

Abstract
In vitro selected ribozymes are promising tools for site‐specific labeling of RNA. Previously known nucleic acid catalysts attached fluorescently labeled adenosine or guanosine derivatives through 2′,5′‐branched phosphodiester bonds to the RNA of interest. Herein, we report new ribozymes that use orthogonal substrates, derived from the antiviral drug tenofovir, and attach bioorthogonal functional groups, as well as affinity handles and fluorescent reporter units through a hydrolytically more stable phosphonate ester linkage. The tenofovir transferase ribozymes were identified by in vitro selection and are orthogonal to nucleotide transferase ribozymes. As genetically encodable functional RNAs, these ribozymes may be developed for potential cellular applications. The orthogonal ribozymes addressed desired target sites in large RNAs in vitro, as shown by fluorescent labeling of E. coli 16S and 23S rRNAs in total cellular RNA.
Keywords: antiviral nucleoside analogues, in vitro selection, ribozymes, site-specific RNA labeling, tenofovir
A strategy for illuminating RNA with fluorescent derivatives of the antiviral acyclic nucleoside phosphonate Tenofovir is presented. Tenofovir transferase ribozymes were identified by in vitro selection and are shown to be orthogonal to nucleotidyl transferase ribozymes, thus enabling site‐specific dual labeling of RNA.
Site‐specific labeling of RNA by covalent attachment of a bioorthogonal functional group or fluorescent reporter is of fundamental importance for biochemical and biophysical studies and in the areas of cellular and synthetic biology.1 Non‐covalent labeling of RNA has been pursued by fluorogen‐activating aptamers that are inserted into the RNA of interest as large fluorescent tags.2 Covalent labeling with small organic fluorophores through bioorthogonal linkages has several advantages. While incorporation of modified nucleotides by solid‐phase synthesis is limited to short RNAs, several strategies are known to post‐synthetically address natural RNAs at terminal or internal positions.3 Selected examples include ribozymes for 3′‐terminal labeling,4 protein enzymes for labeling of the cap structure,5 and RNA–protein complexes to address specific nucleotides in a defined sequence context.6
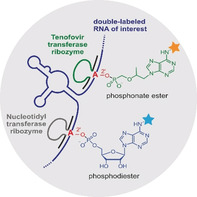
Nucleic acid catalysts, that is, ribozymes and deoxyribozymes can be engineered for any particular sequence and have successfully been used to address internal target sites for various in vitro applications.7 Recently, we reported the FH14 ribozyme to attach N 6‐labeled adenosine to specific internal 2′‐OH groups through 2′,5′‐phosphodiester bonds.8 This ribozyme was identified by direct in vitro selection with N 6‐modified ATP as substrate and a structured RNA library with a bulged adenosine as target site for labeling. The resulting ribozymes proved specific, efficient and generally applicable even in the context of cellular RNA. However, several difficulties arise when FH14 and related ribozymes are considered for potential future cellular applications. N 6‐modified ATP analogues would compete with cellular ATP and ATP‐dependent enzymatic processes, and result in ribozyme‐independent background labeling of RNA by polymerase incorporation.9 Moreover, the resulting 2′,5′‐phosphodiester linkage in the labeled RNA is easily cleaved by natural debranching enzymes.10 We sought to address these issues of lacking orthogonality and vulnerability to debranching enzymes by attachment of non‐natural nucleotide analogues. Specifically, we considered tenofovir diphosphate as a ribozyme substrate. Tenofovir is an FDA‐approved acyclic nucleotide phosphonate that is used as an antiviral drug against human immunodeficiency virus (HIV) and hepatitis B virus (HBV) infections.11 It is administered as a lipophilic cell‐permeable prodrug, in which the negatively charged phosphonate is masked as an isopropyl methyl carbonate ester (tenofovir disoproxil fumarate, TDF),12 or phosphonamidate (tenofovir alafenamide, TAF).13 Upon release by esterases, tenofovir is phosphorylated by cellular nucleotide kinases to tenofovir diphosphate, which is the active drug metabolite that acts as an obligatory chain‐terminator of viral polymerases such as retroviral reverse transcriptase.14 Tenofovir exhibits low cytotoxicity, is a poor substrate for mammalian polymerases,15 and would also be much less efficiently incorporated into RNA compared to ATP analogues. Ribozyme‐catalyzed ligation of tenofovir‐diphosphate to an internal 2′‐OH of any RNA of interest would result in a branched phosphonate ester linkage (Figure 1), which is expected to be more resistant against enzymatic hydrolysis (debranching) compared to a natural phosphodiester linkage. Tenofovir analogues could be equipped with bioorthogonal handles or fluorescent reporters, and thus represent attractive substrates for ribozyme‐catalyzed RNA labeling and visualization.
Figure 1.
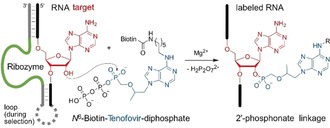
Ribozyme‐catalyzed site‐specific labeling of an RNA target using N 6‐biotinylated tenofovir diphosphate (biotin‐Ten‐DP), forming a phosphonate ester linkage with the 2′‐OH of the bulged adenosine (red) that is located in between two Watson–Crick base‐paired regions with the ribozyme's binding arms (grey). The RNA target sequence used for in vitro selection was 5′‐GGACAUACUGAGCCUUCAA.
The previously identified adenylyl‐transferase ribozyme FH148 was tested with tenofovir diphosphate, but it was not able to use the acyclic adenosine derivative as a substrate for RNA labeling (Figure S1). Therefore, we used in vitro selection from a structured RNA pool containing 40 random nucleotides to identify new ribozymes for site‐specific labeling of RNA with bioorthogonal derivatives of antiviral nucleoside analogues (the in vitro selection scheme is shown in the Supporting Information, Figure S2). We found several tenofovir‐transferase ribozymes (TTRs) that form a phosphonate ester with the 2′‐OH of the target adenosine using N 6‐modified tenofovir diphosphate as substrate (Figure 1).
Using Watson–Crick base‐paired binding arms, the ribozymes target a bulged adenosine in the RNA of interest and catalyze labeling of its 2′‐OH group. A biotinylated tenofovir substrate was used for a bead‐based in vitro selection strategy through streptavidin‐coated magnetic beads, in analogy to the previously established procedure (with N 6‐biotin‐ATP that resulted in FH ribozymes).8 The N 6‐biotinylated tenofovir diphosphate (biotin‐Ten‐DP) was synthesized from 6‐chloropurine in 7 steps (Scheme S1), in analogy to other N 6‐substituted [N‐(2‐phosphonomethoxy)alkyl adenines,16 and was used at a concentration of 200 μm with 20 mm Mg2+ at pH 7.4. The RNA library was partially labeled with Lucifer yellow at its 3′‐terminal ribose, and the progress of the in vitro selection was monitored by measuring fluorescence of eluted RNA after each selection round (Figure 2 a). The incubation time was gradually reduced after round 7 to enrich faster ribozymes. The pool was subjected to cloning after 12 rounds of selection, and Sanger sequencing identified two ribozymes, named FJ1 and FJ8. In addition, the round 7 and round 12 pools were analyzed by Illumina sequencing.17 Comparison of resulting sequence families (Figure 2 b and Figure S3) showed that FJ1 and FJ8 families were most enriched (with log2 ratios of 8.7 and 9.7, respectively), and FJC9 was identified as an additional unrelated ribozyme candidate (log2 ratio of 4.3) that proved worthwhile of more detailed investigation.18 FJ1 and FJ8 share some sequence segments (Figure 2 c), although their predicted secondary structures are quite different (Figure S4).
Figure 2.
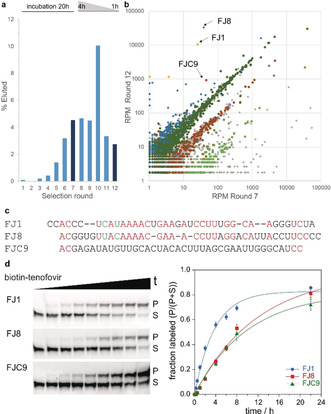
a) Progress of in vitro selection monitored by fluorescence of eluted fraction. Round 7 and round 12 libraries were analyzed by sequencing. b) Abundance of individual sequences in reads per million (RPM) compared in R7 and R12. Sequence clusters with Levenshtein edit distance <4 are indicated in same color. See detailed legend in Figure S3. c) Sequences of ribozymes FJ1, FJ8, and FJC9 (manually aligned). d) Trans activity assay with biotin‐Ten‐DP and 3′‐fluorescein‐labeled 19‐nt RNA substrate GGACAUACUGAGCCUUCAA. k obs=0.27 (FJ1), 0.09 (FJ8), 0.10 (FJC9) h−1.
All three ribozymes FJ1, FJ8, and FJC9 showed high trans activity (Figure 2 d), yielding circa 80 % biotin‐tenofovir‐labeled RNA after overnight incubation. The phosphonate ester linkage in the FJ1‐labeled RNA product was confirmed by mass spectrometry (Figure S5), and the labeling site at the expected 2′‐OH of A10 was revealed by the gel shift of the RNase T1 digestion products and alkaline hydrolysis products (Figure 3 a and Figure S6). Moreover, upon incubation with yeast debranching enzyme Dbr1, the phosphonate linkage in the FJ1 product was significantly more stable compared to the phosphodiester produced by the FH14 ribozyme with N 6‐biotin ATP. Under the in vitro conditions with highly active recombinant Dbr1 enzyme,10 the half‐life was 4–8‐fold increased (Figure 3 b).
Figure 3.
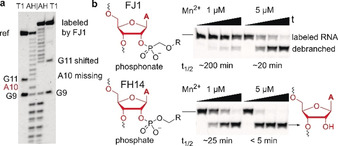
a) Alkaline hydrolysis and RNaseT1 digestion pattern of reference RNA and FJ1‐labeling product confirm the labeling site at A10 (in 5′‐fluorescein‐GACAUACUGAGCCUUCAAAUA). b) Debranching assay with recombinant Dbr1 comparing FJ1 product (top) and FH14 product (bottom). Time points are 0, 15, 30, and 60 min.
RNA labeling with biotin by FJ ribozymes may serve for enrichment of specific RNAs by affinity pulldown, and biotinylated RNA could be stained indirectly with fluorescently labeled streptavidin or anti‐biotin antibodies.19 However, direct fluorescent labeling with small organic fluorophores would be significantly preferred for fluorescence spectroscopy or RNA imaging. In this respect, fluorescent tenofovir analogues or derivatives with bioorthogonal functional groups for conjugation of fluorophores by established techniques are desirable as ribozyme substrates. Therefore, the respective N 6‐hexynyl and N 6‐azidohexyl‐modified tenofovir diphosphates were synthesized and used for the attachment of fluorescein or Cy5 fluorophores through Cu‐catalyzed azide–alkyne cycloaddition (Scheme S2). Unmodified tenofovir diphosphate (Ten‐DP) was tested as a reference.20 Gratifyingly, all three FJ ribozymes tolerated various modifications at the N 6 position of the adenine heterocycle and produced good yields of phosphonate‐labeled RNA (Figure 4, Figure S7). FJC9 was the slowest variant, but it also yielded circa 80 % labeled product after overnight incubation. The fluorescent substrates FAM‐Ten‐DP and Cy5‐Ten‐DP (Scheme S2) were tested with FJ1 and FJC9, and resulted in 40–60 % fluorescently labeled RNA after 22 h.
Figure 4.
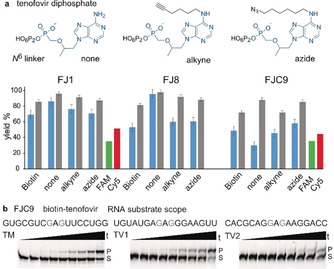
a) Structures of unmodified tenofovir diphosphate, and analogues with N 6‐hexynyl (alkyne) and N 6‐azidohexyl (azide) linkers. Yields of labeled RNA substrate sequence in trans activity assay after 8 h (blue) and 22 h (grey) with biotin‐tenofovir and analogues shown; mean of triplicates with standard error. Yields with fluorophore labeled tenofovir analogues after 22 h. b) Labeling of mutated target RNAs with FJC9 and biotin‐tenofovir to explore RNA substrate scope of FJC9.
Next, the RNA substrate scope of the tenofovir transferase ribozymes was investigated. Transition and transversion mutations were introduced in the parent RNA target, and compensatory mutations in the ribozyme binding arms to maintain Watson–Crick base pairing, in order to explore the potential for general and broad applications of FJ ribozymes. FJC9 turned out as a versatile ribozyme that tolerated all tested sequence variants, including six variants of the original GAG sequence context, in which the G nucleotides were individually changed to each of the other three nucleotides (Figure 4 b and Figure S8). In contrast, the FJ1 and FJ8 ribozymes were less tolerant to mutations, since neither of the fully transition‐ or transversion‐mutated substrates was accepted. Stepwise expansion of the parent sequence beyond the GAG motif determined the substrate sequence requirement for FJ1 as RAGCY (Figure S9). Single mismatches between RNA target and ribozyme binding arms next to the labeling sites significantly reduced the labeling efficiency, indicating little propensity for off‐target labeling (Figure S10).
Direct TTR‐mediated RNA labeling was then demonstrated with FJC9 for the 120nt long E. coli 5S rRNA by specific attachment of FAM‐tenofovir to 5S rRNA. In the context of total cellular RNA, the only fluorescein‐labeled product was detected at the desired size (total cellular RNA was then visualized by staining with Sybr gold, Figure 5 a). Although the sequence demand of FJ1 is a bit more restrictive, the scope is still general enough to find multiple possible target sites in large RNAs, as demonstrated for labeling of E. coli 16S and 23S rRNA with Cy5‐tenofovir (Figure 5 b). Six target sites were chosen according to the preferred sequence motif (Figure S12), and the corresponding FJ1 ribozymes produced Cy5‐labeled bands at the expected positions upon analysis by agarose gel electrophoresis and imaging in the Cy5 channel. Moreover, the FJ1 and FH14 ribozymes are truly orthogonal ribozymes without cross reactivity (Figure S11), which can be used in a single reaction for installation of two different fluorophores on two different target sites within one RNA substrate (Figure 5 c). Alternatively, two different RNAs can be addressed simultaneously in a single reaction, also in the context of total cellular RNA. This is demonstrated for labeling of 23S rRNA with Cy5‐Ten‐DP by FJ1‐23S‐A1572 and of 16S rRNA with FAM‐ATP by FH14‐16S‐A412 (Figure 5 d, and vice versa in Figure S13). The labeling sites in 16S and 23S rRNA were confirmed by primer extension assays with superscript reverse transcriptase, which showed a clear stop signal at the expected positions one nucleotide before the 2′‐labeled target site (Figure 5 e and Figure S14).
Figure 5.
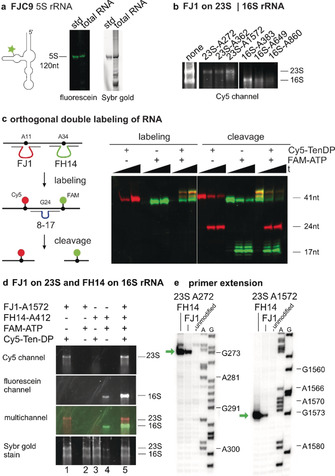
a) Specific labeling of 5S rRNA in total E. coli RNA by FJC9 and FAM‐Ten‐DP. “std” is in vitro transcribed 5S rRNA as size standard. b) Labeling of E. coli 16S and 23S rRNA with FJ1 and Cy5‐Ten‐DP at six different target sites. Gel images show Cy5 channel. For reference, the input RNA is shown to the left (stained with Sybr gold). c) Orthogonal labeling of a 41‐nt RNA transcript with FJ1 and FH14 ribozymes, and analysis by cleavage with an 8–17 deoxyribozyme. The multi‐channel image shows Cy5‐labeled RNA in red, fluorescein‐labeled RNA in green, and the double‐labeled RNA in yellow. d) Orthogonal labeling of E. coli 16S and 23S rRNA with FJ1 and FH14. Lanes 1 and 4 contain matched ribozymes and substrates, lanes 2 and 3 are negative controls with mismatched ribozyme, substrate combination showing orthogonality, and lane 5 contains both ribozymes and both substrates. e) Primer extension assay confirms labeling sites of both FJ1 and FH14. Exemplarily shown for A272 and A1572 on 23S rRNA. Additional sites in Figure S13.
In summary, we have established RNA‐catalyzed labeling of RNA using the antiviral nucleoside analogue tenofovir as scaffold for bioorthogonal ribozyme substrates. This work also demonstrates the reliability of the nucleotide transferase in vitro selection strategy and the design of the structured RNA pool for addressing a predetermined nucleotide in the target RNA for covalent labeling. Moreover, the analysis of the enriched library by Illumina sequencing revealed additional ribozyme variants with greater generality that would have been missed by traditional Sanger sequencing. Importantly, the resulting phosphonate linkages proved more stable towards yeast debranching enzyme (Dbr1) compared to the previously used natural phosphate linkages. Other chemical modification such as thiophosphates or thiophosphonates could further increase the half‐lives.10 In addition, it is worthwhile to continue searching for other ribozymes that can form bioorthogonal linkages completely resistant to enzymatic hydrolysis. The currently available toolbox of orthogonal ribozymes allows the installation of FRET pairs on a single RNA, and to address multiple different RNAs in one experiment with different labels, simply by the corresponding design of the Watson–Crick binding arms of FH and FJ ribozymes. It is therefore expected that these ribozymes will find applications for labeling of RNA in vitro and may be further improved for future cellular applications.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the ERC‐CoG illumizymes (682586 to C.H.). A. Hoskins (University of Wisconsin) is gratefully acknowledged for providing recombinant Dbr1 enzyme.
M. Ghaem Maghami, S. Dey, A.-K. Lenz, C. Höbartner, Angew. Chem. Int. Ed. 2020, 59, 9335.
References
- 1.
- 1a. Holstein J. M., Rentmeister A., Methods 2016, 98, 18–25; [DOI] [PubMed] [Google Scholar]
- 1b. Hanspach G., Trucks S., Hengesbach M., RNA Biol. 2019, 16, 1119–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Trachman R. J., Ferre-D'Amare A. R., Q. Rev. Biophys. 2019, 52, e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Muthmann N., Hartstock K., Rentmeister A., Wiley Interdiscip. Rev. RNA 2020, 11, e1561. [DOI] [PubMed] [Google Scholar]
- 4. Samanta B., Horning D. P., Joyce G. F., Nucleic Acids Res. 2018, 46, e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schulz D., Holstein J. M., Rentmeister A., Angew. Chem. Int. Ed. 2013, 52, 7874–7878; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8028–8032. [Google Scholar]
- 6.
- 6a. Tomkuvienė M., Clouet-d'Orval B., Černiauskas I., Weinhold E., Klimašauskas S., Nucleic Acids Res. 2012, 40, 6765–6773; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Alexander S. C., Busby K. N., Cole C. M., Zhou C. Y., Devaraj N. K., J. Am. Chem. Soc. 2015, 137, 12756–12759; [DOI] [PubMed] [Google Scholar]
- 6c. Li F., Dong J., Hu X., Gong W., Li J., Shen J., Tian H., Wang J., Angew. Chem. Int. Ed. 2015, 54, 4597–4602; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4680–4685. [Google Scholar]
- 7.
- 7a. Welz R., Bossmann K., Klug C., Schmidt C., Fritz H. J., Müller S., Angew. Chem. Int. Ed. 2003, 42, 2424–2427; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 2526–2530; [Google Scholar]
- 7b. Büttner L., Javadi-Zarnaghi F., Höbartner C., J. Am. Chem. Soc. 2014, 136, 8131–8137; [DOI] [PubMed] [Google Scholar]
- 7c. Büttner L., Seikowski J., Wawrzyniak K., Ochmann A., Höbartner C., Bioorg. Med. Chem. 2013, 21, 6171–6180. [DOI] [PubMed] [Google Scholar]
- 8. Ghaem Maghami M., Scheitl C. P. M., Höbartner C., J. Am. Chem. Soc. 2019, 141, 19546–19549. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Grammel M., Hang H., Conrad N. K., ChemBioChem 2012, 13, 1112–1115; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Martin G., Keller W., RNA 1998, 4, 226–230. [PMC free article] [PubMed] [Google Scholar]
- 10. Carrocci T. J., Lohe L., Ashton M. J., Höbartner C., Hoskins A. A., Chem. Commun. 2017, 53, 11992–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. De Clercq E., Holy A., Nat. Rev. Drug Discov. 2005, 4, 928–940; [DOI] [PubMed] [Google Scholar]
- 11b. Pradere U., Garnier-Amblard E. C., Coats S. J., Amblard F., Schinazi R. F., Chem. Rev. 2014, 114, 9154–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gallant J. E., Deresinski S., Clin. Infect. Dis. 2003, 37, 944–950. [DOI] [PubMed] [Google Scholar]
- 13. Ray A. S., Fordyce M. W., Hitchcock M. J., Antiviral Res. 2016, 125, 63–70. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Naesens L., Bischofberger N., Augustijns P., Annaert P., van den Mooter G., Arimilli M. N., Kim C. U., De Clercq E., Antimicrob. Agents Chemother. 1998, 42, 1568–1573; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. van Hemert F. J., Berkhout B., Zaaijer H. L., PLoS One 2014, 9, e106324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cihlar T., Birkus G., Greenwalt D. E., Hitchcock M. J., Antiviral Res. 2002, 54, 37–45. [DOI] [PubMed] [Google Scholar]
- 16. Holý A., Votruba I., Tloušťová E., Masojídková M., Collect. Czech. Chem. C 2001, 66, 1545–1592. [Google Scholar]
- 17.Deep sequencing libraries were prepared from cDNA of rounds 7 and 12 by PCR using an 8-nt UMI as described in the Supporting Information. Illumina sequencing was performed at the Core Unit Systems Medicine at the University of Würzburg. Analysis of the sequencing data was performed using fastaptamer perl scripts: Alam K. K., Chang J. L., Burke D. H., Mol. Ther. Nucleic Acids 2015, 4, e230.25734917 [Google Scholar]
- 18.FJ is our laboratory nomenclature for the tenofovir transferase ribozyme selection, and the numbers 1 and 8 relate to arbitrary clone numbers for Sanger sequencing. C in FJC refers to sequence cluster identified in NGS data; C9 is the 9th cluster in the output generated by fastaptamer_cluster. See the Supporting Information for more details.
- 19. Miller B. R., Wei T., Fields C. J., Sheng P., Xie M., RNA 2018, 24, 1871–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The tenofovir analogues synthesized in this study were used as racemic mixtures. The comparison with commercially available N 6-unmodified (R)-tenofovir diphosphate revealed similar activity of FJ1 with enantiomerically pure and racemic tenofovir diphosphate substrate (Figure S15).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary