

Summary
Alzheimer's disease (AD) displays a long asymptomatic stage before dementia. We characterize AD stage-associated molecular networks by profiling 14,513 proteins and 34,173 phosphosites in human brain with mass spectrometry, highlighting 173 protein changes in 17 pathways. The altered proteins are validated in two independent cohorts, showing partial RNA dependency. Comparisons of brain tissue and cerebrospinal fluid proteomes reveal biomarker candidates. Combining with 5xFAD mouse analysis, we determine 15 Aβ-correlated proteins (e.g., MDK, NTN1, SMOC1, SLIT2 and HTRA1). 5xFAD shows a proteomic signature similar to symptomatic AD but exhibits activation of autophagy and interferon response, and lacks human-specific deleterious events, such as downregulation of neurotrophic factors and synaptic proteins. Multi-omics integration prioritizes AD-related molecules and pathways, including amyloid cascade, inflammation, complement, WNT signaling, TGFβ/BMP signaling, lipid metabolism, iron homeostasis, and membrane transport. Some Aβ-correlated proteins are colocalized with amyloid plaques. Thus, the multilayer omics approach identifies protein networks during AD progression.
Keywords: Alzheimer’s disease, TMT, mass spectrometry, proteomics, proteome, phosphoproteome, transcriptome, RNA-seq, genomics, genome, brain tissue, cerebrospinal fluid, biomarker, systems biology, multi-omics, disease network, mouse model, 5xFAD, Aβ, Tau, APOE, NTN1, Netrin, UNC5C, MDK, SMOC1, ICAM1, complement
Graphical Abstract
eTOC blurb
By mass spectrometry-based proteomics and integrated multi-omics, Bai et al. reveal novel proteins and molecular networks during Alzheimer’s disease progression and validate the proteins in the 5xFAD mice. Further comparisons of brain tissue and cerebrospinal fluid proteomes identify biomarker candidates.
Introduction
Alzheimer's disease (AD) is the most common type of dementia, imposing a substantial burden to patients and the healthcare (Alzheimer's, 2015). Genetic analysis of AD patients revealed three causative genes (APP, PSEN1 and PSEN2), several risk genes (APOE, TREM2, and UNC5C) (Colonna and Wang, 2016; Guerreiro et al., 2013; Wetzel-Smith et al., 2014), and more than 20 low-risk genetic loci (Kunkle et al., 2019; Lambert et al., 2013). Biochemical studies discovered pathological hallmarks of Aβ plaques and Tau neurofibrillary tangles in AD brain (Hyman et al., 2012). The resulting amyloid cascade and Tau hypotheses (Ballatore et al., 2007; Hardy and Selkoe, 2002) become central to AD research. A large number of animal models have been developed to test these hypotheses, but cannot fully recapitulate AD symptoms (LaFerla and Green, 2012; Sasaguri et al., 2017). Current targeted therapeutic strategies have failed to slow down cognitive decline in AD (Graham et al., 2017; Huang and Mucke, 2012), raising concerns about these hypotheses and calling for a broad understanding of molecular and cellular components in AD brain, at both asymptomatic and symptomatic stages during the disease progression (De Strooper and Karran, 2016).
The advent of next-generation sequencing technologies for DNAs, RNAs and proteins (Aebersold and Mann, 2016; Altelaar et al., 2013; Metzker, 2010), provides an unprecedented opportunity to thoroughly characterize human specimens to elucidate disease networks and novel therapeutic targets. Large-scale genomic analysis of AD is in progress (https://www.niagads.org), and transcriptomic analysis constructed potential disease networks (e.g. microglia-specific module) (Miller et al., 2008; Zhang et al., 2013), and identified cell type specific alterations in AD (Mathys et al., 2019). The transcriptome, however, is often not an accurate indicator of protein abundance (Liu et al., 2016), especially in complex brain tissue (Sharma et al., 2015). For example, abnormal aggregation of APP-cleaved Aβ peptide and phospho-Tau highlights substantial deregulation of posttranslational events in AD (Taylor et al., 2002). Analysis of proteome in AD and during aging has been attempted (Seyfried et al., 2017; Wingo et al., 2019). We performed a comprehensive analysis of the aggregated proteome, detecting the deposition of U1 small nuclear ribonucleoprotein (U1 snRNP) and a deficit of RNA splicing in AD brain (Bai et al., 2013; Lutz and Peng, 2018). The study of aggregated subproteome motivates us to revisit the whole proteome during the development of Alzheimer’s disease.
Here, we present a new paradigm of analyzing 14,513 proteins (12,018 genes) and 34,173 phosphosites (46,612 phosphopeptides) in different disease stages of AD, using the latest multiplexed tandem-mass-tag (TMT) method and two-dimensional liquid chromatography-tandem mass spectrometry (LC/LC-MS/MS) (Bai et al., 2017; Niu et al., 2017; Tan et al., 2017; Wang et al., 2019). This ultra-deep analysis is further validated in two independent AD cohorts, and in the 5xFAD mouse model. Protein changes are only partially consistent with the corresponding RNA levels. Comparison of the human and mouse proteomes defines their resemblance and difference. Finally, we integrate multi-omics datasets to prioritize altered proteins/genes and pathways. A number of top-listed proteins emerge with the appearance of amyloid plaques. In summary, these results provide a rich resource of molecular alterations in AD and establish a crucial framework of disease networks/pathways.
Results
Deep Proteomic and Phosphoproteomic Profiling of AD Progression
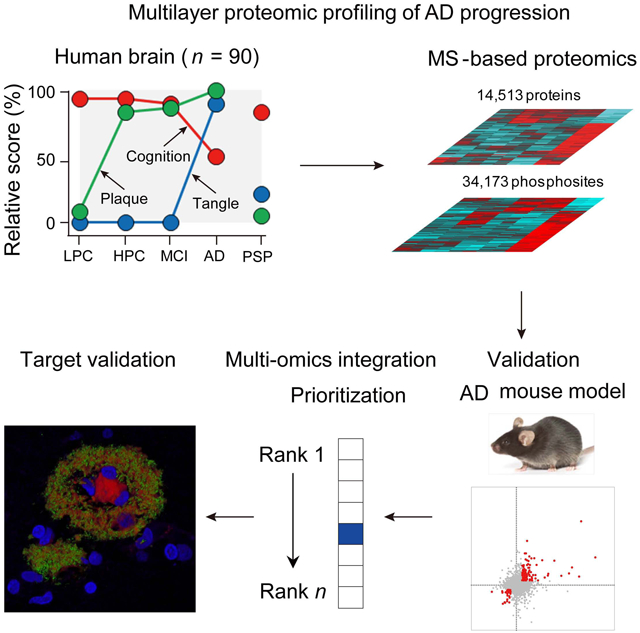
We developed a three-phase strategy to discover, validate and integrate multi-omics AD data (Figure 1A). As clinical proteomic studies are often affected by confounding factors, such as postmortem interval (PMI) (Beach et al., 2015), we performed a survey of mouse brain samples with PMI of 0, 3, and 12 hours. Only 364 (2.6%) out of 14,007 proteins were changed among different PMIs, whereas 4,736 (16.5%) out of 28,699 phosphopeptides were altered in the comparisons (Data S1A and S1B). Thus we diminished the impact of confounding factors by removing PMI-affected proteins and phosphoproteins, selecting well-characterized specimens with very short PMI (3.0 ± 0.8 h) in the discovery study (Table 1 and Data S2A) (Beach et al., 2015), implementing additional quality control metrics to discard samples compromised by protein degradation (Figure S1), and correcting for cell-type composition bias during data analysis (see STAR method).
Figure 1. Whole proteome profiling of AD progression reveals disease stage-dependent proteins/pathways.

(A) Strategy for multilayer profiling of proteome and phosphoproteome in AD cases and the 5xFAD mouse model, and multi-omics data integration. The y-axis shows the relative pathology scores for cognition, plaque, and tangle.
(B) Summary of temporal profiling of five groups of human brain tissue, with each group containing two independent pools. Each pool contained samples from 9 cases.
(C) MS data showing the Aβ level in each group. The relative TMT intensities of the Aβ peptide are shown.
(D) Clustering analysis of the proteomics dataset based on the changed proteins. The TMT intensities (log2 Z-score transformed) of each protein (rows) across five disease groups (columns) are indicated in a colored scale.
(E) Bioinformatics pipeline for identifying DE proteins followed by clustering and module analyses.
(F) Three major clusters (WPC1-3) of DE proteins defined by the WGCNA program. Each line represent one protein. The intensity of each protein is log2 Z-score transformed (mean-centered and scaled by standard deviation). The line color indicates the degree of pattern correlation between each protein and eigenvalue of the cluster. Proteins discussed in the text are shown.
(G) Enriched pathways in the WPC proteins by Fisher’s Exact Test. Significantly enriched pathways (FDR < 20%) are shown.
(H) Detected PPI modules in WPCs.
(I) Validation of selected DE proteins by western blotting with the loading control (tubulin).
Table 1.
Characteristics of human subjects used in this study.
| Banner Sun pooled samples (n = 90) | Banner Sun individual samples (n = 49) | Mount Sinai individual samples (n = 62) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Characteristic | LPC (n = 18) |
HPC (n = 18) |
MCI (n = 18) |
AD (n = 18) |
PSP (n = 18) |
LPC (n = 12) |
HPC (n = 6) |
MCI (n = 6) |
AD (n = 15) |
PSP (n = 10) |
Control (n = 23) |
AD (n = 39) |
| Age (yr) | ||||||||||||
| Mean ± SD | 83.3±8.0 | 87.8±5.1 | 86.4±6.4 | 75.7±8.7 | 84.8±11.4 | 82.9±7.4 | 87.5±2.7 | 84.0±8.2 | 78.0±8.6 | 83.1±9.5 | 81±10.4 | 84±10.1 |
| Range | 69–95 | 78–97 | 74–98 | 60–87 | 59–99 | 73–95 | 84–91 | 74–93 | 60–87 | 64–94 | 61-98 | 62-103 |
| Gender | ||||||||||||
| Male | 14 | 7 | 9 | 5 | 11 | 9 | 1 | 3 | 6 | 5 | 12 | 12 |
| Female | 4 | 11 | 9 | 13 | 7 | 3 | 5 | 3 | 9 | 5 | 11 | 27 |
| PMI (hr) | ||||||||||||
| Mean ± SD | 2.8±0.6 | 2.9±0.8 | 3.1±0.9 | 3.3±1.0 | 2.9±0.6 | 2.6±0.5 | 3.0±1.1 | 3.4±1.0 | 3.1±0.9 | 2.6±0.2 | 9.5±6.3 | 6.4±4.4 |
| Range | 1.5–4.1 | 1.5–4.3 | 2.0–5.0 | 2.0–5.3 | 2.3–4.3 | 1.5–3.3 | 1.5–4.3 | 2.5–5.0 | 2.0–5.3 | 2.3–2.8 | 1.4-20.3 | 2.0-20.7 |
| Braak stage | ||||||||||||
| Mean ± SD | 2.2±0.8 | 3.5±0.5 | 3.6±0.9 | 5.6±0.5 | 3.4±1.1 | 2.0±0.9 | 3.5±0.5 | 3.7±0.8 | 5.5±0.5 | 3.7±1.3 | 1.9±0.8 | 5.4±1.2 |
| Range | 1–3 | 3–4 | 2–6 | 5–6 | 2–5 | 1–3 | 3–4 | 2–4 | 5–6 | 2–5 | 0-3 | 2-6 |
Using the multiplexed tandem-mass-tag (TMT) method and two-dimensional liquid chromatography-tandem mass spectrometry (LC/LC-MS/MS) (Bai et al., 2017; Pagala et al., 2015), we started with frontal cortical samples of 100 human cases, and selected 90 samples of high quality for profiling whole proteome and phosphoproteome (Figure 1B and Figure S1, Table 1 and Data S2A) in 5 equal groups: (i) controls with low pathology of plaques and tangles (LPC), (ii) controls with high Aβ pathology but no detectable cognitive defects (HPC) (Nelson et al., 2012), (iii) mild cognitive impairment (MCI) with Aβ pathology and a slight but measurable defect in cognition (Hyman et al., 2012), (iv) late stage AD with high pathology scores of plaques and tangles, and (v) progressive supranuclear palsy (PSP), another neurodegenerative disorder of tauopathy (Williams and Lees, 2009). A pooling strategy with replicates (Bai et al., 2013) was designed to simplify the analysis, dividing the 90 samples into 10 pools (Figures S1 and S2). Exhaustive LC/LC-MS/MS improved quantitative accuracy and proteome coverage (Niu et al., 2017), quantifying 14,513 proteins (12,018 genes, false discovery rate (FDR) < 1%, Data S2B), and 34,173 phosphosites (46,612 phosphopeptides, FDR < 1%, Data S2C) (Figure 1B and Figures S3A-S3C). The MS-measured Aβ peptide level is largely consistent with the results of plaque pathology in the 5 groups of samples (Figure 1C). The data quality is further supported by high reproducibility of replicates in clustering analysis (Figure 1D), principal component analysis (Figure S3D), and correlation analysis (Figure S3E, R2 > 0.97 between replicates).
To explore proteome dynamics during AD progression, we used a stepwise pipeline (Figure 1E) to define 173 differentially expressed (DE) proteins (FDR < 20%) in 17 pathways, adapted the weighted gene correlation network analysis (WGCNA) (Langfelder and Horvath, 2008) to cluster these DE proteins, and interpreted the clusters with protein-protein interaction (PPI) network. Three whole proteome clusters (termed WPC) were identified from the analysis (Figure 1F), whereas a small number of proteins (n = 8) did not follow any of these clusters (Data S3A). WPC1 (n = 50) displays an early increase in HPC and MCI and a continuous accumulation in AD, including Aβ, TREM2 and CLU. These proteins are enriched in 12 pathways of amyloid cascade, WNT signaling, TGFβ/BMP signaling, G protein signaling, integrin pathway, innate immunity, adaptive immunity, cytoskeleton and extracellular matrix, membrane transport, lipid metabolism, protein folding and degradation, and synaptic function (Figure 1G). WPC2 proteins (n = 88) are relatively stable in the stages of HPC and MCI, but show a marked increase when progressing to AD, including MAPT (tau) and complement components. WPC2 recapitulates all enriched pathways in WPC1, and contains three additional pathways of cytokine signaling, complement and coagulation, and iron homeostasis (Figure 1G). In contrast, WPC3 (n = 27) shows no obvious change in the HPC and MCI, followed by a notable decrease in AD, identifying two more pathways of neurotrophic factor signaling and mitochondrial function (Figure 1G). Furthermore, we integrated the DE proteins in each WPC pattern with the protein-protein interaction network to define 11 functional modules, providing the evidence of protein-protein interaction to support these enrichment pathways (Figure 1H and Data S3B). Importantly, among the 173 DE proteins in AD, 163 (94%) proteins show no significant changes in the PSP brain displaying tauopathy but missing amyloid plaques, indicative of a high degree of AD specificity (Data S3A).
As the majority of these altered proteins are novel, we validated the changes of 11 proteins by western blotting in the pooled AD samples (Figure 1I), and performed two additional large-scale validation analyses by TMT-LC/LC-MS/MS. First, we profiled 49 individual human brain samples from the same Banner Sun cohort used for the pooled study (Data S2A, Figure 2A), identifying 12,650 proteins across all samples (FDR < 1%). Out of the 173 DE proteins identified in the pooled analysis, 121 were re-detected in the individual samples and divided into the WPC1-3 clusters. Consistently, 117 (97%) proteins displayed similar patterns during AD progression (Figures S4A and S4B). Second, we profiled 80 brain cortical samples from an independent Mount Sinai cohort (PMI 7.5 ± 5.4 h) and accepted the proteomic results from 62 cases (23 normal control cases, and 39 AD cases, Data S4A, Figure 2B), identifying 12,147 proteins shared by all samples. Among the 117 DE proteins found in the Banner Sun cohort, 84 were reidentified in the Mount Sinai cohort, in which 58 (69%) showed consistent changes in AD (Figures 2C and 2D, Data S4B). Thus, the two extensive TMT validation analyses strongly support reproducibility of our deep proteomics studies.
Figure 2. Validation of AD-related proteins by profiling individual human samples in both Banner Sun and Mount Sinai cohorts.

(A) Analysis of Banner Sun individual cohort. A total of 49 Banner Sun individual cases were measured with TMT-LC/LC-MS/MS in 5 batches.
(B) Analysis of 80 human cases from Mount Sinai cohort. After removing outliers, 62 cases (23 controls and 39 AD cases) were analyzed.
(C) Bioinformatics pipeline for validating DE proteins by individual cases from two cohorts.
(D) Validated DE proteins in three datasets (Banner Sun pooled samples, Banner Sun individual samples, and Mount Sinai individual samples). Each protein is represented by a colored box after log2 Z-score conversion. The heat map on the far right indicates consistent (up or down) or inconsistent changes between proteome and transcriptome.
(E) Analysis of CSF samples from Banner Sun cohort. After removing outliers, 5 controls and 8 AD cases were analyzed.
(F) Consistent protein changes of 8 proteins in AD brain tissue and CSF.
To investigate the mechanisms underlying the protein changes in AD, we compared our AD proteome datasets with a reported large-scale AD transcriptome dataset (Allen et al., 2016). Among the 58 altered proteins in AD, almost all decreased proteins (89%, 8 out of 9 proteins) are associated with decreased RNA levels, but only 55% (27 out of 49 proteins) of increased proteins link to upregulated transcripts (Figure 2D, Data S4B), suggesting that the remaining 45% of increased proteins are regulated by posttranscriptional mechanisms.
To examine if these protein changes in the AD brain tissue are detectable as potential biomarkers in cerebrospinal fluid (CSF), we performed deep proteomics profiling of CSF samples (n = 13, 5 control and 8 AD cases, Figure 2E) and quantified 5,940 proteins (Data S4C). The analysis detected 49 proteins from the 58 proteins changed in AD brain tissue, in which a list of 8 proteins are consistently changed in the CSF samples (p < 0.05, Figure 2F, Data S4D). In the list, 4 proteins (GPNMB, SMOC1, NPTX2 and VGF) have previously been reported in AD CSF (Dayon et al., 2018; Huttenrauch et al., 2018; Llano et al., 2019; Xiao et al., 2017), and the other 4 proteins (SLC39A12, SLC5A3, SLIT2 and STEAP3) are novel biomarker candidates.
Phosphoproteome-derived Kinase Activities in AD
The phosphoproteome profiling identified 34,173 phosphosites (46,612 phosphopeptides) on 7,083 phosphoproteins (Data S2C, Figure S5A), and as expected microtubule-associated Tau is the most elevated phosphoprotein in AD (Figure 3A). There are 25,698 (75%) pS, 7,216 (21%) pT, and 1,259 (4%) pY sites (Figure 3B). Reproducibility of replicates is verified by clustering analysis (Figure 3C), principal component analysis (Figure S5B), and correlation analysis (Figure S5C, R2 > 0.75 between replicates). In the clustering analysis, the AD group shows the largest distance from other groups, suggesting that AD harbors the most dramatic change in phosphoproteome.
Figure 3. Profiling of protein phosphorylation events in AD.

(A) Measurements of Tau phosphorylation by MS. The relative TMT intensities of a phosphor-Tau peptide in five disease groups are shown.
(B) Distribution of phospho-Ser/Thr/Tyr in the identified phosphosites.
(C) Clustering analysis of the altered phosphopeptides in human samples. The TMT intensities (log2 Z-score transformed) of phosphopeptides (rows) across five disease groups (columns) are indicated in a colored scale.
(D) Bioinformatics pipeline for identifying DE phosphopeptides, clustering analysis, and kinase activity analysis.
(E) Four major phosphoproteome clusters (PPC1-4) of DE phosphopeptides by WGCNA. Each line represents one phosphopeptide. The line color indicates the degree of pattern correlation between each phosphopeptide and eigenvalue of the cluster. Phosphoproteins discussed in the text are shown.
(F) Enriched pathways among the PPC proteins by Fisher’s Exact Test. Significantly enriched pathways (FDR < 20%) are shown.
(G) Overlap of DE proteome and phosphoproteome.
(H) Kinase activities computed from related substrates by the IKAP algorithm, with the absolute abundance of kinase family members shown in a black to white gradient.
(I-J) Substrate phosphorylation levels of specific kinases in four groups of AD samples. The levels of each phosphosite in the four disease groups (LPC, HPC, MCI and AD) are represented by boxes filled with red gradients. FC: fold change.
(K) Potential activation of the MAPK pathway based on MS measurements of the whole proteome and phosphoproteome. The levels of each protein and its phosphosites in the four groups of human samples (LPC, HPC, MCI and AD) are represented by boxes filled with red and blue gradients, respectively. The missing values are shown in white boxes.
Similar to the whole proteome analysis, the stepwise computational pipeline was used to identify 873 DE phosphopeptides (FDR < 20%, 398 proteins, Figure 3D) into four major phosphopeptide clusters (PPC1-4, Figure 3E), whereas a small subset of phosphopeptides (n = 22) did not fit into any of these clusters (Data S5A). PPC1 (n = 39) shows a steady increase during AD development. PPC2 (n = 372) is stable in the HPC and MCI samples, and dramatically increases in AD, reminiscent of the pathology of neurofibrillary tangles. Notably, PPC2 contains 89 phosphopeptides from Tau (56 phosphosites, Data S5B), and 67 from osteopontin (SPP1), a glycoprotein regulating diverse biological processes (e.g. immune response) (Wang and Denhardt, 2008). PPC3 (n = 104) increases slightly in the HPC and MCI cases, and reduces in AD. PPC4 displays the downregulation of phosphorylation in the HPC and MCI, followed by restoration in AD. Together, the PPC1-4 patterns demonstrate that protein phosphorylation is highly dynamic and actively remodeled at different stages of AD.
We next examined the phosphoprotein pathways enriched in each PPC pattern (Figure 3F), revealing two new pathways of splicing dysfunction and calcium hypotheses in AD. Strikingly, there are only 11 proteins overlapped between the altered phosphoproteome (n = 398) and whole proteome (n = 173) (Figure 3G), indicating that the phosphoproteome analysis represents a new layer of molecular regulation, complementing the landscape of the whole proteome.
We then sought to derive kinase activities in the samples according to the levels of phosphorylated substrates. The IKAP machine learning algorithm (Mischnik et al., 2016) deduced the activities of 186 kinases, of which 28 are possibly changed, primarily in AD (Figure 3H). For example, the CDK kinase activity could be computed from its known substrates of MAPT, APP, LMNA, VIM, TLN1 and SQSTM1 (Figure 3I), and the GSK3 kinase activity was similarly inferred (Figure 3J). In total, the ABL, CDK, CAMK, GSK, MAP3K, MAPK, PKC, and TTBK families, and 6 other kinases of PBK, FES, FYN, MARK2, PKA (PRKACA), and PRKDC were likely activated in AD. Remarkably, this list essentially covers all previously known Tau kinases (n = ~11) (Wang and Mandelkow, 2016).
Finally, we examined the levels of the possibly activated 28 kinases and related family members in the whole proteome and phosphoproteome. Although the vast majority of kinases (except MAP3K6) do not change at the whole protein levels (Data S2B), some of the MAP kinase cascades exhibit a marked increase in phosphorylation, including four sequentially activated families: MAP4K (e.g. MAP4K4), MAP3K (e.g. ARAF and MAP3K12), MAP2K (e.g. MAP2K6), and MAPK (e.g. MAPK1/ERK2) (Figure 3K) (Keshet and Seger, 2010). Moreover, the dual specificity protein phosphatase 6 (DUSP6), a negative regulator of MAPK signaling, is significantly reduced in AD (Figure 3K). Collectively, these multilevel proteomic analyses suggest the activation of MAP kinase signaling that is implicated in AD pathogenesis (Kim and Choi, 2010).
Aβ-correlated Protein Alterations and Human-Mouse Comparisons
To explore molecular events induced by Aβ accumulation, we identified a list of 28 Aβ-correlated DE proteins in the whole proteome datasets from both Banner Sun and Mount Sinai cohorts (FDR < 10%, Figures 4A-4C). The Aβ level is based on its fully tryptic peptide (LVFFAEDVGSNK, Figure 4A), increasing ~7-8-fold from low pathology control cases to AD, which correlates well with the plaque pathology in brain tissue. Since this peptide can be generated from either Aβ or the full length APP, we examined the absolute abundance of Aβ and APP by corresponding spectral counts. While this Aβ-derived peptide has 35 spectral counts, APP has more than 30 tryptic peptides outside the Aβ region, which have only 5 average spectral counts (Data S2B), indicating that Aβ is much more abundant than APP in the samples. Moreover, MS quantification of the non-Aβ APP peptides shows no significant change in AD. Thus, the selected peptide is a surrogate for Aβ, which was also previously used for Aβ quantification by mass spectrometry (Seyfried et al., 2017).
Figure 4. Aβ-correlated proteins in AD and the 5xFAD mouse model.

(A) Measurement of Aβ by MS based on its tryptic peptide (red).
(B) Correlation analysis of Aβ and DE proteins in 121 human cases. The degree of correlation was assessed by Pearson correlation coefficients and corresponding p values. Proteins were ranked by correlation coefficient values (p < 0.001).
(C) An example protein correlates well with Aβ. The MS measured intensities in samples were Z-transformed (i.e. mean-centered and scaled by standard deviation).
(D) Analysis of whole proteome in 5xFAD (3, 6, 12 months of age).
(E) human Aβ-correlated proteins validated in 5xFAD, represented by colored boxes after Z score conversion. The heat map on the far right indicates consistent and inconsistent changes between proteome and transcriptome in the mice.
See also Data S6.
We then evaluated the cause and effect relationships of the Aβ-correlated proteins in the 5xFAD model (Oakley et al., 2006) by analyzing a total of 16,128 proteins (11,573 proteins across all samples, FDR < 1%) in the cortex of 5xFAD and wild type (WT) mice (3, 6 and 12 months of age, Figure 4D, Data S6A). In the 28 proteins, 23 were identified in all mice, and 15 (65%) are consistently increased in the animals (Figure 4E, Data S6B), including MDK, NTN1, CTHRC1, NTN3, SMOC1, SFRP1, OLFML3, SLIT2, HTRA1, FLT1, SLIT3, CLU, ICAM1, LSP1, and C4B. Among the 15 proteins, only 4 (27%, OLFML3, CLU, ICAM1 and C4B) proteins are linked to RNA upregulation (Figure 4E) based on RNA-seq analysis (Data S6C), indicating that most Aβ-correlated proteins in 5xFAD are regulated by posttranscriptional mechanisms.
Genetically modified mice cannot fully mimic AD and are often viewed as a model of AD asymptomatic stage (LaFerla and Green, 2012). To test this idea, we compared the mouse DE proteome (5xFAD/WT) with different human group DE proteome (HPC/LPC, MCI/LPC, and AD/LPC, Figure 5A). Interestingly, 5xFAD (12 months of age) shares 97, 89, and 169 protein alterations with HPC, MCI and AD, respectively, The results implicate that 5xFAD has the most similar proteome to the late stage of AD, but not the early stage of AD as previously anticipated (Ashe and Zahs, 2010).
Figure 5. Human-mouse proteomic comparison.

(A) Whole proteome comparison between human cases and 5xFAD. The human HPC, MCI, and AD data were normalized by LPC, while the 5xFAD results were normalized by WT. Each dot represents one protein, and the color shows the dot density. Proteins with consistent changes are shown in the up-right and down-left corners separated by red cutoff lines.
(B) Examples of enriched pathways for human-specific DE proteins. The value of fold-change is indicated by a colored scale.
(C) Examples of activated pathways for 5xFAD-specific DE proteins.
See also Data S6.
Further examination of the DE proteomes in AD cases and 5xFAD mice reveals 37 human-specific, and 69 5xFAD-specific DE proteins (Data S6D and S6E). In the human-specific DE proteins, some synaptic proteins (e.g. CAMKK2 and NPTX2) and neuroprotective factors (e.g. VGF and BDNF) are significantly reduced in AD, but not in the mice (Figure 5B). Intriguingly, the 5xFAD-specific DE proteins might render different response to Aβ insult. For example, lysosome/autophagy pathway proteins (e.g. LAMP1 and HEXB) are highly enriched and increased (Figure 5C). Autophagy is essential to clear damaged organelles and aggregated proteins; and enhanced autophagy might reduce AD-related pathology and reverse memory deficit in mice (Nixon, 2013). Defective autophagy is usually accompanied by the accumulation of the SQSTM1/p62 protein (Moscat and Diaz-Meco, 2009). Indeed, p62 is accumulated in AD, but not in 5xFAD. Moreover, interferon response is markedly increased in 5xFAD but not in AD (Figure 5C). Interferons is a class of cytokines with profound neuroprotective or neurotoxic properties, which are dependent on relative interferon levels and cellular context (Schwartz and Deczkowska, 2016). For example, lack of neuronal interferon-β signaling in mice was reported to block late-stage autophagy, resulting in sporadic Lewy body accumulation and dementia (Ejlerskov et al., 2015). Thus, the species-specific DE proteins may represent different magnitude of proteome remodeling under the pathological conditions in human and mice.
Pathway Ranking and Top-ranked Proteins in Amyloid Plaque Pathology
Integration of genomic data and gene expression information has proven powerful for determining potential causative pathways in Alzheimer’s disease (Zhang et al., 2013). We extended this concept by combining all major dimensional AD datasets to rank genes/proteins and pathways, using order statistics (Aerts et al., 2006) and gene set enrichment analysis (GSEA) (Subramanian et al., 2005) (Figure 6A). We included 7 datasets: MCI whole proteome, AD whole proteome, phosphoproteome, and aggregated proteome (Bai et al., 2013; Lutz and Peng, 2018), genome-wide association analysis (Lambert et al., 2013), transcriptome (Allen et al., 2016), and protein-protein interactome (Huttlin et al., 2017). The integrative protein ranking is based on all 7 datasets (Figure 6B, Data S7A), with APP, APOE, and MAPT as the top 3 of the list, consistent with our current understanding of AD pathogenesis. We then prioritized signaling pathways by GSEA, identifying 16 pathways (FDR < 20%, Figure 6C), collectively classified into 4 major categories of amyloid and Tau pathways, inflammation, growth and development, as well as metabolism and membrane transport. Moreover, subcellular localization analysis indicates that the high-ranked proteins are exceedingly enriched in secreted subproteome (secretome) and cell surface subproteome (FDR < 10%, Figure 6D). For instance, in the top 200 proteins (Data S7B), 42% (n = 83) are secretory proteins; and 20% (n = 40) are cell surface proteins. Moreover, other secretory locations, such as vesicle lumen, secretory granule lumen, and Golgi lumen, are also enriched in this analysis. The results suggest that the secreted and cell surface subproteomes are particularly perturbed in AD, which might have a strong impact on cell communication.
Figure 6. Prioritization of proteins/genes in AD by multi-omics.

(A) Strategy for ranking proteins and pathways. The proteins were first sorted by integrated order statistics, and were then used for pathway ranking by gene set enrichment analysis.
(B) The ranking list by combining 7 datasets. In the interactome, each protein was scored by its distance to the proteins encoded by known AD genes (APP, PSEN1/2, APOE, TREM2, and UNC5C). The integrative ranks of top 40 proteins are shown by boxes of two color gradient, while missing values are indicated by white boxes.
(C) Pathway enrichment by GSEA, categorized into four groups. The bar-code plot indicates the position of the proteins on the sorted rank.
(D) Protein enrichment by subcellular localization.
See also Data S7.
We then examined if highly ranked, Aβ-correlated proteins are associated with amyloid plaque pathology. Indeed, immunohistochemistry experiments show that MDK, NTN1, SMOC1, and ICAM1 are obviously accumulated in some plaque structures of AD, compared to normal control cases (Figure 7A). When co-stained with Aβ in immunofluorescence analysis, MDK and NTN1 are highly colocalized with Aβ plaques, while SMOC1 and ICAM1 are partially colocalized in the plaque areas (Figure 7B). In addition, we also analyzed complement protein C4B in human cases and identified both amyloidopathy and angiopathy (Figure S6), supporting that the elevation of C4B is unlikely due to blood contamination in human postmortem brain samples. In the 5xFAD model, the plaque localization of MDK, NTN1, SMOC1 and ICAM1 is recapitulated in the brain (Figures 7C and 7D), and the accumulation of these proteins is validated by Western blotting (Figures 7E). Finally, we explored if these proteins physically interact with Aβ using an affinity binding assay. The four highly purified recombinant proteins, biotin-labeled Aβ40 and a scrambled control peptide were obtained for the assay (Figure 7F). Clearly, MDK and NTN1 directly binds to the Aβ40 column, but not to the control column, while the other two proteins (SMOC1 and ICAM1) show no binding to Aβ40 in this assay (Figure 7G). Collectively, these results indicate these Aβ-correlated proteins are colocalized with Aβ in the amyloid plaques, with MDK and NTN1 showing direct Aβ-binding, implicating that they may function together in the AD brain.
Figure 7. Characterization of selected protein targets in AD and 5xFAD.

(A) Immunohistochemistry in human brain sections of control and AD cases (frontal cortex). Immunoreactivity was developed with DAB chromogen (brown), with counterstaining of hematoxylin for nuclei (blue). Scale bar: 50 μm.
(B) Immunofluorescence labeling in AD brain sections of the selected proteins (red), Aβ (green) and nuclei (blue by DAPI). Scale bar: 50 μm.
(C) Immunohistochemistry of the selected proteins in WT and 5xFAD mice (12 month old). Scale bar = 50 μm.
(D) Immunofluorescence labeling of selected proteins (red), Aβ (green) and nuclei (blue by DAPI) in 5xFAD cortex (12 month old). Scale bar: 50 μm.
(E) Western blotting in WT and 5xFAD samples (12 month old, hippocampus).
(F) Silver staining of Aβ40 and scramble (Ctl) peptides and human recombinant proteins.
(G) Affinity binding assay between Aβ40 and the recombinant proteins. Aβ40 and scramble (Ctl) peptides were biotinylated and bound to streptavidin beads packed in mini-columns. Individual recombinant proteins were loaded on the columns, followed by wash and elution. The input and eluate were analyzed by western blotting.
See also Figure S6.
Discussion
Achievement of Genome-wide Brain Proteomics Data in AD
Our deep, multilayer proteomic analyses reveal highly confident lists of altered key proteins and phosphoproteins in both AD and the related 5xFAD model, and prioritize molecular alterations by multi-omics datasets with a comprehensive computational pipeline, providing novel insights into the proteins/pathways associated with AD. For example, when the 58 DE proteins were searched in PubMed database, only 13 (23%) proteins were reported in at least 10 AD-related papers (Figure S7A). The acquisition of such rich proteomic information is largely attributed to the access to high quality AD brain tissue (Beach et al., 2015) and the strength of the optimized mass spectrometry-based proteomics platform, including multiplex TMT isobaric labeling (Niu et al., 2017), extensive long gradient LC/LC coupled with high resolution MS/MS analysis (Wang et al., 2015), a refined phosphopeptide enrichment method (Tan et al., 2015), and a newly developed tag-based database JUMP search program (Li et al., 2016; Wang et al., 2014). As regulatory proteins are central players in cells but often exist at low abundance, the high coverage of proteome has become extremely important to uncover those proteins. In addition, the collection of multilayer proteome datasets, such as whole proteome, phosphoproteome, and aggregated subproteome (Bai et al., 2013), moves toward a more complete view of proteotype (defined as the state of a proteome associated with a specific phenotype) (Aebersold and Mann, 2016) to interpret Alzheimer’s disease phenotypes.
The protein level in the AD brain tissue is controlled by the mechanisms of transcription, translation and protein degradation (Liu et al., 2016). Out of the 58 DE proteins detected in AD, 35 proteins are linked to corresponding RNA alterations, and 23 proteins appear to be independent of transcriptional regulation (Figure 2D), in which 9 proteins (MDK, NTN1, CTHRC1, SMOC1, SFRP1, SLIT2, HTRA1, SLIT3, and LSP1) are highly correlated with Aβ in AD cases (Figure 4). Consistently, these 9 proteins are also demonstrated to increase in the brain of 5xFAD mice by proteome profiling, and their related RNA levels do not display upregulation with statistical significance in the RNA-seq analysis. Further experiments show the plaque localization of MDK, NTN1 and SMOC1 in human and the mouse model, and direct Aβ-binding of MDK and NTN1 (Figure 7). Additional evidence comes from a recent proteomics study of amyloid plaques isolated by laser-capture microdissection (Xiong et al., 2019), confirming 5 proteins (MDK, NTN1, SMOC1, SLIT2, and HTRA1) in the plaque areas. Therefore, the protein level in AD brain is regulated by both transcriptional and posttranslational mechanisms, which are not mutually exclusive. It is possible that plaque association slows down the turnover of some proteins, which at least partially contributes to their accumulation in AD.
Compensatory Protective Feedback at the Asymptomatic Stage of AD
Clinically, there is a long, prodromal stage in AD prior to the irreversibly progressive decline of cognition (Hyman et al., 2012). A compelling concept is that initial Aβ accumulation may induce a compensatory feedback that is protective to attenuate the toxic Aβ insult, whereas a vicious feedforward may require additional toxic factors to override the protection, leading to irreversible degeneration (De Strooper and Karran, 2016). In this conceptual frame, we discuss potential roles of the identified proteins in AD by our deep MS analysis (Figure S7B).
Our proteomic analyses identify multiple protein patterns (WPC1-3), dependent on different disease stages. Only WPC1 displays early upregulation in the prodromal AD (HPC and MCI). For example, NTN1/Netrin-1 and NTN3/Netrin-3 are highly correlated with Aβ in human, and are induced in the 5xFAD mice. It has been reported that Netrin-1 overexpression reduces the Aβ level in an AD mouse model (PDAβPPSwe/Ind), and intracerebroventricular injection of Netrin-1 enhances working memory in the mice (Spilman et al., 2016). Interestingly, the Netrin-1 receptor UNC5C is an AD risk gene (Wetzel-Smith et al., 2014). Aberrant UNC5C has been reported to activate cell death signaling in cell culture, but netrin-1 may bind to UNC5C on the cell surface to suppress UNC5C-induced cell death (Hashimoto et al., 2016). Midkine is also highly correlated with Aβ in human cases and in the 5xFAD mice and it is proposed to antagonize Aβ cytotoxicity (Herradon and Perez-Garcia, 2014).
Surprisingly, we identified in WPC1 a large number of development pathway proteins, including WNT5A, WNT5B, RSPO2, GPC5, and SULF2 in WNT signaling, as well as BMP3 and SMOC1 in TGFβ/BMP signaling. Although the functions of these proteins are largely dependent on cellular context, WNT signaling is generally viewed as a protective mechanism for synaptic integrity and memory maintenance (Inestrosa and Varela-Nallar, 2014). For example, WNT5A stimulates the formation of dendrite spines and enhances synaptic plasticity in mammalian brain (Varela-Nallar et al., 2010). Recent genetic studies support the notion that WNT signaling is critical for synaptic function and neuronal viability, and defective WNT signaling exacerbates plaque pathology in mice (Liu et al., 2014). In microglia, WNT signaling may be activated by the AD risk gene TREM2 to enhance microglial viability (Zheng et al., 2017). Consistently, we observed the upregulation of OLFML3, a highly enriched marker for microglia (Crotti and Ransohoff, 2016). In contrast, BMP signaling activation appears to be detrimental by impairing neurogenesis in APP transgenic mice (Crews et al., 2010). Interestingly, both BMP3 and SMOC1 are negative regulators of BMP signaling (Bragdon et al., 2011), implying that repression of BMP signaling might play a positive role in maintaining neurogenesis in AD.
Increase of Detrimental Factors and Decrease of Protective Factors at the Symptomatic Stage of AD
During the transition from MCI to AD, our proteomic analysis uncovers an upregulated protein pattern (WPC2) and a downregulated pattern (WPC3), which may contain numerous dysfunctional pathways to promote cognitive impairment and cell death. WPC2 includes several complement proteins (e.g. C1R, C1S, C3, C4A and C4B). The complement system functions with microglia to trim excess synapses during neurodevelopment, but can be inappropriately activated to prune synapses in AD-related mouse models (Dejanovic et al., 2018; Hong et al., 2016). C3 knockout mice exhibit less hippocampal synapse loss during normal aging. Further genetic crossing analysis support that C3 deficiency protects the mice against synapse loss and memory decline in the APP/PS1 mice (Shi et al., 2017). Thus the increased activity of the complement system may accelerate the development of AD symptoms.
WPC2 also contains numerous cytoskeletal proteins, as exemplified by tau that is hyperphosphorylated and aggregated to contribute to neurodegeneration (Ballatore et al., 2007). Aβ is proposed to initiate the conversion of tau from the normal to toxic form, which mediates the Aβ toxicity at the synapse (Bloom, 2014). Moreover, the toxic tau may cause defect in axonal transport along microtubules (Wang and Mandelkow, 2016), during which iron export can be damaged, resulting in iron accumulation (Lei et al., 2012). Indeed, clinical magnetic resonance imaging suggests that iron accumulates in AD brain (Raven et al., 2013). In WPC2, we found significant upregulation of five proteins in iron homeostasis: LTF (a major iron-binding transport protein), BDH2 (synthesizing siderophore that binds iron), STEAP3 (transferrin-dependent iron uptake), and TSPO (transporting porphyrins and heme) (Ganz, 2013). The excess iron metabolism may cause the accumulation of lipid peroxidation products and reactive oxygen species, eventually leading to cell death. This new form of programmed cell death has recently been termed ferroptosis, different from apoptosis, necrosis and necroptosis (Stockwell et al., 2017). It is possible that the iron-mediated ferroptosis plays a negative role in cell survival, contributing to neurodegeneration in AD.
In contrast, WPC3 proteins decrease in the AD brain. WPC3 contains several positive neuroprotective factors, such as BDNF, VGF, NRN1, CRH and ADCYAP1. Interestingly, these neurotrophic factors are assembled together around the “hub” BDNF in the PPI network, indicating that they function in the same pathway. Multiple in vitro and in vivo studies uniformly suggest their roles in neuroprotection (Han et al., 2014; Lin et al., 2015; Lu et al., 2013; Pedersen et al., 2001). The decrease of these neuroprotective factors may also lead to disease progression.
In summary, our temporal proteomics data support the balance and antagonization of positive and negative activities at different stages of AD. This work also illustrates the ability of deep proteomics technologies to complement genomics and transcriptomics in AD research. The comprehensive multi-omics measurements reveal crucial molecular networks/pathways in AD. The AD and mouse model comparison identifies molecular similarity and discrepancy in a global scale, providing evidence to explain the translational gap between the mouse models and clinical trials. The rich data are anticipated to stimulate subsequent hypothesis-driven research, provide potential CSF biomarkers, deepen our mechanistic understanding of AD pathogenesis, and promote novel strategies for diagnosing and treating this devastating disease.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Junmin Peng (junmin.peng@stjude.org). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Postmortem Brain Tissue and Cerebrospinal Fluid Samples
Human postmortem brain tissue samples (frontal gyrus) were provided by the Brain and Body Donation Program at Banner Sun Health Research Institute. Clinical and pathological diagnoses were based on the established criteria (Beach et al., 2015). A total of 100 tissue cases (Data S2A) were used for protein quality control analysis, 90 of which were finally selected for the pooled explorative proteomics study. In addition, 49 individual cases (Data S2A) were used for the validation proteomics study. Moreover, 20 CSF samples were used for the pilot biomarker study, in which 13 were used in the final dataset. In addition, the other independent cohort (80 cases, in which 62 cases were used in the final dataset; Data S4A) was provided by the Alzheimer's Disease Research Center at Icahn School of Medicine at Mount Sinai, with well characterized criteria for clinical and pathological diagnoses (Wang et al., 2018).
Mouse Brain Tissue
Heterozygous 5xFAD+/− transgenic mice that overexpress familial AD mutant forms of human APP (the Swedish mutation, K670N/M671L; the Florida mutation, I716V; and the London mutation, V717I) and PS1 (M146L, L286V) transgenes under the transcriptional control of the neuron-specific mouse Thy-1 promoter (JAX stock #006554), and littermate controls were used. Mice of 3, 6, and 12 months were used and were of both sexes. Cortical brain tissue samples were dissected rapidly, frozen in liquid nitrogen, and stored at −80° C. Mice were bred and maintained in a specific pathogen free facility in the Animal Resource Center at St. Jude Children’s Research Hospital. All animal protocols were approved by the Institutional Animal Care and Use Committee. Brain tissue samples were dissected rapidly, frozen in liquid nitrogen, and stored at −80° C before analysis.
METHOD DETAILS
Protein Extraction and Protein Quantification
Human or mouse brain samples were lysed and the protein concentrations were quantified as previously described (Bai et al., 2017). The frozen samples were weighed and homogenized in the lysis buffer (50 mM HEPES, pH 8.5, 8 M urea, and 0.5% sodium deoxycholate, 100 μl buffer per 10 mg tissue) with 1x PhosSTOP phosphatase inhibitor cocktail (Sigma-Aldrich). The human CSF samples were extracted in the same buffer. Protein concentration was measured by the BCA assay (Thermo Fisher) and then confirmed by Coomassie-stained short SDS gels.
Sample Quality and Pooling
One major challenge in studying human postmortem brain tissue is that the specimens are usually associated with large biological variations (e.g. age and gender), which are further confounded by a wide range of uncontrollable factors (Hynd et al., 2003), such as environmental elements (e.g. diet, lifestyle), antemortem and pharmacological treatments, end-of-life illnesses, postmortem interval (PMI), and tissue storage conditions. To reduce these biological variations, we followed a previously reported pooling strategy (Bai et al., 2013), in which we successfully identified AD-specific U1 snRNP pathology in aggregated proteome. The pooling strategy averages biological variations, increases statistical power, and reduces workload (Allison et al., 2006). Simulation analysis indicates that 10 cases per pool can reach >90% statistical power (Figure S2). The main caveat of pooling is that samples of poor quality (e.g. degraded or contaminated) may affect the data of the entire pool. To address this problem, we implemented the following steps to improve quality of pooled samples: (i) to select well-stored brain specimens diagnosed by consensus clinical and neuropathological criteria, without large bias in race, age and gender; (ii) to obtain brain tissue with very short PMI (3.0 ± 0.8 h) for the discovery study; and use mouse brain samples to test the effect of PMI on the stability of proteome and phosphoproteome; (iii) to examine lysed brain samples with SDS gels to confirm protein integrity prior to pooling (10 out of 100 samples excluded by this step; Figure S1); (iv) to facilitate data analysis by incorporating samples during disease progression (e.g. LPC, HPC, MCI and AD), as well as other neurodegenerative cases (e.g. PSP with tauopathy); (v) to correct individual protein abundance affected by the cell type composition change in different samples by a multivariate linear model; and (vi) to validate proteins of interest in individual cases, and in an independent cohort.
Protein Digestion and Tandem-Mass-Tag (TMT) Labeling
The analysis was performed with a previously optimized protocol (Bai et al., 2017; Pagala et al., 2015). For whole proteome and phosphoproteome profiling, quantified protein samples (~1 mg in the lysis buffer with 8 M urea) for each TMT channel were proteolyzed with Lys-C (Wako, 1:100 w/w) at 21 °C for 2 h, diluted by 4-fold to reduce urea to 2 M for the addition of trypsin (Promega, 1:50 w/w) to continue the digestion at 21 °C overnight. The insoluble debris was kept in the lysates for the recovery of insoluble proteins. The digestion was terminated by the addition of 1% trifluoroacetic acid. After centrifugation, the supernatant was desalted with the Sep-Pak C18 cartridge (Waters), and then dried by Speedvac. Each sample was resuspended in 50 mM HEPES (pH 8.5) for TMT labeling, and then mixed equally, followed by desalting for the subsequent fractionation. For whole proteome analysis alone, 0.1 mg protein per sample was used.
Extensive Two Dimensional Liquid Chromatography-Tandem Mass Spectrometry (LC/LC-MS/MS)
The TMT labeled samples were fractionated by offline basic pH reverse phase LC, and each of these fractions was analyzed by the acidic pH reverse phase LC-MS/MS (Wang et al., 2015; Xu et al., 2009). Considering that unmodified peptides have different elution profiles from phosphopeptides, we performed two offline LC runs: 5% of the sample for whole proteome (2-3 hr gradient, 40-120 concatenated fractions) and 95% for phosphoproteome (~1 hr gradient, 40-60 fractions) on an XBridge C18 column (3.5 μm particle size, 4.6 mm x 25 cm, Waters; buffer A: 10 mM ammonium formate, pH 8.0; buffer B: 95% acetonitrile, 10 mM ammonium formate, pH 8.0) (Bai et al., 2017).
In the acidic pH LC-MS/MS analysis, each fraction was run sequentially on a column (75 μm x 20-40 cm for the whole proteome, 50 μm x ~30 cm for phosphoproteome, 1.9 μm C18 resin from Dr. Maisch GmbH, 65 °C to reduce backpressure) interfaced with an Q Exactive HF Orbitrap or Fusion MS (Thermo Fisher). Peptides were eluted by a 2-3 hr gradient (buffer A: 0.2% formic acid, 5% DMSO; buffer B: buffer A plus 65% acetonitrile). MS settings included the MS1 scan (410-1600 m/z, 60,000 or 120,000 resolution, 1 x 106 AGC and 50 ms maximal ion time) and 20 data-dependent MS2 scans (fixed first mass of 120 m/z, 60,000 resolution, 1 x 105 AGC, 100-150 ms maximal ion time, HCD, 35-38% normalized collision energy, ~1.0 m/z isolation window with 0.3 m/z offset, and ~15 s dynamic exclusion). For the phosphoproteome analysis, ~1.5 m/z isolation window was used to improve sensitivity, with 31% stepped normalized collision energy.
Phosphopeptide Enrichment
The analysis was carried out by TiO2 beads (GL sciences) as previously reported (Tan et al., 2015). The offline LC fractionated peptides (~0.15 mg in each fraction) were dried and redissolved in the binding buffer (65% acetonitrile, 2% TFA, and 1 mM KH2PO4). TiO2 beads (0.6 mg) were washed twice with the washing buffer (65% acetonitrile, 0.1% TFA), incubated with the peptide fraction at 21 °C for 20 min. The TiO2 beads were washed twice and packed into a C18 StageTip (Thermo Fisher), followed by the elution of phosphopeptide by the basic pH buffer (15% NH4OH, and 40% acetonitrile). The eluates were dried and dissolved in 5% formic acid for LC-MS/MS analysis.
Identification of Proteins and Phosphopeptides by Database Search with JUMP Software
The computational processing of identification was performed with the JUMP search engine to improve the sensitivity and specificity (Wang et al., 2014). In general, commercially available software include two major categories of algorithms: pattern-based scoring (e.g. SEQUEST and MASCOT) and tag-based scoring (e.g. PEAKS). JUMP combines pattern scoring with de novo tag scoring, displaying significant improvement in peptide-spectrum match (PSM) scoring (Wang et al., 2014). In this study, JUMP was used to search MS/MS raw data against a composite target/decoy database (Peng et al., 2003) to evaluate FDR. All original target protein sequences were reversed to generate a decoy database that was concatenated to the target database. FDR in the target database was estimated by the number of decoy matches (nd) and the number of target matches (nt), according to the equation (FDR = nd/nt), assuming mismatches in the target database were the same as in the decoy database. The protein database was generated by combining downloaded Swiss-Prot, TrEMBL, and UCSC databases and removing redundancy (human: 83,955 entries; mouse: 59,423 entries), followed by concatenation with a decoy database. Major parameters included precursor and product ion mass tolerance (± 15 ppm), full trypticity, static mass shift for the TMT tags (+229.16293) and carbamidomethyl modification of 57.02146 on cysteine, dynamic mass shift for Met oxidation (+15.99491) and Ser/Thr/Tyr phosphorylation (+79.96633), maximal missed cleavage (n = 2), and maximal modification sites (n = 3). Putative PSMs were filtered by mass accuracy and then grouped by precursor ion charge state and filtered by JUMP-based matching scores (Jscore and ΔJn) to reduce FDR below 1% for proteins during the whole proteome analysis or 1% for phosphopeptides during the phosphoproteome analysis. If one peptide could be generated from multiple homologous proteins, based on the rule of parsimony, the peptide was assigned to the canonical protein form in the manually curated Swiss-Prot database. If no canonical form was defined, the peptide was assigned to the protein with the highest PSM number.
Phosphosite Localization by the JUMPl Program
To evaluate the confidence of assigned individual phosphosites in peptides, we generated the JUMPl program using the concept of the phosphoRS algorithm (Taus et al., 2011) to calculate phosphosite localization scores (Lscore, 0-100%) for each PSM. In addition to the PSM Lscores, all PSMs were aligned to protein sequences to generate protein Lscores for individual phosphosites. If multiple PSMs are identified for one specific phosphosite, the highest PSM Lscore is accepted as the protein Lscore. As random assignment in PSMs containing ambiguous phosphosites often leads to an excessively large number of protein phosphosites, we implemented numerous rules to alleviate this problem: (i) in one PSM, if phosphosites can be clearly distinguished by Lscores (e.g. the gap of the 1st and 2nd PSM Lscores > 10% for a singly phosphorylated peptide), the top site is selected. (ii) If not, we examined the phosphosites in the corresponding proteins to select the sites with the top protein Lscore. This allows the PSMs of low quality to borrow information derived from the PSMs of high quality. (iii) If neither PSM Lscores nor protein Lscores are distinguishable, a heuristic priority is given to phosphosites with the following order of occurrence: SP-motif, S, T and Y. If none of the rules above can sort putative phosphosites, we sort these PSMs by JUMP Jscores, and then preferentially select protein phosphosites that are previously matched by other PSMs of high Jscores.
Proteomics Coverage Analysis
To integrate proteomic and genomic data, we downloaded human brain transcriptome from The Human Protein Atlas (https://www.proteinatlas.org/humanproteome/brain) (Uhlen et al., 2015), We extracted 14,548 genes expressed in brain from all human protein-coding genes (n = 19,628 genes). To align human proteome with transcriptome, we used the UCSC annotation table (http://hgdownload.cse.ucsc.edu/downloads.html#human) to align UniProt IDs to official gene symbols. If multiple UniProt IDs match to gene symbols, we choose the UniProt ID of the most abundant protein.
Mouse Brain RNA-seq Analysis
Total RNA was isolated from tissue with TRIzol (Invitrogen) and libraries were prepared using Stranded mRNA Library Prep Kit (Illumina): double stranded cDNA fragments were ligated with Illumina paired end adaptors, followed by size selection (~200 bp) and libraries were analyzed by Illumina HiSeq 2000 sequencing systems. Using BWA (0.5.10) aligner, reads were aligned to multiple databases, including the mouse reference genome (MGSCv37), transcriptome (RefSeq and AceView) and all possible combinations of RefSeq exons. Read count for each gene was obtained with HT-seq, and the gene-level fragments per kilobase of transcript per million (FPKM) values were computed.
Immunostaining of Human and Mouse Brain Tissue
The staining was performed essentially as previously reported (Bai et al., 2013). Briefly, brain tissue samples were fixed with 10% formalin, gradual dehydrated, and embedded in paraffin. 7 μm sections were deparaffinized with CitriSolv Hybrid (5 and 10 min) followed with rehydration with isopropanol (5 min x 3). Slides were then rinsed with running tap water (5 min) and distilled water (1 min). Antigen retrieval was performed with 10 mM citric buffer (pH 6.0) with boiled water bath (20 min) and cool down to 21°C. Slides were then rinsed with running tap water (5 min), and distilled water (1min), and were blocked the endogenous peroxidase activity by 1% H2O2 in PBS buffer (10 min). After rinsing by PBS, sections were then blocked with 10% donkey serum in PBS with 0.3% triton X-100 (PBST) for 30 min at 21 °C. Primary antibodies were diluted with PBST plus 2% BSA and 0.2% skim milk (PBST-B) as follow: MDK (1:100, R&D Systems, Cat# AF-258-PB), NTN1 (1:100, Abcam, ab126729), human SMOC1 (1:50, MilliporeSigma, Cat# SAB2701398), mouse SMOC1 (1:50, Thermo Fisher Scientific, Cat#PA5-47754), human ICAM1(1:50, Sigma, Cat# HPA002126), mouse ICAM1(1:100, Abcam, Cat#ab179707), human C4B (1:50, Abcam, Cat# ab181241). Sections were then incubated with diluted primary antibody at 4 °C overnight followed with the biotinylated secondary antibody and ABC reaction according to the instruction of Vectastain ABC kit (Vector Laboratories). After washing with PBS, the staining was developed by 3,3-diaminobenzidine (DAB) solution (Vector Laboratories) or Cyanine 3 tyramide (PerkinElmer) for 2-10 min. For double-immunofluorescence staining with Aβ, sections were incubated with Aβ Ab (82E1,1:100, Immuno-Biological Laboratories Co., Cat# 10323) followed with secondary antibody conjugated with Alexa flour 647 (1:500, Jackson ImmunoResearch Laboratories) and DAPI (1 ng/ml, Thermo Fisher Scientific) for nuclei counter staining. For immunofluorescence staining in human brain sections, the endogenous fluorescence was eliminated by Sudan Black B staining (0.1% in 70% ethanol) for 2 min followed by PBS wash. The slides were mounted with Prolong gold antifade mountant. Fluorescence images were captured by Zeiss LSM 780 confocal microscopy. For the bright field, slides were mounted by Cytoseal 60 (Electron Microscopy Science), and imaged by Zeiss Axioscan.Z1.
Aβ Affinity Binding Assay
Biotinylated Aβ40 peptide and scrambled control peptide (Anaspec), and recombinant proteins (R&D Systems, Novus Biologicals, and eBioscience) was purchased. Scrambled Aβ peptides were biotinylated using EZ-Link NHS-Biotin (Thermo Scientific) and tested by binding to streptavidin sepharose beads (GE Healthcare). The purity of these peptides and recombinant proteins were confirmed by silver-stained SDS gels. During the binding assay, streptavidin beads (1V = 1 μl bed volume) were used to bind Aβ and scrambled control (5 μg per peptide) followed by cleaning with 20V of wash buffer (0.3 x PBS and 0.01% triton X-100). Each peptide-coated column was loaded with one recombinant protein (0.1 μg/μl, ~1 μg protein), incubated on ice for 30 min, washed with 20V wash buffer, then eluted with 30V of elution buffer (50 mM HEPES, 2% SDS, pH 8.5) at 95°C. The inputs (10-30 n g) and eluates (~20%) were analyzed by western blotting with the specific antibodies.
QUANTIFICATION AND STATISTICAL ANALYSIS
TMT-based Peptide/Protein Quantification by JUMP Software Suite
The analysis was performed in the following steps, as previously reported with modifications (Niu et al., 2017): (i) extracting TMT reporter ion intensities of each PSM; (ii) correcting the raw intensities based on isotopic distribution of each labeling reagent (e.g. TMT126 generates 91.8%, 7.9% and 0.3% of 126, 127, 128 m/z ions, respectively); (iii) excluding PSMs of very low intensities (e.g. minimum intensity of 1,000 and median intensity of 5,000); (iv) removing sample loading bias by normalization with the trimmed median intensity of all PSMs; (v) calculating the mean-centered intensities across samples (e.g. relative intensities between each sample and the mean), (vi) summarizing protein or phosphopeptide relative intensities by averaging related PSMs; (vii) finally deriving protein or phosphopeptide absolute intensities by multiplying the relative intensities by the grand-mean of three most highly abundant PSMs. In addition, we performed y1-ion based correction of TMT data.
Covariate Analysis for Correcting Cell Type Composition Bias
As the multivariate linear model is commonly used to correct for covariate effects of gene expression (Miller et al., 2013; Zhang et al., 2013), we used a similar method to correct individual protein abundance by the cell type composition, including neurons, astrocytes, microglia, and oligodendrocytes (Figure S8A). Cell type protein markers were selected based on published cell type resolved mouse proteome (Sharma et al., 2015) by the following criteria: i) considering mouse-human homologs; ii) selecting cell type enrichment > 5 fold (except 3 fold for oligodendrocytes due to limited markers) (Figure S8B). The change of each cell type was estimated as the median of protein abundance across the cell type markers (Figure S8C, left panel). Before cell type correction, the AD samples showed a slight increase of microglia and astrocyte, and a slight decrease of neuron and oligodendrocyte, consistent with the previous proteomics analysis (Seyfried et al., 2017). To correct for cell type effect, each protein (log-transformed TMT intensity) was fitted to a linear regression model (requiring ∣β∣ < 1) against the estimated cell type change, and then adjusted as the residuals plus the intercept (Figure S8C and D). The corrected proteomes were used for all downstream analyses (e.g., differential expression, network analysis etc.).
Estimation of Measurement Variations
The measurement variation was based on the replicated measurements of the samples. The relative expression (i.e. ratio) between replicates for each protein was calculated. The ratios of all proteins from the replicates were fitted with a Gaussian distribution to estimate expected mean and standard deviation.
Differential Expression Analysis of Proteins and Phosphopeptides and False Discovery Rate Evaluation
DE events were defined by identifying proteins/peptides with intergroup variance significantly larger than intragroup variance using ANOVA (analysis of variance) as described previously (Tan et al., 2017). Specifically, we applied a four-step procedure to define DE events as follows: (i) to calculate F statistic for each protein (defined as the ratio of intragroup variance relative to intergroup variance); (ii) to apply a threshold of p value (10%); (iii) to capture functionally meaningful changes, we further filtered the results by the magnitude of change (z score > 2) in one comparison between AD and other groups and manually examined all proteins, accepting the final list of DE proteins; (iv) to estimate the final FDR of the accepted DE proteins (p < 10% plus z score > 2), we performed permutation analysis (n = 1,000 permutations) following a reported procedure (Xie et al., 2005) and found that the estimated FDR below 20%. In addition, DE phosphopeptides were identified by the same method.
Pathway Enrichment by Gene Ontology, KEGG and Hallmark Databases
Pathway enrichment analysis was carried out to infer functional groups of proteins that were enriched in a given dataset. The analysis was performed using Fisher’s exact test (p value) with the BH correction for multiple testing (BH FDR). Enriched pathways with FDR < 20% or lower were considered statistically significant.
Principal Component Analysis
Principal component analysis (PCA) was used to visualize the differences among human disease groups, mouse models, and two corresponding wild-type controls, as well as human-mouse cross-species comparisons. Relative expression of all proteins was used as features of PCA. The pairwise Euclidean distance between features was calculated. PCA was performed using the package prcomp, an R statistical analysis package (version 3.4.0) (Ihaka and Gentleman, 1996).
Hierarchical Clustering
Hierarchical clustering of differentially expressed (DE) proteins and phosphopeptides was performed to determine the differences among disease groups. Hierarchical clustering was carried out using the heatmap.2 function in R statistical analysis package (version 3.4.0) (Ihaka and Gentleman, 1996). The clustering for heatmap.2 was obtained with Ward's algorithm using Euclidean distance. Disease groups were clustered and visualized with heat-maps based on the relative expression profiles.
Weighted Gene Correlation Network Analysis (WGCNA)
The analysis was carried out with the WGCNA R package (Langfelder and Horvath, 2008). To define whole proteome clusters (i.e. WPCs), only the DE proteins were considered. Pearson correlation matrix (with direction, i.e. for building signed correlation network) was calculated using the disease samples (i.e. LPC, HPC, MCI, and AD), and an adjacency matrix was calculated by raising the correlation matrix to a power of 16 using the scale-free topology criterion (Zhang and Horvath, 2005) with modifications. Correlation clusters were defined by hybrid dynamic tree-cutting method (Langfelder and Horvath, 2008) with the minimum height for merging modules at 0.2 and manually merged if necessary. For each cluster, a consensus trend was calculated based on the first principal component (i.e. eigengene). Proteins were assigned to the most correlated cluster (i.e. WPC) with a cutoff of Pearson correlation coefficient (R) at least 0.7. Phosphoproteome clusters (PPCs) were also defined by the same procedure.
Protein-Protein Interaction (PPI) Network Analysis
Proteins in each WPC were superimposed onto a composite PPI database by combining STRING (v10) (Szklarczyk et al., 2015), BioPlex (Huttlin et al., 2017), and InWeb_IM (Li et al., 2017). The BioPlex database was developed by the method of affinity purification and mass spectrometry, whereas the STRING and InWeb contain information from various sources. Due to this heterogeneity of PPI interactions, the STRING and InWeb databases were further filtered by the edge score to ensure high quality, with the following rules: (i) only edges with evidence of physical interactions (e.g. through co-IP or yeast two-hybrid) were considered; (ii) edges of high confidence, as filtered by the edge score, with the cutoff determined by best fitting the log-log degree distribution using the scale free criteria (Barabasi and Oltvai, 2004). The finally accepted STRING and InWeb databases were combined with BioPlex to construct a composite PPI database, which includes 18,515 proteins and 469,993 PPI connections. Modules in each protein cluster were defined by a two-step procedure: (i) only retaining PPI edges if both nodes (i.e. the two connected proteins) from proteins in the same cluster; (ii) calculating a topologically overlapping matrix (Ravasz et al., 2002) for the PPI network, and modularizing such a network into individual modules by the hybrid dynamic tree-cutting method (Langfelder and Horvath, 2008). Modules were annotated using three pathway databases, including Gene Ontology (GO), KEGG and Hallmark by Fisher’s exact test, and visualized by Cytoscape (Shannon et al., 2003).
Multi-omics-based Prioritization of Genes/Proteins and Pathways
To integrate multiple types of datasets for prioritizing genes/proteins in AD, we implemented a gene/protein ranking framework based on order statistics (Aerts et al., 2006; Zhang et al., 2012), which combined N different sets of protein/gene rankings to output one protein/gene ranking. A total of 7 individual datasets of protein/gene ranking were considered for this analysis:
Protein dysregulation in MCI, ranked by the absolute value of log2(MCI/LPC)
Protein dysregulation in AD, ranked by the absolute value of log2(AD/LPC)
Phosphoprotein dysregulation in AD, ranked by the absolute value of log2(AD/LPC)
Insoluble protein dysregulation in AD, ranked by p values of AD/control (Bai et al., 2013)
Risk loci identified by GWAS, based on a meta-analysis of 74,046 individuals provided by The International Genomics of Alzheimer's Project (Lambert et al., 2013), and summarized into gene level using MAGMA (Katsouras et al., 2015). This GWAS data was directly obtained from a previous paper (Seyfried et al., 2017), in which genes were sorted by p values.
Transcriptome data from a previously reported dataset from hundreds of human cases (Allen et al., 2016).
Interaction strength or closeness (defined by network distance in the PPI network) to known AD genes: 3 causal genes (APP, PSEN1/2) plus 3 risk genes (APOE, TREM2, UNC5C) (Scheltens et al., 2016): the shortest paths between each protein/gene and any AD proteins/genes were computed to obtain the minimum distance and the most strongly connected AD protein/gene.
The rank of protein/gene was normalized by the total number of genes/proteins in the dataset, and only protein/genes with at least four data sources were used. Then, the integrative protein ranking was generated by the framework of order statistics (Wilks, 1948). For each protein/gene, the Q statistic was theoretically based on
| (1) |
Where ri is the rank of each data source i, N is the total number of data sources. In practice, the Q value was calculated using a more efficient algorithm (Aerts et al., 2006):
| (2) |
| (3) |
To estimate empirical p values, we used a permutation analysis (Aerts et al., 2006; Zhang et al., 2012), in which the ranks of each data source were randomly permutated (n = 1,000 times of permutations) to derive null Q values. Empirical p values were derived from the estimated null distribution, followed by multiple testing correction using the BH method (Benjamini and Y., 1995).
To summarize the integrative protein/gene ranking into pathway ranking, we identified dysregulated pathways by GSEA (Subramanian et al., 2005). The p value and FDR were derived by permuting gene sets (n = 1,000 permutations) in three pathway databases: GO, KEGG, and Hallmark (all downloaded from the Molecular Signature Database) (Subramanian et al., 2005). Pathways enriched with FDR < 20% were shown.
DATA AND CODE AVAILABILITY
Human proteomics data used in this study are available via the AD Knowledge Portal (https://adknowledgeportal.synapse.org). Additional information can be found via the following links: the Mount Sinai Brain Bank study (https://adknowledgeportal.synapse.org/Explore/Studies?Study=syn3159438) and the StJude_Banner study (https://adknowledgeportal.synapse.org/Explore/Studies?Study=syn21592732). In addition, mouse and pooled AD proteomics data are available in the PRIDE database (http://www.proteomexchange.org) with accession numbers of PXD007974 (mouse), PXD018590 (mouse), and PXD007985 (human). An interactive website (https://penglab.shinyapps.io/ADprofile) was generated to search for individual protein in the AD pooled dataset.
Supplementary Material
Data S1: Whole proteome and phosphoproteome profiling of mouse brain tissues with different postmortem interval (PMI) by TMT-LC/LC-MS/MS, related to Figure 1 and STAR methods.
Data S2: Whole proteome and phosphoproteome profiling of AD postmortem human brain tissues by TMT-LC/LC-MS/MS, related to Figure 1 and Figure 3.
Data S3: Analysis of whole proteome profiling of AD progression reveals disease stage-dependent proteins and pathways, related to Figure 1.
Data S4: Validation of AD-related proteins by profiling individual human samples in both Banner Sun and Mount Sinai cohorts, and deep proteome profiling of cerebrospinal fluid (CSF) samples from Banner Sun cohort, related to Figure 2.
Data S5: Analysis of protein phosphorylation events in AD, related to Figure 3.
Data S6: Aβ-correlated proteins in AD and human-mouse proteomic comparison, related to Figure 4 and Figure 5.
Data S7: Prioritization of proteins/genes in AD by multi-omics datasets, related to Figure 6.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Aβ (6E10) | BioLegend | Cat# 803001, RRID:AB_2564653 |
| Anti-Aβ (82E1) | Immuno-biological laboratories |
Cat#010323, RRID:AB_1540460 |
| Anti-C4B | Abcam | Cat# ab181241, RRID:AB_2814770 |
| Anti-CEP63 | Abcam | Cat# ab175090 |
| Anti-ICAM1 | Cell Signaling | Cat# 4915, RRID:AB_2280018 |
| Anti-ICAM1 | MilliporeSigma | Cat#HPA002126, RRID:AB_1078470 |
| Anti-ICAM1 | Abcam | Cat#Ab179707, RRID:AB_2814769 |
| Anti-MDK | Thermo Fisher Scientific | Cat# PA5-28823, RRID:AB_2546299 |
| Anti-MDK | R&D Systems | Cat# AF-258-PB, RRID:AB_2143400 |
| Anti-MDK | Abcam | Cat# Ab45151, RRID:AB_2274158 |
| Anti-NTN1 | MilliporeSigma | Cat# MABN1154 |
| Anti-NTN1 | Abcam | Cat# ab126729, RRID:AB_11131145 |
| Anti-NeuN | MilliporeSigma | Cat# MAB377, RRID:AB_2298772 |
| Anti-OLFML3 | Thermo Fisher Scientific | Cat# PA5-49313, RRID:AB_2634767 |
| Anti-PCSK1 | MilliporeSigma | Cat#WH0005122M2, RRID:AB_1843131 |
| Anti-phospho-Tau (AT8) | Thermo Fisher Scientific | Cat# MN1020B, RRID:AB_223648 |
| Anti-RGS4 | Abcam | Cat# ab9964, RRID:AB_296731 |
| Anti-SMOC1 | MilliporeSigma | Cat# SAB2701398, RRID:AB_2814768 |
| Anti-SMOC1 | Thermo Fisher Scientific | Cat# PA5-47754, RRID:AB_2606919 |
| Anti-SQSTM1 | Cell Signaling | Cat# 13121, RRID:AB_2750574 |
| Anti-VGF | Abcam | Cat# ab69989, RRID:AB_1271455 |
| Anti-β tubulin | Developmental Studies Hybridoma Bank | Cat# E7, RRID:AB_528499 |
| Biological Samples | ||
| Human brain and CSF samples of postmortem Alzheimer’s disease and other controls (LPC, HPC, MCI, AD, and PSP) | Brain and Body Donation Program at Banner Sun Health Research Institute |
https://www.bannerhealth.com |
| Human brain samples of postmortem Alzheimer’s disease and controls | The Mount Sinai/JJ Peters VA Medical Center Brain Bank (MSBB–Mount Sinai NIH Neurobiobank) |
https://icahn.mssm.edu/research/nih-brain-tissue-repository |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Aβ40 peptide | Anaspec | Cat# AS-23512 |
| Scrambled peptides | Anaspec | Cat# AS-25383 |
| MDK protein | R&D Systems | Cat# 258-MD-050 |
| NTN1 protein | R&D Systems | Cat# 6419N1025 |
| SMOC1 protein | Novus Biologicals, LLC | Cat# 6074-SM-050 |
| ICAM1 protein | eBioscience | Cat# BMS313 |
| TO-PRO-3 Iodide (642/661) | Thermo Fisher Scientific | Cat# T3605 |
| TMT reagents | Thermo Fisher Scientific | Cat# 90110 |
| Deposited Data | ||
| Interactive proteome data | This paper | https://penglab.shinyapps.io/ADprofile/ |
| Proteomics data of 5xFAD mice and AD pooled samples in the Banner Sun cohort | This paper | PPRIDE database (www.proteomexchange.org): with accession numbers of PXD007974 (mouse), PXD018590 (mouse), and PXD007985 (human). |
| Proteomics data of AD individual cases in the Banner Sun and Mount Sinai cohorts | This paper | The AD Knowledge Portal (https://adknowledgeportal.synapse.org): https://doi.org/10.7303/syn21638690 https://doi.org/10.7303/syn21347564 |
| Human brain transcriptome | Allen et al., 2016 | https://doi.org/10.7303/syn3163039 |
| 5xFAD mouse brain transcriptome | This paper | (https://www.ncbi.nlm.nih.gov/geo/) GSE140286 |
| Experimental Models: Organisms/Strains | ||
| Mouse: 5xFAD Tg6799 | The Jackson Laboratory | Cat# 34848-JAX |
| Mouse: C57BL/6J | The Jackson Laboratory | Cat# 000664 |
| Software and Algorithms | ||
| JUMP | Wang et al., 2014 | http://www.stjuderesearch.org/site/lab/peng |
| WGCNA | Langfelder et al., 2008 | https://labs.genetics.ucla.edu/horvath/CoexpressionNetwork/Rpackages/WGCNA/ |
| R | R Foundation for Statistical Computing | https://www.r-project.org/ |
| javaGSEA | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
| IKAP | Mischnik et al., 2016 | https://github.com/marcel-mischnik/IKAP.git |
Highlights.
Deep profiling of proteome and phosphoproteome in AD progression
Validation of protein alterations in two independent AD cohorts
Identification of Aβ-induced protein changes in AD and the 5xFAD mouse model
Prioritization of proteins and pathways in AD by multi-omics integration
Acknowledgements
We thank all other members in the lab and center for technical support and discussion.
Funding: This work was partially supported by National Institutes of Health grants R01AG047928 (J.P.), R01AG053987 (J.P.), R01GM114260 (J.P.), R01AG064909 (G.Y. and J.P.), RF1AG057440 (B.Z. and V.H.), R01NS079796 (G.Y.), U24NS072026 (T.G.B.), P30AG019610 (T.G.B.), Arizona Department of Health Services (contract 211002) (T.G.B.), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001) (T.G.B.), U01AG046170 (V.H. and B.Z.), RF1 AG057440 (V.H., B.Z. and J.P.), R01AG057907 (V.H. and B.Z.), and ALSAC (American Lebanese Syrian Associated Charities). The MS analysis was performed in the Center of Proteomics and Metabolomics at St. Jude Children’s Research Hospital, partially supported by NIH Cancer Center Support Grant (P30CA021765).
The results published here are available via the AD Knowledge Portal (https://adknowledgeportal.synapse.org). The AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the Accelerating Medicines Partnership (AMP-AD) Target Discovery Program and other National Institute on Aging (NIA)-supported programs to enable open-science practices and accelerate translational learning. Data are available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.synapse.org/DataAccess/Instructions).
Transcriptome data were provided by the following source: the Mayo Clinic Alzheimer’s Disease Genetic Studies, led by Dr. Nilufer Taner and Dr. Steven G. Younkin, Mayo Clinic, Jacksonville, FL using samples from the Mayo Clinic Study of Aging, the Mayo Clinic Alzheimer’s Disease Research Center, and the Mayo Clinic Brain Bank. Data collection was supported through funding by NIA grants P50 AG016574, R01 AG032990, U01 AG046139, R01 AG018023, U01 AG006576, U01 AG006786, R01 AG025711, R01 AG017216, R01 AG003949, NINDS grant R01 NS080820, CurePSP Foundation, and support from Mayo Foundation. Study data also includes samples collected through the Banner Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona, led by Dr. Thomas G. Beach with support shown above.
Abbreviations:
- AD
Alzheimer’s disease
- MS
mass spectrometry
- TMT
tandem mass tags
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aebersold R, and Mann M (2016). Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355. [DOI] [PubMed] [Google Scholar]
- Aerts S, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, Tranchevent LC, De Moor B, Marynen P, Hassan B, et al. (2006). Gene prioritization through genomic data fusion. Nat Biotechnol 24, 537–544. [DOI] [PubMed] [Google Scholar]
- Allen M, Carrasquillo MM, Funk C, Heavner BD, Zou F, Younkin CS, Burgess JD, Chai HS, Crook J, Eddy JA, et al. (2016). Human whole genome genotype and transcriptome data for Alzheimer's and other neurodegenerative diseases. Sci Data 3, 160089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison DB, Cui X, Page GP, and Sabripour M (2006). Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet 7, 55–65. [DOI] [PubMed] [Google Scholar]
- Altelaar AF, Munoz J, and Heck AJ (2013). Next-generation proteomics: towards an integrative view of proteome dynamics. Nat Rev Genet 14, 35–48. [DOI] [PubMed] [Google Scholar]
- Alzheimer's A (2015). 2015 Alzheimer's disease facts and figures. Alzheimers Dement 11, 332–384. [DOI] [PubMed] [Google Scholar]
- Ashe KH, and Zahs KR (2010). Probing the biology of Alzheimer's disease in mice. Neuron 66, 631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B, Hales CM, Chen PC, Gozal Y, Dammer EB, Fritz JJ, Wang X, Xia Q, Duong DM, Street C, et al. (2013). U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer's disease. Proc Natl Acad Sci U S A 110, 16562–16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B, Tan H, Pagala VR, High AA, Ichhaporis VP, Hendershot L, and Peng J (2017). Deep Profiling of Proteome and Phosphoproteome by Isobaric Labeling, Extensive Liquid Chromatography, and Mass Spectrometry. Methods Enzymol 585, 377–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, and Trojanowski JQ (2007). Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci 8, 663–672. [DOI] [PubMed] [Google Scholar]
- Barabasi AL, and Oltvai ZN (2004). Network biology: understanding the cell's functional organization. Nat Rev Genet 5, 101–113. [DOI] [PubMed] [Google Scholar]
- Beach TG, Adler CH, Sue LI, Serrano G, Shill HA, Walker DG, Lue L, Roher AE, Dugger BN, Maarouf C, et al. (2015). Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology 35, 354–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Y. H (1995). Controlling the False Discovery Rate: a practival andpowerful approach to multiple testing. J R Stat Soc Series B 57, 289–300. [Google Scholar]
- Bloom GS (2014). Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71, 505–508. [DOI] [PubMed] [Google Scholar]
- Bragdon B, Moseychuk O, Saldanha S, King D, Julian J, and Nohe A (2011). Bone morphogenetic proteins: a critical review. Cell Signal 23, 609–620. [DOI] [PubMed] [Google Scholar]
- Colonna M, and Wang Y (2016). TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci 17, 201–207. [DOI] [PubMed] [Google Scholar]
- Crews L, Adame A, Patrick C, DeLaney A, Pham E, Rockenstein E, Hansen L, and Masliah E (2010). Increased BMP6 Levels in the Brains of Alzheimer's Disease Patients and APP Transgenic Mice Are Accompanied by Impaired Neurogenesis. J Neurosci 30, 12252–12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti A, and Ransohoff RM (2016). Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity 44, 505–515. [DOI] [PubMed] [Google Scholar]
- Dayon L, Nunez Galindo A, Wojcik J, Cominetti O, Corthesy J, Oikonomidi A, Henry H, Kussmann M, Migliavacca E, Severin I, et al. (2018). Alzheimer disease pathology and the cerebrospinal fluid proteome. Alzheimers Res Ther 10, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, and Karran E (2016). The Cellular Phase of Alzheimer's Disease. Cell 164, 603–615. [DOI] [PubMed] [Google Scholar]
- Dejanovic B, Huntley MA, De Maziere A, Meilandt WJ, Wu T, Srinivasan K, Jiang Z, Gandham V, Friedman BA, Ngu H, et al. (2018). Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron 100, 1322–1336 e1327. [DOI] [PubMed] [Google Scholar]
- Ejlerskov P, Hultberg JG, Wang J, Carlsson R, Ambjorn M, Kuss M, Liu Y, Porcu G, Kolkova K, Friis Rundsten C, et al. (2015). Lack of Neuronal IFN-beta-IFNAR Causes Lewy Body- and Parkinson's Disease-like Dementia. Cell 163, 324–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz T (2013). Systemic iron homeostasis. Physiol Rev 93, 1721–1741. [DOI] [PubMed] [Google Scholar]
- Graham WV, Bonito-Oliva A, and Sakmar TP (2017). Update on Alzheimer's Disease Therapy and Prevention Strategies. Annu Rev Med 68, 413–430. [DOI] [PubMed] [Google Scholar]
- Guerreiro R, Bras J, and Hardy J (2013). SnapShot: genetics of Alzheimer's disease. Cell 155, 968–968 e961. [DOI] [PubMed] [Google Scholar]
- Han P, Tang Z, Yin J, Maalouf M, Beach TG, Reiman EM, and Shi J (2014). Pituitary adenylate cyclase-activating polypeptide protects against beta-amyloid toxicity. Neurobiol Aging 35, 2064–2071. [DOI] [PubMed] [Google Scholar]
- Hardy J, and Selkoe DJ (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Toyama Y, Kusakari S, Nawa M, and Matsuoka M (2016). An Alzheimer Disease-linked Rare Mutation Potentiates Netrin Receptor Uncoordinated-5C-induced Signaling That Merges with Amyloid beta Precursor Protein Signaling. J Biol Chem 291, 12282–12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herradon G, and Perez-Garcia C (2014). Targeting midkine and pleiotrophin signalling pathways in addiction and neurodegenerative disorders: recent progress and perspectives. Br J Pharmacol 171, 837–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, and Mucke L (2012). Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenrauch M, Ogorek I, Klafki H, Otto M, Stadelmann C, Weggen S, Wiltfang J, and Wirths O (2018). Glycoprotein NMB: a novel Alzheimer's disease associated marker expressed in a subset of activated microglia. Acta Neuropathol Commun 6, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, et al. (2012). National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynd MR, Lewohl JM, Scott HL, and Dodd PR (2003). Biochemical and molecular studies using human autopsy brain tissue. J Neurochem 85, 543–562. [DOI] [PubMed] [Google Scholar]
- Ihaka R, and Gentleman R (1996). R: a language for data analysis and graphics. J Comput Graph Stat 5, 299–314. [Google Scholar]
- Inestrosa NC, and Varela-Nallar L (2014). Wnt signaling in the nervous system and in Alzheimer's disease. J Mol Cell Biol 6, 64–74. [DOI] [PubMed] [Google Scholar]
- Katsouras I, Zhao D, Spijkman MJ, Li M, Blom PW, de Leeuw DM, and Asadi K (2015). Controlling the on/off current ratio of ferroelectric field-effect transistors. Sci Rep 5, 12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshet Y, and Seger R (2010). The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol Biol 661, 3–38. [DOI] [PubMed] [Google Scholar]
- Kim EK, and Choi EJ (2010). Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta 1802, 396–405.20079433 [Google Scholar]
- Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, et al. (2019). Genetic meta-analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet 51, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM, and Green KN (2012). Animal models of Alzheimer disease. Cold Spring Harb Perspect Med 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, Wong BX, Adlard PA, Cherny RA, Lam LQ, et al. (2012). Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med 18, 291–295. [DOI] [PubMed] [Google Scholar]
- Li T, Wernersson R, Hansen RB, Horn H, Mercer J, Slodkowicz G, Workman CT, Rigina O, Rapacki K, Staerfeldt HH, et al. (2017). A scored human protein-protein interaction network to catalyze genomic interpretation. Nat Methods 14, 61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang X, Cho JH, Shaw TI, Wu Z, Bai B, Wang H, Zhou S, Beach TG, Wu G, et al. (2016). JUMPg: An Integrative Proteogenomics Pipeline Identifying Unannotated Proteins in Human Brain and Cancer Cells. J Proteome Res 15, 2309–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WJ, Jiang C, Sadahiro M, Bozdagi O, Vulchanova L, Alberini CM, and Salton SR (2015). VGF and Its C-Terminal Peptide TLQP-62 Regulate Memory Formation in Hippocampus via a BDNF-TrkB-Dependent Mechanism. J Neurosci 35, 10343–10356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Tsai CW, Deak F, Rogers J, Penuliar M, Sung YM, Maher JN, Fu Y, Li X, Xu H, et al. (2014). Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer's disease. Neuron 84, 63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Beyer A, and Aebersold R (2016). On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 165, 535–550. [DOI] [PubMed] [Google Scholar]
- Llano DA, Devanarayan P, Devanarayan V, and Alzheimer's Disease Neuroimaging, I. (2019). VGF in Cerebrospinal Fluid Combined With Conventional Biomarkers Enhances Prediction of Conversion From MCI to AD. Alzheimer Dis Assoc Disord. [DOI] [PubMed] [Google Scholar]
- Lu B, Nagappan G, Guan X, Nathan PJ, and Wren P (2013). BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat Rev Neurosci 14, 401–416.23674053 [Google Scholar]
- Lutz BM, and Peng J (2018). Deep Profiling of the Aggregated Proteome in Alzheimer's Disease: From Pathology to Disease Mechanisms. Proteomes 6, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng ZY, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang XQ, et al. (2019). Single-cell transcriptomic analysis of Alzheimer's disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzker ML (2010). Sequencing technologies - the next generation. Nat Rev Genet 11, 31–46. [DOI] [PubMed] [Google Scholar]
- Miller JA, Oldham MC, and Geschwind DH (2008). A systems level analysis of transcriptional changes in Alzheimer's disease and normal aging. J Neurosci 28, 1410–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Woltjer RL, Goodenbour JM, Horvath S, and Geschwind DH (2013). Genes and pathways underlying regional and cell type changes in Alzheimer's disease. Genome Med 5, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischnik M, Sacco F, Cox J, Schneider HC, Schafer M, Hendlich M, Crowther D, Mann M, and Klabunde T (2016). IKAP: A heuristic framework for inference of kinase activities from Phosphoproteomics data. Bioinformatics 32, 424–431. [DOI] [PubMed] [Google Scholar]
- Moscat J, and Diaz-Meco MT (2009). p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 137, 1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, Castellani RJ, Crain BJ, Davies P, Del Tredici K, et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol 71, 362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu M, Cho JH, Kodali K, Pagala V, High AA, Wang H, Wu Z, Li Y, Bi W, Zhang H, et al. (2017). Extensive Peptide Fractionation and y1 Ion-Based Interference Detection Method for Enabling Accurate Quantification by Isobaric Labeling and Mass Spectrometry. Anal Chem 89, 2956–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA (2013). The role of autophagy in neurodegenerative disease. Nat Med 19, 983–997. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26, 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagala VR, High AA, Wang X, Tan H, Kodali K, Mishra A, Kavdia K, Xu Y, Wu Z, and Peng J (2015). Quantitative protein analysis by mass spectrometry. Methods Mol Biol 1278, 281–305. [DOI] [PubMed] [Google Scholar]
- Pedersen WA, McCullers D, Culmsee C, Haughey NJ, Herman JP, and Mattson MP (2001). Corticotropin-releasing hormone protects neurons against insults relevant to the pathogenesis of Alzheimer's disease. Neurobiol Dis 8, 492–503. [DOI] [PubMed] [Google Scholar]
- Peng J, Elias JE, Thoreen CC, Licklider LJ, and Gygi SP (2003). Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res 2, 43–50. [DOI] [PubMed] [Google Scholar]
- Ravasz E, Somera AL, Mongru DA, Oltvai ZN, and Barabasi AL (2002). Hierarchical organization of modularity in metabolic networks. Science 297, 1551–1555. [DOI] [PubMed] [Google Scholar]
- Raven EP, Lu PH, Tishler TA, Heydari P, and Bartzokis G (2013). Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer's disease detected in vivo with magnetic resonance imaging. J Alzheimers Dis 37, 127–136. [DOI] [PubMed] [Google Scholar]
- Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, De Strooper B, Hardy J, Vassar R, Winblad B, and Saido TC (2017). APP mouse models for Alzheimer's disease preclinical studies. EMBO J 36, 2473–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, and Van der Flier WM (2016). Alzheimer's disease. Lancet 388, 505–517. [DOI] [PubMed] [Google Scholar]
- Schwartz M, and Deczkowska A (2016). Neurological Disease as a Failure of Brain-Immune Crosstalk: The Multiple Faces of Neuroinflammation. Trends Immunol 37, 668–679. [DOI] [PubMed] [Google Scholar]
- Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, Deng Q, Nguyen T, Hales CM, Wingo T, et al. (2017). A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer's Disease. Cell Syst 4, 60–72 e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch UK, Philips MA, et al. (2015). Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci 18, 1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi QQ, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B, and Lemere CA (2017). Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilman PR, Corset V, Gorostiza O, Poksay KS, Galvan V, Zhang J, Rao R, Peters-Libeu C, Vincelette J, McGeehan A, et al. (2016). Netrin-1 Interrupts Amyloid-beta Amplification, Increases sAbetaPPalpha in vitro and in vivo, and Improves Cognition in a Mouse Model of Alzheimer's Disease. J Alzheimers Dis 52, 223–242. [DOI] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, et al. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H, Wu Z, Wang H, Bai B, Li Y, Wang X, Zhai B, Beach TG, and Peng J (2015). Refined phosphopeptide enrichment by phosphate additive and the analysis of human brain phosphoproteome. Proteomics 15, 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H, Yang K, Li Y, Shaw TI, Wang Y, Blanco DB, Wang X, Cho JH, Wang H, Rankin S, et al. (2017). Integrative Proteomics and Phosphoproteomics Profiling Reveals Dynamic Signaling Networks and Bioenergetics Pathways Underlying T Cell Activation. Immunity 46, 488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taus T, Kocher T, Pichler P, Paschke C, Schmidt A, Henrich C, and Mechtler K (2011). Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res 10, 5354–5362. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, and Fischbeck KH (2002). Toxic proteins in neurodegenerative disease. Science 296, 1991–1995. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Varela-Nallar L, Alfaro IE, Serrano FG, Parodi J, and Inestrosa NC (2010). Wingless-type family member 5A (Wnt-5a) stimulates synaptic differentiation and function of glutamatergic synapses. Proc Natl Acad Sci U S A 107, 21164–21169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Diaz AK, Shaw TI, Li Y, Niu M, Cho JH, Paugh BS, Zhang Y, Sifford J, Bai B, et al. (2019). Deep multiomics profiling of brain tumors identifies signaling networks downstream of cancer driver genes. Nat Commun 10, 3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang Y, Li Y, Bai B, Wang X, Tan H, Liu T, Beach TG, Peng J, and Wu Z (2015). Systematic optimization of long gradient chromatography mass spectrometry for deep analysis of brain proteome. J Proteome Res 14, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KX, and Denhardt DT (2008). Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev 19, 333–345. [DOI] [PubMed] [Google Scholar]
- Wang M, Beckmann ND, Roussos P, Wang E, Zhou X, Wang Q, Ming C, Neff R, Ma W, Fullard JF, et al. (2018). The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer's disease. Sci Data 5, 180185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li Y, Wu Z, Wang H, Tan H, and Peng J (2014). JUMP: a tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol Cell Proteomics 13, 3663–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, and Mandelkow E (2016). Tau in physiology and pathology. Nat Rev Neurosci 17, 5–21. [DOI] [PubMed] [Google Scholar]
- Wetzel-Smith MK, Hunkapiller J, Bhangale TR, Srinivasan K, Maloney JA, Atwal JK, Sa SM, Yaylaoglu MB, Foreman O, Ortmann W, et al. (2014). A rare mutation in UNC5C predisposes to late-onset Alzheimer's disease and increases neuronal cell death. Nat Med 20, 1452–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilks SS (1948). Order statistics. Bull Amer Math Soc 54, 6–50. [Google Scholar]
- Williams DR, and Lees AJ (2009). Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol 8, 270–279. [DOI] [PubMed] [Google Scholar]
- Wingo AP, Dammer EB, Breen MS, Logsdon BA, Duong DM, Troncosco JC, Thambisetty M, Beach TG, Serrano GE, Reiman EM, et al. (2019). Large-scale proteomic analysis of human brain identifies proteins associated with cognitive trajectory in advanced age. Nat Commun 10, 1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao MF, Xu D, Craig MT, Pelkey KA, Chien CC, Shi Y, Zhang J, Resnick S, Pletnikova O, Salmon D, et al. (2017). NPTX2 and cognitive dysfunction in Alzheimer's Disease. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Pan W, and Khodursky AB (2005). A note on using permutation-based false discovery rate estimates to compare different analysis methods for microarray data. Bioinformatics 21, 4280–4288. [DOI] [PubMed] [Google Scholar]
- Xiong F, Ge W, and Ma C (2019). Quantitative proteomics reveals distinct composition of amyloid plaques in Alzheimer's disease. Alzheimers Dement 15, 429–440. [DOI] [PubMed] [Google Scholar]
- Xu P, Duong DM, and Peng JM (2009). Systematical Optimization of Reverse-Phase Chromatography for Shotgun Proteomics. J Proteome Res 8, 3944–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, et al. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell 153, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, and Horvath S (2005). A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4, Article17. [DOI] [PubMed] [Google Scholar]
- Zhang J, Benavente CA, McEvoy J, Flores-Otero J, Ding L, Chen X, Ulyanov A, Wu G, Wilson M, Wang J, et al. (2012). A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 481, 329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Jia L, Liu CC, Rong Z, Zhong L, Yang L, Chen XF, Fryer JD, Wang X, Zhang YW, et al. (2017). TrEm2 Promotes Microglial Survival by Activating Wnt/beta-Catenin Pathway. J Neurosci 37, 1772–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Whole proteome and phosphoproteome profiling of mouse brain tissues with different postmortem interval (PMI) by TMT-LC/LC-MS/MS, related to Figure 1 and STAR methods.
Data S2: Whole proteome and phosphoproteome profiling of AD postmortem human brain tissues by TMT-LC/LC-MS/MS, related to Figure 1 and Figure 3.
Data S3: Analysis of whole proteome profiling of AD progression reveals disease stage-dependent proteins and pathways, related to Figure 1.
Data S4: Validation of AD-related proteins by profiling individual human samples in both Banner Sun and Mount Sinai cohorts, and deep proteome profiling of cerebrospinal fluid (CSF) samples from Banner Sun cohort, related to Figure 2.
Data S5: Analysis of protein phosphorylation events in AD, related to Figure 3.
Data S6: Aβ-correlated proteins in AD and human-mouse proteomic comparison, related to Figure 4 and Figure 5.
Data S7: Prioritization of proteins/genes in AD by multi-omics datasets, related to Figure 6.
Data Availability Statement
Human proteomics data used in this study are available via the AD Knowledge Portal (https://adknowledgeportal.synapse.org). Additional information can be found via the following links: the Mount Sinai Brain Bank study (https://adknowledgeportal.synapse.org/Explore/Studies?Study=syn3159438) and the StJude_Banner study (https://adknowledgeportal.synapse.org/Explore/Studies?Study=syn21592732). In addition, mouse and pooled AD proteomics data are available in the PRIDE database (http://www.proteomexchange.org) with accession numbers of PXD007974 (mouse), PXD018590 (mouse), and PXD007985 (human). An interactive website (https://penglab.shinyapps.io/ADprofile) was generated to search for individual protein in the AD pooled dataset.