev D. A., Seo Y. J., Ordoukhanian P., Romesberg F. E., J. Am. Chem. Soc. 2009, 131, 14620–14621; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7d. Leconte A. M., Romesberg F. E., Nat. Methods 2006, 3, 667–668; [DOI] [PubMed] [Google Scholar]
- 7e. Hirao I., Mitsui T., Kimoto M., Yokoyama S., J. Am. Chem. Soc. 2007, 129, 15549–15555; [DOI] [PubMed] [Google Scholar]
- 7f. Mitsui T., Kimoto M., Harada Y., Yokoyama S., Hirao I., J. Am. Chem. Soc. 2005, 127, 8652–8658; [DOI] [PubMed] [Google Scholar]
- 7g. Hirao I., Harada Y., Kimoto M., Mitsui T., Fujiwara T., Yokoyama S., J. Am. Chem. Soc. 2004, 126, 13298–13305. [DOI] [PubMed] [Google Scholar]
- 8. Li L., Degardin M., Lavergne T., Malyshev D. A., Dhami K., Ordoukhanian P., Romesberg F. E., J. Am. Chem. Soc. 2014, 136, 826–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Lavergne T., Lamichhane R., Malyshev D. A., Li Z., Li L., Sperling E., Williamson J. R., Millar D. P., Romesberg F. E., ACS Chem. Biol. 2016, 11, 1347–1353; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Seo Y. J., Malyshev D. A., Lavergne T., Ordoukhanian P., Romesberg F. E., J. Am. Chem. Soc. 2011, 133, 19878–19888; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Someya T., Ando A., Kimoto M., Hirao I., Nucleic Acids Res. 2015, 43, 6665–6676; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Ishizuka T., Kimoto M., Sato A., Hirao I., Chem. Commun. 2012, 48, 10835–10837; [DOI] [PubMed] [Google Scholar]
- 9e. Kimoto M., Sato A., Kawai R., Yokoyama S., Hirao I., Nucleic Acids Symp. Ser. 2009, 53, 73–74; [DOI] [PubMed] [Google Scholar]
- 9f. Moriyama K., Kimoto M., Mitsui T., Yokoyama S., Hirao I., Nucleic Acids Res. 2005, 33, e129; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9g. Eggert F., Kulikov K., Domnick C., Leifels P., Kath-Schorr S., Methods 2017, 120, 17–27; [DOI] [PubMed] [Google Scholar]
- 9h. Eggert F., Kath-Schorr S., Chem. Commun. 2016, 52, 7284–7287; [DOI] [PubMed] [Google Scholar]
- 9i. Domnick C., Eggert F., Kath-Schorr S., Chem. Commun. 2015, 51, 8253–8256; [DOI] [PubMed] [Google Scholar]
- 9j. Domnick C., Hagelueken G., Eggert F., Schiemann O., Kath-Schorr S., Org. Biomol. Chem. 2019, 17, 1805–1808. [DOI] [PubMed] [Google Scholar]
- 10. McCown P. J., Winkler W. C., Breaker R. R., Methods Mol. Biol. 2012, 848, 113–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Betz K., Malyshev D. A., Lavergne T., Welte W., Diederichs K., Dwyer T. J., Ordoukhanian P., Romesberg F. E., Marx A., Nat. Chem. Biol. 2012, 8, 612–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berendsen H. J. C., van der Spoel D., van Drunen R., Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar]
- 13. Best R. R. B., Zhu X., Shim J., Lopes P. E., Mittal J., Feig M., A. D. Mackerell, Jr. , J. Chem. Theory Comput. 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cochrane J. C., Lipchock S. V., Smith K. D., Strobel S. A., Biochemistry 2009, 48, 3239–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Klein D. J., Ferre-D′Amare A. R., Methods Mol. Biol. 2009, 540, 129–139; [DOI] [PubMed] [Google Scholar]
- 15b. Klein D. J., Wilkinson S. R., Been M. D., Ferre-D′Amare A. R., J. Mol. Biol. 2007, 373, 178–189; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Cochrane J. C., Lipchock S. V., Strobel S. A., Chem. Biol. 2007, 14, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gholamalipour Y., Mudiyanselage A. K., Martin C. T., Nucleic Acids Res. 2018, 46, 9253–9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Fang R., Moss W. N., Rutenberg-Schoenberg M., Simon M. D., PLoS Genet. 2015, 11, e1005668; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Pintacuda G., Young A. N., Cerase A., Front. Mol. Biosci. 2017, 4, 90; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17c. Liu F., Somarowthu S., Pyle A. M., Nat. Chem. Biol. 2017, 13, 282–289; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17d. Maenner S., Blaud M., Fouillen L., Savoye A., Marchand V., Dubois A., Sanglier-Cianferani S., Van Dorsselaer A., Clerc P., Avner P., Visvikis A., Branlant C., PLoS Biol. 2010, 8, e1000276; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17e. Lu Z., Zhang Q. C., Lee B., Flynn R. A., Smith M. A., Robinson J. T., Davidovich C., Gooding A. R., Goodrich K. J., Mattick J. S., Mesirov J. P., Cech T. R., Chang H. Y., Cell 2016, 165, 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y., Ptacin J. L., Fischer E. C., Aerni H. R., Caffaro C. E., San Jose K., Feldman A. W., Turner C. R., Romesberg F. E., Nature 2017, 551, 644–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary