

Abstract
Equilibrative nucleoside transporters (ENTs) transport nucleosides across the blood-testis barrier (BTB). ENTs are of interest to study the disposition of nucleoside reverse-transcriptase inhibitors (NRTIs) in the human male genital tract because of their similarity in structure to nucleosides. HeLa S3 cells express ENT1 and ENT2 and were used to compare relative interactions of these transporters with selected NRTIs. Inhibition of [3H]uridine uptake by NBMPR was biphasic, with IC50 values of 11.3 nM for ENT1 and 9.6 μM for ENT2. Uptake measured with 100 nM NBMPR represented ENT2-mediated transport; subtracting that from total uptake represented ENT1-mediated transport. The kinetics of ENT1- and ENT2-mediated [3H]uridine uptake revealed no difference in Jmax (16.53 and 30.40 pmol cm−2 min−1) and an eightfold difference in Kt (13.6 and 108.9 μM). The resulting fivefold difference in intrinsic clearance (Jmax/Kt) for ENT1- and ENT2 transport accounted for observed inhibition of [3H]uridine uptake by 100 nM NBMPR. Millimolar concentrations of the NRTIs emtricitabine, didanosine, lamivudine, stavudine, tenofovir disoproxil, and zalcitabine had no effect on ENT transport activity, whereas abacavir, entecavir, and zidovudine inhibited both transporters with IC50 values of ∼200 µM, 2.5 mM, and 2 mM, respectively. Using liquid chromatography-tandem mass spectrometry and [3H] compounds, the data suggest that entecavir is an ENT substrate, abacavir is an ENT inhibitor, and zidovudine uptake is carrier-mediated, although not an ENT substrate. These data show that HeLa S3 cells can be used to explore complex transporter selectivity and are an adequate model for studying ENTs present at the BTB.
SIGNIFICANCE STATEMENT
This study characterizes an in vitro model using S-[(4-nitrophenyl)methyl]-6-thioinosine to differentiate between equilibrative nucleoside transporter (ENT) 1- and ENT2-mediated uridine transport in HeLa cells. This provides a method to assess the influence of nucleoside reverse-transcriptase inhibitors on natively expressed transporter function. Determining substrate selectivity of the ENTs in HeLa cells can be effectively translated into the activity of these transporters in Sertoli cells that comprise the blood-testis barrier, thereby assisting targeted drug development of compounds capable of circumventing the blood-testis barrier.
Introduction
Equilibrative nucleoside transporters (ENTs) are sodium-independent uniporters responsible for the transport of nucleosides and nucleobases across cell membranes (Baldwin et al., 2004; Molina-Arcas et al., 2009; Young et al., 2013; Boswell-Casteel and Hays, 2017). The ENTs are physiologically important ubiquitously expressed proteins that provide nucleosides for DNA and RNA synthesis ( Young et al., 2013; Uhlén et al., 2015; Huang et al., 2017). Plagemann and colleagues first characterized nucleoside transport in cultured mammalian cells, including HeLa cells (Plagemann and Shea, 1971; Plagemann and Erbe, 1972; Plagemann et al., 1978). Uridine transport in HeLa cells was subsequently shown to display both 6-S-[(4-nitrophenyl)methyl]-6-thioinosine (NBMPR)-sensitive and -insensitive components, leading to suggestions that ligand interaction with nucleoside transport may involve a single transporter with multiple binding sites (Dahlig-Harley et al., 1981; Plagemann and Wohlhueter, 1984).
Later studies showed two isoforms of these transporters, ENT1 and ENT2. These isoforms are distinguished by their distinct sensitivities for NBMPR; nanomolar concentrations effectively block ENT1 activity (NBMPR-sensitive), whereas ENT2 inhibition requires micromolar concentrations (NBMPR-insensitive) (Griffith and Jarvis, 1996; Griffiths et al., 1997a,b; Young et al., 2013; Huang et al., 2017). The cloning of ENT1 (Griffiths et al., 1997a) and ENT2 (Griffiths et al., 1997b; Crawford et al., 1998) established the distinct molecular identities of the NBMPR-sensitive (high affinity; ENT1) and -insensitive (low affinity; ENT2) components of equilibrative nucleoside transport in mammalian cells, and heterologous expression of cloned ENTs results in differential inhibition of transport indicative of ENT1 and ENT2 activity (Ward et al., 2000; Sundaram et al., 2001; Yao et al., 2001; Tang et al., 2016; Huang et al., 2017). These two transporters are of particular interest in studying the disposition of nucleoside reverse-transcriptase inhibitors (NRTIs) because of the structural similarity between these compounds and endogenous nucleosides. NRTIs prevent the conversion of viral RNA into double-stranded DNA and are currently used to treat infections caused by pathogens such as the human immunodeficiency virus (HIV) or hepatitis B virus (Matthews, 2006; Lucas and Nelson, 2015; Lok et al., 2017).
Because HIV and hepatitis B virus are sexually transmitted, it is crucial for these compounds to reach therapeutic concentrations within the male reproductive system (Trépo et al., 2014; Lucas and Nelson, 2015). The blood-testis barrier (BTB), formed by tight junctions between Sertoli cells within seminiferous tubules, protects developing germ cells from endogenous and exogenous agents. As a result, the BTB is an obstacle for NRTIs to reach the lumen of the seminiferous tubules (Miller and Cherrington, 2018). However, there is evidence that some NRTIs, including lamivudine, zidovudine, didanosine, and tenofovir disoproxil, are detectable in the seminal plasma of HIV-1–positive individuals treated with these therapies (Pereira et al., 1999; Lowe et al., 2007). Passage of these compounds across the Sertoli cell epithelium presumably involves the sequential activity of a basal uptake transporter and an apical efflux transporter (Klein et al., 2013; Miller and Cherrington, 2018). ENT1 is localized to the basal membrane and ENT2 is localized to the apical membrane of Sertoli cells in rat and human testis (Klein et al., 2013), which creates a possible route of entry into the lumen of the seminiferous tubule for ENT substrates. Furthermore, uridine transport across rat Sertoli cell monolayers displays a sensitivity to NBMPR that is consistent with expression of rat ent1 at the basal membrane and expression of rat ent2 at the apical membrane (Klein et al., 2013). Uridine is a well-established substrate for human ENTs and can be used to study the impact of various compounds on ENT activity (Ward et al., 2000; Kato et al., 2005; Klein et al., 2013).
The present study uses HeLa S3 (“HeLa”) cells, which endogenously express functional ENT1 and ENT2, as a robust in vitro model of natively expressed ENT transport activity. Using NBMPR to distinguish between contribution of ENT1 and ENT2 to observed activity, these cells were subsequently used to assess the impact of nine clinically relevant NRTIs on ENT-mediated uridine transport. The data suggest that some, but not all, NRTIs modulate uridine uptake through substrate-transport competition. Entecavir is shown to be a substrate for both ENT1 and ENT2, whereas abacavir is a nontransported inhibitor of the ENTs. Although mediated zidovudine was observed, transport does not appear to involve either ENT.
Materials and Methods
[3H]Uridine (specific activity 38 Ci/mmol) was purchased from Perkin-Elmer (Waltham, MA). [3H]Abacavir and [3H]zidovudine (specific activities 0.1 and 12.3 Ci/mmol, respectively) were purchased from Moravek (Brea, CA). NBMPR was purchased from Tocris (Bristol, UK). Uridine, guanosine, adenosine, inosine, thymidine, and cytidine were purchased from Sigma-Aldrich (St. Louis, MO). Abacavir sulfate, emtricitabine, entecavir hydrate, didanosine, lamivudine, stavudine, tenofovir disoproxil, zalcitabine, and zidovudine were purchased from Cayman Chemical (Ann Arbor, MI). Optima LC/MS-grade methanol, acetonitrile, and formic acid were purchased from ThermoFisher Scientific (Hampton, NH).
Cell Culture.
HeLa S3 cells were purchased from American Type Culture Collection (Catalog number CCL-2.2; Manassas, VA) and propagated in Ham’s F-12 media (Sigma-Aldrich) supplemented with 2.5 g/l sodium bicarbonate, 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. When 80%–90% confluent, cells were subcultured using the American Type Culture Collection protocol and maintained in a humidified 5% CO2 incubator at 37°C.
Reverse-Transcription Polymerase Chain Reaction.
Total RNA was isolated from cells using a Qiagen RNeasy Mini Kit (Qiagen, Hilden, Germany), and first-strand cDNA synthesis was carried out with Invitrogen Cells-to-cDNA II Kit using random decamers (Invitrogen, Carlsbad, CA). Specific primers for human ENT1 and ENT2 were designed and synthesized by Integrated DNA Technologies (Coralville, IA). The primers for hENT1 were 5′-CATGACAACCAGTCACCAGCC-3′ (5′-primer) and 5′-GCACACAATTGCCCGGAACAG-3′ (3′-primer). The primers for hENT2 were 5′-CATTACATGGCGCGAGGAGAC-3′ (5′-primer) and 5′-GCAGAGCAGCGCCTTGAACAG-3′ (3′-primer). Polymerase chain reaction (PCR) amplification of ENT1 and ENT2 were carried out using Apex 2X Taq Master Mix (Genessee Scientific, San Diego, CA) with the cycling conditions as follows: initial denaturation at 94°C for 3 minutes followed by 35 cycles of denaturation at 94°C for 45 seconds, annealing at 57°C for 45 seconds and extension for 2 minutes at 72°C, and a final extension at 72°C for 10 minutes. The PCR products were checked by gel electrophoresis using a 1% agarose gel with ethidium bromide staining. To validate primer specificity, the products were excised and purified from the gel before being sent for Sanger sequencing by Eton Biosciences. Full-length plasmids were used as a positive control. The sequences were aligned with the control plasmid sequences obtained from GenBank for ENT1 (Accession number AF190884) and ENT2 (Accession number AF034102).
Transport Experiments with Radiolabeled Compounds.
Transport experiments were performed as previously described with minor modifications (Severance et al., 2017; Sandoval et al., 2018). Briefly, HeLa cells (250,000 cells/ml) were plated into Nunc MicroWell 96-well optical bottom plates (ThermoFisher Scientific, Waltham, MA) at 200 μl/well and grown to confluence. Cell culture media was aspirated, and cells were washed three times with 300 μl of room-temperature Waymouth’s Buffer (WB; 2.5 mM CaCl 2H2O, 28 mM D-glucose, 13 mM HEPES, 135 mM NaCl, 1.2 mM MgCl, 0.8 mM MgSO4 7H2O, pH 7.4), using a Biotek 405 LS Microplate Washer (BioTek, Winooski, VT). Cells were incubated with 50 μl of transport buffer dispensed with an Integra VIAFLO 96-well multichannel pipette (Integra Lifesciences, Plainsboro, NJ), containing WB, approximately 20 nM [3H]uridine, and additional compounds as noted in the figure legends. After selected time intervals, transport was halted by washing three times with 300 μl of ice-cold WB. Following the washes, 200 μl of MicroScint-20 scintillation cocktail (Perkin-Elmer) was added per well, and the plates were sealed with microplate film (USA Scientific, Ocala, FL) and incubated at room temperature for 2 hours before measurements were taken. Total accumulated radioactivity was determined by using a Wallac 1450 MicroBeta TriLux liquid scintillation counter (Perkin-Elmer).
Transport Experiments with Compounds for LC-MS/MS.
For transport on 48-well plates, cells were plated at 300,000 cells/ml by using 500 μl and grown to confluence. Cells were washed three times with 400 μl of room-temperature WB, followed by an incubation with 150 μl of transport buffer containing approximately 20 nM [3H]uridine, 60 nM [3H]zidovudine, or 10 μM [3H]abacavir and either 100 or 500 μM of unlabeled compound, depending on the experiment. Transport was terminated after 4 minutes by rinsing cells three times with 400 μl ice-cold WB. After terminating transport, 1:1 MeOH/ACN (v/v) with internal standard (500 ng/ml) was added and incubated overnight at 4°C. A calibration curve was generated in wells without cells for each test compound, ranging from 62.5 to 500 ng/ml with internal standard (500 ng/ml) in 1:1 MeOH/ACN (v/v) and incubated overnight. Samples were dried the following day and resuspended in 1:1 MeOH/ACN/H2O with 0.1% formic acid and shaken on ice for 30 minutes. Samples were collected and centrifuged for 1 hour at 10,000g in a tabletop centrifuge at 4°C. The supernatant was collected, and samples were placed into 96-well plates for LC-MS/MS analysis.
LC-MS/MS Quantification of Analytes.
A Shimadzu Prominence HPLC system (Shimadzu, Kyoto, Japan) coupled to a SCIEX QTRAP 4500 mass spectrometer (SCIEX, Framingham, MA) was used for LC-MS/MS analysis. Abacavir and entecavir were detected in positive ion mode with 5.5 kV ion spray voltage and 80 V DP. Zidovudine, uridine, and stavudine were detected in negative ion mode with −4.5 kV ion spray voltage and −60 V DP. Source gas 1 and 2 and curtain gas were set to 20, 40, and 20, respectively, and entrance and collision cell exit potentials were both set to 10 V for both polarities. Entecavir served as the internal standard for abacavir, and vice versa. Stavudine served as the internal standard for uridine and zidovudine. Two microliters of each sample was injected onto a Luna Omega polar C18 column (50 × 2.1 mm with 1.6 μM beads; Phenomenex, Torrance, CA). The mobile phases were H2O with 0.1% formic acid (A) and acetonitrile with 0.1% formic acid (B). A 7.5-minute gradient was run for abacavir and entecavir with a flow rate of 0.3 ml/min as follows: 5% B for 2 minutes, 5%–90% B over 5 minutes, 90% B for 2 minutes, and then re-equilibration at 5% B for 0.5 minutes. Analytes were detected by multiple reaction monitoring by the following mass transitions: 287.1 > 191.1 for abacavir (27 eV collisions) and 278.1 > 152.1 for entecavir (25 eV collisions).
A 7.5-minute gradient was run for uridine, stavudine, and zidovudine with a flow rate of 0.25 ml/min as follows: 0% B for 2 minutes, 0%–80% B over 5 minutes, 80% B for 2 minutes, and then re-equilibration at 0% B for 0.5 minutes. Analytes were detected by multiple reaction monitoring by the following mass transitions: 243.0 > 110.0 for uridine (collision energy of −20 V), 223.0 > 125.0 for stavudine (−15 eV collisions), and 226.0 > 223.0 for zidovudine (−14 eV collisions).
Data Analysis.
All experiments were performed with cells cultured from different passages and with multiple replicates (three to six) with an “n” as indicated in the figure legends. Transport was typically expressed in mols per centimeter squared of confluent cell surface area (∼0.04 mg of cell protein per centimeter squared) [3H]Uridine uptake in the presence of endogenous nucleosides and NRTIs was represented as a percentage of [3H]uridine uptake in the absence of NRTIs or NBMPR. GraphPad Prism 7 (GraphPad Software, San Diego, CA) was used to calculate all statistics and generate kinetic parameters. Data reported are represented by mean ± S.D. unless otherwise noted. An unpaired t test was used to calculate the statistical difference between kinetic parameters (P < 0.05). An extra-sum of squares F test was used to calculate the statistical difference between IC50 values generated from best-fit lines for endogenous nucleosides (P < 0.05). Data in Table 2 for the endogenous nucleoside IC50 values are presented as mean ± S.E.M. Two-way analysis of variance with Bonferonni’s correction for multiple comparisons was performed for studies using NRTIs (P < 0.05). One-way analysis of variance with Bonferonni’s correction for multiple comparisons was performed for additional studies on uridine, abacavir, entecavir, and zidovudine (P < 0.05).
TABLE 2.
Summary of IC50 values of endogenous nucleosides on [3H]uridine uptake in HeLa cells
An extra-sum of squares F test (P < 0.05) was done on the best fit lines generated from the nonlinear regression IC50 (eq. 3) to compare generated IC50 values on GraphPad Prism 7. A statistically significant difference between the IC50 values for inosine and cytidine was found (P = 0.0137 and P < 0.0001). No statistical difference was observed for adenosine (P = 0.8384), guanosine (P = 0.7952), and thymidine (P = 0.0574). All values are reported as mean ± S.E.M. of triplicate values (n = 3).
| Nucleoside | IC50 (ENT1) (μM) | IC50 (ENT2) (μM) |
|---|---|---|
| Adenosine | 59.17 ± 20.92 | 67.65 ± 34.77 |
| Guanosine | 297.60 ± 179.60 | 398.40 ± 311.80 |
| Thymidine | 621.50 ± 272.60 | 159.10 ± 85.37 |
| Inosine | 312.80 ± 179.10* | 32.03 ± 17.69* |
| Cytidine | 9.44 ± 1.50* | 144.0 ± 80.91* |
The LC-MS/MS data were analyzed using Analyst software (SCIEX). Peak areas were converted into concentrations (nanograms per milliliter) based on normalized calibration curves of internal standards run for each set of samples. The concentrations were then converted to pmol per centimeter squared for ease of comparison with data from radiolabeled compound transport experiments. Each of these experiments was performed in duplicate with an n = 3.
Results
mRNA Expression of Human ENT1 and ENT2.
Figure 1 shows the PCR-amplified DNA products for ENT1 and ENT2 in HeLa cells and full-length plasmids as positive controls run on an agarose gel stained with ethidium bromide. Excised product bands were sent for Sanger sequencing (data not shown) to validate primer specificity and mRNA expression of the ENTs. Analysis of the PCR-amplified products from total HeLa cell RNA shows that ENT1 and ENT2 are transcriptionally expressed when compared with the positive controls and aligned sequences from GenBank. The smaller product bands found in lane 4 were confirmed to be degraded or truncated ENT2 mRNA by sequence alignment. Subsequent experiments demonstrate the functionality of these transporters.
Fig. 1.

Reverse-transcription PCR showing mRNA expression of human ENT1 and ENT2 in HeLa cells. Analysis of PCR-amplified DNA products for ENT1 and ENT2 from HeLa S3 cells and positive control full-length plasmids by means of agarose gel electrophoresis and stained with ethidium bromide. Product bands were excised and sent for Sanger sequencing. L: DNA ladder. (1) ENT1 amplified from total HeLa S3 cDNA. (2) ENT1 amplified from a control ENT1 plasmid. (3) ENT2 amplified from total HeLa S3 cDNA. (4) ENT2 amplified from a control ENT2 plasmid.
Inhibition of [3H]Uridine Uptake by NBMPR.
Figure 2 shows the accumulation of 20 nM [3H]uridine in HeLa cells over 7 minutes. Net accumulation is effectively linear over this period and reduced by >95% with the addition of 5 mM unlabeled uridine, suggesting that virtually all uridine uptake is carrier-mediated. The addition of a 100-nM concentration of the ENT1-selective inhibitor, NBMPR, reduced total uridine uptake by about 80%. The addition of 100 μM eliminated the remaining transport. These data suggest that ENT1 supports 80% of the carrier-mediated uridine transport, whereas ENT2 supports the remaining 20% in these cells.
Fig. 2.

Seven-minute time course of [3H]uridine uptake in HeLa cells. [3H]Uridine uptake is effectively linear through 7 minutes and approximately 80% blocked by 100 nM NBMPR and >95% blocked by 5 mM uridine and 100 µM NBMPR. The best fit line represents a one-phase association. All values are reported as mean ± S.D. (n = 3).
[3H]Uridine uptake measured in HeLa cells in the presence of increasing concentrations of NBMPR (0–100 μM) illustrates the distinct inhibitory profiles of both transporters. Inhibition is dose-dependent (Fig. 3) and biphasic, including both high affinity and low affinity elements. A two-site inhibition model adequately describes the results:
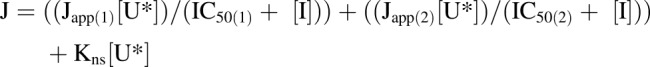 |
(1) |
where J is total transport of radiolabeled uridine (U*), IC50(1) is the concentration of NBMPR that blocks 50% of “NBMPR-sensitive” uridine transport, and IC50(2) is the concentration of NBMPR (“I”) that blocks 50% of “NBMPR-insensitive” uridine transport. Japp(1) and Japp(2) are constants that reflect the Jmax for mediated uridine transport, the respective NBMPR IC50 ratios, and the Michaelis constants for ENT1 and ENT2 uridine transport (Groves et al., 1994). Kns is a first-order constant that reflects the nonsaturable component of total uptake of radiolabeled uridine (including diffusion, nonspecific surface binding, and incomplete rinsing). The IC50 values for NBMPR inhibition from four separate experiments for ENT1 and ENT2 were 11.33 ± 2.85 nM and 9.60 ± 4.47 μM (Fig. 3). Based on observations reported in previous studies, the high and low affinity IC50 values reflect inhibition of ENT1 and ENT2, respectively.
Fig. 3.

Inhibition of [3H]uridine uptake by increasing concentrations of NBMPR in HeLa cells. The experiments were conducted with approximately 20 nM [3H]uridine and terminated at 2 minutes. A best-fit line was fitted using a two-site inhibition model described in eq. 1. Data points are reported as mean ± S.D. of six replicate values (n = 3).
Kinetics of Uridine Transport.
Because [3H]uridine uptake is effectively linear for 7 minutes, data after 2 minutes estimated the initial unidirectional rate of uridine influx for subsequent kinetic analyses. Increasing concentrations of nonradiolabeled uridine coincubated with 20 nM [3H]uridine in HeLa cells evaluate the kinetic profile of uridine transport (Fig. 4). Kinetic parameters determined using total uptake of radiolabeled uridine included transporter-mediated and nonsaturable components. Uridine uptake measured in the presence of 100 nM NBMPR represents ENT2-mediated transport and total uridine transport minus the ENT2-mediated component represents ENT1-mediated transport. The kinetics of each component are adequately described by Michaelis-Menten kinetics:
| (2) |
where J is total uridine transport, Jmax(1 or 2) is maximal uridine transport (pmol per centimeter squared minute) for ENT1 or ENT2, and Kt(1 or 2) is the Michaelis constant (nonsaturable component) for ENT1 or ENT2. There is no significant difference in Jmax values for the ENT1 and ENT2 components of total uridine transport in HeLa cells (P = 0.0699, 16.53 ± 3.44 vs. 30.40 ± 5.27 pmol cm−2 min−1, respectively). However, there is an eightfold difference in Kt (P < 0.0001, 13.60 ± 3.07 vs. 108.9 ± 7.87 μM, respectively) for ENT1- and ENT2-mediated transport, respectively (Fig. 4; Table 1). The intrinsic clearance (Jmax/Kt) of uridine by ENT1 is approximately fivefold higher (P < 0.0001, 1.24 ± 0.04 μl cm-2 min-1 vs. 0.27 ± 0.03 μl cm-2 min-1) than that for ENT2, thereby accounting for the observable difference in NBMPR-sensitive (ENT1) and -insensitive (ENT2) transport of 20 nM [3H]uridine in Fig. 1.
Fig. 4.

Michaelis-Menten kinetics of ENT-mediated uridine uptake in HeLa cells. Data were collected after 2 minutes. The experiments were conducted with approximately 20 nM [3H]uridine in the presence of increasing concentrations of uridine. Total uridine uptake (A), ENT1-specific uptake (B), and ENT2-specific uptake (C). ENT2-specific uptake was determined with the addition of 100 nM NBMPR, ENT1-specific uptake was determined by subtracting ENT2-specific uptake from total uptake. Best-fit lines were fitted to determine relevant values using GraphPad Prism 7. The red dashed lines in (A) represent predicted total uptake and nonspecific uptake determined using the kinetic parameters for ENT1- and ENT2-specific uptake. Data points in graphs are reported as mean ± S.D. of six replicates (n = 3).
TABLE 1.
Relevant kinetic parameters of uridine uptake in HeLa cells
An unpaired t test was used to calculate kinetic parameters generated from three separate experiments (P < 0.05). A statistically significant difference between Kt and intrinsic clearance for ENT1- and ENT2-specific uridine uptake was found (P < 0.0001 for both). No statistical difference was observed for Jmax (P = 0.0699). All values are reported as mean ± S.D. of six replicate values (n = 3). * indicates P < 0.05
| ENT1-Specific Uridine Uptake | ENT2-Specific Uridine Uptake | |
|---|---|---|
| Jmax (pmol cm−2 min−1) | 16.53 ± 6.88 | 30.40 ± 10.55 |
| Kt (µM) | 13.60 ± 6.15* | 108.9 ± 15.74* |
| Intrinsic clearance Jmax/ Kt (μl cm−2 min−1) | 1.24 ± 0.07* | 0.28 ± 0.06* |
Inhibition of [3H]Uridine Uptake by Endogenous Nucleosides.
[3H]Uridine uptake measured with increasing concentrations of adenosine, guanosine, cytidine, inosine, or thymidine in the presence or absence of 100 nM NBMPR showed the differing inhibitory profiles of nucleosides for both transporters (Fig. 5). The following equation describes the results:
| (3) |
where U represents [3H]uridine (approximately 20 nM) and N represents endogenous nucleoside concentration. This equation generates a best fit line and IC50 values for each nucleoside. An extra-sum of squares F test compared these values (P < 0.05). There was no significant difference between the IC50 values for ENT1 and ENT2 of adenosine (59.17 ± 20.92 and 67.65 ± 34.77 μM, P = 0.8384), guanosine (297.60 ± 179.60 and 398.40 ± 311.80 μM, P = 0.7952), and thymidine (621.50 ± 272.60 and 159.10 ± 85.37 μM, P = 0.0574). There was a significant difference in IC50 values for ENT1 and ENT2 with inosine (312.80 ± 179.10 and 32.03 ± 17.69 μM, P = 0.0137) and cytidine (312.80 ± 179.10 and 32.03 ± 17.69 μM, P value < 0.0001) (Table 2).
Fig. 5.

Inhibition of [3H]uridine uptake in HeLa cells by endogenous nucleosides. IC50 analysis of adenosine (A), guanosine (B), and thymidine (C), inosine (D), and cytidine (E). Total [3H]uridine uptake (black open circle), ENT1-mediated [3H]uridine uptake (blue diamond), and ENT2-mediated [3H]uridine uptake (red triangle) were measured in the presence of endogenous nucleosides. ENT2 mediated uptake was taken as that occurring in the presence of 100 nM NBMPR. ENT1 data points were determined by subtracting calculated ENT2 uptake from total uptake and fitting the data to a single component IC50 equation. Best-fit lines were created to determine relevant values using GraphPad Prism 7. Increasing concentrations of each compound were incubated with approximately 20 nM [3H]uridine or 20 nM [3H]uridine and 100 nM NBMPR. Data points in the graph are reported as mean ± S.D. of triplicate values (n = 3).
Inhibition of [3H]Uridine Uptake by NRTIs.
The effect of nine NRTIs on [3H]uridine uptake in the presence or absence of 100 nM NBMPR (Fig. 6) explored the relative effects of NRTIs on ENT1- and ENT2-mediated transport. The inhibition of uridine uptake by 100 nM NBMPR (control) suggests that approximately 80% of total uridine transport is mediated by ENT1. Moreover, the remaining (ENT2-mediated) transport is eliminated by the addition of either 100 µM NBMPR or 5 mM unlabeled uridine (data not shown). Only abacavir, entecavir, and zidovudine significantly inhibit [3H]uridine uptake (Fig. 6, A, B, and C). Two-way ANOVA with Bonferroni’s correction for multiple comparisons showed that in the absence of NBMPR, 1 mM entecavir and zidovudine block 29% and 24% of [3H]uridine uptake (P = 0.0205 and P = 0.0316), respectively. Notably, abacavir blocks 46% of [3H]uridine uptake at 100 µM, 72% at 300 µM, and 90% at 1 mM (P < 0.0001 for each concentration). In the presence of 100 nM NBMPR, 1 mM abacavir blocks an additional 18% of the remaining [3H]uridine uptake (P = 0.0164), indicating a significant interaction of abacavir with ENT2. Figure 7 shows the IC50 values for these three compounds for inhibition of ENT1 and ENT2. Previous NRTI inhibition studies and solubility determined concentration ranges. A single component IC50 equation for [3H]uridine uptake in the presence or absence of 100 nM NBMPR (ENT2) plus the NRTI of interest generated IC50 values for ENT2-mediated uridine transport. Subtracting the ENT2-mediated component of uridine accumulation from total accumulation yielded IC50 values for ENT1-mediated [3H]uridine uptake. There is a significant difference in the IC50 values for inhibition of ENT1- and ENT2-mediated [3H]uridine uptake for abacavir and zidovudine but not for entecavir (Table 3). The IC50 values for NRTI inhibition of ENT1- and ENT2-mediated [3H]uridine uptake for abacavir are 110 ± 10 and 220 ± 10 µM (P < 0.001), respectively. For zidovudine, IC50 values are higher for ENT1 than ENT2 (2.54 ± 0.15 and 1.64 ± 0.21 mM, P = 0.0235), respectively. For entecavir, the IC50 values are 3.05 ± 0.9 and 2.07 ± 0.17 mM (P = 0.3479).
Fig. 6.

Inhibition of ENT-mediated [3H]uridine uptake in HeLa cells by nine NRTIs. The experiments were terminated after 2 minutes. All graphs are represented by relative uptake as a percentage of control ([3H]uridine only). Three concentrations of abacavir (A), entecavir (B), zidovudine (C), didanosine (D), zalcitabine (E), emtricitabine (F), tenofovir disoproxil (G), stavudine (H), and lamivudine (I) were incubated with approximately 20 nM [3H]uridine or 20 nM [3H]uridine and 100 nM NBMPR as indicated on the x-axis. Significance between control without NBMPR and the NRTIs is indicated as *P < 0.05; ***P < 0.0001. Significance between control with 100 nM NMBPR and the NRTIs is indicated as †P < 0.05. Two-way analysis of variance with Bonferonni’s correction for multiple comparisons was used for statistical significance analysis using GraphPad Prism 7. Data in graphs are reported as mean ± S.D. of four replicate values (n = 3).
Fig. 7.

IC50 analysis of abacavir (A), entecavir (B), and zidovudine (C). Total [3H]uridine uptake (black open circle), ENT1-mediated [3H]uridine uptake (blue diamond), and ENT2-mediated [3H]uridine uptake (red triangle) was measured in the presence of increasing concentrations of NRTIs. ENT2-mediated uptake was determined with the addition of 100 nM NBMPR. ENT1 data points were determined by subtracting ENT2 uptake from total uptake and fitting the data to a single component IC50 equation. Best-fit lines were created to determine relevant values using GraphPad Prism 7. Increasing concentrations of each compound were incubated with approximately 20 nM [3H]uridine or 20 nM [3H]uridine and 100 nM NBMPR. Data points in the graph are reported as mean ± S.D. of six replicate values (n = 3).
TABLE 3.
Summary of IC50 values of abacavir, entecavir, and zidovudine for inhibition of [3H]uridine uptake
A single component IC50 equation was used to generate values for total uptake and ENT2-mediated uptake (+100 nM NBMPR), and the ENT1 component was generated by subtracting the ENT2-mediated component from total uptake. An unpaired t test was used to determine statistical significance. All values are reported as mean ± S.D. of six replicate values (n = 3).
| NRTI | IC50 (ENT1) (mM) | IC50 (ENT2) (mM) |
|---|---|---|
| Abacavir | 0.11 ± 0.02* | 0.22 ± 0.01* |
| Entecavir | 3.05 ± 1.56 | 2.07 ± 0.29 |
| Zidovudine | 2.54 ± 0.26* | 1.64 ± 0.36* |
Inhibition of Uridine and NRTI uptake with NBMPR.
The transport of uridine, abacavir, entecavir, and zidovudine is directly examined by using LC-MS/MS and/or radiolabeled substrate to determine the concentration of accumulated compounds in HeLa cells in the absence or presence of NBMPR. A 4-minute incubation time in single wells of 48-well plates (well within the linear range of uridine uptake noted in Fig. 2) was used to obtain a large enough sample for LC-MS/MS analysis (Fig. 8). One-way ANOVA with Bonferonni’s multiple comparisons test showed a statistically significant decrease in unlabeled uridine and entecavir uptake in the presence of 100 µM NBMPR (P = 0.0065 and P = 0.0201, respectively). [3H]Uridine uptake significantly decreases in the presence of both 100 nM and 100 µM NBMPR (P < 0.0001). For zidovudine, NBMPR did not significantly change accumulation of zidovudine in LC-MS/MS analysis (P = 0.4902) or by tracking accumulation of [3H]zidovudine. However, accumulation of [3H]zidovudine significantly decreased in the presence of unlabeled zidovudine (P = 0.0206). Interestingly, NBMPR did not inhibit abacavir accumulation in LC-MS/MS analysis (P = 0.9658). Furthermore, NBMPR, unlabeled abacavir, or unlabeled uridine failed to influence accumulation of the [3H]abacavir (P = 0.8671).
Fig. 8.

Inhibition of uptake of uridine (A), abacavir (B), zidovudine (C), and entecavir (D) by NBMPR using radiolabeled substrates detected by a liquid scintillation counter and unlabeled substrates detected by LC-MS/MS. All data are reported as percentage of control. One-way ANOVA with Bonferonni’s multiple comparison correction was used to determine statistical significance. Data in graphs are reported as mean ± S.D. of duplicate values (n = 3).
Discussion
Using NBMPR to differentiate between ENT1- and ENT2-mediated transport in HeLa cells provides a method for studying the influence of NRTIs on the function of these distinct processes. Figures 1–5 and Tables 1 and 2 characterize our model system for assessing the interaction of NRTIs with the ENTs. Reverse-transcription PCR analysis confirmed the mRNA expression of human ENT1 and ENT2 in HeLa cells (Fig. 1). Functional assessment of these transporters shows that uridine transport in HeLa cells consists of two components, distinguishable by their sensitivity to inhibition by NBMPR. One hundred nanomolars NBMPR blocks approximately 80% of mediated [3H]uridine (20 nM) transport, whereas 100 µM NBMPR blocks the remaining 20% (Fig. 3).These high and low affinity interactions resolve as two distinct IC50 values (11 nM vs. 9 µM) that correlate well with early reports of NBPMR inhibition of native uridine transport in cultured mammalian cells (Plagemann and Roth, 1969; Plagemann and Wohlhueter, 1984) and with more recent studies with heterologously expressed ENT1 and ENT2 (Ward et al., 2000; Sundaram et al., 2001; Yao et al., 2001). NBMPR enables studying the kinetics of uridine transport in HeLa cells. Although the maximal rate of ENT2-mediated transport is modestly larger than that supported by ENT1, the significantly higher apparent affinity of ENT1 for uridine results in an ENT1-supported intrinsic clearance in HeLa cells that is fivefold greater than that supported by ENT2. This observation also quantitatively accounts for the extent of inhibition of uridine transport by 100 nM NBMPR (Fig. 3).
The kinetic values for uridine transport and inhibition of ENT1 and ENT2 by NBMPR are similar to those previously reported. The IC50 values ENT1 and ENT2 were similar to the values for nucleoside transport mediated both by cloned ENTs (0.4–150 nM and 2.8–10 µM, respectively) (Griffiths et al., 1997a,b; Ward et al., 2000; Huang et al., 2017) and in native mammalian cells (1–10 nM and 1–10 µM, respectively) (Dahlig-Harley et al., 1981). In studies that measure the nucleoside transport kinetics by ENT1 and ENT2 heterologously expressed in Xenopus oocytes (Mackey et al., 1999) or porcine kidney cells (Ward, 2000European Medicines Agency Committee for Human Medicinal Products, 2017), the Kt values for uridine transport are approximately 0.2–0.4 mM. However, in this study, the apparent Kt values are 13.6 and 108.9 µM for ENT1 and ENT2, respectively.
The apparent discrepancy may be a result of the measurements in the presented study that reflect activity of native ENT-mediated transport in human cells, and these values are well within the range of values for Na+-independent native uridine transport in various mammalian cell types, including K562 cells, Novikoff rat hepatoma cells, and mouse lymphocytic leukemia cell lines (10–300 µM) (Plagemann et al., 1978; Plagemann and Wohlhueter, 1984; Boleti et al., 1997), which involve some combination of ENT1 and ENT2 activity. There is variability in the kinetics reported in the literature. However, the characterization of these values for the ENTs has not always focused on natively expressed human systems. More research on natively expressing human systems is needed to understand the complexity of human transporters to use this data for in vivo predictions on drug disposition and pharmacokinetics.
It is worth noting that the relative expression of ENT1 versus ENT2 in different lines of HeLa cells can be markedly different. Indeed, some cell lines appear to express differing levels of “NBPMR-sensitive” transport and “NBMPR-insensitive” transport (Lauzon and Paterson, 1977; Dahlig-Harley et al., 1981). The cell line employed in this study (HeLa S3) expresses both ENT1 and ENT2 (Figs. 1 and 2) and displays kinetically distinguishable transport activity. Taken together, these observations show that NBMPR provides a means to examine the independent kinetic contributions to uridine transport by natively expressed ENT1 and ENT2 activity in HeLa cells, thereby providing a means to assess the distinct influence of NRTIs on each of these processes. There is great value in a system that promotes high-throughput assessment of transport activity without the complexity that arises from limitations with the use of primary cultures of native cells and the polarized expression of ENTs in Sertoli cells.
The endogenous nucleosides adenosine, guanosine, thymidine, cytidine, and inosine showed differing levels of inhibition of ENT-mediated transport in HeLa cells. Guanosine and adenosine show no significant difference in IC50 values on [3H]uridine uptake by ENT1 and ENT2. Although not statistically significant, thymidine appears to have a higher affinity for ENT2 (Table 2). Inosine also shows significantly higher affinity for ENT2, whereas cytidine shows significantly higher affinity for ENT1. The values reported here for the endogenous nucleosides are similar to values reported by Ward et al. (2000), except for cytidine, which is found to have higher affinity for both ENT1 and ENT2 (ENT1: 9 µM vs. 580 µM and ENT2: 144 µM vs. 5 mM).
Nine NRTIs and their involvement with the inhibition of uridine uptake into HeLa cells assessed the interaction of NRTIs in ENT-mediated transport in human cells (Fig. 6). Abacavir, entecavir, and zidovudine inhibit 50% of total uridine uptake at concentrations of 129 µM, 2.23 mM, and 2.39 mM, respectively. Notably, these concentrations are higher than the maximum plasma concentrations (Cmax) found in patients taking these drugs. Abacavir has a Cmax of 2.23–15.3 µM (Hervey and Perry, 2000), zidovudine has a Cmax of 1.5–10 µM (Langtry and Campoli-Richards, 1989), and entecavir has a Cmax of 15–30 pM (Baraclude, 2005; Matthews, 2006). Consequently, the ratios of Cmax to IC50 are all well below 0.1 for entecavir and zidovudine, initially suggested by the Giacomini et al. (2010) and subsequently supported by both the * indicates P < 0.05 and the European Medicines Agency Committee for Human Medicinal Products, 2012, European Medicines Agency Committee for Human Medicinal Products, 2017 as the cutoff to identify potential perpetrators of unwanted drug-drug interactions (DDIs). However, the ratio of Cmax to IC50 is just above 0.1 for abacavir, suggesting that it may contribute to unwanted DDIs. It is unlikely that any of the tested NRTIs, except for abacavir, represent a potential source of unwanted DDIs with ENTs. If abacavir or another ENT inhibitor is taken in conjunction with an ENT substrate, there is potential for increased exposure to the ENT substrate, which may result in toxicity. Therefore, it is important to continue to examine the substrate selectivity profile of ENT1 and ENT2 to identify potential DDIs with ENT substrates and inhibitors. This area remains largely unexplored. There is a clear interaction with abacavir and the ENTs that should be noted, and identifying other interactions will be essential for generating sufficient evidence on potential DDIs with the ENTs to warrant inclusion in regulatory guidelines.
Both guanosine analogs tested, abacavir and entecavir, showed inhibition of uridine uptake. However, guanosine itself showed modest inhibition of uridine uptake. Additional nucleoside analogs need to be tested to have a more complete representation of ENT selectivity. In addition to the data presented, screening a larger library of nucleoside analogs would contribute to the development of a pharmacophore for both ENT1 and ENT2. Targeted drug development of ENT substrates could improve the treatment of viral infections and cancer within the male genital tract. Predicting the ability of compounds to cross the BTB would also be possible using substrate selectivity data and computer modeling, which could result in a more effective drug development process and patient treatment.
The possibility that abacavir, entecavir, and zidovudine may be inhibitors rather than substrates is explored using LC-MS/MS and transport of radiolabeled compounds. Whereas NBMPR significantly reduced entecavir uptake, it did not reduce abacavir and zidovudine uptake into HeLa cells, consistent with the conclusion that their interactions with ENTs may be restricted to inhibition. [3H]Zidovudine uptake was inhibited by 5 mM zidovudine, indicating that it enters HeLa cells by a carrier-mediated process, but NBMPR failed to significantly reduce zidovudine uptake determined by either LC-MS/MS or accumulation of the radiolabeled substrate, suggesting its entry into HeLa cells may involve a non–ENT-mediated pathway.
There is conflicting evidence in the literature about the role of ENTs in the transport of the three aforementioned compounds. In BeWo cells and plasma membrane vesicles from human placental tissue, abacavir uptake was observed to be ENT1-mediated (Cerveny et al., 2018). Another study concluded that abacavir uptake in human erythrocytes and human T-lymphoblastoid CD4+ CEM cells is carrier-independent (Mahony et al., 2004). Similarly, there are also conflicting views on the transport for zidovudine. Whereas transplacental passage of zidovudine appears to be independent of the ENTs (Karbanova et al., 2017), other studies showed that zidovudine is transported by recombinant hENT2 expressed in Xenopus laevis oocytes (Yao et al., 2001). Another study suggested that zidovudine is a substrate for hENT2 but a nontransported inhibitor of ENT1 (Molina-Arcas et al., 2009). With respect to the NRTIs that had no inhibitory impact on uridine transport in HeLa cells, Molina-Arcas et al. (2009) reported that didanosine and zalcitabine are substrates for ENTs. Another study concluded that entecavir and emtricitabine uptake is inhibited by NBMPR in BeWo cells (Ma et al., 2017). It is worth stressing that although our data did not show any inhibition of [3H]uridine uptake by didanosine, emtricitabine, lamivudine, stavudine, tenofovir disoproxil, or zalcitabine, it is possible that higher concentrations (i.e., >5 mM) may reveal a low affinity interaction that could result in net transport.
These data demonstrate the complexity of substrate selectivity between ENT1 and ENT2. Based on previous studies that localized ENT1 and ENT2 in different tissues and the crystal structure of ENT1 with adenosine reuptake inhibitors such as NBMPR, understanding the structural selectivity between the human ENTs would be an asset for targeted drug development of compounds that use these transporters (http://www.proteinatlas.org; Uhlén et al., 2015; Boswell-Casteel and Hays, 2017; Wright and Lee, 2019). In rat and human testes, ENT1 is localized to the basal membrane, whereas ENT2 is localized to the apical membrane of Sertoli cells (Klein et al., 2013). This transepithelial transport pathway provides a mechanism of circumventing the BTB and would be an ideal target in drug development for diseases in the testis. Designing compounds that are substrates for both ENT1 and ENT2 has the potential to eliminate a viral sanctuary site, reduce the rate of testicular relapse in certain cancers, and create effective male contraceptives (Miller and Cherrington, 2018). In summary, we describe a robust in vitro model for studying ENT-mediated transport that can be effectively translated into the investigation of transport by the same proteins in Sertoli cells comprising the BTB.
Abbreviations
- BTB
blood-testis barrier
- Cmax
maximum plasma concentration
- DDI
drug-drug interaction
- ENT
equilibrative nucleoside transporter
- HIV
human immunodeficiency virus
- LC
liquid chromatography
- MS/MS
tandem mass spectrometry
- NBMPR
S-[(4-nitrophenyl)methyl]-6-thioinosine
- NRTI
nucleoside reverse transcriptase inhibitor
- PCR
polymerase chain reaction
- WB
Waymouth’s buffer
Authorship Contributions
Participated in research design: Miller, Hau, Jilek, Wright, Cherrington.
Conducted Experiments: Miller, Hau, Jilek, Morales.
Performed data analysis: Miller, Hau, Jilek, Morales, Wright.
Wrote or contributed to the writing of the manuscript: Miller, Hau, Jilek, Morales, Wright, Cherrington.
Footnotes
This work was supported by the National Institutes of General Medical Sciences [R01GM123643], [R01GM129777] and the National Institutes of Health Training in Environmental Toxicology of Human Diseases [2T32ES007091-36A1].
References
- Baldwin SA, Beal PR, Yao SYM, King AE, Cass CE, Young JD. (2004) The equilibrative nucleoside transporter family, SLC29. Pflugers Arch 447:735–743. [DOI] [PubMed] [Google Scholar]
- Baraclude (2005) Package insert. Bristol-Myers Squibb, New York.
- Boleti H, Coe IR, Baldwin SA, Young JD, Cass CE. (1997) Molecular identification of the equilibrative NBMPR-sensitive (es) nucleoside transporter and demonstration of an equilibrative NBMPR-insensitive (ei) transport activity in human erythroleukemia (K562) cells. Neuropharmacology 36:1167–1179. [DOI] [PubMed] [Google Scholar]
- Boswell-Casteel RC, Hays FA. (2017) Equilibrative nucleoside transporters-A review. Nucleosides Nucleotides Nucleic Acids 36:7–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerveny L, Ptackova Z, Ceckova M, Karahoda R, Karbanova S, Jiraskova L, Greenwood SL, Glazier JD, Staud F. (2018) Equilibrative nucleoside transporter 1 (ENT1, SLC29A1) facilitates transfer of the antiretroviral drug abacavir across the placenta. Drug Metab Dispos 46:1817–1826. [DOI] [PubMed] [Google Scholar]
- Crawford CR, Patel DH, Naeve C, Belt JA. (1998) Cloning of the human equilibrative, nitrobenzylmercaptopurine riboside (NBMPR)-insensitive nucleoside transporter ei by functional expression in a transport-deficient cell line. J Biol Chem 273:5288–5293. [DOI] [PubMed] [Google Scholar]
- Dahlig-Harley E, Eilam Y, Paterson ARP, Cass CE. (1981) Binding of nitrobenzylthioinosine to high-affinity sites on the nucleoside-transport mechanism of HeLa cells. Biochem J 200:295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Medicines Agency Committee for Human Medicinal Products (2012). Guideline on the investigation of drug interactions. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf.
- European Medicines Agency Committee for Human Medicinal Products (2017)Guideline on the investigation of drug interactions. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, et al. International Transporter Consortium (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith DA, Jarvis SM. (1996) Nucleoside and nucleobase transport systems of mammalian cells. Biochim Biophys Acta 1286:153–181. [DOI] [PubMed] [Google Scholar]
- Griffiths M, Beaumont N, Yao SYM, Sundaram M, Boumah CE, Davies A, Kwong FYP, Coe I, Cass CE, Young JD, et al. (1997a) Cloning of a human nucleoside transporter implicated in the cellular uptake of adenosine and chemotherapeutic drugs. Nat Med 3:89–93. [DOI] [PubMed] [Google Scholar]
- Griffiths M, Yao SY, Abidi F, Phillips SE, Cass CE, Young JD, Baldwin SA. (1997b) Molecular cloning and characterization of a nitrobenzylthioinosine-insensitive (ei) equilibrative nucleoside transporter from human placenta. Biochem J 328:739–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves CE, Evans KK, Dantzler WH, Wright SH. (1994) Peritubular organic cation transport in isolated rabbit proximal tubules. Am J Physiol 266:F450–F458. [DOI] [PubMed] [Google Scholar]
- Hervey PS, Perry CM. (2000) Abacavir: a review of its clinical potential in patients with HIV infection. Drugs 60:447–479. [DOI] [PubMed] [Google Scholar]
- Huang W, Zeng X, Shi Y, Liu M. (2017) Functional characterization of human equilibrative nucleoside transporter 1. Protein Cell 8:284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbanova S, Cerveny L, Ceckova M, Ptackova Z, Jiraskova L, Greenwood S, Staud F. (2017) Role of nucleoside transporters in transplacental pharmacokinetics of nucleoside reverse transcriptase inhibitors zidovudine and emtricitabine. Placenta 60:86–92. [DOI] [PubMed] [Google Scholar]
- Kato R, Maeda T, Akaike T, Tamai I. (2005) Nucleoside transport at the blood-testis barrier studied with primary-cultured sertoli cells. J Pharmacol Exp Ther 312:601–608. [DOI] [PubMed] [Google Scholar]
- Klein DM, Evans KK, Hardwick RN, Dantzler WH, Wright SH, Cherrington NJ. (2013) Basolateral uptake of nucleosides by Sertoli cells is mediated primarily by equilibrative nucleoside transporter 1. J Pharmacol Exp Ther 346:121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langtry HD, Campoli-Richards DM. (1989) Zidovudine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs 37:408–450. [DOI] [PubMed] [Google Scholar]
- Lauzon GJ, Paterson AR. (1977) Binding of the nucleoside transport inhibitor nitrobenzylthioinosine to HeLa cells. Mol Pharmacol 13:883–891. [PubMed] [Google Scholar]
- Lok AS, Zoulim F, Dusheiko G, Ghany MG. (2017) Hepatitis B cure: from discovery to regulatory approval. J Hepatol 67:847–861. [DOI] [PubMed] [Google Scholar]
- Lowe SH, van Leeuwen E, Droste JAH, van der Veen F, Reiss P, Lange JMA, Burger DM, Repping S, Prins JM. (2007) Semen quality and drug concentrations in seminal plasma of patients using a didanosine or didanosine plus tenofovir containing antiretroviral regimen. Ther Drug Monit 29:566–570. [DOI] [PubMed] [Google Scholar]
- Lucas S, Nelson AM. (2015) HIV and the spectrum of human disease. J Pathol 235:229–241. [DOI] [PubMed] [Google Scholar]
- Ma Z, Yang X, Jiang T, Bai M, Zheng C, Zeng S, Sun D, Jiang H. (2017) Multiple SLC and ABC transporters contribute to the placental transfer of entecavir. Drug Metab Dispos 45:269–278. [DOI] [PubMed] [Google Scholar]
- Mackey JR, Yao SYM, Smith KM, Karpinski E, Baldwin SA, Cass CE, Young JD. (1999) Gemcitabine transport in xenopus oocytes expressing recombinant plasma membrane mammalian nucleoside transporters. J Natl Cancer Inst 91:1876–1881. [DOI] [PubMed] [Google Scholar]
- Mahony WB, Domin BA, Daluge SM, Zimmerman TP. (2004) Membrane permeation characteristics of abacavir in human erythrocytes and human T-lymphoblastoid CD4+ CEM cells: comparison with (-)-carbovir. Biochem Pharmacol 68:1797–1805. [DOI] [PubMed] [Google Scholar]
- Matthews SJ. (2006) Entecavir for the treatment of chronic hepatitis B virus infection. Clin Ther 28:184–203. [DOI] [PubMed] [Google Scholar]
- Miller SR, Cherrington NJ. (2018) Transepithelial transport across the blood-testis barrier. Reproduction 156:R187–R194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Arcas M, Casado FJ, Pastor-Anglada M. (2009) Nucleoside transporter proteins. Curr Vasc Pharmacol 7:426–434. [DOI] [PubMed] [Google Scholar]
- Pereira AS, Kashuba AD, Fiscus SA, Hall JE, Tidwell RR, Troiani L, Dunn JA, Eron JJ, Jr., Cohen MS. (1999) Nucleoside analogues achieve high concentrations in seminal plasma: relationship between drug concentration and virus burden. J Infect Dis 180:2039–2043. [DOI] [PubMed] [Google Scholar]
- Plagemann PGW, Erbe J. (1972) Thymidine transport by cultured Novikoff hepatoma cells and uptake by simple diffusion and relationship to incorporation into deoxyribonucleic acid. J Cell Biol 55:161–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagemann PGW, Marz R, Wohlhueter RM. (1978) Uridine transport in Novikoff rat hepatoma cells and other cell lines and its relationship to uridine phosphorylation and phosphorolysis. J Cell Physiol 97:49–72. [DOI] [PubMed] [Google Scholar]
- Plagemann PGW, Roth MF. (1969) Permeation as the rate-limiting step in the phosphorylation of uridine and choline and their incorporation into macromolecules by Novikoff hepatoma cells. Competitive inhibition by phenethyl alcohol, persantin, and adenosine. Biochemistry 8:4782–4789. [DOI] [PubMed] [Google Scholar]
- Plagemann PGW, Shea MA. (1971) Transport as the rate-limiting step in the incorporation of uridine into mengovirus ribonucleic acid in Novikoff rat hepatoma cells. J Virol 7:137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagemann PGW, Wohlhueter RM. (1984) Nucleoside transport in cultured mammalian cells. Multiple forms with different sensitivity to inhibition by nitrobenzylthioinosine or hypoxanthine. Biochim Biophys Acta 773:39–52. [DOI] [PubMed] [Google Scholar]
- Sandoval PJ, Zorn KM, Clark AM, Ekins S, Wright SH. (2018) Assessment of substrate-dependent ligand interactions at the organic cation transporter OCT2 using six model substrates. Mol Pharmacol 94:1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severance AC, Sandoval PJ, Wright SH. (2017) Correlation between apparent substrate affinity and OCT2 transport turnover. J Pharmacol Exp Ther 362:405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram M, Yao SYM, Ingram JC, Berry ZA, Abidi F, Cass CE, Baldwin SA, Young JD. (2001) Topology of a human equilibrative, nitrobenzylthioinosine (NBMPR)-sensitive nucleoside transporter (hENT1) implicated in the cellular uptake of adenosine and anti-cancer drugs. J Biol Chem 276:45270–45275. [DOI] [PubMed] [Google Scholar]
- Tang PCT, Yang C, Li RW-S, Lee SM-Y, Hoi MP-M, Chan S-W, Kwan Y-W, Tse C-M, Leung GP-H. (2016) Inhibition of human equilibrative nucleoside transporters by 4-((4-(2-fluorophenyl)piperazin-1-yl)methyl)-6-imino-N-(naphthalen-2-yl)-1,3,5-triazin-2-amine. Eur J Pharmacol 791:544–551. [DOI] [PubMed] [Google Scholar]
- Trépo C, Chan HLY, Lok A. (2014) Hepatitis B virus infection. Lancet 384:2053–2063. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015) Proteomics. Tissue-based map of the human proteome. Science 347:1260419. [DOI] [PubMed] [Google Scholar]
- U.S Department of Health And Human Services (2017) Available from: https://www.fda.gov/media/108130/downloadIn Vitro Metabolism and Transporter-Mediated Drug-Drug Interaction Studies Guidance for Industry Draft Guidance pp 1–45, Food and Drug Administration, Center for Drug Evaluation and Research. [Google Scholar]
- Ward JL, Sherali A, Mo ZP, Tse CM. (2000) Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells. Ent2 exhibits a low affinity for guanosine and cytidine but a high affinity for inosine. J Biol Chem 275:8375–8381. [DOI] [PubMed] [Google Scholar]
- Wright NJ, Lee SY. (2019) Structures of human ENT1 in complex with adenosine reuptake inhibitors. Nat Struct Mol Biol 26:599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao SY, Ng AM, Sundaram M, Cass CE, Baldwin SA, Young JD. (2001) Transport of antiviral 3′-deoxy-nucleoside drugs by recombinant human and rat equilibrative, nitrobenzylthioinosine (NBMPR)-insensitive (ENT2) nucleoside transporter proteins produced in Xenopus oocytes. Mol Membr Biol 18:161–167. [DOI] [PubMed] [Google Scholar]
- Young JD, Yao SYM, Baldwin JM, Cass CE, Baldwin SA. (2013) The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol Aspects Med 34:529–547. [DOI] [PubMed] [Google Scholar]