

Abstract
Aim
A Japanese individual with schizophrenia harboring a novel exonic deletion in RELN was recently identified by genome‐wide copy‐number variation analysis. Thus, the present study aimed to generate and analyze a model mouse to clarify whether Reln deficiency is associated with the pathogenesis of schizophrenia.
Methods
A mouse line with a novel RELN exonic deletion (Reln‐del) was established using the CRISPR/Cas9 method to elucidate the underlying molecular mechanism. Subsequently, general behavioral tests and histopathological examinations of the model mice were conducted and phenotypic analysis of the cerebellar granule cell migration was performed.
Results
The phenotype of homozygous Reln‐del mice was similar to that of reeler mice with cerebellar atrophy, dysplasia of the cerebral layers, and abrogated protein levels of cerebral reelin. The expression of reelin in heterozygous Reln‐del mice was approximately half of that in wild‐type mice. Conversely, behavioral analyses in heterozygous Reln‐del mice without cerebellar atrophy or dysplasia showed abnormal social novelty in the three‐chamber social interaction test. In vitro reaggregation formation and neuronal migration were severely altered in the cerebellar cultures of homozygous Reln‐del mice.
Conclusion
The present results in novel Reln‐del mice modeled after our patient with a novel exonic deletion in RELN are expected to contribute to the development of reelin‐based therapies for schizophrenia.
Reelin is a large extracellular matrix protein expressed in many brain regions.1 During developmental stages, reelin is secreted primarily by Cajal–Retzius cells and plays a crucial role in neuronal migration and layer formation in the cerebral cortex2, 3; it is also required in cerebellar development.4 In the adult brain, reelin is produced by GABAergic interneurons and contributes to synaptic plasticity, dendritic morphology, and cognitive function.5, 6, 7 Previous studies reported lissencephaly and cerebellar hypoplasia in patients carrying RELN mutations; this phenotype is similar to that observed in reeler mice.8, 9 The full‐length reelin protein is approximately 400 kDa in size, with eight repeated domains known as reelin repeats and is cleaved by proteases, such as disintegrin and metalloproteinase, with thrombospondin motifs 3 at two specific sites10: one site between reelin repeats 2 and 3 (N‐t site) and another site between reelin repeats 6 and 7 (C‐t site). In previous studies, two Reln‐mutant mice were used to evaluate reelin function. Jackson Reln homozygous mice (Relnrl‐J) have a 150‐kB genomic deletion, whereas Orleans Reln homozygous mice (Relnrl‐Orl) produce a transcript with a 220‐bp deletion.11, 12 Reelin protein is not produced in Relnrl‐J mice,5 whereas Relnrl‐Orl mice produce a non‐secreting protein partially cleaved at the C‐terminal.13, 14, 15 Little reelin signaling induced reversed cortical layering and cerebellar atrophy in both Relnrl‐J and Relnrl‐Orl mice. Furthermore, heterozygous Relnrl‐J (Relnrl‐J/+) mice showed memory dysfunction,5, 16 anxiety, and pre‐pulse inhibition (PPI). Additionally, we recently reported that heterozygous Relnrl‐Orl (Relnrl‐Orl/+) mice showed dysfunction in sociability, locomotor coordination, and anxiety.17 Reelin plays an important role in the cortex and hippocampus, which contribute significantly to memory formation and retention.7, 16, 18
Genetic studies suggest that RELN is associated with psychiatric disorders, including schizophrenia and autism spectrum disorder.19, 20 Specifically, rare variants of RELN, including de novo or rare missense variants and an exonic deletion of RELN, were identified as risk factors for schizophrenia.21, 22, 23 Our recent genome‐wide copy‐number variation analysis using a high‐resolution array in a cohort comprising mainly Japanese subjects identified a schizophrenic patient with a novel exonic deletion in RELN.24 We further confirmed that the relative amount of reelin in serum was lower in this subject17 and established induced pluripotent stem cells (iPSC) from the subject.25, 26, 27 Several clinical studies suggest that reduced reelin in the brain and peripheral blood might be associated with mental disorders.28, 29, 30, 31
Behavioral changes in Reln‐mutant mice, reported by numerous studies, exhibit schizophrenia‐like phenotypes (Table 1)5, 31, 32, 33, 34; however, insights into the biological mechanisms during brain development due to reelin reduction in humans remain limited. One clue regarding this issue comes from our recent quantitative single‐cell time‐lapse analysis of neuronal differentiation and migration using Reln‐del iPSC revealing the developmental significance of reelin in migrating directionality.25 To examine directional abnormality in neuronal migration observed in human RELN‐deleted iPSC in vivo, we generated a mouse line in the C57BL/6J strain harboring the specific Reln deletion identified by us in a Japanese subject with schizophrenia, and named the line Reln‐del mice. Our behavioral analyses revealed abnormal sociability in heterozygous Reln‐del mice. This novel Reln‐del mouse model may be valuable for understanding reelin signaling, and for studies on the pathological mechanisms of schizophrenia via the reelin deletion.
Table 1.
Comparison of behavioral changes in Reln mutant mice
| Sawahata et al., 2019 (Present study) | Sobue et al. (2018)17 | Lalonde et al. (200433 | Salinger et al. (200334 | Qiu et al. (2006)5 | Sakai et al. (2016)31 | |
|---|---|---|---|---|---|---|
| Mutant mice strains (Background) | Reln‐del (C57BL/6) | Orleans hetero (BALB/C) | Orleans homo (BALB/C) | Jackson hetero (B6C3Fe) | Jackson hetero (B6C3Fe) | ⊿C‐KI (C57BL/6) |
| Age, sex | 10–19 weeks, male/female | 10–15 weeks, male/female | 12 weeks, male | 10 weeks, male | 6 weeks, male | 11 weeks, male |
| Stationary beams | NA | NA | ↓ | NA | NA | NA |
| Acoustic responsiveness | NA | NA | NA | NA | NA | NA |
| Wire hang latency | NA | NA | NA | NA | NA | ↓ |
| Locomotor activity | = | = | ↑ | NA | NA | NA |
| Open field | = | ↑ | ↑ | = | = | ↑ |
| Elevated plus maze | = | = | ↓ | NA | = | ↑ |
| Tail suspension test | NA | NA | NA | NA | NA | = |
| Porsolt forced swim test | NA | NA | NA | NA | NA | = |
| Y‐maze | = | = | NA | NA | NA | = |
| Barnes maze test | NA | NA | NA | NA | NA | = |
| T‐maze | NA | NA | = | NA | NA | ↓ (Working memory) |
| Novel object recognition | = | = | NA | ↓ | NA | NA |
| Social interaction test | ↓ (Social novelty) | ↓ | NA | = | NA | ↓ |
| Rotarod test | = | ↓ | ↓ | NA | = | ↑ |
| Fear conditioning test | = | = | NA | = | ↓ (Context) | = |
| Water maze test | NA | NA | ↓ | NA | = | NA |
| Prepulse inhibition | = | ↑ (Acoustic response) | NA | = | ↓ (82 dB) | = |
| MK801‐induced hyperlocomotion | NA | = | NA | NA | NA | NA |
| METH‐induced hyperlocomotion | NA | ↓ | NA | NA | NA | NA |
↑, higher than wild‐type; ↓, lower than wild‐type; =, no difference; NA, not applicable.
Methods
CRISPR/Cas plasmid for Reln deletion
A pair of oligo DNAs (Invitrogen, Carlsbad, CA, USA) corresponding to Reln guide RNA was hybridized and ligated using T4 DNA ligase (TOYOBO, Osaka, Japan) into linearized pSpCas9(BB)‐2A‐GFP (PX458) plasmid (Addgene plasmid # 48138; http://n2t.net/addgene:48138; RRID:Addgene_48138) digested with BbsI (NEB, Ipswich, MA, USA) as previously described.35, 36, 37 DNA primers are listed in Table S2.
Animal experiments
All research and animal care procedures were approved by the Nagoya University Animal Care and Use Committee. Mice were housed in groups of maximum six animals per cage and maintained on a regular 12‐h light/dark cycle (09:00 to 21:00 hours light period) at a constant 23°C temperature. Food and water were available ad libitum. Heterozygous Reln‐del mice were generated by intercrossing Reln‐del males and C57BL/6J females. Littermate WT C57BL/6J mice were used as controls, and 10–19‐week‐old Reln‐del and WT mice were used in all experiments. All animal protocols were approved by the Animal Care and Use Committee of Nagoya University Graduate School of Medicine; in addition, the Principles for the Care and Use of Laboratory Animals, which were approved by the Japanese Pharmacological Society, and the National Institutes of Health Guide for the Care and Use of Laboratory Animals were followed.
Other materials and methods are described in Supplementary materials.
Results
Generation of Reln‐del mice
We previously identified a schizophrenia subject harboring a partial RELN deletion in chr 7 (102919640–102930809, NCBI36/hg18), which includes the region encompassing exons 52–58 encoding reelin repeats 7–8 (Fig. 1a).17, 24 We generated a genetically modified mouse corresponding to this deletion in RELN by genome editing using the CRISPR/Cas9 method; the strain was C57BL/6J, the standard murine line for general behavioral analyses. The Reln gene structure between human and mouse is highly conserved. First, we designed 2 CRISPR‐targeted guide RNAs and a donor DNA for inserting a FLAG‐tag sequence and a stop codon in exon 52 to generate the Reln‐del mouse model (Fig. 1b,c). When two guide RNAs were transiently transfected into the mouse neuroblastoma cell line Neuro2a, we found that the #4 guide RNA showed the strongest cleavage activity by the T7E1 assay (Fig. 1d). We examined the possibility of the off‐target effect of the #4 guide RNA using the CRISPR Design tool supported by MIT (http://crispr.mit.edu/) and confirmed that there were no pseudo‐sequences due to a mismatch of less than three base pairs in the whole mouse coding regions (Table S1). The #4 guide RNA was chemically synthesized as a Crispr‐RNA, and tracr‐RNA, Cas9 protein, and the donor DNA were co‐microinjected into 171 fertilized eggs in C57BL/6J mice bred at Charles River, which were cultured up to the two‐cell stage. Next, we transplanted 119 eggs into the oviducts of pseudopregnant mice, and 18 littermates were born. As a result, we confirmed that six knock‐in mice were obtained as designed by polymerase chain reaction (PCR) cloning (Fig. 1e).
Figure 1.
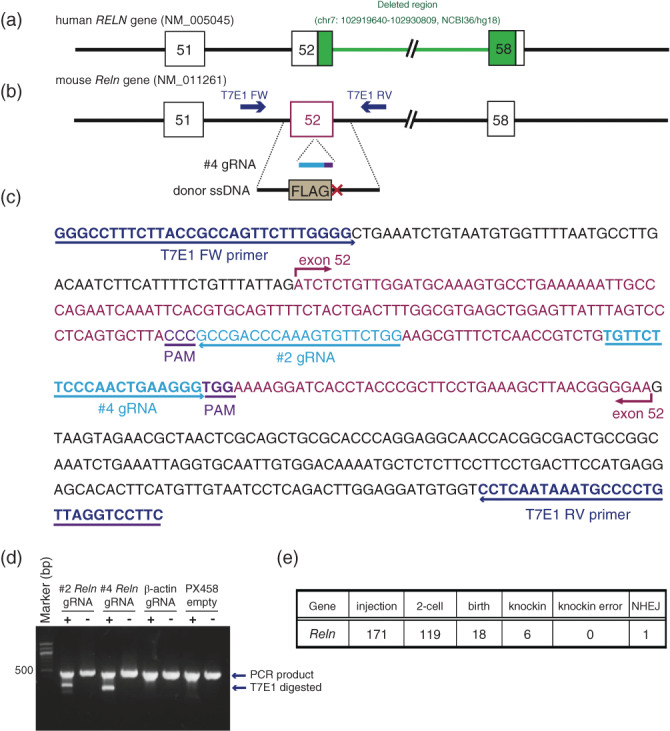
Generation of Reln‐del mice by CRISPR/Cas9. Schematic RELN gene structure in (a) human and (b) mouse. (a) The genomic region (exon 52 to 58) that was deleted in the Japanese subject with schizophrenia is shown in green.24 (b) CRISPR/Cas9 strategy for generating the model mouse with Reln deletion as shown in (a). Guide RNAs (gRNA) and single‐stranded DNA (ssDNA) for insertion of a stop codon in exon 52 of Reln gene. (c) Sequence around mouse Reln exon 52 and design of gRNA for CRISPR target and primers to perform the T7 endonuclease I (T7EI) assay for cleavage activity. (d) Results of the T7EI assay. The amount of shortened DNA shows cleavage activity by T7EI. (e) Summary of the results following injection of the CRISPR mixture into fertilized C57BL/6J egg pronucleus.
Validation of Reln‐del mice
Screening of the generated mice by conventional genotyping was achieved with three primers designed to distinguish wild‐type (WT) and mutant (Reln‐del) alleles by agarose gel electrophoresis (Fig. 2a,b, Table S2). Homozygous Reln‐del mice were born normally and exhibited normal growth until 21 days after birth. However, Reln‐del mice exhibited phenotypic similarities to staggerer and other reeler mice (Fig. 2c, Movie S1).
Figure 2.
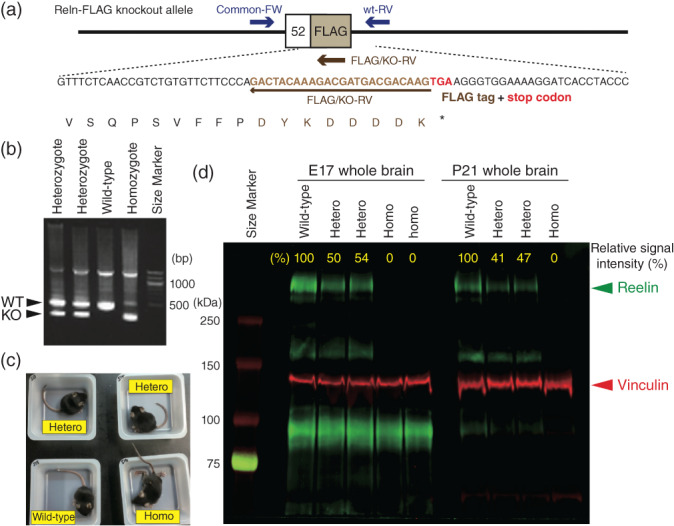
Validation of Reln‐del mice. (a) Primer design for genotyping Reln‐del mice. (b) Polymerase chain reaction for genotyping Reln‐del mice. Larger bands indicate wild‐type (WT) allele, and smaller bands indicate mutant allele. (c) Appearance of Reln‐del mouse 21 days after birth. Homozygous Reln‐del mice staggered (see also Supplementary Movie S1). (d) Immunoblot analysis to confirm reelin deletion in Reln‐del mouse brain. Fifty‐microgram total lysate from the whole brain of postnatal day 21 mice were loaded in each well. The signal was detected by an Odyssey imaging platform. The molecular weight of full‐length reelin protein is approximately 388 kDa, which is indicated by the green signal. The internal control vinculin (117kDa) is indicated by the red signal. The expression level of full‐length reelin relative to that of vinculin was calculated by the software attached to Odyssey and is shown in yellow font.
We also validated the reelin protein expression level in the novel Reln‐del mice by immunoblotting (Fig. 2d). In embryonic day 17 and postnatal day 21, the expression of reelin in heterozygous Reln‐del mice was approximately half the reelin expression in WT mice; reelin was detected barely in homozygous Reln‐del mice in those ages. Expression of vinculin as an internal control was comparable between the genotypes. The specific reelin antibody used in the current study could not detect lower molecular‐weight bands by immunoblotting in homozygous Reln‐del mice, indicating that they were null mutants with no truncated reelin.
Histological analysis of Reln‐del mice
We investigated histological changes in Reln‐del mice. Hematoxylin/eosin‐stained brain slices of homozygous Reln‐del mice showed severe brain malformation (Fig. 3). Specifically, cerebellar atrophy, enlargement of the cerebral ventricles (Fig. 3a–c) and cerebral dysplasia (Fig. 3d–f), which were one of the typical reeler phenotypes38, 39, 40, 41 were remarkable. In addition, we observed hippocampal malformation and disruption of the dentate gyrus and granule layer (Fig. 3g–i). On the other hand, heterozygous Reln‐del mice did not show severe brain malformation like homozygous Reln‐del mice.
Figure 3.
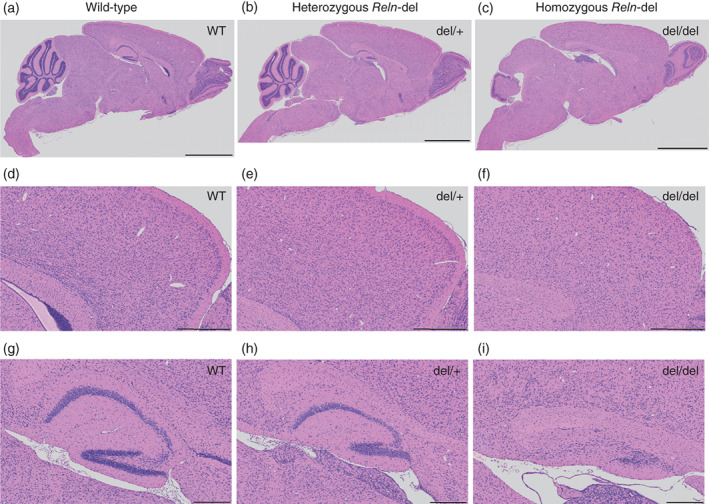
Histological analysis of brain in Reln‐del mice. (a–c) Hematoxylin/eosin (HE)‐stained sagittal sections of whole brain in (a) wild‐type (WT) mice, (b) heterozygous Reln‐del mice, and (c) homozygous Reln‐del mice. Scale bars: 2 mm. (d–f) HE‐stained cortical sections, highly magnified from those presented in (a–c). Scale bars: 500 μm. (g–i) HE‐stained hippocampal sections, highly magnified from those presented in (a–c). Scale bars: 200 μm. All sections are from 2–3‐week‐old mice.
Behavioral analyses of Reln‐del mice
Reln mutation is a risk factor in several mental disorders, such as schizophrenia and autism spectrum disorder. Therefore, we performed global behavioral analyses using the following 10 behavioral tests to determine whether Reln‐del mice were a relevant model for schizophrenia: locomotor, rotarod, open field, elevated plus maze (EPM), social interaction, marble burying, PPI, Y‐maze, novel object recognition, and fear conditioning tests.
We used heterozygous Reln‐del mice for all behavioral analyses, because nearly all homozygous Reln‐del mice were hypoplastic and had difficulty in reaching the adult stage. Additionally, the original Japanese subject that was the basis of our studies harbored a heterozygous Reln deletion.17
First, we evaluated the general locomotor function in heterozygous Reln‐del mice, because homozygous mice showed severe incoordination and cerebellar atrophy, which may change motor, emotional, and cognitive function. Heterozygous Reln‐del mice showed normal locomotor activity and coordinated movement in the locomotor and the rotarod tests (Fig. 4a–d). These data show that we could evaluate emotional and cognitive functions of heterozygous Reln‐del mice with normal motor function.
Figure 4.
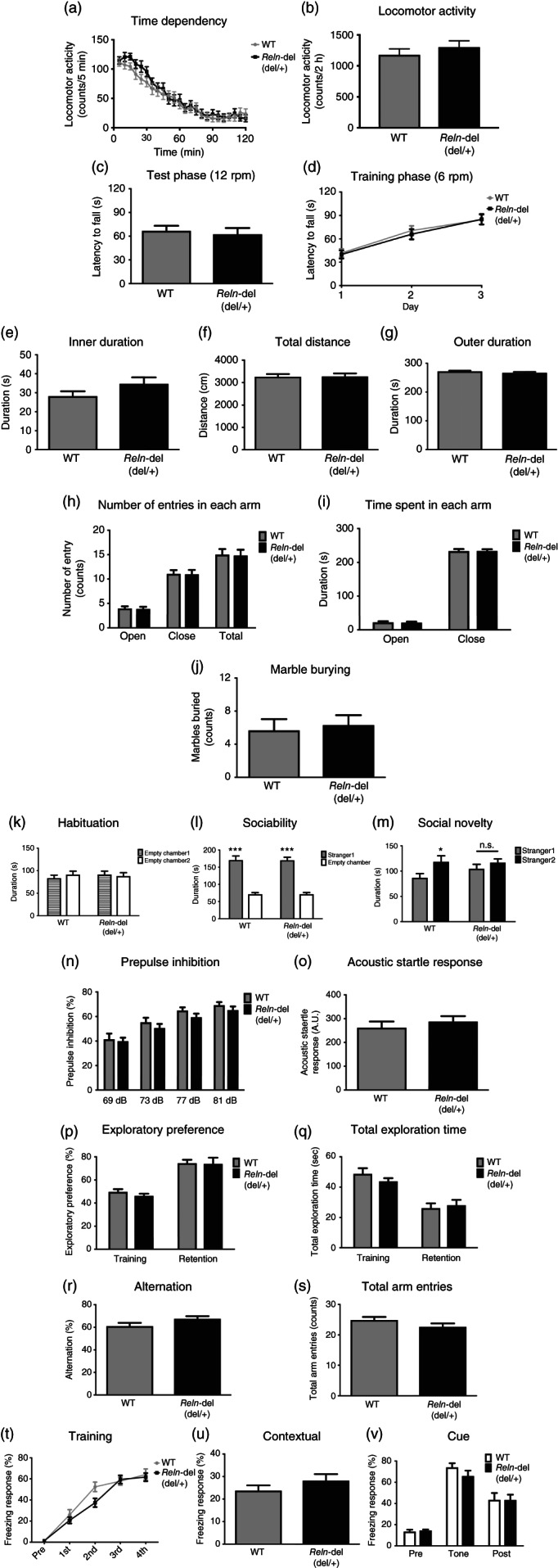
Behavioral analyses in heterozygous Reln‐del mice. Performance in the (a,b) locomotor activity, (c,d) rotarod, (e–g) open field, (h,i) elevated plus maze, (j) marble‐burying, (n,o) pre‐pulse inhibition, (p,q) novel object recognition, (r,s) Y‐maze, and (t–v) fear‐conditioning tests. Data represent the mean ± standard error of the mean (SEM; n = 24 for wild‐type [WT] mice, n = 26 for Reln‐del mice in Fig. 4a,b; n = 24 for WT mice, n = 26 for Reln‐del mice in Fig. 4c,d; n = 24 for WT mice, n = 27 for Reln‐del mice in Fig. 4e–g; n = 24 for WT mice, n = 25 for Reln‐del mice in Fig. 4f,i; n = 12 for WT mice, n = 13 for Reln‐del mice in Fig. 4j; n = 24 for WT mice, n = 26 for Reln‐del mice in Fig. 4n,o; n = 12 for WT mice, n = 13 for Reln‐del mice in Fig. 4p,q; n = 22 for WT mice, n = 22 for Reln‐del mice in Fig. 4r,s; n = 12 for WT mice, n = 13 for Reln‐del mice in Fig. 4t–v). (k–m) Performance in the three‐chambered social interaction test: (k) habituation, (l) sociability, and (m) social novelty sessions. Data represent the mean ± SEM (n = 18–20 for WT mice, n = 20 for Reln‐del mice). ***P < 0.001 between empty chamber and stranger 1 or *P < 0.05 between stranger 1 and stranger 2 (two‐way analysis of variance with post‐hoc Sidak's multiple comparisons test).
A previous study has reported that heterozygous reeler mice have reduced anxiety.42 Therefore, we evaluated anxiety in the open field and EPM tests. Typically, WT mice spent more time in the periphery than the center of the circle in the open field test and preferred the closed arms compared with the open arms in the EPM test, given that the center of the circle and the open arm were sources of anxiety in mice. Importantly, there were no significant differences between the WT and Reln‐del mice in both tests (Fig. 4e–i). To investigate anxiety and irrational behavior, we performed the marble‐burying test and found that Reln‐del mice showed normal behavior (Fig. 4j).
Impairments of social behavior and sensorimotor gating function are the main symptoms of schizophrenia. To examine these points of heterozygous Reln‐del mice, we performed the three‐chambered social interaction and the PPI tests. There were no significant differences in habituation or sociability sessions between the WT and heterozygous Reln‐del mice (Fig. 4k,l). In the social novelty session, WT mice significantly preferred the novel mouse (stranger 2) than the familiar mouse (stranger 1). Conversely, heterozygous Reln‐del mice showed the same interest in both strangers 1 and 2 (Fig. 4m). In the PPI test, heterozygous Reln‐del mice exhibited a phenotype similar to that of WT mice (Fig. 4n,o). The difference may have caused low attention to stranger mice, but not anxiety to the strangers and some kind of recognition, as the levels of WT and heterozygous Reln‐del mice were not different in open field (Fig. 4e–g), EPM (Fig. 4h,i), or novel object recognition tests (Fig. 4p,q). Further in heterozygous Reln‐del mice, Reln‐del mice exhibited no significant phenotypes in memory function in the Y‐maze and fear‐conditioning test (Fig. 4r–v).
Cerebellar granule cell migration in Reln‐del mice
We performed the time‐lapse analysis to assess migration in cerebellar granule cells isolated from Reln‐del mice and conducted biochemical analysis of reelin signaling as well. Reelin binding to its target receptor, such as apolipoprotein E receptor 2 and very‐low‐density‐lipoprotein receptor (VLDLR), leads to the phosphorylation of the adaptor protein Dab1.43 We prepared cerebellar granule neurons from postnatal WT and Reln‐del mice for reaggregation, which is often used to study neuronal migration,44 and conducted a 3‐day time‐lapse analysis (Fig. 5, [Link], [Link]). Then, we performed automatic recognition and quantification of the cell body and neurite length (Fig. S1). When comparing the cerebellar granule neuronal cultures of the WT and heterozygous Reln‐del mutant mice, both the average distance covered during neuronal migration from the reaggregates (WT: 97.69 μm [72 cells]; heterozygous Reln‐del: 91.87 μm [73 cells]) and the average neurite length (WT: 21.41 μm [10 views]; heterozygous Reln‐del: 16.82 μm [10 views]) were not significantly different (Fig. S1); however, reaggregates from homozygous Reln‐del mice were affected by cerebellar hypoplasia and showed severe structural defects compared with those from both WT and heterozygous Reln‐del mice; therefore, homozygous Reln‐del mice were difficult to quantify (Fig. 5c, Movie S4). Next, we analyzed the reelin signaling pathway in these cerebellar neuronal reaggregation cultures. First, we confirmed that reelin was highly expressed in WT cultures and that the reelin protein expression level was reduced in cultures from heterozygous Reln‐del mice (Fig. 5d), whereas the expression levels of the internal control vinculin and the dopaminergic neuronal marker tyrosine hydroxylase were unchanged (Fig. S2a). Finally, we examined the reelin signaling quantitatively by analyzing the relative expression levels of p‐Dab1 and total Dab1 (Figs. S2b,c) and found that the phosphorylation of Dab1 was likely impaired in cultures from heterozygous Reln‐del mice.
Figure 5.
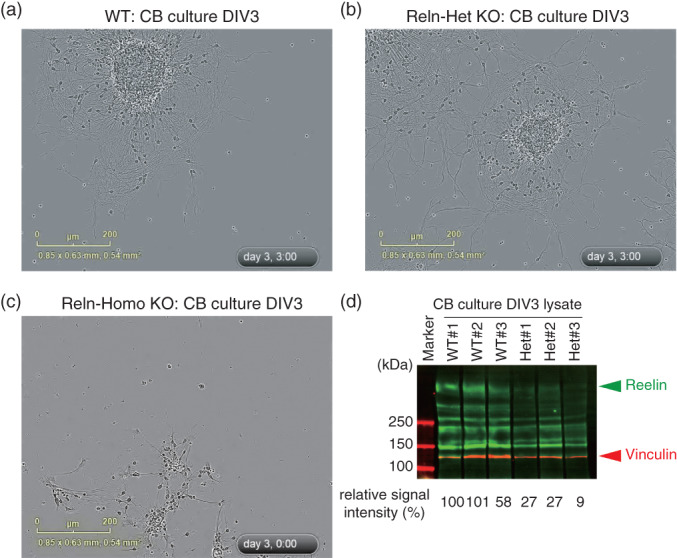
Reln‐del cerebellar reaggregation culture. (a–c) Time‐lapse movies of cerebellar reaggregation cultures during days 0–3. Final point of each movie in cultures from (a) wild‐type (WT), (b) heterozygous Reln‐del, and (c) homozygous Reln‐del mice. Full movies are in [Link], [Link]. (d) Immunoblot analysis of Reln‐del cerebellar reaggregation cultures at day 3. The immunoblots were probed with antibodies against reelin.
Discussion
Disorders of neuronal migration in humans result from the disruption of the normal movement of neurons from their original site of birth to their final destination during early development.45 Mutations of many genes are involved in neuronal migration disorders and include LIS1 and DCX in classical lissencephaly spectrum, TUBA1A in microlissencephaly with agenesis of the corpus callosum, and RELN and VLDLR in lissencephaly with cerebellar hypoplasia.8, 45 Genetic studies on rare variants implicate that RELN is associated with schizophrenia.19, 46, 47 Furthermore, our copy‐number variation analysis identified a subject with schizophrenia who harbored a novel exonic deletion in RELN.24 As reelin plays important roles in various pathways from development to postnatal function, Reln‐del mice are expected to be a reasonable schizophrenia model that can reveal the pathogenic mechanisms underlying schizophrenia. Future studies can facilitate accumulation of sufficient genetic data and clinical phenotypes in humans for elucidation of the molecular mechanisms of schizophrenia caused by a decrease in reelin expression.
In the current study, we generated a novel Reln‐del mouse model to mimic the Japanese subject with schizophrenia harboring the specific exonic deletion in RELN and compared it with the findings of previous studies on other reeler‐associated models, including Orleans reeler mice (Table 1). Specifically, we designed a Reln‐del mutant allele carrying mutations to code a FLAG‐tag and stop codon in exon 52, corresponding to the amino acid residues within the seventh and eighth reelin repeats (Fig. 1). The homozygous Reln‐del mice exhibited the typical phenotypes of the staggerer as well as the classical reeler mice (Fig. 2c).38 Immunoblotting of whole‐brain lysates using an anti‐reelin antibody recognizing the N‐terminal region of reelin revealed that the endogenous reelin level, which was barely detectable in homozygous Reln‐del brain lysates, was approximately half of that in WT mice in heterozygous Reln‐del mice (Fig. 2d). In contrast, Orleans reeler mice have a transcriptional reelin product with a 220‐bp deletion within the seventh and eighth reelin repeats and potentially express a truncated reelin.11, 12 Our novel Reln‐del mice more faithfully reflect the Japanese subject with the RELN deletion compared with our previous study using Orleans reeler mice.17 In addition, we generated the Reln‐del mice in the C57BL/6J strain that is commonly used for behavior studies in schizophrenia models, distinct from Orleans reeler mice, which were maintained in the Balb/c strain. Differences of inbred strains and/or reelin products might underlie differences in the behavioral phenotypes between the mouse models (Table 1).
Histological analysis showed cerebellar atrophy, hippocampal dysgenesis, and cerebral dysplasia in homozygous Reln‐del (Fig. 3). These phenotypes are similar to those of both Jackson and Orleans reeler homozygous mice.38, 39, 40, 41 On the other hand, we found that heterozygous Reln‐del mice did not show severe brain malformation like homozygous Reln‐del mice (Fig. 3). These results are similar to those reported by previous other studies.38, 41
As the results of global behavioral analyses, we found abnormal social novelty in heterozygous Reln‐del mice (Fig. 4), indicating that heterozygous Reln‐del mice partially mimicked the Japanese subject with schizophrenia who harbors the same RELN mutation. Several studies reported that Relnrl‐J/+ mice showed abnormal behaviors, such as PPI deficits,5, 48 unusual anxiety,42 and reduced cognition and sociability.5, 16 These differences may be associated with the novel RELN mutation in heterozygous Reln‐del mice. However, the expression pattern of reelin protein in heterozygous Reln‐del was similar to that of Relnrl‐J mice based on our immunoblot analysis, which indicate that the reelin‐deficient phenotype might be minimally affected by the growth environment or genetic background. It was reported that reelin related with neuronal plasticity and spine formation.7 Most heterozygous reelin mutant mice, including our mice, showed abnormal behaviors without clear histological changes.5, 17, 31, 33, 34 These kinds of abnormal behaviors may be caused by minimal changes, such as neuronal plasticity and spine formation. Approximately half the levels of reelin may be sufficient for normal neuronal development in the critical developmental stages because reelin signaling includes autoinhibition systems, such as Dab1 reduction. However, reelin is invaluable in the adult stage and has some neuronal function because expression levels are less than those observed in the developmental stage and distribution is different. These phenomena suggest that there is a lack of gene‐dose dependency in mice with Reln‐del.
A study in humans showed that patients with lissencephaly carrying RELN mutations in splicing recognition sites also exhibited cerebellar hypoplasia due to a decrease in full‐length reelin,8 in agreement with the features such as altered neuronal differentiation and migration observed in our in vitro phenotypes.44 Specifically, we showed severe structural defects in homozygous Reln‐del cultures (Fig. 5c and Movie S4), which closely associate with cerebellar hypoplasia,8 indicating a potentially reliable lissencephaly model. Conversely, neurons in heterozygous Reln‐del cultures are expected to be more likely a good model of schizophrenia based on genetic studies.17, 24 Neurons in heterozygous Reln‐del cultures showed an unmarked phenotype of migration abnormality, but there were no differences in gene dose between neurite migration and elongation in vitro (Fig. 5a,b, Fig. S1, and [Link], [Link]).
The iPSC technology is a promising tool for creating a new generation of pathophysiological assays for in vitro drug screening, which allows time‐course analysis of neurons from patient‐derived iPSC that are not readily available in human studies, and recent studies utilizing iPSC facilitate the interrogation of molecular pathways and cellular dynamics in humans.49 Nonetheless, the pathogenesis of schizophrenia remains poorly understood, and analysis in animal models is necessary for understanding the neural circuits and tissue structures in schizophrenia. Using reliable and predictive mouse models is essential to understand the neurobiological basis of schizophrenia and the development of novel drugs with improved therapeutic efficacy.50
Our previous trajectory analysis of the highly homologous neurons, such as tyrosine‐hydroxylase‐positive neurons from iPSC, is one of the best ways to understand the mechanisms of microstructural defects in the brains of subjects with schizophrenia.25, 51 Cerebellar neurons from homozygous Reln‐del mice showed a very severe migration defect that was similar to the outgrowth abnormality of tyrosine‐hydroxylase‐positive neurons derived from iPSC of the Japanese subject with Reln‐del. In our in vitro analysis, the cell bodies and neurites of the cerebellar reaggregates were too fine, and the population was too dense, which hindered the measurement of the area of each cell body and the trajectory analysis. Overcoming these technical issues will allow for more precise comparisons of neuronal dynamics and morphology in these cultures with those of human iPSC‐derived neurons. In vitro analyses alone are insufficient to elucidate the molecular pathology of individuals by behavioral analyses of schizophrenia model mice. Advantages of highly homologous differentiation of human iPSC into specific neuronal subtypes will significantly aid in consideration of in vitro analyses, such as reelin signaling during migrating, probably promoting the development of reelin‐related therapies.
In conclusion, our in vitro and in vivo data indicate that the novel Reln‐del mice, using CRISPR/Cas9, which partially mimicked schizophrenia‐like behavior, provide a novel platform for elucidation of the pathogenesis underlying schizophrenia and the development of reelin‐based therapies.
Disclosure statement
The authors have no conflicts of interest to declare.
Author contributions
M.S. and D.M. wrote the main text and prepared most of the figures. D.M. generated the Reln‐del mice. M.S. performed most of the behavioral and histological experiments. D.M., Y.A., H.K., I.K., K.K., A.S., E.S., M.S., A.K., R.I., B.A., H.K., and K.I. performed molecular and cellular biological experiments. All other authors commented on and refined the manuscript. N.O., K.Y., T.N., T.N., and K.K. supervised the overall project. All authors have carefully read the paper and approved the final manuscript.
Supporting information
Table S1. Mutations in coding regions at putative off‐target loci containing up to 4 bp mismatches for Reln target are listed. This list was calculated by following CRISPR design tool: http://crispr.mit.edu/. Mismatches compared to on‐target sequence are shown in red. PAM sequences are labeled in green.
Table S2. DNA primers used in this study.
Figure S1. Quantitative analysis of cerebellar reaggregation culture (refer to Fig. 5). (a–c) Cell body (green) and neurite (magenta) were automatically recognized by NeuroTrack software from the data captured by time‐lapse imaging with IncuCyte Zoom [(a) WT, (b) heterozygous Reln‐del, and (c) homozygous Reln‐del]. (d) Measurements of the distance of the cell body (recognized by NeuroTrack) from the aggregate. The migration distances of 72 cells in the WT and 73 cells in the heterozygous Reln‐del were measured; no significant difference was found when the measurements were compared by t‐test using Prism8 software. In the homozygous Reln‐del, the aggregate was hardly formed, and it was therefore impossible to measure any migration distance. (e) The length of neurites recognized by NeuroTrack was quantified. In WT and heterozygous Reln‐del, the lengths of neurites in 10 fields were compared by multiple t‐tests using Prism8 software and no significant difference was found.
Figure S2. Reln‐del cerebellar reaggregation culture (refer to Fig. 5). Immunoblot analysis of Reln‐del cerebellar reaggregation cultures at Day 3. The immunoblots were probed with antibodies against (a) tyrosine hydroxylase (TH), (b) disabled 1 (DAB1) (pY232), and (c) phospho‐DAB1(p‐DAB) together with antibody against glyceraldehyde 3‐phosphate dehydrogenase as an internal control and quantified by the Odyssey detection system.
Movie S1. Behaviors of Reln‐del mice (refer to Fig. 2c).
Movie S2. Migrations of neurons from WT cerebellar aggregation culture (refer to Fig. 5a).
Movie S3. Migrations of neurons from heterozygous Reln‐del cerebellar aggregation culture (refer to Fig. 5b).
Movie S4. Migrations of neurons from homozygous Reln‐del cerebellar aggregation culture (refer to Fig. 5c).
Appendix S1. Supporting information.
Acknowledgments
We thank the Division for Research of Laboratory Animals; the Division for Medical Research Engineering for animal care and use; and the Division of Psychiatry and Brain and Mind Research Center. We also thank Enago (www.enago.jp) for the English‐language review. This study was supported by AMED under Grants JP19dm0107087, JP19dm0207005h, JP19ak0101113, JP19dk0307081; Japan Society for the Promotion of Science (JSPS) KAKENHI Grants 23110506, 23700443, 25110715, 25460284, 17H04252, and 18H04040; the Private University Research Branding Project from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT). Innovative Areas ‘Glial assembly: a new regulatory machinery of brain function and disorders’; Nagoya University Hospital Funding for Clinical Research; The NAKAJIMA Foundation, and the SENSHIN Medical Research Foundation.
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1. Alcantara S, Ruiz M, D'Arcangelo G et al Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J. Neurosci. 1998; 18: 7779–7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ogawa M, Miyata T, Nakajima K et al The reeler gene‐associated antigen on Cajal‐Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron 1995; 14: 899–912. [DOI] [PubMed] [Google Scholar]
- 3. Tissir F, Goffinet AM. Reelin and brain development. Nat. Rev. Neurosci. 2003; 4: 496–505. [DOI] [PubMed] [Google Scholar]
- 4. Mariani J, Crepel F, Mikoshiba K, Changeux JP, Sotelo C. Anatomical, physiological and biochemical studies of the cerebellum from Reeler mutant mouse. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1977; 281: 1–28. [DOI] [PubMed] [Google Scholar]
- 5. Qiu S, Korwek KM, Pratt‐Davis AR, Peters M, Bergman MY, Weeber EJ. Cognitive disruption and altered hippocampus synaptic function in Reelin haploinsufficient mice. Neurobiol. Learn. Mem. 2006; 85: 228–242. [DOI] [PubMed] [Google Scholar]
- 6. Herz J, Chen Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat. Rev. Neurosci. 2006; 7: 850–859. [DOI] [PubMed] [Google Scholar]
- 7. Rogers JT, Rusiana I, Trotter J et al Reelin supplementation enhances cognitive ability, synaptic plasticity, and dendritic spine density. Learn. Mem. 2011; 18: 558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hong SE, Shugart YY, Huang DT et al Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 2000; 26: 93–96. [DOI] [PubMed] [Google Scholar]
- 9. Hirota Y, Nakajima K. Control of neuronal migration and aggregation by Reelin Signaling in the developing cerebral cortex. Front. Cell Dev. Biol. 2017; 5: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ogino H, Hisanaga A, Kohno T et al Secreted metalloproteinase ADAMTS‐3 inactivates Reelin. J. Neurosci. 2017; 37: 3181–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bar I, Lambert De Rouvroit C, Royaux I et al A YAC contig containing the reeler locus with preliminary characterization of candidate gene fragments. Genomics 1995; 26: 543–549. [DOI] [PubMed] [Google Scholar]
- 12. Hirotsune S, Takahara T, Sasaki N et al The reeler gene encodes a protein with an EGF‐like motif expressed by pioneer neurons. Nat. Genet. 1995; 10: 77–83. [DOI] [PubMed] [Google Scholar]
- 13. Takahara T, Ohsumi T, Kuromitsu J et al Dysfunction of the Orleans reeler gene arising from exon skipping due to transposition of a full‐length copy of an active L1 sequence into the skipped exon. Hum. Mol. Genet. 1996; 5: 989–993. [DOI] [PubMed] [Google Scholar]
- 14. de Bergeyck V, Nakajima K, Lambert de Rouvroit C et al A truncated Reelin protein is produced but not secreted in the ‘Orleans’ reeler mutation (Reln[rl‐Orl]). Brain Res. Mol. Brain Res. 1997; 50: 85–90. [DOI] [PubMed] [Google Scholar]
- 15. Derer P, Derer M, Goffinet A. Axonal secretion of Reelin by Cajal‐Retzius cells: Evidence from comparison of normal and Reln(Orl) mutant mice. J. Comp. Neurol. 2001; 440: 136–143. [DOI] [PubMed] [Google Scholar]
- 16. Rogers JT, Zhao L, Trotter JH et al Reelin supplementation recovers sensorimotor gating, synaptic plasticity and associative learning deficits in the heterozygous reeler mouse. J. Psychopharmacol. 2013; 27: 386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sobue A, Kushima I, Nagai T et al Genetic and animal model analyses reveal the pathogenic role of a novel deletion of RELN in schizophrenia. Sci. Rep. 2018; 8: 13046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ishii K, Nagai T, Hirota Y et al Reelin has a preventive effect on phencyclidine‐induced cognitive and sensory‐motor gating deficits. Neurosci. Res. 2015; 96: 30–36. [DOI] [PubMed] [Google Scholar]
- 19. Folsom TD, Fatemi SH. The involvement of Reelin in neurodevelopmental disorders. Neuropharmacology 2013; 68: 122–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ishii K, Kubo KI, Nakajima K. Reelin and neuropsychiatric disorders. Front. Cell. Neurosci. 2016; 10: 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fromer M, Pocklington AJ, Kavanagh DH et al De novo mutations in schizophrenia implicate synaptic networks. Nature 2014; 506: 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou Z, Hu Z, Zhang L et al Identification of RELN variation p.Thr3192Ser in a Chinese family with schizophrenia. Sci. Rep. 2016; 6: 24327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Costain G, Lionel AC, Merico D et al Pathogenic rare copy number variants in community‐based schizophrenia suggest a potential role for clinical microarrays. Hum. Mol. Genet. 2013; 22: 4485–4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kushima I, Aleksic B, Nakatochi M et al High‐resolution copy number variation analysis of schizophrenia in Japan. Mol. Psychiatry 2017; 22: 430–440. [DOI] [PubMed] [Google Scholar]
- 25. Arioka Y, Shishido E, Kubo H et al Single‐cell trajectory analysis of human homogenous neurons carrying a rare RELN variant. Transl. Psychiatry 2018; 8: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arioka Y, Kushima I, Kubo H, Mori D, Ozaki N. Induced pluripotent stem cells derived from a schizophrenia patient with ASTN2 deletion. Stem Cell Res. 2018; 30: 81–84. [DOI] [PubMed] [Google Scholar]
- 27. Arioka Y, Kushima I, Mori D, Ozaki N. Three lines of induced pluripotent stem cells derived from a 15q11.2‐q13.1 duplication syndrome patient. Stem Cell Res. 2018; 31: 240–243. [DOI] [PubMed] [Google Scholar]
- 28. Guidotti A, Auta J, Davis JM et al Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: A postmortem brain study. Arch. Gen. Psychiatry 2000; 57: 1061–1069. [DOI] [PubMed] [Google Scholar]
- 29. Fatemi SH, Stary JM, Earle JA, Araghi‐Niknam M, Eagan E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr. Res. 2005; 72: 109–122. [DOI] [PubMed] [Google Scholar]
- 30. Eastwood SL, Harrison PJ. Cellular basis of reduced cortical reelin expression in schizophrenia. Am. J. Psychiatry 2006; 163: 540–542. [DOI] [PubMed] [Google Scholar]
- 31. Sakai K, Shoji H, Kohno T, Miyakawa T, Hattori M. Mice that lack the C‐terminal region of Reelin exhibit behavioral abnormalities related to neuropsychiatric disorders. Sci. Rep. 2016; 6: 28636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sobue A, Ito N, Nagai T et al Astroglial major histocompatibility complex class I following immune activation leads to behavioral and neuropathological changes. Glia 2018; 66: 1034–1052. [DOI] [PubMed] [Google Scholar]
- 33. Lalonde R, Hayzoun K, Derer M, Mariani J, Strazielle C. Neurobehavioral evaluation of Reln‐rl‐orl mutant mice and correlations with cytochrome oxidase activity. Neurosci. Res. 2004; 49: 297–305. [DOI] [PubMed] [Google Scholar]
- 34. Salinger WL, Ladrow P, Wheeler C. Behavioral phenotype of the reeler mutant mouse: Effects of RELN gene dosage and social isolation. Behav. Neurosci. 2003; 117: 1257–1275. [DOI] [PubMed] [Google Scholar]
- 35. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR‐Cas9 system. Nat. Protoc. 2013; 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cong L, Ran FA, Cox D et al Multiplex genome engineering using CRISPR/Cas systems. Science 2013; 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aida T, Chiyo K, Usami T et al Cloning‐free CRISPR/Cas system facilitates functional cassette knock‐in in mice. Genome Biol. 2015; 16: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hamburgh M. Analysis of the postnatal developmental effects of "Reeler," a neurological mutation in mice. A study in developmental genetics. Dev. Biol. 1963; 8: 165–185. [DOI] [PubMed] [Google Scholar]
- 39. Katsuyama Y, Terashima T. Developmental anatomy of reeler mutant mouse. Dev. Growth Differ. 2009; 51: 271–286. [DOI] [PubMed] [Google Scholar]
- 40. Guy J, Staiger JF. The functioning of a cortex without layers. Front. Neuroanat. 2017; 11: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Duveau V, Madhusudan A, Caleo M, Knuesel I, Fritschy JM. Impaired reelin processing and secretion by Cajal‐Retzius cells contributes to granule cell dispersion in a mouse model of temporal lobe epilepsy. Hippocampus 2011; 21: 935–944. [DOI] [PubMed] [Google Scholar]
- 42. Ognibene E, Adriani W, Granstrem O, Pieretti S, Laviola G. Impulsivity‐anxiety‐related behavior and profiles of morphine‐induced analgesia in heterozygous reeler mice. Brain Res. 2007; 1131: 173–180. [DOI] [PubMed] [Google Scholar]
- 43. Benhayon D, Magdaleno S, Curran T. Binding of purified Reelin to ApoER2 and VLDLR mediates tyrosine phosphorylation of Disabled‐1. Brain Res. Mol. Brain Res. 2003; 112: 33–45. [DOI] [PubMed] [Google Scholar]
- 44. Toyo‐Oka K, Sasaki S, Yano Y et al Recruitment of katanin p60 by phosphorylated NDEL1, an LIS1 interacting protein, is essential for mitotic cell division and neuronal migration. Hum. Mol. Genet. 2005; 14: 3113–3128. [DOI] [PubMed] [Google Scholar]
- 45. Kato M. Genotype‐phenotype correlation in neuronal migration disorders and cortical dysplasias. Front. Neurosci. 2015; 9: 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li W, Guo X, Xiao S. Evaluating the relationship between reelin gene variants (rs7341475 and rs262355) and schizophrenia: A meta‐analysis. Neurosci. Lett. 2015; 609: 42–47. [DOI] [PubMed] [Google Scholar]
- 47. Wang Z, Hong Y, Zou L et al Reelin gene variants and risk of autism spectrum disorders: An integrated meta‐analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014; 165B: 192–200. [DOI] [PubMed] [Google Scholar]
- 48. Tueting P, Costa E, Dwivedi Y et al The phenotypic characteristics of heterozygous reeler mouse. Neuroreport 1999; 10: 1329–1334. [DOI] [PubMed] [Google Scholar]
- 49. Falk A, Heine VM, Harwood AJ et al Modeling psychiatric disorders: From genomic findings to cellular phenotypes. Mol. Psychiatry 2016; 21: 1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jones CA, Watson DJ, Fone KC. Animal models of schizophrenia. Br. J. Pharmacol. 2011; 164: 1162–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Arioka Y, Hirata A, Kushima I, Aleksic B, Mori D, Ozaki N. Characterization of a schizophrenia patient with a rare RELN deletion by combining genomic and patient‐derived cell analyses. Schizophr. Res. 2019. 10.1016/j.schres.2019.10.038 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Mutations in coding regions at putative off‐target loci containing up to 4 bp mismatches for Reln target are listed. This list was calculated by following CRISPR design tool: http://crispr.mit.edu/. Mismatches compared to on‐target sequence are shown in red. PAM sequences are labeled in green.
Table S2. DNA primers used in this study.
Figure S1. Quantitative analysis of cerebellar reaggregation culture (refer to Fig. 5). (a–c) Cell body (green) and neurite (magenta) were automatically recognized by NeuroTrack software from the data captured by time‐lapse imaging with IncuCyte Zoom [(a) WT, (b) heterozygous Reln‐del, and (c) homozygous Reln‐del]. (d) Measurements of the distance of the cell body (recognized by NeuroTrack) from the aggregate. The migration distances of 72 cells in the WT and 73 cells in the heterozygous Reln‐del were measured; no significant difference was found when the measurements were compared by t‐test using Prism8 software. In the homozygous Reln‐del, the aggregate was hardly formed, and it was therefore impossible to measure any migration distance. (e) The length of neurites recognized by NeuroTrack was quantified. In WT and heterozygous Reln‐del, the lengths of neurites in 10 fields were compared by multiple t‐tests using Prism8 software and no significant difference was found.
Figure S2. Reln‐del cerebellar reaggregation culture (refer to Fig. 5). Immunoblot analysis of Reln‐del cerebellar reaggregation cultures at Day 3. The immunoblots were probed with antibodies against (a) tyrosine hydroxylase (TH), (b) disabled 1 (DAB1) (pY232), and (c) phospho‐DAB1(p‐DAB) together with antibody against glyceraldehyde 3‐phosphate dehydrogenase as an internal control and quantified by the Odyssey detection system.
Movie S1. Behaviors of Reln‐del mice (refer to Fig. 2c).
Movie S2. Migrations of neurons from WT cerebellar aggregation culture (refer to Fig. 5a).
Movie S3. Migrations of neurons from heterozygous Reln‐del cerebellar aggregation culture (refer to Fig. 5b).
Movie S4. Migrations of neurons from homozygous Reln‐del cerebellar aggregation culture (refer to Fig. 5c).
Appendix S1. Supporting information.