

Abstract
Fragile X syndrome (FXS) is the most common genetic form of intellectual disability caused by a CGG repeat expansion in the 5′‐UTR of the Fragile X mental retardation gene FMR1, triggering epigenetic silencing and the subsequent absence of the protein, FMRP. Reactivation of FMR1 represents an attractive therapeutic strategy targeting the genetic root cause of FXS. However, largely missing in the FXS field is an understanding of how much FMR1 reactivation is required to rescue FMRP‐dependent mutant phenotypes. Here, we utilize FXS patient‐derived excitatory neurons to model FXS in vitro and confirm that the absence of FMRP leads to neuronal hyperactivity. We further determined the levels of FMRP and the percentage of FMRP‐positive cells necessary to correct this phenotype utilizing a mixed and mosaic neuronal culture system and a combination of CRISPR, antisense and expression technologies to titrate FMRP in FXS and WT neurons. Our data demonstrate that restoration of greater than 5% of overall FMRP expression levels or greater than 20% FMRP‐expressing neurons in a mosaic pattern is sufficient to normalize a FMRP‐dependent, hyperactive phenotype in FXS iPSC‐derived neurons.
Keywords: Cas9, CGG repeats, CRISPR, FMR1 reactivation, FMRP, Fragile X syndrome, hyperactivity
Previous studies have demonstrated that near full restoration of FMRP levels in Fragile X syndrome iPSC‐derived neuronal models can be achieved using gene editing tools; however, the minimum level of FMRP expression needed to normalize functional differences has not been established. In this study, we have confirmed a spontaneous, electrically hyperactive FXS phenotype in multiple iPSC‐derived neuronal lines. We then used CRISPR gene editing, cell mixing models, ASO‐mediated knockdown of FMR1 and targeted demethylation of CGG repeats to demonstrate that partial expression of FMRP can correct the hyperactive phenotype in iPSC‐derived FXS neurons.

Abbreviations
- ASO
antisense oligonucleotide
- ESC
embryonic stem cell
- FXS
Fragile X syndrome
- HCS
high‐content screening
- iPSC
induced pluripotent stem cell
- KO
knockout
- MEA
multielectrode array
- MOI
multiplicity of infection
- NBM
neurobasal media
- NGN2
Neurogenin‐2
- PEI
polyethylenimine
- PFA
paraformaldehyde
- qRT‐PCR
quantitative reverse transcription polymerase chain reaction
- RPM
rotations per minute
- UTR
untranslated region
- WT
wild type
1. INTRODUCTION
Fragile X syndrome (FXS) is a neurodevelopmental disorder caused by a CGG repeat expansion in the 5′‐UTR of the Fragile X mental retardation gene, FMR1 (Verkerk et al., 1991). Expansions of >200 repeats can lead to hypermethylation of the CGG repeats and CpG islands in the upstream promoter region. This hypermethylation results in heterochromatin formation and silencing of the FMR1 transcript, thereby preventing FMRP protein production (Fu et al., 1991; Pieretti et al., 1991). FMRP is an RNA‐binding protein (Ashley, Wilkinson, Reines, & Warren, 1993; Brown et al., 2001) that is highly expressed in neurons (Devys, Lutz, Rouyer, Bellocq, & Mandel, 1993; Feng et al., 1997) where it plays a key role in regulating local activity‐dependent synaptic translation (Weiler et al., 1997). The absence of FMRP affects both synaptic formation and maturation (Comery et al., 1997), as well as different forms of synaptic and homeostatic plasticity (Deng, Sojka, & Klyachko, 2011; Huber, Gallagher, Warren, & Bear, 2002; Soden & Chen, 2010; Zhang et al., 2018). A potential manifestation of this abnormal synaptic function is increased excitability in FXS neurons (Contractor, Klyachko, & Portera‐Cailliau, 2015). For example, increased seizure susceptibility has been observed in both FXS patients (Musumeci et al., 1999) and Fmr1 knockout mice (Musumeci et al., 2000). Moreover, studies in Fmr1 knockout mice showing aberrant ion channel expression and function (Deng et al., 2013; Gross, Yao, Pong, Jeromin, & Bassell, 2011; Zhang et al., 2012; Zhu et al., 2018), altered intrinsic neuronal properties (Gibson, Bartley, Hays, & Huber, 2008; Zhang et al., 2016) and augmented network activity (Gibson et al., 2008; Gonçalves, Anstey, Golshani, & Portera‐Cailliau, 2013) all demonstrate FMRP‐dependent effects on neuronal hyperexcitability. The Fmr1 knockout mice have been a good model for understanding the signaling pathways, pathophysiology and behavioral phenotypes associated with FXS. However, disease‐modifying therapeutics from mouse models have not translated well to the clinic (Berry‐Kravis et al., 2017).
Fragile X syndrome patient‐derived iPSCs represent an alternative model system to identify potential strategies for FMR1 reactivation, rather than targeting downstream pathways. FXS iPSCs retain expanded CGG repeats, promoter hypermethylation and FMR1 silencing after reprogramming (Sheridan et al., 2011; Urbach, Bar‐Nur, Daley, & Benvenisty, 2010) and have been used to demonstrate that removal of the expanded CGG repeat region leads to demethylation of the FMR1 promoter and reactivation of FMR1 (Park, Halevy, Lee, Sung, & Lee, 2015; Xie et al., 2016). Additionally, recent studies have shown that removal of the CGG repeat region in FXS iPSC‐derived neurons not only fully restores FMRP levels, but also normalizes a hyperexcitable phenotype (Liu et al., 2018), as well as rescues synaptic scaling deficits (Zhang et al., 2018). Moreover, demethylation of the extended CGG could also restore FMR1 levels and attenuate increased spontaneous activity in FXS iPSC‐derived neurons (Liu et al., 2018). While it has been demonstrated that near complete restoration of FMR1 levels was able to normalize a hyperexcitable phenotype, there has not yet been a systematic assessment to determine if partial restoration is sufficient to correct this phenotype.
In this study, we utilize two different isogenic iPSC pairs to confirm that the absence of FMRP leads to neuronal hyperactivity using multielectrode arrays (MEAs). We used orthogonal gene expression technologies to titrate the levels of FMRP expression in excitatory human neurons. We then determined the levels of FMRP necessary to correct this phenotype by two separate means: First, the proportion of FMRP‐positive neurons needed throughout the culture, and second, the total amount of FMRP required as a percentage of control levels. Our data demonstrate that partial restoration of FMRP is sufficient to normalize hyperactivity in FXS iPSC‐derived neurons.
2. MATERIALS AND METHODS
2.1. iPSC culture
WT isogenic control and FXS iPSC lines used in this study are listed in Table 1. iPSCs were cultured on Geltrex (Thermo Fisher Scientific, A1413302) coated flasks in StemFlex™ medium (Thermo Fisher Scientific, A3349401) and fed with fresh media every other day. Cells were passaged using ReLeSR™ (STEMCELL Technology, 05873) and tested for mycoplasma and karyotypic abnormalities. iPSCs were cryopreserved in CryoStem™ hPSC Freezing Medium (Biological industries, 05‐710‐1E). To recover the iPSCs, vials were thawed in a 37°C water bath for 2 min until a small piece of ice was left in the vial. 1 ml thawing media (StemFlex with 1× RevitaCell (Thermo Fisher Scientific, A2644501)) was added dropwise and then transferred to a 50 ml conical containing 10 ml thawing media. Cryovials were rinsed once with 1 ml thaw media. Cells were spun at 200 g for 5 min at room temperature, and the supernatant aspirated. Cells were resuspended in 10 ml thawing media and plated. Mycoplasma tests were conducted by Cell Line Genomics once a big batch of iPS cells got biobanked, and karyotyping analysis was performed by WiCell ~ every 10 passages. Use of FXS iPSCs has been reviewed and approved by Emory Internal Review Board on August 3, 2012, # CR8_IRB00001764 (IBR approval).
Table 1.
List of iPSC lines used in the study
2.2. FMR1 KO ISOGENIC IPSC ENGINEERING
To generate FMR1 KO iPSCs, Epi49 WT iPSCs (Thermo Fisher Scientific, A18945) were cultured in Geltrex coated T75 flask. The day before electroporation, cells were fed with fresh StemFlex™ medium with 1× RevitaCell supplements. Cells were dissociated with 5 ml Accutase™ cell dissociation reagent (STEMCELL technology, 07920). After washing once with PBS, cells were resuspended in Resuspension buffer R (Neon™ Transfection System 100 μL Kit, Invitrogen, 10431915) to a final cell density ~ 108/ml. Transfer 60 µg Cas9‐2A‐GFP‐FMR1 gRNA plasmid (with the gRNA sequence 5′‐TATTATAACCTACAGGAGGT‐3′ targeting against exon 3) into the dissociated iPSCs and electroporate with the following program: Pulse voltage 1,100 V; Pulse width 30ms; Pulse number 1; Cell density at 1 × 108 cells/ml. After electroporation, cells were plated into a Geltrex coated T75 flask using StemFlex™ medium with 1× RevitaCell supplements. On day 3 post‐electroporation, cells were dissociated with Accutase for FACS sorting to enrich the GFP + population, and replated onto Geltrex coated 10 cm Petri dish at ~5,000 cells/plate. Fluorescence‐activated flow cytometry cell sorting were performed using FACS Aria sorter at Whitehead Institute for Biomedical Research FACS core facility. GFP fluorescent signal was gated at 103 artificial fluorescence units (AFU), non‐electroporated control cells were used as a control. When recovery sorted iPSCs, 1× RevitaCell was supplemented in the StemFlex medium to enhance the cell viability. iPSC cell colonies were hand‐picked using a Nikon stereomicroscope equipped with 37°C thermo plate and expanded in StemFlex medium from 96 wells to 12 wells, and further expanded for cryopreservation. Two primers HW15 (5′‐actatattgccgttatgtcccactcag‐3′) and HW16 (5′‐taagatgagttagtcaaaagcacgtgtc‐3′) were used to generate the PCR amplicon that flanks the gRNA target region in exon 3 of FMR1. Based on sequencing results, clone A2 has homozygous frameshift mutation leading to a premature stop codon for FMRP protein with the following truncated peptide sequence MEELVVEVRGSNGAFYKAFVKDVHEDSITVAFENNWQPDRQIPFHDVRFRL* (Figure 6a).
Figure 6.
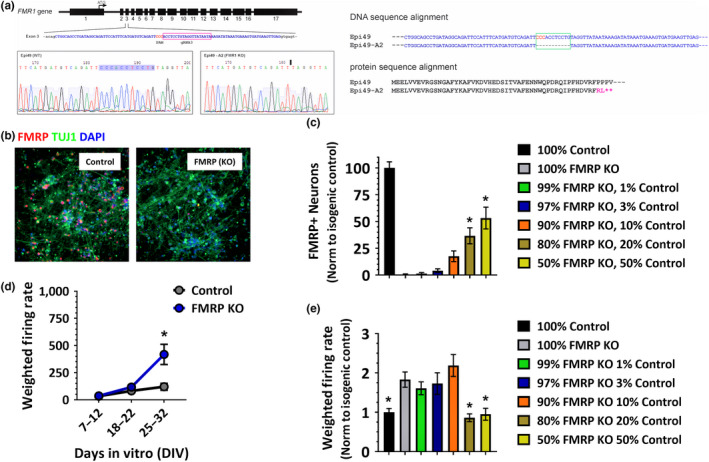
FMRP knockout neurons shown increased spontaneous activity that is normalized via partial addition of FMRP expression neurons. (a) Generation of FMR1 KO iPSC by CRISPR/Cas9‐mediated genetic editing from a WT iPSC line (Epi49). Left, a gRNA was designed to target the exon 3 of FMR1 locus from 3′ to 5′ ends. Amplicon sequence identified Clone A2 with homozygous deletion in the gRNA targeting region. The deleted sequence and the new ligation joint were highlighted in the sequence profiles of WT and the A2 clone. Right, DNA and protein sequence alignments of the WT and FMR1 KO iPSC indicate that the FMR1 KO clone produces an N‐terminal truncated polypeptide. Asterisks denote stop codons. (b) Immunofluorescent images from control WT iPSC‐derived neurons (left panel) and neurons in which FMRP has been knocked out. Blue—nuclei, green—β‐Tubulin and red—FMRP. (c) Increased spontaneous activity as measures by multi‐well MEAs shows that increased neuronal activity in FMRP KO neurons begins to manifest after 3 weeks in culture. (d) Quantification of FMRP‐positive neurons in the different FMRP mosaic cultures shows increasing FMRP‐positive neurons with increasing percentages of WT neurons mixed into FMRP KO cultures. Data normalized to control cultures. (e) Quantification of spontaneous activity recorded on MEAs three weeks post‐plating demonstrates reduced neuronal activity in FMRP KO neuronal cultures in which greater than 20% FMRP‐expressing neurons are present. Error bars represent ± SEM [Colour figure can be viewed at wileyonlinelibrary.com]
2.3. NEURONAL DIFFERENTIATION OF IPSC USING NGN2 METHOD
NGN2 neurons were derived from human WT and FXS iPSCs according to a modified method described by Zhang et al. (2013). Briefly, WT and FXS iPS cells were cultured on Geltrex in StemFlex™ medium and dissociated with TrypLE™ (Thermo Fisher, 12605010) as single cells. Cells were infected with pTet‐O‐NGN2‐2A‐Puro virus (1 × 109 IFU/ml) in suspension at MOI ~ 5 and plated in StemFlex with 1× RevitaCell. Infected cells were cultured and expanded on Geltrex in StemFlex media. On differentiation day 0, cells were dissociated with TrypLE™ and replated in StemFlex with RevitaCell and 2 μg/ml Doxycycline (Fisher Scientific, BP2653‐1) to induce TetO gene expression. Media was changed 24 hr post‐induction to DMEM/F12 (Thermo Fisher, 11320033) with 1× N2 supplement (Thermo Fisher, 17502048), 1× Glutamax (Thermo Fisher, 35050061), 0.3% D‐(+)Glucose (Sigma‐Aldrich, G8769), 2 μg/ml Doxycycline and 3.34 μg/ml Puromycin (Thermo Fisher, A1113808) to start selection. 48 hr post‐induction, the media was changed to differentiation media containing 1× B27 supplement (Thermo Fisher, 17504044). Neurons were dissociated on day 3 with room temperature StemPro™ Accutase™ Cell Dissociation Reagent (Thermo Fisher, A1110501) and either immediately replated into PDL and Laminin (Thermo Fisher, 23017‐015) coated plates in NBM media (Neurobasal™ Media (Thermo Fisher, 21103049) with 1× MEM NEAA (Thermo Fisher, 11140050), 1× Glutamax, 0.3% D‐(+)‐Glucose, 2 μg/ml Doxycycline, 3.34 μg/ml Puromycin, 1× B27 supplement, and 10 ng/ml rhBDNF (R&D Systems, 248‐BDB/CF)) and rhGDNF (R&D Systems, 212‐GD)) or cryopreserved in Nalgene cryogenic vials (Thermo Scientific) at densities ranging from 5 million to 10 million per 1 ml. The day following plating, fresh NBM was added to the cultures. Half media changes were conducted weekly.
2.4. qRT‐PCR
Cells were lysed using RealTime ready cell lysis buffer (Roche Life Science, 7248431001) with 1:80 diluted RNAse inhibitor (Sigma, 3335402001) and DNAseI (Thermo Fisher Scientific, AM2222). 96‐well plates were lysed by aspirating the media the cells and adding 50 μl lysis buffer per well. The plates were shaken at 1,250 RPM for 25 min. The lysate was mixed with a pipette 10 times and checked for lysis under the microscope. Plates were frozen for at least 15 min at −80°C. Lysates were thawed on ice and diluted 1:5 with nuclease‐free water. 2.25 μl of diluted total lysate was used in a PreAmp reaction with the FMR1‐FAM and POP4‐ABY TaqMan assays (5 μl PreAmp Master Mix (Applied Biosystems 4488593), 2.5 μl 0.2× diluted FMR1 and POP4 assays, 0.25 μl 40× TaqMan RT enzyme mix (Applied Biosystems A36107C), and 2.25 μl lysate). The preamp reaction was run for 12 cycles in an Applied Biosystems QS7 machine. PreAmp reactions were diluted 1:5 and 2 μl used in a qPCR reaction with the same two TaqMan assays (5 μl TaqMan Fast Advanced Master Mix (Applied Biosystems 4444557), 0.5 μl 20× FMR1‐FAM TaqMan assay, 0.5 μl 20× POP4‐ABY TaqMan assay, 2 μl Nuclease‐Free water) and run on a QuantStudio 7 (Thermo Fisher Scientific). Specific probe information is given in Table 2.
Table 2.
List of qPCR probes used in the study
| Taqman probe name | Vendor | Assay ID | Cat. No. |
|---|---|---|---|
| FAM‐FMR1 | Thermo Fisher Scientific | Hs00924547_m1 | 4,331,182 |
| POP4‐ABY | Thermo Fisher Scientific | Hs00198357_m1qsy | CCU002NR |
2.5. Immunofluorescence and imaging analysis
Neurons were fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature. Cells were permeabilized with PBST (1× PBS solution with 0.5% Triton X‐100) before blocking with 5% BSA in PBST. Cells were then incubated with appropriately diluted primary antibodies in PBST with 1% BSA for 1 hr at room temperature or overnight at 4°C. After washing with PBST for 3 × 10 min at room temperature, cells were incubated with fluorophore labeled secondary antibodies and DAPI. Cells were washed three times with PBST at room temperature and subjected to imaging with a 20× objective using CellInsight CX7 High‐Content Screening (HCS) Platform in wide‐field mode (Thermo Fisher Scientific). Images were analyzed using a modified “Neurite Detection Assay” as part of the HCS analysis suite of tools. Briefly, neuronal bodies were identified by staining with the neuron‐specific β‐III tubulin marker using nuclei as seeds. Neurites were then detected via β‐III tubulin staining originated from identified neuronal bodies. Total FMRP levels were quantified by using the FMRP intensity within a qualified neuronal body for all cells within a well. FMRP‐positive neurons were quantified by dividing the number of cell bodies with a FMRP intensity greater than a background level by the total number of neurons in the well. Background FMRP levels were empirically determined for each plate by quantifying the average and standard deviation of FMRP intensity in FXS neurons. A neuron was considered to be FMRP positive if the FMRP intensity was greater than the background level plus 3 standard deviations. Data were plotted with Prism GraphPad for statistical analysis. For imaging‐based quantification, unless otherwise specified, >10 representative images were quantified, and data were plotted as mean ± SD. Dilution for antibodies used in this study is listed in Table 3.
Table 3.
List of antibodies used for immunofluorescence experiments
| Antibodies | Vendor | Cat. No. | Dilution |
|---|---|---|---|
| Mouse anti‐FMRP (5C2) | BioLegend | 834701 | 1:200 |
| Mouse anti‐Cas9 (7A9‐3A3) | Active Motif | 61577 | 1: 100 |
| Chicken anti‐TUJ1 | AVES | TUJ89947984 | 1:500 |
| DAPI | Sigma‐Aldrich | 10236276001 | 1:2,000 |
| Donkey anti‐Mouse IgG (H + L) | Invitrogen | A‐21203 | 1:2,000 |
| Goat anti‐Chicken IgY (H + L) | Invitrogen | A‐11039 | 1:2,000 |
| Donkey anti‐Mouse IgG (H + L) | Invitrogen | A‐31571 | 1:2,000 |
2.6. MEA and data analysis
Of 48‐well multielectrode array plates containing an array 16 embedded gold electrodes were used for all MEA experiments (Cat. #M768‐KAP; Axion BioSystems, Atlanta, GA). The day before cell plating, MEA wells were coated with a 0.07% polyethylenimine (PEI) solution (Sigma, St. Louis, MO) for 2 hr in a 37°C cell‐culture incubator. Plates were rinsed with sterile, deionized water four times and air‐dried in a biological safety cabinet overnight. Neurons were cultured in BrainPhys medium (STEMCELL Technology) supplemented with 1× N‐2 Supplement (Thermo Fisher), 1 μg/ml Laminin (Sigma‐Aldrich), Neural Supplement B (Cellular Dynamics, M1029), Nervous System Supplement (Cellular Dynamics, M1031) and 1× Penicillin–Streptomycin (Thermo Fisher). On the day of plating, a 10 µl drop of complete BrainPhys media containing 80 μg/ml Laminin was placed directly in the center of each well over the MEA electrodes. The plates were then incubated for at least 1 hr at 37°C. A 10 µl drop of cell suspension was then plating directly into the 10 µl drop of complete BrainPhys media containing 80 μg/ml Laminin, for a final volume of 20 µl and final density of 75,000 neurons per well. The cells were allowed to attach to the substrate in a cell‐culture incubator for 1 hr, and then, 500 μl culture medium was added to the wells. Cells were grown at 37°C in a humidified 5% CO2/95% O2 incubator. To maintain the culture, a 50% medium change with fresh 37°C culture medium was performed every 3–4 days.
Spontaneous neuronal activity, including burst rate and firing rate, were recorded every day using the Axion Maestro MEA platform (Axion Biosystems). 5‐min recordings were performed every day before or after dCas9‐Tet1/CGG sgRNA, FMR1 mRNA and FMR1 ASO treatments. For all MEA experiments, at least three separate cell platings, with a range of 4–8 wells per condition per plating were performed. Only active electrodes, defined as >5 spikes per minute, and active wells, defined as at least 3 active electrodes, were used for analysis. Spike detection was performed by using a spike threshold of >6 standard deviations from the root mean square of the electrode noise. A weighted firing rate average was calculated by multiplying the average spike rate, in spikes per minute, of all active electrodes within the well by the number of active electrodes. As an example, a well with 10 active electrodes whose average spike rate over the 5‐min recording was 25 spikes per minute would result in an weighted firing rate of 250. Detected spikes were sorted and analyzed using a custom MATLAB program (Newberry et al., 2016) and presented as mean ± SD, and are considered statistically significant when p < .05 by paired Student's t test or one‐way ANOVA, where appropriate.
2.7. FMR1 mRNA transfection
FMR1 mRNA was designed with full substitution of pseudo‐U and ARCA capped to enhance the stability and translation efficiency. The mRNA was synthesized by TriLink BioTechnologies (L‐6009) and purified with Silica membrane with enzymatical polyadenylation and Dnase treatment. mRNA transfection was performed using Lipofectamine™ MessengerMAX™ Transfection Reagent (Thermo Fisher Scientific, LMRNA003) according to manufacturer's instruction. Briefly for transfection of each well, various amounts of mRNAs were diluted in 15 μl OptiMEM and combine with prediluted 15 μl 2× Lipofectamine MessengerMAX reagent (0.5 μl/well) in equal volume of OptiMEM. Transfectant was fully mixed by vortexing and incubation at RT for 5 min. 30 μl transfection cocktail was added into each well of the neuronal culture for transfection. Change 75% of the media 16 hr after transfection. A synthetic CleanCap™ EGFP mRNA (TriLink, L‐7201) was used as a transfection positive control in this study.
2.8. dCas9‐Tet1 construct and lentiviral transduction
The Fuw‐dCas9‐Tet1CD‐P2A‐BFP (Addgene, 108245), Fuw‐dCas9‐dTet1CD‐P2A‐BFP (Addgene, 108246) and CGG sgRNA‐mCherry co‐expression plasmids were gifts from Rudolf Jaenisch lab at Whitehead Institute, MIT, Cambridge, MA. The FMR1 CGG sgRNA expression construct was cloned by inserting annealed oligos into modified pgRNA plasmid (Addgene plasmid: 44,248) with AarI site (Liu et al., 2018). All constructs were sequenced before lentiviral packaging and production at 1 × 109 IFU/ml by Alstem LLC. For lentiviral transduction, neurons were infected with combination of dCas9‐Tet1CD/CGG sgRNA and dCas9‐dTet1CD/CGG sgRNA at various MOI, and media were changed 16 hr post‐transduction.
2.9. FMR1 Antisense Oligonucleotide (ASO) treatment
Two FMR1 ASOs were designed and synthesized by EXIQON (FMR1 ASO #1: Product sequence: ACAGATCTTAGACGTT, Cat. No. LG00199842; FMR1 ASO #2: Product sequence: GGAATAAGAATTACGG, Cat. No. LG00199843). For FMR1 ASO titration, various amount of FMR1 ASOs (10 nM to 1 μM) were added into WT neurons cultured on PDL/Laminin‐coated plate for imaging analysis or PEI/Laminin‐coated MEA plates for electrophysiological analysis.
2.10. Statistical analysis
Data from different cell lines and treatment conditions were compared using appropriate statistical tests (i.e., unpaired t test, one‐way ANOVA, Dunnett's multiple comparison test, two‐way ANOVA, Sidak's multiple comparison test) and are considered statistically significant when p < .05. The specific statistical test used for all data comparisons throughout the manuscript is indicated after the data is presented. Data are presented as mean ± SD.
3. RESULTS
3.1. Quantification of FMRP expression in isogenic pair of iPSC‐derived neurons
In this study, we utilized an isogenic pair of iPSCs in which the expression level of FMRP was the differentiating variable. This pair was generated by identifying clonal lines following CRISPR‐mediated removal of the CGG repeat region from the FMR1 gene of a human FXS iPSC line harboring a >450 CGG repeat expansion (Xie et al., 2016). We have previously confirmed both the lack of FMR1 expression and increased spontaneous activity in these FXS iPSC‐derived cortical excitatory neurons as compared to CRISPR‐edited isogenic control (Liu et al., 2018). Figure 1a shows representative immunofluorescent images from both the FXS iPSC‐derived neurons (SW_FXS) and isogenic control neurons (SW_C1_2) at 28 days post‐plating demonstrating robust FMRP staining in cell bodies of control neurons compared to no FMRP staining in the FXS iPSC‐derived neurons. Figure 1a also demonstrates that both cell lines display multiple neuronal markers such as the neuronal class III β‐tubulin marker, the dendritic marker MAP2 and neuronal marker NeuN. FMRP levels are quantified in Figure 1b by the percentage of FMRP + neurons, with close to 80% of C1_2 neurons positive for FMRP, and around 1% of FXS neurons positive for FMRP (C1_2: 79.2 ± 15.4%, n = 18 wells; FXS: 1.2 ± 1.3%, n = 24 wells).
Figure 1.

iPSC‐derived neurons from a Fragile X patient do not express FMRP and show increased spontaneous activity. (a) Immunofluorescent images from FXS iPSC‐derived neurons (FXS) and a CRIPSR/Cas9‐corrected isogenic control (C1_2) stained for nuclei (blue) FMRP (red) and neuronal markers (green) demonstrate no FMRP staining FXS neurons (lower 3 panels), compared to robust FMRP staining in the cell soma for corrected C1_2 neurons (upper 3 panels). The neuronal markers (green) used are β‐Tubulin (first column), MAP2 (second column) and NeuN (third column). (b) Quantification of FMRP‐positive neurons for both FXS and C1_2 neurons demonstrates that the majority of corrected C1_2 neurons are positive for FMRP, whereas FXS neurons are not. (c) Increased spontaneous activity as measures by multi‐well MEAs shows that increased FXS neuronal activity begins to manifest after 3 weeks in culture. Example MEA traces from 3 separate electrodes for both C1_2 neuronal cultures (d) and FXS neuronal cultures (e) demonstrate both tonic and burst activity patterns, as well as higher overall spontaneous activity in FXS cultures. Error bars represent ± SEM [Colour figure can be viewed at wileyonlinelibrary.com]
3.2. Absence of FMRP results in increased spontaneous activity
To confirm published work demonstrating increased spontaneous activity in excitatory FXS iPSC‐derived neurons, we compared the levels of spontaneous electrical activity for each iPSC‐derived neuronal line using multi‐well MEA plates. The level of spontaneous activity was quantified using an average weighted firing rate per well, which was calculated as a product of the spontaneous spikes per minute and the number of active electrodes in the well. Figure 1c shows the spontaneous activity for both lines of iPSC‐derived neurons over the course of seven weeks. These data demonstrate that neurons lacking FMRP expression begin to show increased spontaneous activity after 3 weeks which is maintained throughout the remaining time in culture. Cumulative data from the fourth week show a significantly higher weighted firing rate for FXS iPSC‐derived neurons when compared to the corrected isogenic control (FXS: 760.5 ± 918.0, n = 120 wells, C1_2: 406.0 ± 254.6, n = 118 wells, *p < .01, two‐way ANOVA as compared to respective time point of C1_2 condition, Sidak's multiple comparison test).
The following experiments were carried out to address two similar but distinct ways to quantify FMRP expression. The first method, quantifying the number of FMRP‐positive neurons in a neuronal network within a well, was addressed by creating mixed cultures of FMRP‐positive and FMRP‐negative neurons. This was achieved by mixing FXS and isogenic neurons together at certain ratios and by transfecting FXS neurons with FMR1 mRNA. Since the FXS neurons did not transfect equally, this resulted in a binary classification of neurons that either transiently expressed high levels of FMRP, and neurons that were not transfected and therefore did not express FMRP. The second method, quantifying the total FMRP expression in all neurons within a well, was addressed by using methods that would either increase FMRP levels across FXS neuronal culture, such as transduction with the dCas9‐Tet1 construct, or uniformly decrease FMRP levels across isogenic control neuronal cultures by using varying concentration of an FMR1 ASO.
3.3. Mixed neuronal culture determines the level of FMRP mosaicism for functional restoration
We next sought to determine how many FMRP‐positive neurons would be needed to normalize the increased spontaneous activity seen in FXS neuronal cultures. To do this, we generated mixed cultures of FMRP‐expressing (C1_2) and FMRP‐non‐expressing (FXS) neurons by titrating specific ratios of C1_2 neurons into FXS cultures at the day of plating. This paradigm allowed for the generation of a panel of FMRP mixed cultures that mimic neuronal environments in FXS mosaic male patients and female carriers. Figure 2a shows representative images of from 100% C1_2 and 100% FXS neuronal cultures, along with FXS cultures in which 1% and 20% C1_2 neurons have been mixed with 99% and 80% FXS neurons, respectively. The percentage of C1_2 neurons for each of these mixed cultures is quantified by ICC in Figure 2b, showing increasing amounts of FMRP‐positive neurons as the ratio of C1_2 to FXS neurons is increased. When the level of spontaneous activity was measured for the different mixed‐ratio cultures, a significant increase in spontaneous activity was seen in 100% FXS cultures as compared to 100% C1_2 (100% C1_2: 1.00 ± 0.97, n = 21 wells; 100% FXS: 5.38 ± 6.01, n = 30 wells; *p < .01, one‐way ANOVA as compared to 100% FXS condition, Dunnett's multiple comparison test). As C1_2 neurons were titrated into FXS cultures, only FXS cultures in which 20% or greater C1_2 neurons are present showed significant decreases in spontaneous activity when compared to the 100% FXS cultures. Cultures which had 20% or greater C1_2 neurons demonstrated spontaneous activity levels comparable to 100% C1_2 levels, suggesting that at least 20% FMRP‐expressing neurons are needed to normalize increased spontaneous activity seen in FXS neuronal cultures (100% C1_2: 1.00 ± 0.97, n = 21 wells; 20% C1_2: 1.25 ± 1.17, n = 28 wells, 50% C1_2: 1.53 ± 1.30, n = 26 wells, p > .9, one‐way ANOVA as compared to 100% FXS condition, Dunnett's multiple comparison test).
Figure 2.
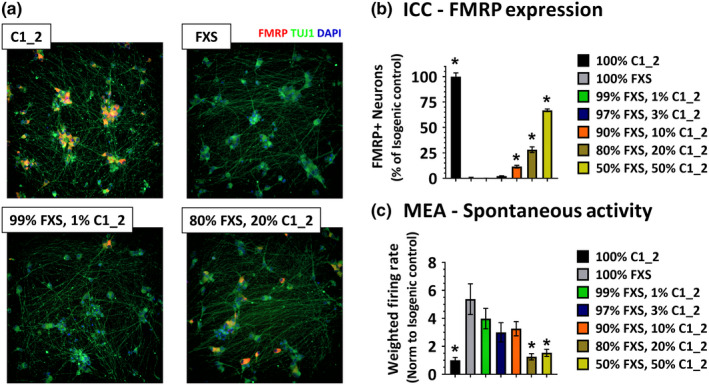
Mixing FMRP‐positive and FMRP‐negative neurons results in a FMRP mosaic culture. (a) Immunofluorescent images from C1_2 neurons (upper left panel) and FXS neurons (upper right panel) in which 1% (lower left panel) and 20% FMRP‐positive neurons (lower right panel) have been mixed. Blue—nuclei, green—β‐Tubulin and red—FMRP. (b) Quantification of FMRP‐positive neurons in the different FMRP mosaic cultures shows increasing FMRP‐positive neurons with increasing percentages of C1_2 neurons mixed into FXS cultures. Data normalized to C1_2 cultures. (c) Quantification of spontaneous activity recorded on MEAs three weeks post‐plating demonstrates reduced neuronal activity in FXS neuronal cultures in which greater than 20% FMRP‐expressing neurons are present. Error bars represent ± SEM [Colour figure can be viewed at wileyonlinelibrary.com]
3.4. Transient transfection of WT FMR1 mRNA results in mosaic FMRP expression pattern to rescue FXS functional phenotype
To further explore the levels of FMRP‐positive neurons needed to normalize FXS neuronal hyperactivity, we transfected varying amounts of WT human FMR1 mRNA into FXS cultures. Due to the stochastic nature of mRNA uptake during transfection, this experimental paradigm produced a FMRP mixed culture similar to that seen with cell mixing. Representative images of C1_2, FXS and FXS plus 8 or 16 ng of FMR1 mRNA are shown in Figure 3a. The lower two panels demonstrate the stochastic nature of FMRP expression 24 hr post‐transfection in which neurons are either FMRP positive or FMRP negative, with the number of FMRP‐expressing neurons dependent upon the amount of transfected FMR1 mRNA. The percentage of FMRP‐expressing neurons is quantified in Figure 3b, showing that as the amount of FMR1 mRNA is increased, the percentage of FMRP + neurons also increases, with a maximal level of around 25% FMRP‐positive neurons achieved 24 hr after transfecting. This FMRP expression at 24 hr represented the peak expression levels, with levels beginning to decrease 48–72 hr later. Due to the transient nature of FMRP expression following FMR1 mRNA transfection, the greatest effect on spontaneous activity was seen 24–32 hr post‐transfection (Figure 3c). Here, the data from 3 recording sessions at 24, 28 and 32 hr post‐transfection were averaged and normalized to a pre‐transfection baseline recording. From these data, a significant difference in spontaneous activity between C1_2 and FXS is seen during this post‐transfection time period. In addition, only FXS cultures that were transfected with 24ng of FMR1 mRNA demonstrated a significant reduction in spontaneous activity compared to the mock‐treated FXS neuronal cultures (FXS: 2.5 ± 2.2, n = 123; FXS + 24ng FMR1 mRNA: 1.6 ± 2.8, n = 153, *p < .05, one‐way ANOVA as compared to FXS condition, Dunnett's multiple comparison test). This level of FMR1 mRNA corresponded to 24.6 ± 5.5% FMRP‐positive neurons (n = 6 wells, *p < .05, one‐way ANOVA as compared to FXS condition). These data support the conclusion that >20% FMRP‐expressing neurons normalize the increased spontaneous activity seen in FXS cortical neuronal cultures.
Figure 3.
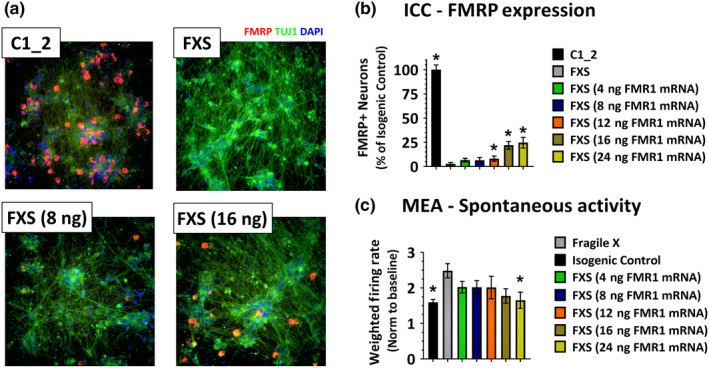
Transfection of FMR1 mRNA into FXS cultures results in a transient FMRP mosaic culture. (a) Immunofluorescent images from C1_2 neurons (upper left panel) and FXS neurons (upper right panel) in which 8ng FMR1 mRNA (lower left panel) and 16ng FMR1 mRNA (lower right panel) have been transiently transfected. Blue—nuclei, green—β‐Tubulin and red—FMRP. (b) Quantification of FMRP‐positive neurons in cultures 24 hr following transfection with varying amounts of FMR1 mRNA shows an increasing number of FMRP‐positive neurons with increasing amounts of FMR1 mRNA. Data normalized to C1_2 cultures. (c) Quantification of spontaneous activity, normalized to a pre‐transfection baseline, recorded on MEAs beginning 24hr post‐transfection of varying amounts of FMR1 mRNA, shows reduced neuronal activity in FXS neuronal cultures in which greater than 25% FMRP‐expressing neurons are present. Error bars represent ± SEM [Colour figure can be viewed at wileyonlinelibrary.com]
3.5. Titration of FMR1 ASO leads to varying reduced levels of FMRP reduction in WT neurons results in increased spontaneous activity
After establishing that >20% FMRP‐expressing neurons were needed to normalize the increased spontaneous activity seen in FXS cultures, we next sought to determine the overall expression levels of FMRP, rather than the number of highly expressing FMRP neurons, that are required to normalize FXS spontaneous activity. To achieve this, we first attempted to uniformly lower FMR1 levels across the culture using antisense oligonucleotides (ASOs) targeted to the FMR1 mRNA in C1_2 neurons (Figure 4). Figure 4a demonstrates the level of both FMR1 and FMRP following a 2 week treatment with two different FMR1‐targeting ASOs. Here, a robust knockdown of FMR1 is seen following treatment with both ASOs at 1 µM (ASO #1:28.8 ± 1.8% of control, n = 3; ASO #2:26.6 ± 2.8% of control, n = 3). An even greater reduction in FMRP was observed following ASO treatment, with ASO #1 producing around a 93% reduction in FMRP and ASO #2 producing around a 96% reduction (ASO #1:7.0 ± 0.5% of control, n = 3; ASO #2:4.4 ± 2.0% of control, n = 3). When the spontaneous activity of C1_2 cultures was recorded 2 weeks post‐ASO treatment, only cultures treated with 1 µM ASO #2 showed a significant increase in spontaneous activity as compared to C1_2 cultures treated with a non‐targeting control ASO. Cultures treated with 1 µM ASO #1 did not show a significant increase in spontaneous activity, suggesting that 7% FMRP expression within the culture was sufficient to prevent the increased spontaneous activity, whereas the 4% FMRP expression produced with 1 µM ASO #2 was a great enough reduction to induce increase spontaneous activity (Figure 4b; C1_2 + 1 µM Control ASO: 57.0 ± 50.0, n = 21; C1_2 + 1 µM ASO #1:90.8 ± 92.5, n = 9; C1_2 + 1 µM ASO #2:241.4 ± 188.8, n = 12; *p < .05, one‐way ANOVA as compared to C1_2 + 1 µM Control ASO, Dunnett's multiple comparison test). These data suggest that while >20% FMRP‐expressing neurons are needed to normalize increased spontaneous activity in FMRP mosaic cultures, a lower level of FMRP expression across all neurons in a culture may be enough to normalize increased spontaneous activity due to the absence of FMRP.
Figure 4.

ASO‐mediated reduction of FMR1 and FMRP in FMRP expression neurons results in increased spontaneous activity. (a) Concentration response curves for relative FMR1 expression (first column) and FMRP expression (second column) following a two‐week treatment with varying concentrations of two different ASOs shows a robust, concentration‐dependent knockdown of both FMR1 and FMRP. (b) Quantification of spontaneous activity recorded on MEAs two weeks post‐ASO treatment demonstrates increased activity only in C1_2 cultures in which greater than 95% FMRP knockdown is achieved, as denoted by the bar labeled 4%. Error bars represent ± SEM [Colour figure can be viewed at wileyonlinelibrary.com]
3.6. Titration of dCas9‐Tet1/CGG sgRNA leads to increasing levels of FMRP in FXS neurons and functional rescue
In addition to knocking down FMRP in FMRP‐expressing C1_2 neurons, we also attempted to reactivate FMRP in FXS neurons. To this end, we utilized a catalytically inactive Cas9 protein fused to the methylcytosine dioxygenase Tet1 as targeted DNA demethylation editing machinery. We have previously shown that this construct, when targeted to the CGG repeat region of the FMR1 gene, can demethylate this hypermethylated region and reactivate FMR1, resulting in FMRP expression in multiple FXS iPSCs and neurons (Liu et al., 2018). Here, we transduced iPSC‐derived FXS neurons with low titers of the dCas9‐Tet1 construct to produce partial reactivation of FMR1 (Figure 5). Figure 5a depicts representative images from FXS, C1_2 and FXS neurons transduced with 1 MOI of the dCas9‐Tet1 construct. These images show no FMRP expression in the FXS neurons, robust FMRP expression in the C1_2 neurons, and low expression of FMRP in the majority of FXS neurons following transduction with dCas9‐Tet1/CGG gRNA lentiviruses at 1 MOI. The quantification of FMRP intensity, normalized to C1_2 is shown in Figure 5b. Here, FXS neurons show less than 1% FMRP intensity as compared to C1_2 neurons (C1_2: 100 ± 14.9%, n = 6 wells; FXS:0.9 ± 0.7%, n = 6 wells). A non‐significant increase in FMRP intensity is seen in both FXS neurons transduced with a high MOI of a catalytically inactive Tet1 (dCas9‐dTet1) control and a low MOI of the active dCas9‐Tet1 construct (5 MOI dCas9‐dTet1: 3.4 ± 3.8%, n = 6 wells; p = .4; 0.5 MOI dCas9‐Tet1: 5.4 ± 2.5%, n = 6 wells; p = .06, one‐way ANOVA as compared to FXS condition, Dunnett's multiple comparison test). A statistically significant increase in FMRP intensity of around 7% was seen in FXS neurons transduced with 1 MOI of the active dCas9‐Tet1 as compared to non‐transduced FXS control neurons (FXS:0.9 ± 0.7%, n = 6 wells; 1 MOI dCas9‐Tet1: 7.1 ± 4.4%, n = 6 wells; *p < .01, one‐way ANOVA as compared to FXS condition, Dunnett's multiple comparison test). When spontaneous activity levels were measured following transfection, both control FXS neurons and FXS neurons transduced with the control dCas9‐dTet1 construct showed statistically significant increases in spontaneous activity as compared to FMRP expression C1_2 neurons, whereas FXS neurons transduced with 0.5 and 1 MOI of the dCas9‐Tet1 construct did not (Figure 5c; C1_2: 65.6 ± 174.5, n = 479 electrodes, FXS: 121.3 ± 272.7, n = 479 electrodes; 5 MOI dCas9‐dTet1: 144.4 ± 308.9, n = 463 electrodes; 0.5 MOI dCas9‐Tet1: 97.0 ± 213.4, n = 451 electrodes; 1 MOI dCas9‐Tet1: 82.7 ± 210.8, n = 449 electrodes; *p < .01, one‐way ANOVA as compared to C1_2 condition, Dunnett's multiple comparison test). These data indicate that >7% FMRP levels are sufficient to normalize increased spontaneous activity in FXS neurons—a similar threshold level identified through our FMR1 ASO experiments in FMRP‐expressing C1_2 neurons in which greater than 95% FMRP reduction (or <5% FMRP levels) was needed to induce an increased spontaneous activity phenotype.
Figure 5.

Partial reactivation of FMRP using a dCas9‐Tet1 construct reduces increased spontaneous activity. (a) Immunofluorescent images from FXS neurons (left panel), C1_2 neurons (middle panel) and FXS neurons 2 weeks post‐transduction of 1 MOI dCas9‐Tet1. Blue—nuclei, green—β‐Tubulin and red—FMRP. (b) Quantification of FMRP intensity for C1_2 neurons and FXS neurons transfected with 5 MOI dCas9‐dTet1, 0.5 MOI or 1 MOI dCas9‐Tet1 shows increase FMRP intensity with 1 MOI dCas9‐Tet1. Data normalized to C1_2. (c) Quantification of spontaneous activity recorded on MEAs two weeks post‐transduction demonstrates reduced spontaneous activity from FXS cultures with greater than 5% of C1_2 FMRP expression. Error bars represent ± SEM [Colour figure can be viewed at wileyonlinelibrary.com]
3.7. Confirmation of neuronal hyperactivity phenotype in genetically engineered FMRP KO neurons
Finally, we utilized a commercially available healthy, normal iPSC line to create a second pair of isogenic iPSC‐derived neurons that either did or did not have detectable levels of FMRP. Here, a FMR1 knockout line (FMRP KO) was created by inserting a premature stop codon in the third exon of the FMR1 gene, which resulted in normal levels of truncated FMR1 mRNA but no expression of FMRP (Figure 6a). Representative immunofluorescent images for this pair of isogenic iPSC‐derived neurons are shown in Figure 6b. In addition, electrical activity recorded from these lines demonstrated an increased spontaneous activity phenotype in FMRP KO neurons that developed after 3 weeks in culture as compared to wild type (WT), a similar timeframe seen with the first pair of isogenic neurons we profiled (Figure 6c; FMRP KO: 418.2 ± 427.9, n = 21 wells, WT: 120.1 ± 123.4, n = 15 wells, *p < .01, two‐way ANOVA as compared to the respective time point of the control condition, Sidak's multiple comparison test). Moreover, similar results were obtained when we performed a cell mixing experiment in which WT neurons were titrated into a culture of FMRP KO cells. These results are quantified in Figure 6d showing increasing amounts of FMRP‐positive neurons as an increasing percentage of FMRP expression control neurons are titrated into FMRP KO cultures. A statistically significant increase in FMRP‐positive neurons is seen in both 80% FMRP KO/ 20% Control (36.6 ± 18.3%, n = 6 wells; *p < .01, one‐way ANOVA as compared to 100% FMRP KO condition, Dunnett's multiple comparison test) and 50% FMRP KO/50% Control (53.2 ± 24.8%, n = 6 wells; *p < .01, one‐way ANOVA as compared to 100% FMRP KO condition, Dunnett's multiple comparison test). When spontaneous activity levels were recorded from these cultures, only cultures with 20% or greater FMRP‐expressing neurons show a significant reduction in spontaneous activity levels as compared to the 100% FMRP KO neurons (Figure 6e; 100% control: 1.0 ± 0.6, n = 32 wells; 100% FMRP KO: 1.8 ± 1.1, n = 31 wells; 80% FMRP KO/ 20% control: 0.9 ± 0.3, n = 12 wells; 50% FMRP KO/ 50% control: 1.0 ± 0.5, n = 12 wells; *p < .05, one‐way ANOVA as compared to 100% FMRP KO condition, Dunnett's multiple comparison test). These data support an FMRP‐dependent increased spontaneous activity phenotype, as well as confirm a threshold level of >20% FMRP‐expressing neurons needed in a FMRP mosaic culture to normalize increased spontaneous neuronal activity.
4. DISCUSSION
In this study, we have confirmed previous reports of increased spontaneous activity in FXS patient‐derived excitatory neurons differentiated from iPSCs using two isogenic pairs of cell lines in which the presence or absence of FMRP is the dependent variable. In addition, we have demonstrated that this increased neuronal activity can be normalized through partial restoration of FMRP‐expressing neurons within the neuronal network. Our results suggest that the number of FMRP‐expressing neurons within mosaic cultures in which neurons are either expressing FMRP or not, needs to be greater than 20% in order to normalize the increased spontaneous activity. In addition, our results suggest that the overall level of FMRP expression throughout all neurons within the culture needs to be greater than 5% of normal FMRP levels in order to restore the level of spontaneous activity to control levels. These data help to establish initial threshold levels for potential therapeutics aimed at either fully reactivating FMR1 in a subset of neurons or partially reactivating FMR1 across large neuronal populations.
We utilized two different paradigms to generate FMRP mosaicism in our iPSC‐derived neuronal cultures—titrating varying percentages of FMRP‐expressing neurons into FXS cultures and transiently transfecting FXS neurons with different amounts of FMR1 mRNA. These FMRP mosaic cultures were composed of different percentages of FMRP‐positive neurons. Based on these experiments, our data suggest that greater than 20% FMRP‐positive neurons are needed to normalize increased spontaneous neuronal activity. This number is slightly lower than previously reported percentages of FMRP‐positive lymphocytes in FXS patients needed for predicting mental retardation. These cut‐off levels have been reported to range between 28% and 50% (de Graaff et al., 1996; Loesch, Huggins, & Hagerman, 2004; Tassone et al., 1999; Willemsen et al., 1997). Additionally, a case report has been described in which a FXS patient with 22% FMRP‐positive leukocytes presented with normal physical features and a high‐functioning status, compared to his fully affected brother with a typical FXS presentation and no detectable FMRP (Han, Powell, Phalin, & Chehab, 2006). Another study that analyzed FMRP expression in both peripheral blood cells and primary fibroblasts from full mutation and mosaic FXS patients concluded that low, but non‐zero expression of FMRP may be sufficient to impact cognitive function (Pretto et al., 2014). It should be noted that these studies were evaluating FMRP expression in peripheral cells, not in neurons, in which FMRP levels may not correlate. Additionally, there are multiple reports that relate the percentage of FMRP‐positive hair follicles—which, like brain tissue, also originate from the ectoderm during embryonic development—to cognitive function, whereas the percentage of FMRP‐positive lymphocytes does not (Rujito et al., 2010; Willemsen et al., 2003). These clinical reports support the hypothesis that partial restoration of FMRP may be sufficient to normalize the FXS phenotype and that low‐level neuronal FMRP reactivation throughout the brain can positively impact FXS patients. Our study is the first to demonstrate that mixed FMRP cell cultures models can recapitulate the mosaicism observed in some FXS patients, in that similar partial FMRP expression may be sufficient to normalize neuronal activity in the disease.
The use of patient iPSC‐derived neurons as a physiologically relevant model of CNS disease, in conjunction with genome editing techniques to generate isogenic controls, has revolutionized the ability to model the effects of specific disease mutations in a dish (Soldner & Jaenisch, 2018). This especially holds promise for therapeutic approaches in FXS that are focused on reactivation of FMR1 as a disease‐modifying strategy. Currently, there are no animal models of FXS that allow for investigating mechanisms of FMR1 reactivation. The most widely used model, the Fmr1 KO mouse, cannot be used to probe potential approaches targeting the root cause of the disease due to the absence of the Fmr1 gene rather than its inactivation. In addition, attempts to knock‐in CGG repeat expansions in the mouse have not resulted in hypermethylation of the repeat region and silencing of Fmr1 (Berman & Willemsen, 2009; Entezam et al., 2007). Therefore, since FXS patient iPSCs retain expanded CGG repeats, promoter hypermethylation and FMR1 silencing after reprogramming (Sheridan et al., 2011; Urbach et al., 2010), they represent an attractive disease‐relevant model for investigating potential mechanisms for reactivating FMR1. Indeed, proof of concept studies showing reactivation of FMR1 in FXS‐derived iPSCs and differentiated neurons have been demonstrated (Park et al., 2015; Xie et al., 2016). Additionally, several groups are now using FXS patient ESCs and iPSCs to model functional effects in induced FXS human neurons (Achuta et al., 2018; Liu et al., 2018; Telias, Kuznitsov‐Yanovsky, Segal, & Ben‐Yosef, 2015; Telias, Segal, & Ben‐Yosef, 2016; Zhang et al., 2018) that can be corrected with fully restored FMRP levels via either CRISPR/Cas9‐mediated removal of the CGG repeat region (Liu et al., 2018; Zhang et al., 2018) or demethylation of the CGG repeat region (Liu et al., 2018).
The above‐mentioned studies have laid the groundwork for demonstrating that FXS functional phenotypes can be corrected in vitro. Our study is the first to attempt to understand the minimal levels of FMRP needed to normalize a specific functional phenotype—spontaneous network activity using MEAs. As such, potential small molecule therapeutics targeting FMR1 reactivation should aim to reach a critical threshold level of FMRP expression in order increase the likelihood of disease modification in patients. While not designed to activate FMR1, several small molecule therapeutics have been investigated in the clinic and have failed to address various FXS symptoms (Berry‐Kravis et al., 2017). We assessed the ability of some these compounds, along with other compounds that have been shown to reactivate FMR1 in non‐neuronal cells (Kumari & Usdin, 2014; Pietrobono et al., 2002; Tabolacci et al., 2008), to increase the expression of FMR1 in our iPSC‐derived patient neurons. Our results however showed that we were unable to demonstrate significant reactivation of FMR1 following a three‐week treatment with any of the compounds tested (Table 4). The inability of these drugs to address the root cause of the disease, along with the poor translatability of the fmr1 KO mouse model, could have contributed to their lack of efficacy.
Table 4.
Relative expression levels of FMR1 following treatment with compounds previously investigated in the clinic for FXS or shown to upregulate FMR1 in non‐neuronal cells
| FMR1 Relative Expression | |||
|---|---|---|---|
| Treatment | Avg (% Control) | StDev | n |
| Control (DMSO) | 100.00 | 20.51 | 3 |
| FXS (DMSO) | 0.08 | 0.02 | 3 |
| Baclofen | 0.44 | 0.09 | 3 |
| 5‐AzaC | 2.75 | 0.74 | 3 |
| Rimonabant | 0.26 | 0.03 | 3 |
| Lovastatin | 2.58 | 4.54 | 3 |
| Metformin | 0.55 | 0.13 | 3 |
| Mavoglurant | 0.60 | 0.09 | 3 |
| MPEP | 0.22 | 0.01 | 3 |
| Valproic Acid | 0.46 | 0.10 | 3 |
| Minocycline | 0.16 | 0.01 | 3 |
| Splitomicin | 0.08 | 0.07 | 3 |
It should be noted that the data from these experiments should be interpreted carefully due to a few limitations of the study. Firstly, the reactivation of FMRP was carried out in one isogenic line. Future studies should also determine whether this threshold level of FMRP is consistent across multiple patient lines with different CGG repeat expansions and methylation statuses. In addition, the increased spontaneous activity phenotype consistently demonstrated in FXS neuronal culture models is only one functional endpoint. This hyperactivity in vitro can be considered relevant within the context of increased seizure susceptibility in FXS mouse models (Musumeci et al., 2000) and FXS patients (Musumeci et al., 1999); however, it may not be the most relevant functional measurement for other FXS symptoms such as cognitive impairment. It will be important for future work to also elucidate the minimal levels of FMRP required to correct other potentially more‐relevant functional deficits such as synaptic scaling (Zhang et al., 2018).
In summary, we have demonstrated an FMRP‐dependent neuronal phenotype of increased spontaneous activity in FXS iPSC‐derived neurons that can be normalized through partial restoration of FMRP. Our data suggest that either greater than 20% FMRP‐expressing neurons in a mosaic population, or greater than 5% overall FMRP expression levels throughout a neuronal network can be used as thresholds for evaluating potential therapeutic interventions focused on reactivating FMRP in iPSC‐derived neurons from FXS patients.
CONFLICT OF INTEREST
All authors have a financial stake in Fulcrum Therapeutics.
AUTHOR CONTRIBUTIONS
J.G., H.W. and A.C. conceived the idea for this project. J.G. and H.W. designed the experiments and interpreted the data. J.G., H.W., C.N., C.S., V.V., D.Q. and K.J. performed the experiments. R.J. provided dCas9‐Tet1, dCas9‐dTet1 and FMR1 CGG gRNA constructs. S.W. provided the FXS_SW isogenic iPSCs. J.G. and H.W. wrote the manuscript with inputs from all the other authors.
DISCLAIMER
This manuscript is not under consideration for publication elsewhere and only appears as a pre‐print on BioRxiv.org. All authors have agreed for authorship, read and approved the manuscript, and given consent for submission and subsequent publication of the manuscript. All procedures were approved through an internal approval process.
ACKNOWLEDGEMENTS
We thank Drs. Shawn Liu and Rudolf Jaenisch at Whitehead Institute at MIT for dCas9‐Tet1, FRM1 CGG plasmids and pX458 Cas9‐T2A‐GFP gRNA backbone plasmid, Dr. Steve Warren at Emory University for providing FXS_SW isogenic iPSC lines, Dr. Marius Wernig at Stanford University for providing pTet‐O‐NGN2‐2A‐Puro plasmid. We thank members of Fulcrum Therapeutics for discussion and suggestions on the manuscript. R.J. is co‐founder of Fate Therapeutics, Fulcrum Therapeutics and Omega Therapeutics
Graef JD, Wu H, Ng C, et al. Partial FMRP expression is sufficient to normalize neuronal hyperactivity in Fragile X neurons. Eur J Neurosci. 2020;51:2143–2157. 10.1111/ejn.14660
John D. Graef and Hao Wu are equally contributed to this work.
Editor by: Dr. Ania Majewska.
The peer review history for this article is available at https://publons.com/publon/10.1111/ejn.14660
Contributor Information
John D. Graef, Email: jgraef@fulcrumtx.com.
Hao Wu, Email: hwu@fulcrumtx.com.
DATA AVAILABILITY STATEMENT
The raw data that support the findings of this study are available from the corresponding authors (JG and HW), upon request.
REFERENCES
- Achuta, V. , Möykkynen, T. , Peteri, U.‐K. , Turconi, G. , Rivera, C. , Keinänen, K. , & Castrén, M. L. (2018). Functional changes of AMPA responses in human induced pluripotent stem cell–derived neural progenitors in fragile X syndrome. Science Signaling, 11, eaan8784 10.1126/scisignal.aan8784 [DOI] [PubMed] [Google Scholar]
- Ashley, C. , Wilkinson, K. , Reines, D. , & Warren, S. (1993). FMR1 protein: Conserved RNP family domains and selective RNA binding. Science, 262, 563–566. 10.1126/science.7692601 [DOI] [PubMed] [Google Scholar]
- Berman, R. F. , & Willemsen, R. (2009). Mouse models of Fragile X‐associated tremor ataxia. Journal of Investigative Medicine, 57, 837–841. 10.2310/JIM.0b013e3181af59d6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry‐Kravis, E. M. , Lindemann, L. , Jønch, A. E. , Apostol, G. , Bear, M. F. , Carpenter, R. L. , … Jacquemont, S. (2017). Drug development for neurodevelopmental disorders: Lessons learned from fragile X syndrome. Nature Reviews Drug Discovery, 17(4), 280–299. 10.1038/nrd.2017.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, V. , Jin, P. , Ceman, S. , Darnell, J. C. , O’Donnell, W. T. , Tenenbaum, S. A. , … Warren, S. T. (2001) Microarray identification of FMRP‐associated brain mRNAs and altered mRNA translational profiles in Fragile X syndrome. Cell, 107(4), 477–487. 10.1016/S0092-8674(01)00568-2 [DOI] [PubMed] [Google Scholar]
- Comery, T. A. , Harris, J. B. , Willems, P. J. , Oostra, B. A. , Irwin, S. A. , Weiler, I. , & Greenough, W. T. (1997). Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proceedings of the National Academy of Sciences, 94, 5401–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor, A. , Klyachko, V. A. , & Portera‐Cailliau, C. (2015). Altered neuronal and circuit excitability in Fragile X syndrome. Neuron, 87, 699–715. 10.1016/j.neuron.2015.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaff, E. , de Vries, B. B. , Willemsen, R. , van Hemel, J. O. , Mohkamsing, S. , Oostra, B. A. , & van den Ouweland, A. M. (1996). The fragile X phenotype in a mosaic male with a deletion showing expression of the FMR1 protein in 28% of the cells. American Journal of Medical Genetics, 64, 302–308. [DOI] [PubMed] [Google Scholar]
- Deng, P.‐Y. , Rotman, Z. , Blundon, J. A. , Cho, Y. , Cui, J. , Cavalli, V. , … Klyachko, V. A. (2013). FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron, 77, 696–711. 10.1016/j.neuron.2012.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, P.‐Y. , Sojka, D. , & Klyachko, V. A. (2011). Abnormal presynaptic short‐term plasticity and information processing in a mouse model of Fragile X syndrome. Journal of Neuroscience, 31, 10971–10982. 10.1523/JNEUROSCI.2021-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devys, D. , Lutz, Y. , Rouyer, N. , Bellocq, J.‐P. , & Mandel, J.‐L. (1993). The FMR–1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nature Genetics, 4, 335–340. 10.1038/ng0893-335 [DOI] [PubMed] [Google Scholar]
- Entezam, A. , Biacsi, R. , Orrison, B. , Saha, T. , Hoffman, G. E. , Grabczyk, E. , … Usdin, K. (2007). Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene, 395, 125–134. 10.1016/j.gene.2007.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Y. , Gutekunst, C.‐A. , Eberhart, D. E. , Yi, H. , Warren, S. T. , & Hersch, S. M. (1997). Fragile X mental retardation protein: Nucleocytoplasmic shuttling and association with somatodendritic ribosomes. Journal of Neuroscience, 17, 1539–1547. 10.1523/JNEUROSCI.17-05-01539.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y.‐H. , Kuhl, D. , Pizzuti, A. , Pieretti, M. , Sutcliffe, J. S. , Richards, S. , … Caskey, C. T. (1991). Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell, 67, 1047–1058. 10.1016/0092-8674(91)90283-5 [DOI] [PubMed] [Google Scholar]
- Gibson, J. R. , Bartley, A. F. , Hays, S. A. , & Huber, K. M. (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of Fragile X syndrome. Journal of Neurophysiology, 100, 2615–2626. 10.1152/jn.90752.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves, T. J. , Anstey, J. E. , Golshani, P. , & Portera‐Cailliau, C. (2013). Circuit level defects in the developing neocortex of Fragile X mice. Nature Neuroscience, 16, nn.3415 10.1038/nn.3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, C. , Yao, X. , Pong, D. L. , Jeromin, A. , & Bassell, G. J. (2011). Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. Journal of Neuroscience, 31, 5693–5698. 10.1523/JNEUROSCI.6661-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, X. , Powell, B. R. , Phalin, J. L. , & Chehab, F. F. (2006). Mosaicism for a full mutation, premutation, and deletion of the CGG repeats results in 22% FMRP and elevated FMR1 mRNA levels in a high‐functioning fragile X male. American Journal of Medical Genetics Part A, 140A, 1463–1471. 10.1002/ajmg.a.31291 [DOI] [PubMed] [Google Scholar]
- Huber, K. M. , Gallagher, S. M. , Warren, S. T. , & Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proceedings of the National Academy of Sciences, 99, 7746–7750. 10.1073/pnas.122205699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari, D. , & Usdin, K. (2014). Polycomb group complexes are recruited to reactivated FMR1 alleles in Fragile X syndrome in response to FMR1 transcription. Human Molecular Genetics, 23, 6575–6583. 10.1093/hmg/ddu378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. X. , Wu, H. , Krzisch, M. , Wu, X. , Graef, J. , Muffat, J. , … Jaenisch, R. (2018). Rescue of Fragile X syndrome neurons by DNA methylation editing of the FMR1 gene. Cell, 173, 1–14. 10.1016/j.cell.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesch, D. Z. , Huggins, R. M. , & Hagerman, R. J. (2004). Phenotypic variation and FMRP levels in fragile X. Mental Retardation and Developmental Disabilities Research Reviews, 10, 31–41. 10.1002/mrdd.20006 [DOI] [PubMed] [Google Scholar]
- Musumeci, S. A. , Bosco, P. , Calabrese, G. , Bakker, C. , Sarro, G. B. , Elia, M. , … Oostra, B. A. (2000). Audiogenic seizures susceptibility in transgenic mice with Fragile X syndrome. Epilepsia, 41, 19–23. 10.1111/j.1528-1157.2000.tb01499.x [DOI] [PubMed] [Google Scholar]
- Musumeci, S. , Hagerman, R. , Ferri, R. , Bosco, P. , Bernardina, D. B. , Tassinari, C. , … Elia, M. (1999). Epilepsy and EEG findings in males with Fragile X syndrome. Epilepsia, 40, 1092–1099. 10.1111/j.1528-1157.1999.tb00824.x [DOI] [PubMed] [Google Scholar]
- Newberry, K. , Wang, S. , Hoque, N. , Kiss, L. , Ahlijanian, M. K. , Herrington, J. , & Graef, J. D. (2016). Development of a spontaneously active dorsal root ganglia assay using multiwell multielectrode arrays. Journal of Neurophysiology, 115, 3217–3228. 10.1152/jn.01122.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, C. , Halevy, T. , Lee, D. , Sung, J. , & Lee, J. (2015) Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC‐derived neurons. Cell Reports, 13(2), 234–241 10.1016/j.celrep.2015.08.084 [DOI] [PubMed] [Google Scholar]
- Pieretti, M. , Zhang, F. , Fu, Y.‐H. , Warren, S. T. , Oostra, B. A. , Caskey, C. T. , & Nelson, D. L. (1991). Absence of expression of the FMR‐1 gene in Fragile X syndrome. Cell, 66, 817–822. 10.1016/0092-8674(91)90125-I [DOI] [PubMed] [Google Scholar]
- Pietrobono, R. , Pomponi, M. , Tabolacci, E. , Oostra, B. , Chiurazzi, P. , & Neri, G. (2002). Quantitative analysis of DNA demethylation and transcriptional reactivation of the FMR1 gene in fragile X cells treated with 5‐azadeoxycytidine. Nucleic Acids Research, 30, 3278–3285. 10.1093/nar/gkf434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pretto, D. , Yrigollen, C. M. , Tang, H.‐T. , Williamson, J. , Espinal, G. , Iwahashi, C. K. , … Tassone, F. (2014). Clinical and molecular implications of mosaicism in FMR1 full mutations. Frontiers Genetics, 5, 318 10.3389/fgene.2014.00318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rujito, L. , Kustiani, D. , Severijnen, L. , Hanzon, P. , Faradz, S. M. , & Willemsen, R. (2010). Hair root FMRP expression for screening of fragile X full mutation females. UNIVERSA MEDICINA, 30, 11–21. [Google Scholar]
- Sheridan, S. D. , Theriault, K. M. , Reis, S. A. , Zhou, F. , Madison, J. M. , Daheron, L. , … Haggarty, S. J. (2011). Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of Fragile X syndrome. PLoS ONE, 6, e26203 10.1371/journal.pone.0026203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soden, M. E. , & Chen, L. (2010). Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. The Journal of Neuroscience, 30, 16910–16921. 10.1523/JNEUROSCI.3660-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner, F. , & Jaenisch, R. (2018). Stem cells, genome editing, and the path to translational medicine. Cell, 175, 615–632. 10.1016/j.cell.2018.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabolacci, E. , Moscato, U. , Zalfa, F. , Bagni, C. , Chiurazzi, P. , & Neri, G. (2008). Epigenetic analysis reveals a euchromatic configuration in the FMR1 unmethylated full mutations. European Journal of Human Genetics, 16, 1487–1498. 10.1038/ejhg.2008.130 [DOI] [PubMed] [Google Scholar]
- Tassone, F. , Hagerman, R. J. , Iklé, D. N. , Dyer, P. N. , Lampe, M. , Willemsen, R. , … Taylor, A. K. (1999). FMRP expression as a potential prognostic indicator in Fragile X syndrome. American Journal of Medical Genetics, 84, 250–261. [DOI] [PubMed] [Google Scholar]
- Telias, M. , Kuznitsov‐Yanovsky, L. , Segal, M. , & Ben‐Yosef, D. (2015). Functional deficiencies in Fragile X neurons derived from human embryonic stem cells. Journal of Neuroscience, 35, 15295–15306. 10.1523/JNEUROSCI.0317-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telias, M. , Segal, M. , & Ben‐Yosef, D. (2016). Immature responses to GABA in Fragile X neurons derived from human embryonic stem cells. Frontiers in Cellular Neuroscience, 10, 121 10.3389/fncel.2016.00121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbach, A. , Bar‐Nur, O. , Daley, G. Q. , & Benvenisty, N. (2010). Differential modeling of Fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell, 6, 407–411. 10.1016/j.stem.2010.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk, A. , Pieretti, M. , Sutcliffe, J. S. , Fu, Y.‐H. , Kuhl, D. , Pizzuti, A. , … Warren, S. T. (1991). Identification of a gene (FMR‐1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in Fragile X syndrome. Cell, 65, 905–914. 10.1016/0092-8674(91)90397-H [DOI] [PubMed] [Google Scholar]
- Weiler, I. , Irwin, S. A. , Klintsova, A. Y. , Spencer, C. M. , Brazelton, A. D. , Miyashiro, K. , … Greenough, W. T. (1997). Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proceedings of the National Academy of Sciences, 94, 5395–5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen, R. , Smits, A. , Mohkamsing, S. , van Beerendonk, H. , de Haan, A. , de Vries, B. , … Oostra, B. (1997). Rapid antibody test for diagnosing Fragile X syndrome: A validation of the technique. Human Genetics, 99, 308–311. 10.1007/s004390050363 [DOI] [PubMed] [Google Scholar]
- Willemsen, R. , Smits, A. , Severijnen, L.‐A. , Jansen, M. , Jacobs, A. , Bruyn, D. E. , & Oostra, B. (2003). Predictive testing for cognitive functioning in female carriers of the Fragile X syndrome using hair root analysis. Journal of Medical Genetics, 40, 377–379. 10.1136/jmg.40.5.377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, N. , Gong, H. , Suhl, J. A. , Chopra, P. , Wang, T. , & Warren, S. T. (2016). Reactivation of FMR1 by CRISPR/Cas9‐mediated deletion of the expanded CGG‐repeat of the Fragile X Chromosome. PLoS ONE, 11, e0165499 10.1371/journal.pone.0165499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Liang, Z. , Zhu, P. , Li, M. , Yi, Y.‐H. , Liao, W.‐P. , & Su, T. (2016). Altered intrinsic properties and bursting activities of neurons in layer IV of somatosensory cortex from Fmr‐1 knockout mice. Experimental Neurology, 280, 60–69. 10.1016/j.expneurol.2016.03.025 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Brown, M. R. , Hyland, C. , Chen, Y. , Kronengold, J. , Fleming, M. R. , … Kaczmarek, L. K. (2012). Regulation of neuronal excitability by interaction of Fragile X mental retardation protein with slack potassium channels. Journal of Neuroscience, 32, 15318–15327. 10.1523/JNEUROSCI.2162-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Pak, C. , Han, Y. , Ahlenius, H. , Zhang, Z. , Chanda, S. , … Südhof, T. C. (2013). Rapid single‐step induction of functional neurons from human pluripotent stem cells. Neuron, 78, 785–798. 10.1016/j.neuron.2013.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Marro, S. G. , Zhang, Y. , Arendt, K. L. , Patzke, C. , Zhou, B. , … Chen, L. (2018). The Fragile X mutation impairs homeostatic plasticity in human neurons by blocking synaptic retinoic acid signaling. Science Translational Medicine, 10, eaar4338 10.1126/scitranslmed.aar4338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, P. , Li, J. , Zhang, L. , Liang, Z. , Tang, B. , Liao, W.‐P. , … Su, T. (2018). Development‐related aberrations in Kv1.1 α‐subunit exert disruptive effects on bioelectrical activities of neurons in a mouse model of Fragile X syndrome. Progress in Neuro‐Psychopharmacology and Biological Psychiatry, 84, 140–151. 10.1016/j.pnpbp.2018.02.011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data that support the findings of this study are available from the corresponding authors (JG and HW), upon request.