

Abstract
Objective
The pathology of frontotemporal dementia, termed frontotemporal lobar degeneration (FTLD), is characterized by distinct molecular classes of aggregated proteins, the most common being TAR DNA‐binding protein‐43 (TDP‐43), tau, and fused in sarcoma (FUS). With a few exceptions, it is currently not possible to predict the underlying pathology based on the clinical syndrome. In this study, we set out to investigate the relationship between pathological and clinical presentation at single symptom level, including neuropsychiatric features.
Methods
The presence or absence of symptoms from the current clinical guidelines, together with neuropsychiatric features, such as hallucinations and delusions, were scored and compared across pathological groups in a cohort of 150 brain donors.
Results
Our cohort consisted of 68.6% FTLD donors (35.3% TDP‐43, 28% tau, and 5.3% FUS) and 31.3% non‐FTLD donors with a clinical diagnosis of frontotemporal dementia and a different pathological substrate, such as Alzheimer's disease (23%). The presence of hyperorality points to FTLD rather than non‐FTLD pathology (p < 0.001). Within the FTLD group, hallucinations in the initial years of the disease were related to TDP‐43 pathology (p = 0.02), including but not limited to chromosome 9 open reading frame 72 (C9orf72) repeat expansion carriers. The presence of perseverative or compulsive behavior was more common in the TDP‐B and TDP‐C histotypes (p = 0.002).
Interpretation
Our findings indicate that neuropsychiatric features are common in FTLD and form an important indicator of underlying pathology. In order to allow better inclusion of patients in targeted molecular trials, the routine evaluation of patients with frontotemporal dementia should include the presence and nature of neuropsychiatric symptoms. ANN NEUROL 2020;87:950–961
The term frontotemporal dementia (FTD) defines a group of neurodegenerative syndromes with diverse clinical presentations, including the behavioral variant of frontotemporal dementia (bvFTD) and language dominant syndromes, such as primary progressive aphasia (PPA), including the nonfluent/agrammatic variant of PPA (nfPPA), and the semantic variant of PPA (svPPA).1, 2 Other syndromes that are part of this group are characterized by prominent movement symptoms, such as corticobasal syndrome, progressive supranuclear palsy, and FTD with motor neuron disease. Most patients present with mixed behavior, language, and motor symptoms, but are diagnosed based on their most pronounced and first onset of symptoms and/or behavior. The past decade has seen a fast evolution of knowledge on the clinical and genetic features of FTD. In 2011, diagnostic criteria for bvFTD and PPA were revised which improved diagnostic accuracy.1, 2 Mutations in genes, such as chromosome 9 open reading frame 72 (C9orf72), progranulin (GRN), and microtubule associated protein tau (MAPT) have been identified in about 25% of patients with FTD.3 More recently, neuropsychiatric symptoms, such as psychosis and depressed mood, have been recognized to be part of the early clinical presentation of FTD, both in C9orf72 repeat expansion carriers and noncarriers.4, 5, 6 Due to the clinical variability and overlap of symptoms with primary psychiatric diseases and other neurodegenerative diseases, such as Alzheimer's disease, and given the lack of biomarkers, it remains challenging to diagnose FTD accurately in a clinical setting.
The pathology of FTD, termed frontotemporal lobar degeneration (FTLD), is characterized by the unifying macroscopic hallmark of atrophy of the frontal and temporal lobes.7 On a microscopic level, aggregates of distinct types of misfolded proteins can be observed. TAR DNA‐binding protein‐43 (TDP‐43) aggregates occur in approximately 50% of patients, microtubule associated protein tau (MAPT) in 40%, and fused in sarcoma (FUS) aggregates are seen in 5 to 10%.7, 8 C9orf72 and GRN mutations are associated with TDP‐43 aggregation, whereas mutations in MAPT lead to tau aggregation.5 Within the molecular class TDP‐43, 5 different histotypes (A‐E) have been described based on the morphology and distribution of cytoplasmic and neuritic aggregates across brain layers.9 TDP‐C is the predictable histopathology in the majority of patients with svPPA.10 However, in the remaining sporadic patients with FTD, the underlying phenotype can scarcely be predicted based on the clinical phenotype.
Previous efforts to identify clinicopathological correlations in FTD focused on the underlying pathologies of the FTD clinical subgroups.10, 11, 12, 13, 14, 15, 16, 17 When focusing on the 5 core behavioral symptoms of bvFTD, predicting the underlying pathological phenotype remains challenging.18 Here, we set out to investigate clinicopathological correlations at the symptom level in a large FTD cohort encompassing all clinical variants, and include a broader spectrum of symptoms than those that have been incorporated in the consensus clinical criteria, such as neuropsychiatric symptoms.
Materials and Methods
Subjects
Donors were selected from the cohort of The Netherlands Brain Bank. First, records of autopsied donors between 2008 and 2017 were searched for the terms “frontotemporal dementia” and “frontotemporal lobar degeneration.” The resulting reports were reviewed by a neurologist (M.S.) to ascertain whether FTD was a differential clinical diagnosis, and to record the main clinical diagnosis and the pathological diagnosis. Donors were excluded from the analysis when brain tumors or brain infarcts larger than 15mm were reported, as well as lacunar infarcts larger than 3mm in the thalamus or basal ganglia. Low‐level white matter disease in the deep and periventricular white matter was not regarded as an exclusion criterion. This yielded a cohort of 150 donors. All donors were clinically diagnosed with FTD, pathologically diagnosed with FTLD, or diagnosed with both FTD and FTLD. The donors had been evaluated in different clinical settings in the Netherlands, including academic and community hospitals. All donors had provided informed consent for brain donation and the use of their medical records for research according to the ethical guidelines19 (Table 1 and Supplementary Table S1).
Table 1.
Demographic and Pathologic Data of all Subjects and Main Pathological Groups
| All donors | TDP‐43 donors | tau donors | FUS donors | Non‐FTLD donors | |
|---|---|---|---|---|---|
| N | 150 (100%) | 53 (35.3%) | 42 (28%) | 8 (5.3%) | 47 (31.3%) |
| Gender, M:F | 81:69 | 25:28 | 24:18 | 7:1 | 25:22 |
| Age at onset, mean ± SD | 59.31 ± 10.84 | 59.38 ± 9.82 | 58.67 ± 11.85 | 49.38 ± 10.81 | 61.51 ± 10.29 |
| Age at diagnosis, mean ± SD | 62.21 ± 10.41 | 62.09 ± 8.88 | 61.86 ± 11.42 | 51.38 ± 10.36 | 64.49 ± 10.17 |
| Age at death, mean ± SD | 67.69 ± 10.19 | 67.11 ± 8.56 | 67.33 ± 10.68 | 57.25 ± 11.02 | 70.45 ± 10.31 |
| Disease duration, yrs; mean ± SD | 8.37 ± 4.70 | 7.74 ± 4.71 | 8.66 ± 4.88 | 7.88 ± 6.03 | 8.92 ± 4.33 |
| Time to diagnosis, yrs; mean ± SD | 2.89 ± 3.17 | 2.72 ± 3.12 | 3.19 ± 3.45 | 2.0 ± 1.52 | 2.98 ± 3.23 |
| Time to dependency in ADL, yrs; mean ± SD | 4.49 ± 3.22 | 4.16 ± 3.32 | 4.79 ± 3.39 | 2.75 ± 1.67 | 4.91 ± 3.09 |
| Brain weight, gr; mean ± SD | 1101.15 ± 168.25 | 1063.17 ± 167.84 | 1082.14 ± 180.94 | 1124 ± 152.08 | 1157.06 ± 148.07 |
| Thal stage (median; IQR) | 1 (0–3) | 1 (0–3) | 1 (0–1) | 0 (0–0) | 5 (4–5) |
| Braak stage for tau (median; IQR) | 3 (1–5) | 2 (1–2) | NA | 1 (0–2) | 6 (4.75–6) |
| Braak stage for Lewy body (median; IQR) | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0 (0–4) |
Values are expressed as mean ± SD or median ± IQR.
ADL = activities of daily living; FTLD = frontotemporal lobar degeneration; FUS = fused in sarcoma; IQR = interquartile range; NA = not applicable; TDP = TAR DNA‐binding protein.
Assessment of Clinical and Pathological Features
Clinical Data
The majority of donors in our cohort received more than one clinical diagnosis during their life. First, the main clinical diagnosis was identified, based on the recorded diagnosis after complete neurological assessment. Differential diagnoses were also used for analysis as this reflects the clinical complexity of the donors. Next, 4 disease time points were identified and included (1) the year of symptom onset, (2) the year of clinical diagnosis, (3) the year of loss of independency in activities of daily living, and (4) the year of death. Demographic data, age at all disease time points, and duration to reach disease time points from the year of symptom onset were included in the analysis.
The neurological symptoms present in the first 3 years of disease onset were evaluated and scored as present (1) or absent (0). If symptoms were not explicitly mentioned, they were considered absent. The core behavioral symptoms, as defined by the framework of Rascovsky criteria, were scored present if at least one of the subcategories for each symptom was reported.1 These symptoms include disinhibition, apathy or inertia, perseverative or compulsive behavior, and hyperorality. The behavioral symptom “loss of empathy” was not scored, because this was almost never explicitly mentioned, and it is challenging to derive from the clinical records. Language impairments were clearly mentioned in the donors’ clinical history, and were scored according to the current PPA criteria in 1 of the 3 types of aphasia (nfPPA, svPPA, or logopenic variant PPA [lvPPA]).2 Motor signs and symptoms included apraxia (motor and/or ideational), falls, signs of motor neuron disease, and parkinsonism. The neuropsychiatric symptoms hallucinations, delusions, mania, and depression were scored if present in the first 3 years of the disease.20 The medical history of each donor prior to the clinical onset of dementia was reviewed for psychiatric diagnoses. The family history of each donor was assessed with respect to dementia, other neurodegenerative diseases, and psychiatric diseases. The family history was considered positive if at least one first‐degree relative was affected. Information on genetic status was collected from clinical files or from postmortem genetic testing on brain tissue.
Pathological Procedures and Diagnoses
The brain tissue was dissected into anatomically defined structures, in accordance to Brain Net Europe's Code of Conduct for brain banking.21 Briefly, the right hemisphere was fixed in 4% formaldehyde for 4 weeks and dissected into 24 standard regions for diagnostic purposes. The pathological diagnosis was first determined based on guidelines in force at the time of autopsy. As the pathological proteins have been visualized using various antibodies over the past 10 years, all available regions from all donors were immunostained for the main pathological proteins: amyloid‐beta (IC16 antibody; kind gift of Prof Dr Korth, Heinrich Heine University, Düsseldorf, Germany), phosphorylated‐tau (AT8; Pierce Biotechnology, Rockford, IL), and phosphorylated‐TDP43 (pTDP43; Cosmo Bio, Tokyo, Japan). If all abovementioned proteins were not detected or pathological notes suggested FUS pathology, immunohistochemistry for FUS pathology was also performed (HPA008784; Sigma Aldrich, St. Louis, MO). If the clinical and pathological diagnoses were indicative for parkinsonism, additional immunohistochemistry was performed to assess alpha‐synuclein pathology (Zymed; Thermo Fisher Scientific, Bleiswijk, The Netherlands). The final pathological diagnosis was defined based on a combination of the original neuropathological report and on the new evaluation from immunohistochemical stainings. The presence of other pathologies beside the main pathological diagnosis was also taken into consideration. Each donor was staged for amyloid‐beta, tau deposition, and alpha‐synuclein pathology according to the Thal and Braak staging systems.22, 23, 24
FTLD‐TDP histotypes were assessed based on the criteria proposed by Lee et al,9 according to the following definitions. Type A: pathology predominantly in the second layer where both compact neuronal cytoplasmic inclusions and short/small threads are seen. Type B: pathology across all layers where mostly neuronal cytoplasmic inclusions are seen; threads are either absent or few. Type C: long/thick threads are seen; neuronal cytoplasmic inclusions are absent or rare; no white matter involvement. Type D: cat‐eye inclusions across all layers. Type E: granular neuronal cytoplasmic inclusions filling dendrites across all layers; extracellular pathology shows as diffuse granularity; pathology broadly affects the white matter and extends to brainstem. Donors with TDP pathology that did not match the criteria for any TDP subtype were classified as TDP‐U (“unknown”).
Statistical Analysis
One‐way ANOVA with Tukey's post hoc test was used to compare age at all 4 disease time points, disease duration, time to diagnosis, time to dependency in activities of daily living, brain weight, Thal stage, and Braak stages between groups. Pearson's chi‐squared test was used to compare gender distribution and the distribution of donors with a concordant (1) or discordant (0) clinicopathological diagnosis before and after 2011. The presence (1) or absence (0) of signs and symptoms were compared using Pearson's chi‐squared test on 3 group levels: (1) between FTLD and non‐FTLD donors, (2) between the 3 FTLD subtypes: FTLD‐TDP, FTLD‐tau, and FTLD‐FUS, and (3) between the TDP histotypes A through E. A p value of 0.05 was considered significant and corrected for multiple comparisons with the Benjamini‐Hochberg procedure. All statistical analyses were performed using the Statistical Package for the Social Sciences (SPSS version 24 for Windows, Chicago, IL).
Results
Demographics
FUS donors were found to be younger than non‐FTLD donors at disease onset (p = 0.02) and younger than all other separate groups of donors at diagnosis (FUS vs TDP: p = 0.03; FUS vs tau: p = 0.04; FUS vs non‐FTLD: p < 0.01), and death (FUS vs TDP: p = 0.05; FUS vs tau: p = 0.05; and FUS vs non‐FTLD: p < 0.01). No differences were found between the groups of donors with respect to disease duration (p = 0.61) and gender distribution (p = 0.19). The demographic findings in our cohort are in line with previous studies on pathologically confirmed FTD12, 13, 25, 26, 27, 28 (see Table 1).
Donors with TDP‐C pathology have a longer disease duration compared with donors with TDP‐E pathology (p = 0.006). No differences were found between TDP histotypes with respect to gender distribution (p = 0.57; see Table 2).
Table 2.
Demographic and Pathologic Data of Main TDP‐43 Histotypes
| TDP‐A donors | TDP‐B donors | TDP‐C donors | TDP‐E donors | |
|---|---|---|---|---|
| N | 17 | 14 | 13 | 7 |
| Gender, M:F | 7:10 | 6:8 | 6:7 | 5:2 |
| Age at onset, mean ± SD | 60.82 ± 8.58 | 57.29 ± 15.25 | 58.39 ± 5.58 | 62.43 ± 6.32 |
| Age at diagnosis, mean ± SD | 62.59 ± 8.50 | 60.50 ± 13.88 | 61.85 ± 4.12 | 64.57 ± 4.83 |
| Age at death, mean ± SD | 67.71 ± 8.36 | 64.64 ± 12.73 | 68.69 ± 3.40 | 66.14 ± 4.34 |
| Disease duration, yrs; mean ± SD | 6.88 ± 3.79 | 7.36 ± 5.00 | 10.31 ± 3.40 | 3.71 ± 3.40 |
| Time to diagnosis, yrs; mean ± SD | 1.77 ± 2.25 | 3.21 ± 3.56 | 3.46 ± 3.53 | 2.14 ± 3.53 |
| Time to dependency in ADL, yrs; mean ± SD | 3.06 ± 2.91 | 4.54 ± 3.78 | 5.92 ± 2.84 | 2.86 ± 3.67 |
| Brain weight, gr; mean ± SD | 1026.18 ± 153.51 | 1060.29 ± 218.91 | 1039.15 ± 136.76 | 1178 ± 98.91 |
| Thal stage, median; IQR | 0.5 (0–2) | 1 (0–3) | 1 (0.5–2) | 2 (1–3) |
| Braak stage for tau, median; IQR | 2 (0–3.75) | 2 (1–2.25) | 1 (0.5–2) | 1 (0–2) |
| Braak stage for Lewy body, median; IQR | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0 (0–0) |
Values are expressed as mean ± SD or median ± IQR.
ADL = activities of daily living; IQR = interquartile range; TDP = TAR DNA‐binding protein.
Clinical and Pathological Overview
Clinical Diagnoses
The majority of donors in our cohort received FTD as main or differential diagnosis at least once during their disease duration (146/150; 97%). Interestingly, 4 donors that were never diagnosed with FTD had TDP‐43 pathology; 2 donors were clinically diagnosed as having Alzheimer's disease and had underlying TDP‐A pathology, 1 donor had a clinical diagnosis of schizoaffective disorder with underlying TDP‐C pathology, and the fourth donor had a clinical diagnosis of multisystem atrophy with underlying TDP‐E pathology.
After a complete neurological assessment, 115 of 146 donors (79%) who received FTD as a differential clinical diagnosis had it confirmed as their main clinical diagnosis, with a distribution of 77 of 146 bvFTD (53%), 30 of 146 PPA (21%), 4 of 146 corticobasal syndrome (3%), and 4 of 146 progressive supranuclear palsy (3%). In 31 donors (21%), Alzheimer's disease was the main clinical diagnosis (Fig 1 and Supplementary Table S1).
Figure 1.
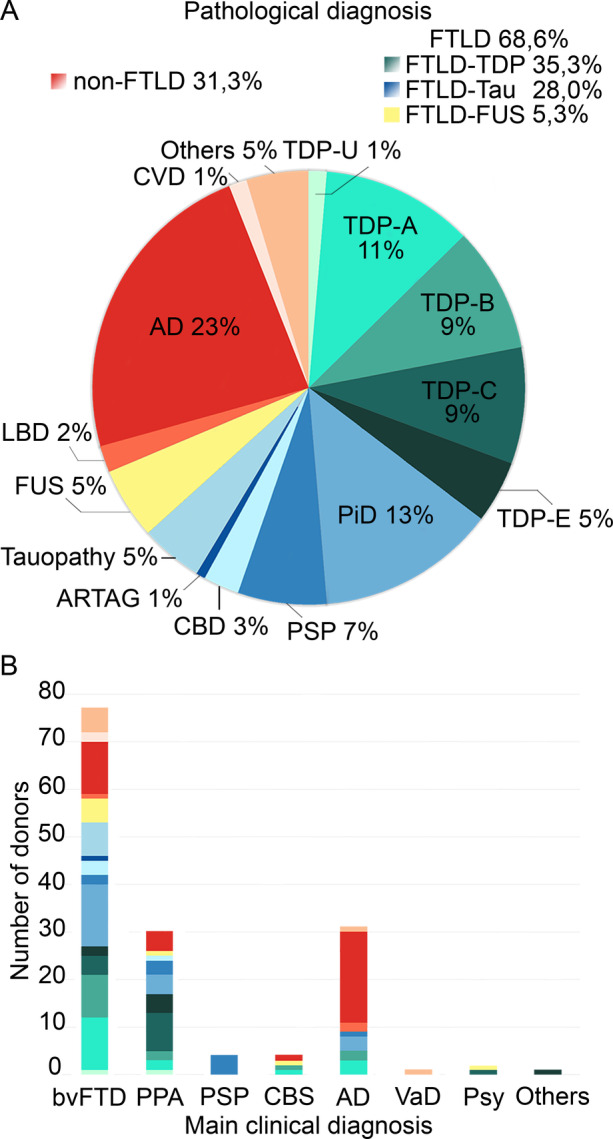
Pathological diagnoses of all donors in the cohort (A) and the distribution of pathology grouped by main clinical diagnosis (B). AD = Alzheimer's disease; ARTAG = aging‐related tau astrogliopathy; CBD = corticobasal degeneration; CBS = corticobasal syndrome; CVD = cerebrovascular disease; FTLD = frontotemporal lobar degeneration; FUS = fused in sarcoma; LBD = Lewy body disease; PiD = Pick's disease; PPA = primary progressive aphasia; PSP = progressive supranuclear palsy; Psy = primary psychiatric disease; TDP = TAR DNA‐binding protein; VaD = vascular dementia.
Psychiatric History and Symptoms
Twelve donors received a clinical differential diagnosis of a primary psychiatric disorder after the clinical onset of dementia. Among those 12 donors, 6 were pathologically diagnosed with FTLD‐TDP, 3 with FTLD‐tau, and 3 with FTLD‐FUS. Primary psychiatric diagnoses in FTLD donors were depression (n = 6), bipolar disorder (n = 3), schizoaffective disorder (n = 1), obsessive–compulsive disorder (n = 1), and post‐traumatic stress disorder (n = 1).
In the medical history of the donors, a psychiatric disorder was reported in 32 donors, who were diagnosed with depression, anxiety, panic disorder, dysthymia, obsessive‐compulsive disorder, psychosis, or substance addiction. Among donors with a psychiatric diagnosis in their medical history, 15 donors (47%) had underlying FTLD‐TDP pathology (28% of all FTLD‐TDP), 9 donors (28%) had FTLD‐tau (21% of all FTLD‐tau), 1 donor (3%) had FTLD‐FUS, 5 donors (16%) had underlying Alzheimer's disease pathology (17% of all Alzheimer's disease donors), and 2 donors (6%) had other pathologies.
A psychiatric disorder in at least one first‐degree relative was reported in 8 non‐FTLD donors (17%) and in 18 FTLD donors (17%). Eleven FTLD donors (61%) with a positive family history had underlying TDP‐43 pathology, 5 donors had tau pathology (27%), and 2 donors had FUS pathology (11%; Fig 2 and Supplementary Table S1).
Figure 2.
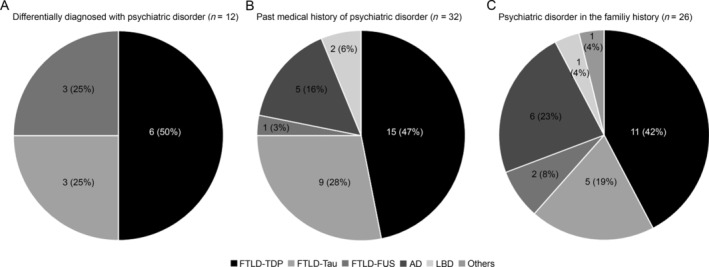
Psychiatric history of donors: proportion of pathological classes among (A) donors differentially diagnosed with a primary psychiatric disorder after the clinical onset of dementia; (B) donors with a medical history of psychiatric disorder; and (C) donors with a family history of primary psychiatric disorder. AD = Alzheimer's disease; FTLD = frontotemporal lobar degeneration; FUS = fused in sarcoma; LBD = Lewy body disease; TDP = TAR DNA‐binding protein.
Pathological Diagnoses
Overall, 103 of 150 donors had FTLD pathology, consisting of 53 TDP‐43, 42 tau, and 8 FUS donors. Among the TDP‐43 donors, 17 (11%) were classified as TDP‐A, 14 (9%) as TDP‐B, 13 (9%) as TDP‐C, 7 (5%) as TDP‐E, and 2 (1%) as TDP‐U. Non‐FTLD donors (n = 47) exhibited Alzheimer's disease pathology (n = 35), cerebrovascular disease pathology (n = 2), Lewy body disease (n = 3), and other pathologies (n = 7; Alexander's disease, spinocerebellar ataxia, post‐anoxic encephalopathy, Wernicke‐Korsakoff disease, and neuronal intranuclear inclusion disease29; see Fig 1).
Statistical analysis revealed a lower brain weight on autopsy in non‐FTLD donors compared with TDP‐43 donors (p = 0.03). As expected, Thal and Braak stage for tau neurofibrillary pathology were significantly higher in the group of non‐FTLD donors compared to TDP‐43 and FUS donors (p < 0.01), and Thal stage was higher in non‐FTLD compared to FTLD‐tau donors (p < 0.01). Braak stage for alpha‐synuclein pathology was also higher in the group of non‐FTLD donors compared to TDP‐43 and tau donors (p < 0.01; see Table 1).
Genetics
Half of FTLD donors in the present study were tested for C9orf72, GRN, and/or MAPT mutations postmortem, on the basis of a suggestive pathology supported by a positive family history for dementia or psychiatric disease. Of the 42 FTLD‐tau donors, 12 were tested for MAPT mutation, 10 were positive, and 2 were negative. Of the 53 FTLD‐TDP donors, 13 were positive for C9orf72 repeat expansion, 6 were positive for GRN mutation, and 20 were negative for both. The remarkably higher prevalence of MAPT mutation carriers in our cohort is consistent with other studies in the Dutch population30 (Table 3).
Table 3.
Genetics of FTLD Donors
| Carrier status | Carriers (n) | Noncarriers (n) | Not tested (n) | |
|---|---|---|---|---|
| FTLD‐tau donors (n = 42) | MAPT | 10 | 2 | 30 |
| FTLD‐TDP donors (n = 53) | C9orf72 | 13 | 20 | 14 |
| GRN | 6 | |||
C9orf72 = chromosome 9 open reading frame 72; FTLD = frontotemporal lobar degeneration; GRN = progranulin; MAPT = microtubule associated protein tau; TDP = TAR DNA‐binding protein.
Clinicopathological Diagnostic Agreement
Clinicopathological diagnostic agreement was calculated as the percentage of main clinical diagnoses that matched the pathological diagnosis in the cohort of 150 donors. The total clinicopathological diagnostic agreement in our cohort was 76%. Among the donors who had been clinically diagnosed before or during 2011, the clinicopathological agreement was 72%, whereas this was significantly higher with 93% (chi‐squared (1) = 5.77; p = 0.02) in donors diagnosed after the implementation of novel diagnostic criteria.
Comparison of Clinical Features Between Pathological Groups
FTLD and non‐FTLD
The clinical hallmark that best discriminated between FTLD and non‐FTLD donors was the presence of hyperorality in the first 3 years from disease presentation (chi‐squared (1) = 24.55; p < 0.001). Hyperorality was reported with a higher frequency in FTLD (60/103; 58%) compared to non‐FTLD donors (7/47; 15%). The prevalence of hyperorality was higher in FTLD‐bvFTD (42/58; 72%) compared to both FTLD‐nfPPA (3/11; 27%) and FTLD‐svPPA donors (4/10; 40%; chi‐squared (2) = 10.36; p < 0.01).
Perseverative or compulsive behavior was also found to be present in a higher proportion of FTLD compared to non‐FTLD cases (63% vs 36%; chi‐squared (1) = 9.45; p = 0.01). The prevalence of a perseverative or compulsive behavior was 70% (7/10) in FTLD‐svPPA, 66% (38/58) in FTLD‐bvFTD, and 55% (6/11) in FTLD‐nfPPA (chi‐squared (2) = 0.64; p = 0.73).
The most reported behavioral symptom in FTLD donors was disinhibition. The prevalence of disinhibition did not differ between FTLD and non‐FTLD donors (73% vs 74%; chi‐squared (1) = 0.045; p = 0.89) and was higher in FTLD‐bvFTD donors (45/58; 78%) compared to FTLD‐nfPPA donors (4/11; 36%; chi‐squared (2) = 7.62; p = 0.22; bvFTD vs nfPPA: p < 0.01; bvFTD vs svPPA: p = 0.60; svPPA vs nfPPA: p = 0.12; Figs 3A and 4).
Figure 3.
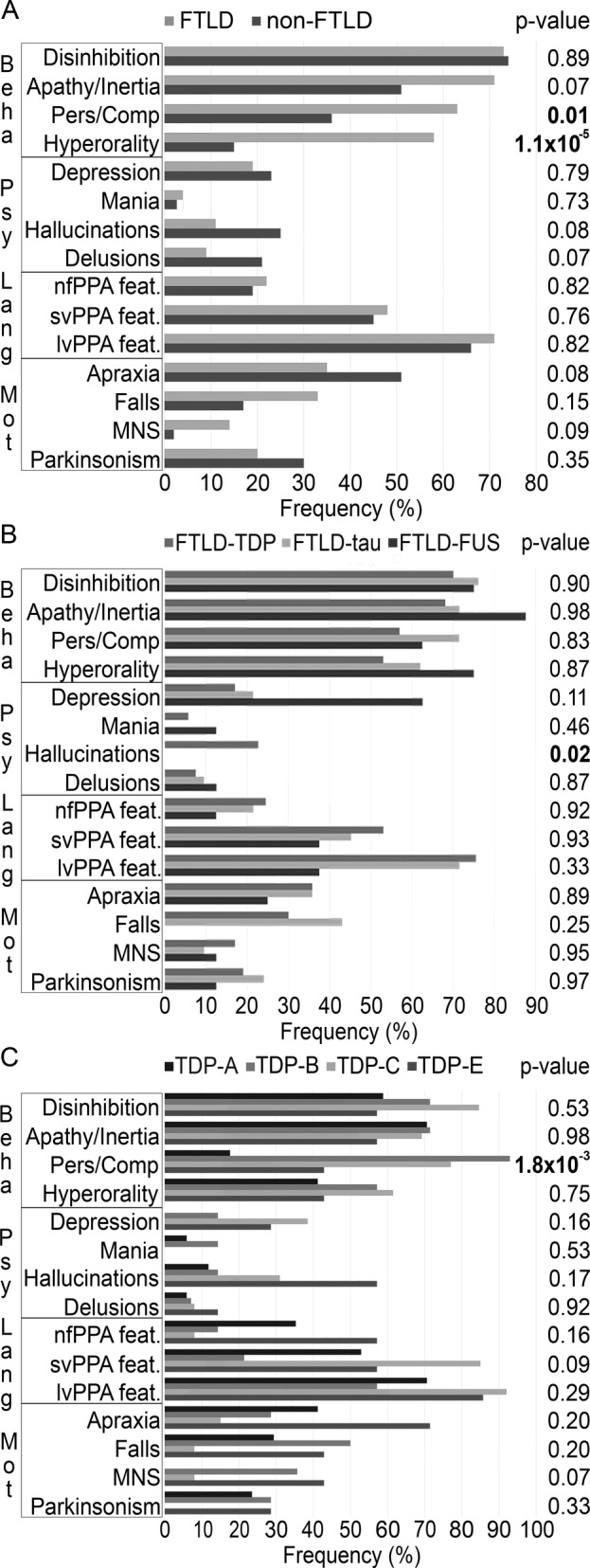
Comparison of symptom frequency between pathological groups of brain donors (A) FTLD versus non‐FTLD donors. (B) TDP‐43 versus tau versus FUS donors. (C) TDP‐43 histotypes (A vs B vs C vs E). Beha = behavioral features; feat = features; FTLD = frontotemporal lobar degeneration; FUS = fused in sarcoma; Lang = language features; lvPPA = logopenic variant of primary progressive aphasia; MNS = signs of motor neuron disease; Mot = motor features; nfPPA = nonfluent/agrammatic variant of primary progressive aphasia; Pers/Comp = perseverative or compulsive behavior; Psy = psychiatric features; svPPA = semantic variant of primary progressive aphasia; TDP = TAR DNA‐binding protein.
Figure 4.
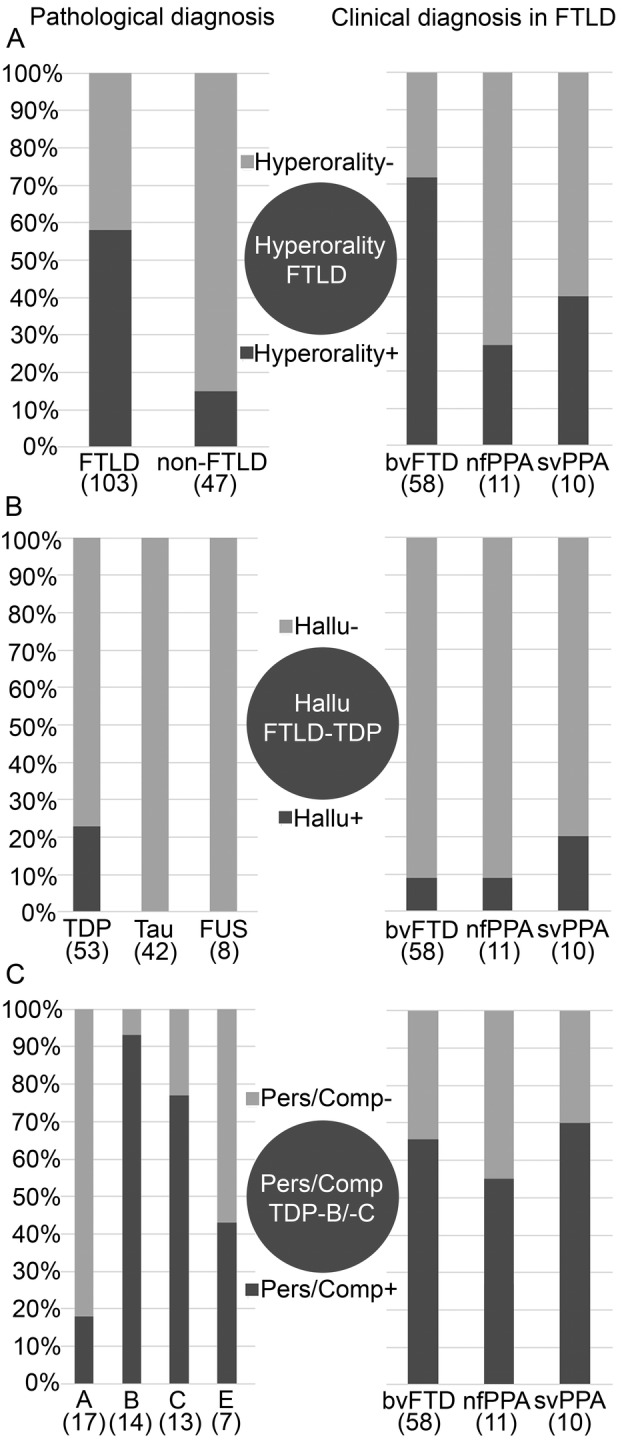
Frequency of symptoms among pathological classes of donors (left) and among main clinical classes of FTLD donors (right) (A) hyperorality, (B) hallucinations, and (C) perseverative or compulsive behavior. The scale shows increasing (A) hyperorality, (B) hallucinations, and (C) perseverative or compulsive behavior. bvFTD = behavioral variant of frontotemporal dementia; FTLD = frontotemporal lobar degeneration; FUS = fused in sarcoma; Hallu = hallucinations; nfPPA = nonfluent/agrammatic variant of primary progressive aphasia; svPPA = semantic variant of primary progressive aphasia; Pers/Comp = perseverative or compulsive behavior; TDP = TAR DNA‐binding protein.
The presence of psychiatric symptoms did not help in discriminating between FTLD and non‐FTLD donors. However, when looking at the nature of hallucinations, visual were prevalent in non‐FTLD donors, whereas 8 of 12 donors presented with visual hallucinations, and in 4 of 12 donors the type of hallucination was not specified. On the other hand, auditory hallucinations were prevalent in FTLD donors, with 7 of 12 being reported with auditory hallucinations, 1 of 12 with visual hallucinations, and 4 of 12 without explicit mention of the nature of the hallucinations.
FTLD‐TDP, FTLD‐Tau, and FTLD‐FUS
No difference was found concerning the presence of behavioral symptoms, when comparing the frequency of clinical features in FTLD‐TDP, FTLD‐tau, and FTLD‐FUS donors in our cohort. In FTLD donors, the prevalence of hallucinations did not differ among bvFTD (5/58; 9%), nfPPA (1/11; 9%) and svPPA (2/10; 20%) donors (chi‐squared (2) = 1.23; p = 0.54). Hallucinations were recorded only in FTLD‐TDP donors and were the only clinical feature that could discriminate between the major FTLD molecular classes (chi‐squared (2) = 12.81; p = 0.02; TDP vs tau chi‐squared (1) = 10.88; p < 0.01; and TDP vs FUS chi‐squared (1) = 2.255; p = 0.13). Hallucinations were reported in 12 donors in our FTLD‐TDP cohort, of which 3 were found to carry a C9orf72 repeat expansion. This indicates that the presence of hallucinations is not exclusive of the C9orf72 phenotype but is more widely associated with TDP‐43 pathology (Figs 3B and 4).
FTLD‐TDP Histotypes
The symptom that most discriminated between FTLD‐TDP subtypes was the presence of a perseverative or compulsive behavior (chi‐squared (3) = 20.75; p = 1.8 × 10−3). Perseverative or compulsive behavior had a lower prevalence in TDP‐A (3/17; 18%) and TDP‐E (3/7; 43%) compared to TDP‐B and TDP‐C donors (13/14; 93% and 10/13; 77%, respectively). Among the donors in these groups, 4 TDP‐A and 9 TDP‐B subjects carried a C9orf72 repeat expansion. Without C9orf72 carriers, the result of the comparison between TDP‐A and TDP‐B donors remained significant (Figs 3C and 4).
Other Pathologies
All regions of the brain were evaluated for presence of other pathological proteins. In FTLD‐TDP donors, tau aggregates were present in the hippocampus in 75% of the donors and amyloid‐beta plaques in the neocortex in 68% of the donors. In donors with Alzheimer's disease, Lewy body co‐pathology in the limbic system was found in 19 donors (19/35; 54%). Noticeably, among these 19 Alzheimer's disease donors with Lewy body co‐pathology, 7 were also found to have TDP‐43 co‐pathology in the limbic system, thus showing 2 co‐pathologies in addition to the main pathological diagnosis. Overall, limbic TDP‐43 co‐pathology was detected in 14 Alzheimer's disease donors (40%) and in 8 FTLD‐tau donors (19%31; see Table 4). Hallucinations were reported in 12 of 47 non‐FTLD donors, mainly Alzheimer's disease donors (8/12). Co‐pathology was common among Alzheimer's disease donors with hallucinations: they presented with Lewy body pathology (n = 4), TDP‐43 pathology (n = 2), or both Lewy body and TDP‐43 pathologies (n = 3). Other non‐FTLD donors with hallucinations were pathologically diagnosed with Lewy body disease (n = 2), spinocerebellar ataxia (n = 1), and neuronal intranuclear inclusion disease (n = 1; Fig 3A and Table 4).
Table 4.
Prevalence of Co‐Pathologies by Pathological Diagnosis
| N | Pathological diagnosis | ||||||
|---|---|---|---|---|---|---|---|
| FTLD‐TDP | FTLD‐TAU | AD | LBD | FUS | |||
| 53 | 42 | 35 | 3 | 8 | |||
| COPATHOLOGY | TDP‐43 pathology | n | 53 | 8 | 14 | 1 | 0 |
| % | 100 | 19 | 40 | 33 | 0 | ||
| FTD‐MND copathology* | n | 7 | NA | NA | NA | 1 | |
| % | 13 | NA | NA | NA | 13 | ||
| Braak stage tau 1–2 | n | 30 | NA | 0 | 2 | 4 | |
| % | 57 | 0 | 67 | 50 | |||
| Braak stage tau 3–4 | n | 8 | NA | 3 | 0 | 0 | |
| % | 15 | 9 | 0 | 0 | |||
| Braak stage tau 5–6 | n | 2 | NA | 32 | 0 | 0 | |
| % | 4 | 91 | 0 | 0 | |||
| Aß plaques Thal 1–2 | n | 22 | 17 | 2 | 3 | 1 | |
| % | 42 | 40 | 6 | 100 | 13 | ||
| Aß plaques Thal 3 | n | 9 | 3 | 1 | 0 | 0 | |
| % | 17 | 7 | 3 | 0 | 0 | ||
| Aß plaques Thal 4–5 | n | 4 | 1 | 32 | 0 | 0 | |
| % | 8 | 2 | 91 | 0 | 0 | ||
| Lewy bodies | n | 1 | 0 | 19 | 3 | 1 | |
| % | 2 | 0 | 54 | 100 | 13 | ||
Aß = amyloid beta; AD = Alzheimer's disease; FTLD = frontotemporal lobar degeneration; FUS = fused in sarcoma; LBD = Lewy body disease; MND = motor neuron disease; NA = not applicable; TDP = TAR DNA‐binding protein.
*Defined by neuronal loss within anterior horn neurons of the spinal cord and in motor nuclei of the brainstem and the presence of phosphorylated TDP‐43 or FUS neuronal cytoplasmic inclusions in lower motor neurons of the spinal cord and in motor nuclei of the brainstem.
Discussion
We studied clinicopathological correlations in a large brain bank cohort of 150 donors and identified several early symptoms that can help to differentiate between underlying pathology. Hyperorality is the clinical feature that could best distinguish underlying FTLD from non‐FTLD pathology. The presence of auditory hallucinations is associated with underlying FTLD‐TDP pathology, and perseverative or compulsive behavior points to FTD‐TDP type B or C histotypes.
Hyperorality Points to FTLD Pathology
Hyperorality is the early behavioral feature that can best improve the ante‐mortem discrimination of FTLD from non‐FTLD pathology. The higher prevalence of hyperorality in pathologically confirmed FTLD donors compared to Alzheimer's disease donors has been described before when focusing on the clinical diagnosis of bvFTD.18 Our work supports the finding from a recent clinical cohort study that hyperorality is detectable not only in bvFTD but also in patients with PPA.32 However, the prevalence of hyperorality was higher in FTLD‐bvFTD compared to FTLD‐PPA donors in our cohort. Our results confirm previous findings that hyperorality is present in the 3 major FTLD subclasses FTLD‐TDP,18, 33 FTLD‐tau,33, 34 and FTLD‐FUS.35 Interestingly, a recent clinicopathological study of bvFTD found that hyperorality was present in FTLD‐tau and FTLD‐FUS donors, but not in FTLD‐TDP donors.36 This result is likely related to the small size of their cohort. Our data support the notion that hyperorality in the early phase of the disease points to FTLD pathology, without further indication on FTLD pathological subclasses.
Hyperorality was included as a core bvFTD diagnostic criteria in 2011,1 and the clinicopathological agreement in our cohort after 2011 increased from 72% to 93%. This indicates that the addition of hyperorality to the diagnostic criteria contributed to improving the clinicopathological agreement.
Disinhibition is reported to be more prevalent in bvFTD,1 however, in our study, we find a similar prevalence of disinhibition between FTLD and non‐FTLD donors. This finding is possibly related to the design of this study, where non‐FTLD donors were included in the cohort because of the differential clinical diagnosis of FTD, which can be prompted by the presence of behavioral symptoms, such as disinhibition. Moreover, in the FTLD‐group, all FTD clinical syndromes were taken into account, so that the higher prevalence of disinhibition in bvFTD was balanced by the lower prevalence among nfPPA.1
Neuropsychiatric Symptoms Aid in Diagnosing FTLD and its Molecular Subclasses
The presence of neuropsychiatric symptoms likely prompted the diagnosis of psychiatric disease that was recorded in 12 FTLD donors in our cohort, half of whom showed TDP‐43 pathology. The medical history and the family history of FTLD donors were enriched with psychiatric diagnoses, particularly for donors with TDP‐43 pathology, showing a connection between TDP‐43 pathology and a psychiatric clinical profile. According to current diagnostic criteria, a primary psychiatric diagnosis is still considered an exclusion criterion for FTD.1, 2 On the contrary, our results and those of other recent studies6, 37, 38 suggest that including more neuropsychiatric symptoms in the clinical assessment might improve diagnostic accuracy.
Hallucinations
The early presence of hallucinations is found in a subset of patients with FTD. This study shows that in the non‐FTLD group, hallucinations were mostly visual and related to the presence of multiple pathologies, mainly alpha‐synuclein pathology. On the other hand, auditory hallucinations are specific for TDP‐43 pathology in our FTLD group, and not exclusively related to the presence of C9orf72 repeat expansion.39, 40, 41 Our finding of a 12% prevalence of hallucinations in FTLD donors is in line with previous studies, where the prevalence of hallucinations varies from 8% to 18% in pathologically confirmed FTD patients.42, 43, 44 In our study, the clinicopathological correlations are based on an extensive pathological screening, taking into account all clinical FTD syndromes, newly characterized disease groups, and the presence of multiple pathologies. Previous studies based on clinicopathological bvFTD cohorts have either found hallucinations in both tau positive and tau negative,42, 43 or when FTLD subclasses were taken into account, the prevalence of hallucinations was higher in the TDP group44, 45 or too infrequent to be compared.18 With regard to the nature of the hallucinations, few studies are available and point either to a higher prevalence of auditory hallucinations46 or to a higher prevalence of visual hallucinations,44 without taking into account multiple pathologies. Additional studies should be done to point out if auditory hallucinations are linked to FTLD‐TDP without concomitant pathologies, as we describe here.
Perseverative or Compulsive Behavior
Perseverative or compulsive behavior was found in a higher proportion of all molecular subclasses of FTLD donors compared to non‐FTLD donors. In the group of FTLD donors, the prevalence of a perseverative or compulsive behavior was not significantly different among svPPA, bvFTD, and nfPPA. A possible explanation for this is that both a perseverative and/or a compulsive behavior were scored for this category,1 and not just compulsive behavior, which is common in svPPA.18, 47 Pathologically, perseverative or compulsive behavior was reported in a significantly higher proportion of TDP‐B (87.5%) and TDP‐C (77%) donors when compared with other histotypes. This outcome was not related to the presence of C9orf72 mutation carriers. Perry et al (2017) found compulsive behavior to be more common in TDP‐C,18 in line with our findings. The reason we also found TDP‐B to show a high proportion of perseverative or compulsive behavior is likely due to the setup of our study, as we included all clinical syndromes of FTD and we used the new expanded classification of TDP A‐E histotypes.9 Overall, we have shown that neuropsychiatric features are common in the early stages of FTD and can aid in the diagnosis of underlying FTLD also with respect to the identification of molecular subclasses and histotypes.
Presence of Other Pathologies
Presence of other pathologies was observed across all pathological diagnoses in our cohort (see Table 4). Although amyloid pathology was mainly found in the neocortex, TDP‐43, tau, and Lewy body pathologies were mostly observed in the hippocampus and in the amygdala. Co‐occurrence of multiple pathologies in patients with dementia is being increasingly reported in autopsy series.48 Growing evidence supports an early involvement of the limbic structures in the genesis of multiple proteinopathies: limbic‐predominant age‐related TDP‐43 encephalopathy31 and Alzheimer's disease with Lewy bodies in the amygdala49 are examples of newly defined pathological entities where multiple pathologies are observed. In our cohort, Lewy bodies were observed in 54% of donors pathologically diagnosed with Alzheimer's disease and clinically diagnosed with FTD. We observed that visual hallucinations were common among Alzheimer's disease donors, especially with co‐occurring Lewy body pathology. This neuropsychiatric clinical profile could explain that donors with Alzheimer's disease pathology received FTD as a clinical diagnosis in our cohort, as also suggested in previous clinicopathological cohorts.42 In other clinicopathological studies, Alzheimer's disease donors with Lewy bodies presented with higher scores on the Neuropsychiatric Inventory compared with Alzheimer's disease donors without Lewy bodies.49 Further studies are needed to explore the relationship between multiple pathologies and the presence of psychiatric symptoms.
Demographics
Between TDP‐43 histotypes, the longest disease duration was observed among TDP‐C donors, as reported in previous works on patients with pathologically confirmed svPPA,50 whereas the shortest disease duration was observed in donors with TDP‐E, a histotype previously associated with a very rapid disease progression.9
Strengths and Limitations
This study represents one of the largest pathologically confirmed cohort of FTLD donors, it includes all FTD clinical syndromes and assesses clinical profiles at the symptom level, including psychiatric features. Our results reinforce the notion that behavioral features, as defined in current bvFTD criteria,1 cannot discern among TDP‐43, tau, and FUS pathologies.18 The strength of this study is that we expand that outcome to a cohort including all FTD clinical variants. Extensive records were available for all donors and allowed a detailed assessment of clinical features, although a symptom like diminished empathy is not explicitly mentioned and, therefore, not scored. Another limitation of this retrospective study is that the majority of the donors did not have standardized forms for the scoring of symptoms available, such as the Neuropsychiatric Inventory. To minimize the selection bias and to reflect as much as possible the heterogenous clinical scenario of everyday practice, donors were included in this study who were clinically diagnosed in different clinical settings, with FTD as either the main or a differential diagnosis. The detailed assessment of pathology, performed by taking into account the evolving knowledge in the field, reprocessing of brain sections to identify the newly characterized subtypes, and the presence of multiple co‐proteinopathies, is a major strength of this study. The replication of our study in another large FTD brain bank cohort could validate our results.
Our work contributes to the growing evidence that neuropsychiatric symptoms could constitute a crucial component of the clinical FTD framework.
Author Contributions
M.S., E.S., D.G., Y.A.L.P., and A.A.D developed the study concept and design. M.S., P.G., Y.T., H.S., J.C.v.S., A.J.M.R., A.D., N.B.B., J.J.M.H., Y.A.L.P., and A.A.D. were involved in data acquisition and analysis. M.S., Y.A.L.P., and A.A.D gave substantial contribution in drafting the manuscript and figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Supplementary Table 1 – Clinical and pathologic data of all donors
Acknowledgments
We would like to thank Betty Tijms for statistical assistance.
Financial support: The European Academy of Neurology supported this work with a research fellowship granted to M. Scarioni. This study was supported by a grant from the Memorabel ZonMW (733050507).
References
- 1. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gorno‐Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol 2019;266:2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galimberti D, Dell'Osso B, Altamura AC, Scarpini E. Psychiatric symptoms in frontotemporal dementia: epidemiology, phenotypes, and differential diagnosis. Biol Psychiatry 2015;78:684–692. [DOI] [PubMed] [Google Scholar]
- 5. Snowden JS, Adams J, Harris J, et al. Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotroph Lateral Scler Front Degener 2015;16:497–505. [DOI] [PubMed] [Google Scholar]
- 6. Gossink FT, Vijverberg EGB, Krudop W, et al. Psychosis in behavioral variant frontotemporal dementia. Neuropsychiatr Dis Treat 2017;13:1099–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mackenzie IR, Neumann M. Molecular neuropathology of frontotemporal dementia: insights into disease mechanisms from postmortem studies. J Neurochem 2016;138:54–70. [DOI] [PubMed] [Google Scholar]
- 8. Seelaar H, Klijnsma KY, de Koning I, et al. Frequency of ubiquitin and FUS‐positive, TDP‐43‐negative frontotemporal lobar degeneration. J Neurol 2010;257:747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee EB, Porta S, Michael Baer G, et al. Expansion of the classification of FTLD‐TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol 2017;134:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rohrer JD, Lashley T, Schott JM, et al. Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain 2011;134:2565–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hodges JR, Davies RR, Xuereb JH, et al. Clinicopathological correlates in frontotemporal dementia. Ann Neurol 2004;56:399–406. [DOI] [PubMed] [Google Scholar]
- 12. Kertesz A, McMonagle P, Blair M, et al. The evolution and pathology of frontotemporal dementia. Brain 2005;128:1996–2005. [DOI] [PubMed] [Google Scholar]
- 13. Shi J, Shaw CL, Du Plessis D, et al. Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol 2005;110:501–512. [DOI] [PubMed] [Google Scholar]
- 14. Forman MS, Farmer J, Johnson JK, et al. Frontotemporal dementia: clinicopathological correlations. Ann Neurol 2006;59:952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Snowden JS, Thompson JC, Stopford CL, et al. The clinical diagnosis of early‐onset dementias: diagnostic accuracy and clinicopathological relationships. Brain 2011;134:2478–2492. [DOI] [PubMed] [Google Scholar]
- 16. Chare L, Hodges JR, Leyton CE, et al. New criteria for frontotemporal dementia syndromes: clinical and pathological diagnostic implications. J Neurol Neurosurg Psychiatry 2014;85:865–870. [DOI] [PubMed] [Google Scholar]
- 17. Spinelli EG, Mandelli ML, Miller ZA, et al. Typical and atypical pathology in primary progressive aphasia variants. Ann Neurol 2017;81:430–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perry DC, Brown JA, Possin KL, et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain 2017;140:3329–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Klioueva NM, Rademaker MC, Dexter DT, et al. BrainNet Europe's code of conduct for brain banking. J Neural Transm 2015;122:937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. American Psychiatric Association . Diagnostic and statistical manual of mental disorders. 5th ed. Washington, DC: American Psychiatric Publishing, 2013. [Google Scholar]
- 21. Alafuzoff I. Minimal neuropathologic diagnosis for brain banking in the normal middle‐aged and aged brain and in neurodegenerative disorders In: Huitinga I, Webster MJ, eds. Brain Banking. Vol 150 Amsterdam: Elsevier, 2018:131‐141. [DOI] [PubMed] [Google Scholar]
- 22. Thal DR, Rüb U, Orantes M, Braak H. Phases of Aβ‐deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 23. Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006;112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Braak H, Del Tredici K, Rüb U, et al. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 2003;24:197–211. [DOI] [PubMed] [Google Scholar]
- 25. Hodges JR, Davies R, Xuereb J, et al. Survival in frontotemporal dementia. Neurology 2003;61:349–354. [DOI] [PubMed] [Google Scholar]
- 26. Johnson JK, Diehl J, Mendez MF, et al. Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol 2005;62:925–930. [DOI] [PubMed] [Google Scholar]
- 27. Knopman DS, Boeve BF, Parisi JE, et al. Antemortem diagnosis of frontotemporal lobar degeneration. Ann Neurol 2005;57:480–488. [DOI] [PubMed] [Google Scholar]
- 28. Urwin H, Josephs KA, Rohrer JD, et al. FUS pathology defines the majority of tau‐ and TDP‐43‐negative frontotemporal lobar degeneration. Acta Neuropathol 2010;120:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cupidi C, Dijkstra AA, Melhem S, et al. Refining the spectrum of neuronal intranuclear inclusion disease: a case report. J Neuropathol Exp Neurol 2019;78:665–670. [DOI] [PubMed] [Google Scholar]
- 30. Rizzu P, Van Swieten JC, Joosse M, et al. High prevalence of mutations in the microtubule‐associated protein tau in a population study of frontotemporal dementia in The Netherlands. Am J Hum Genet 1999;64:414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain 2019;142:1503–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ahmed RM, Irish M, Kam J, et al. Quantifying the eating abnormalities in frontotemporal dementia. JAMA Neurol 2014;71:1540–1546. [DOI] [PubMed] [Google Scholar]
- 33. Piguet O, Petersén A, Yin Ka Lam B, et al. Eating and hypothalamus changes in behavioral‐variant frontotemporal dementia. Ann Neurol 2011;69:312–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu WT, Mandrekar JN, Parisi JE, et al. Clinical features of pathologic subtypes of behavioral‐variant frontotemporal dementia. Arch Neurol 2007;64:1611–1616. [DOI] [PubMed] [Google Scholar]
- 35. Snowden JS, Hu Q, Rollinson S, et al. The most common type of FTLD‐FUS (aFTLD‐U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol 2011;122:99–110. [DOI] [PubMed] [Google Scholar]
- 36. Kobayashi Z, Arai T, Kawakami I, et al. Clinical features of the behavioural variant of frontotemporal dementia that are useful for predicting underlying pathological subtypes of frontotemporal lobar degeneration. Psychogeriatrics 2018;18:307–312. [DOI] [PubMed] [Google Scholar]
- 37. Vijverberg EGB, Gossink F, Krudop W, et al. The diagnostic challenge of the Late‐onset frontal lobe syndrome: clinical predictors for primary psychiatric disorders versus behavioral variant frontotemporal dementia. J Clin Psychiatry 2017;78:e1197–e1203. [DOI] [PubMed] [Google Scholar]
- 38. Ducharme S, Pearl‐Dowler L, Gossink F, et al. The frontotemporal dementia versus primary psychiatric disorder (FTD versus PPD) checklist: a bedside clinical tool to identify behavioral variant FTD in patients with Late‐onset behavioral changes. J Alzheimers Dis 2019;67:113–124. [DOI] [PubMed] [Google Scholar]
- 39. Sha SJ, Takada LT, Rankin KP, et al. Frontotemporal dementia due to C9ORF72 mutations: clinical and imaging features. Neurology 2012;79:1002–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Devenney EM, Ahmed RM, Halliday G, et al. Psychiatric disorders in C9orf72 kindreds. Neurology 2018;91:e1498–e1507. [DOI] [PubMed] [Google Scholar]
- 41. Ducharme S, Bajestan S, Dickerson BC, Voon V. Psychiatric presentations of C9orf72 mutation: what are the diagnostic implications for clinicians? J Neuropsychiatry Clin Neurosci 2017;23:195–205. [DOI] [PubMed] [Google Scholar]
- 42. Mendez MF, Joshi A, Tassniyom K, et al. Clinicopathologic differences among patients with behavioral variant frontotemporal dementia. Neurology 2013;80:561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Léger GC, Banks SJ. Neuropsychiatric symptom profile differs based on pathology in patients with clinically diagnosed behavioral variant frontotemporal dementia. Dement Geriatr Cogn Disord 2014;37:104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Landqvist Waldö M, Gustafson L, Passant U, Englund E. Psychotic symptoms in frontotemporal dementia: a diagnostic dilemma? Int Psychogeriatr 2015;27:531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Velakoulis D, Walterfang M, Mocellin R, et al. Frontotemporal dementia presenting as schizophrenia‐like psychosis in young people: clinicopathological series and review of cases. Br J Pychiatry 2009;194:298–305. [DOI] [PubMed] [Google Scholar]
- 46. Schoder D, Hannequin D, Martinaud O, et al. Morbid risk for schizophrenia in first‐degree relatives of people with frontotemporal dementia. Br J Psychiatry 2010;197:28–35. [DOI] [PubMed] [Google Scholar]
- 47. Rosso SM, Roks G, Stevens M, et al. Complex compulsive behaviour in the temporal variant of frontotemporal dementia. J Neurol 2001;248:965–970. [DOI] [PubMed] [Google Scholar]
- 48. Robinson JL, Lee EB, Xie SX, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age‐related and APOE4‐associated. Brain 2018;141:2181–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chung EJ, Babulal GM, Monsell SE, et al. Clinical features of Alzheimer disease with and without Lewy bodies. JAMA Neurol 2015;72:789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Roberson ED, Hesse JH, Rose KD, et al. Frontotemporal dementia progresses to death faster than Alzheimer disease. Neurology 2005;65:719–725. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 – Clinical and pathologic data of all donors