

Abstract
Acidic organelles and vesicles, such as endosomes, lysosomes, autophagosomes, trans-Golgi network, and synaptic vesicles, are known to play important roles in a broad range of cellular events. To facilitate studying these multifunctional systems, we describe here an acid-brightening fluorescent protein (abFP), which fluoresces strongly at acidic pH, but is almost nonfluorescent at or above physiological pH, making it well suited for imaging molecules residing in acidic microenvironment in live cells. Specifically, a quinoline-containing unnatural amino acid Qui is incorporated into the chromophore of EGFP via genetic code expansion to generate the abFP. When being exposed to acidic environment, protonation of Qui results in a cationic chromophore and fluorescence increase. Protocols are presented to express abFP in E. coli and mammalian cells, and to fluorescently image the endocytosis of δ opioid receptor-abFP fusion protein in mammalian cells. This strategy may be similarly applicable to other fluorescent proteins to enable acidic imaging.
Keywords: Genetic code expansion; unnatural amino acid; L-β-(quinoline-6-yl) alanine, Qui; acid-brightening fluorescent protein (abFP); acidic vesicles and organelles; imaging
1. Introduction
Eukaryotic cells comprise a group of membrane-enclosed organelles with distinct functions. Under physiological conditions, their pH varies greatly. For instance, the pH of cytosol, nucleus, and endoplasmic reticulum are near neutral, while endosomes, lysosomes, autophagosomes, trans-Golgi network, and synaptic vesicles are acidic (Casey, Grinstein, & Orlowski, 2010). These acidic organelles and vesicles are known to play key roles in secretory and endocytic pathways as well as many other cellular events. For example, endosomes sort and deliver cellular materials to various destinations (Maxfield & McGraw, 2004); lysosomes break down and recycle obsolete or un-used biomolecules (Settembre, Fraldi, Medina, & Ballabio, 2013); and synaptic vesicles store and release various neurotransmitters for interneuronal signaling (Sinning & Hubner, 2013). To achieve optimal cellular functions, it is critical to maintain an appropriate pH within individual organelles and vesicles; otherwise, they may lose functions and cause diseases (Casey et al., 2010). Moreover, due to their important roles in pathogen defense (Cossart & Helenius, 2014; Saftig & Klumperman, 2009) and multi-drug resistance in cancer (Zhitomirsky & Assaraf, 2016), these organelles have emerged as a class of attractive therapeutic targets (Bonam et al., 2019; Rajendran, Knolker, & Simons, 2010; Sakhrani & Padh, 2013). Therefore, it is important to understand the multifunctional roles of these organelles and vesicles as well as their relationship with diseases.
Fluorescence microscopy provides precise spatial-temporal resolution and has been widely used in investigating dynamic cellular processes in live cells. To study acidic organelles and vesicles, it would be ideal to have fluorescent pH indicators that are nonfluorescent at physiological pH 7.4 and turn on fluorescence at acidic luminal pH, so that the targeting organelles and vesicles (with acidic pH) can brighten up and create a sharp contrast from the dark background of the cytosol (with neutral pH). For this purpose, a series of small molecule and fluorescent protein (FP)-based fluorescent indicators have been developed. Small molecule pH indicators, which are available in different fluorescent colors, can selectively and effectively label acidic organelles, making them valuable tools (Adie et al., 2003; Asanuma et al., 2014). However, despite these advantages, small molecule indicators generally lack genetic specificity and reversibility of labelling. A genetically encoded indicator can be stably expressed in live cells, allowing for precise location to targeting subcellular destinations and noninvasive imaging overtime. FPs have been engineered and genetically encoded to sense intracellular pH changes (Kneen, Farinas, Li, & Verkman, 1998; Miesenbock, De Angelis, & Rothman, 1998; Y. Shen, Rosendale, Campbell, & Perrais, 2014; Tsien, 2009). However, most monomeric FPs and their derived pH sensors are only fluorescent at neutral or slightly basic pH, and their fluorescence will be quenched in an acidic environment (Lin & Schnitzer, 2016). Due to such inherited limitations, the conventional FP-based sensors are only useful when the organelles and vesicles are exposed to neutral or basic pH, but are not capable of visualizing the intravascular contents, resting vesicles, or intracellular vesicle dynamics.
To address the needs for visualizing acidic organelles and vesicles in live cells, we recently developed an abFP (acid-brightening fluorescent protein) possessing a pH profile contrary to conventional FPs. Through genetic code expansion, a quinoline-containing unnatural amino acid (Uaa) called Qui was genetically incorporated at site 66 of the enhanced green fluorescent protein (EGFP) to form a chromophore with a positive charge. In contrast to conventional FPs, the resultant abFP fluoresces brightly at acidic pH but becomes invisible at and above physiological pH. By fusing the abFP to target receptors, we were able to image ligand-induced endocytosis of opioid receptors locating at acidic lysosomes. In this chapter, we will discuss the genetic encoding of Qui to express abFP in both E. coli and mammalian cells as well as its application in fluorescence imaging of opioid receptors within acidic vesicles.
2. Developing and characterizing Qui-based abFP
We will first discuss the design of Qui, followed by a brief description of its genetic incorporation by evolving Qui-specific tRNA/synthetase pairs. Next, we will discuss the incorporation of Qui into model proteins in both E. coli and mammalian cells. Finally, we will introduce an example of fluorescence imaging of endocytosed receptors using Qui-containing abFP.
2.1. Design of Qui-based abFP
The chromophore of most FPs consists of a tyrosine residue. The protonation-deprotonation equilibrium of the phenolic group of this residue shifts in response to pH changes, leading to pH-dependent spectral characteristics (Fig. 1A). When pH reduces to acidic values, the uncharged phenol species is favored and the phenolate species decreases, resulting in abrupt decline in fluorescence intensity at the excitation wavelengths of anionic phenolate state (Fig. 1B).
Fig. 1.
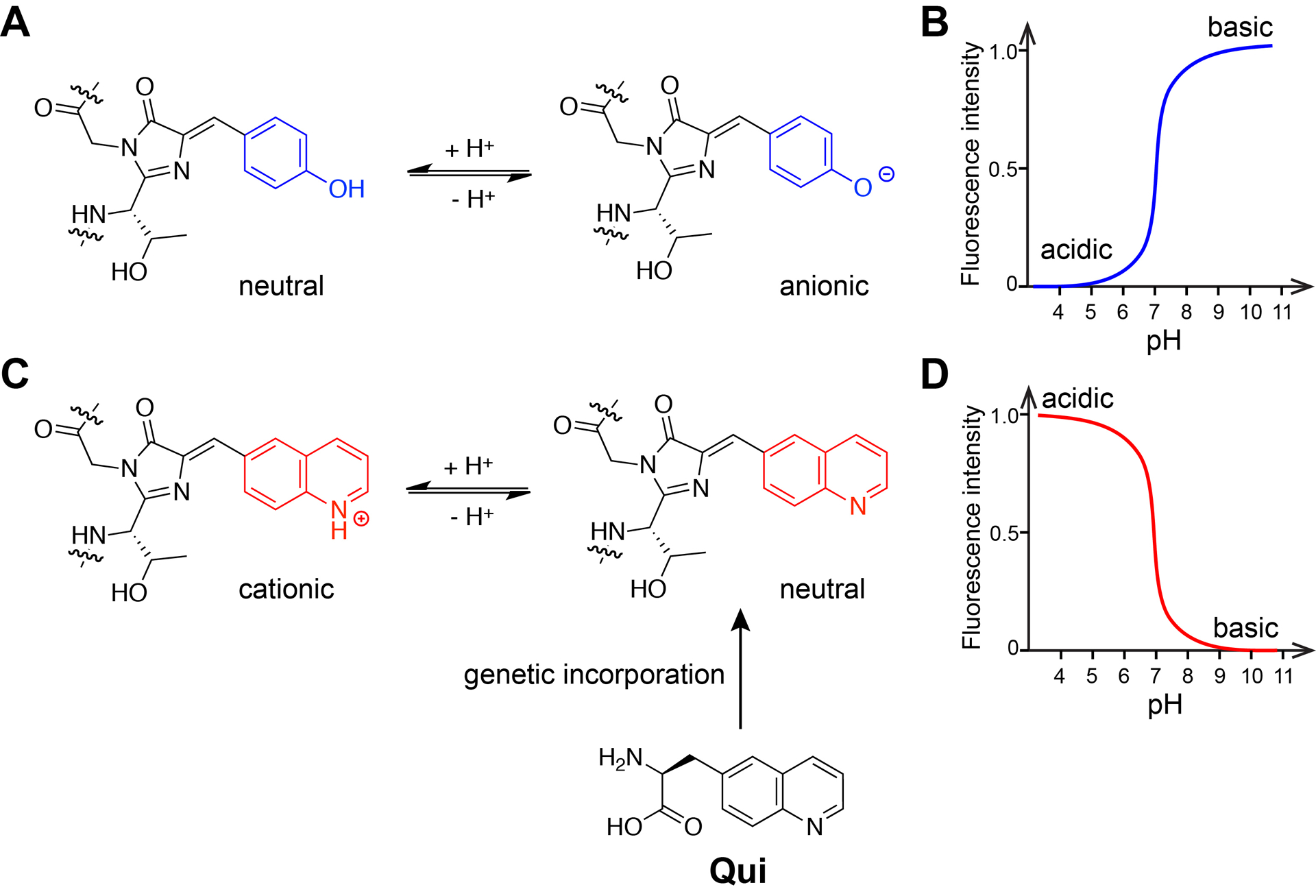
Quinoline-based chromophore to reverse the pH profile of FP. (A) The chromophore of common FPs contains a Tyr residue, which equilibrates between the neutral phenol and anionic phenolate states in response to pH change. (B) The profile of fluorescence intensity vs pH for common FPs. (C) Genetic incorporation of Qui in placement of Tyr will create a quinoline-based chromophore, which will equilibrate between a cationic and a neutral chromophore state. (D) Expected fluorescence intensity vs pH profile for the quinoline-based chromophore. Adapted with permission from Fu, C. et al. (2018). Genetically Encoding Quinoline Reverses Chromophore Charge and Enables Fluorescent Protein Brightening in Acidic Vesicles. Journal of the American Chemical Society, 140, 11058–11066. Copyright (2018) American Chemical Society.
We reasoned whether introduction of a positive charge onto the chromophore in place of the anionic chromophore would reverse the acid-quenching pH profile of common FPs. An acidic pH would shift equilibrium toward cationic state of the chromophore, excitation of which will result in fluorescence increase. We chose quinoline mainly based on two considerations. First, quinoline has a pKa value of 5 to 6 at the nitrogen atom in the conjugated ring for protonation, which is ideal for sensing pH changes in acidic organelles and vesicles. Second, introduction of quinoline into a chromophore should still maintain the strong fluorescence of the resultant FP. Previous studies indicate that most FPs contain a heteroatom at the phenol position in the chromophore (Chudakov, Matz, Lukyanov, & Lukyanov, 2010). We also showed that after removing the heteroatom at this position via unnatural amino acid mutagenesis, GFP significantly lost its fluorescence (L. Wang, Xie, Deniz, & Schultz, 2003). We thus designed a quinoline containing Uaa (L-β-(quinoline-6-yl) alanine, Qui) with a nitrogen atom at the phenol position (para position of β-carbon) (Fig. 1C). We expect that by substitution of tyrosine with Qui into the FP, the re-designed chromophore would establish a new equilibrium between cationic and neutral states under different pH, resulting in an acid-brightening FP (Fig. 1D).
2.2. Genetic encoding of Qui in E. coli
Genetic code expansion has been widely used to incorporate designer Uaas into proteins to afford them with novel functions in live cells and even animals (L. Wang, 2017; L. Wang, Brock, Herberich, & Schultz, 2001; L. Wang & Schultz, 2004). To genetically encode a Uaa into a protein, an orthogonal pair of aminoacyl-tRNA synthetase (aaRS)/tRNA is needed that specifically incorporate this Uaa in response to a unique codon, for example, the TAG amber stop codon.
As Qui is structurally similar to L-3-(2-naphthyl)alanine (Nap), initially we tested whether a previously reported Nap-specific Methanosarcina mazei (Mm) NapRS/tRNAPyl pair was able to incorporate Qui (Lacey, Louie, Noel, & Wang, 2013). Unfortunately, this pair didn’t work for Qui possibly due to the electrostatic effect of the nitrogen atom on the quinoline ring. We then decided to evolve a new aaRS/tRNA pair to be specific for Qui. Multiple Methanosarcina mazei pyrrolysyl-tRNA synthetase (MmPylRS) mutants have been evolved to be specific for bulky phenylalanine analogues, such as a benzoyl group, an azobenzene group, and long alkyl halide chains (Hoppmann et al., 2014; Lacey et al., 2013; Xiang et al., 2014). Based on these examples, we presumed that the bulky quinoline group would be recognized by a MmPylRS mutant. We thus generated a mutant library of MmPylRS through saturation mutagenesis of five residues (N346NNK, C348NNK, V401NNK, W417NNK, and G419NNK) and then subjected it to selections as described before (Takimoto, Dellas, Noel, & Wang, 2011; N. Wang et al., 2018). One MmPylRS mutant, with N346Q, C348S, V401G, and W417T mutations, was identified from the selection and named as MmQuiRS. The MmQuiRS comprises markedly different amino acid sequences in the active site from those of MmNapRS and MmPylRS.
To evaluate the incorporation specificity and efficiency of Qui into proteins in E. coli, we co-expressed the evolved MmQuiRS/tRNAPyl pair with an EGFP gene containing a TAG codon at position 182 (EGFP-182TAG) in E.coli. In the absence of Qui, no full length EGFP was detected; when 1 mM Qui was supplemented in the culture media, full length EGFP was produced with the yield of 3.5 mg/L. The purified full length EGFP-182Qui was further analyzed with electrospray ionization time of flight mass spectrometry (ESI-TOF-MS). Peaks corresponding to Qui incorporation at site 182 were clearly observed, while no peaks corresponding to mis-incorporation of natural amino acids at site 182 were identified. These results indicate that the evolved MmQuiRS/tRNAPyl was able to incorporate Qui into proteins in E. coli with high specificity and efficiency (Fu et al., 2018).
2.3. Expression of abFP in E. coli and characterization of its fluorescence properties
To utilize Qui for pH sensing, we replaced the chromophore forming residue of EGFP, Tyr at position 66, with Qui (Fig. 2A). We co-expressed the EGFP-66TAG gene with the MmQuiRS/tRNAPyl pair in E. coli. When 1 mM Qui was supplemented in the culture media, full length EGFP protein was produced. The purified EGFP-66Qui was analyzed with ESI-TOF-MS, which confirmed that Qui was site specifically incorporated at position 66 of EGFP and a new chromophore was formed in a similar manner as the wild type EGFP (Fu et al., 2018).
Fig. 2.
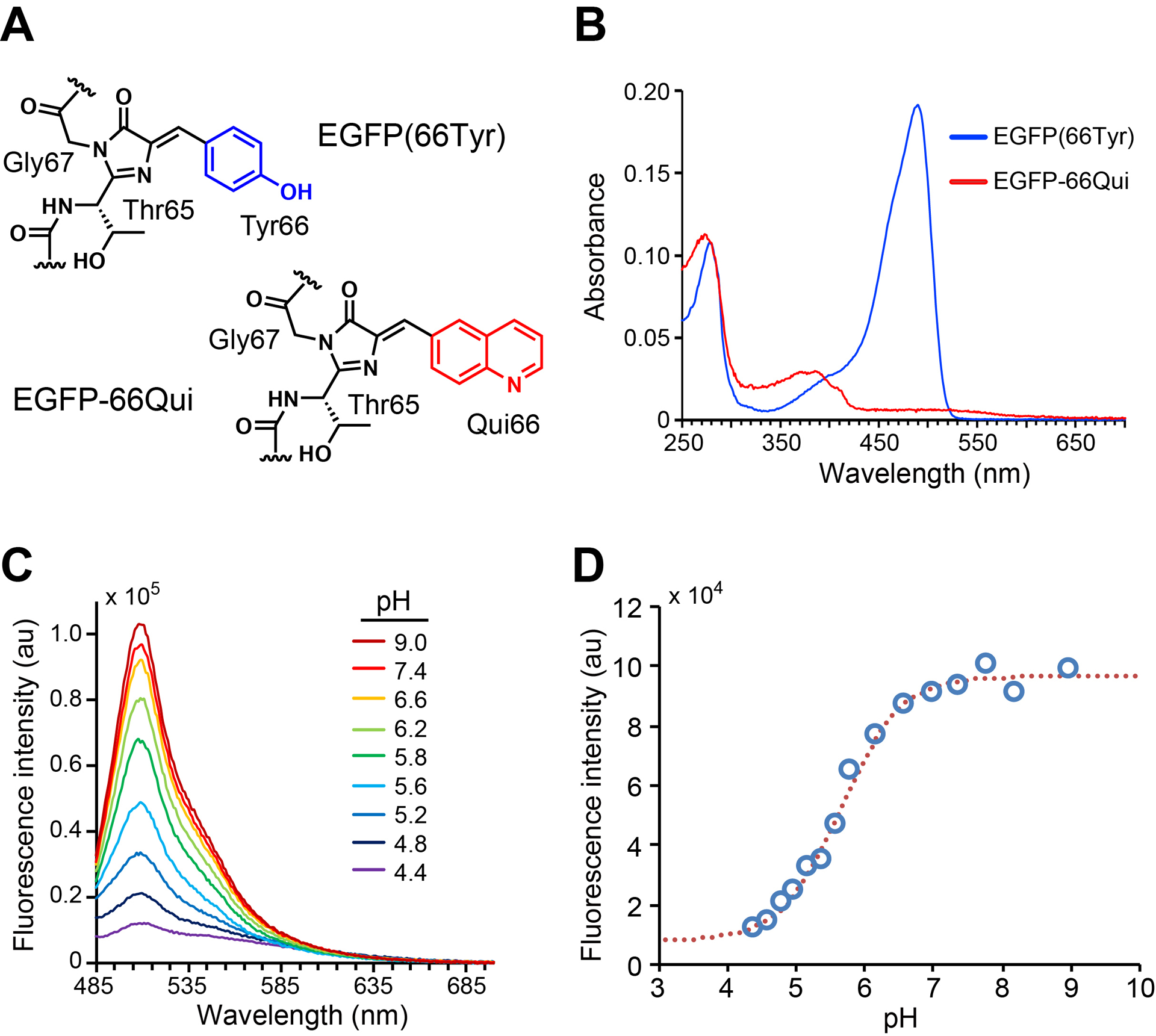
Genetically encode Qui into EGFP chromophore to generate EGFP-66Qui. (A) Chromophore structures of EGFP and EGFP-66Qui. (B) UV–vis absorbance spectra of EGFP and EGFP-66Qui in phosphate buffer (pH 7.4). (C) Fluorescence emission spectra of EGFP-66Qui in buffers with indicated pH values. (D) pH profile of EGFP-66Qui. Fluorescence emission intensity at 505 nm of EGFP-66Qui (measured in C) was plotted against pH. Adapted with permission from Fu, C. et al. (2018). Genetically Encoding Quinoline Reverses Chromophore Charge and Enables Fluorescent Protein Brightening in Acidic Vesicles. Journal of the American Chemical Society, 140, 11058–11066. Copyright (2018) American Chemical Society.
In phosphate buffer with neutral pH, the wild type EGFP(EGFP-66Tyr) had a major absorption peak at 488 nm, while EGFP-66Qui had a major absorption peak at 385 nm and a small absorption at 475 nm (Fig. 2B). Since 385 nm is within the UV region, which is not suitable for live cell imaging, we chose 475 nm as the excitation wavelength for measuring the fluorescence emission spectra of EGFP-66Qui under different pH. Fluorescence emission peaked at 510 nm in aqueous buffer ranging from pH 4.4 to 9. The fluorescence intensity changed in a pH-dependent manner. When pH increased from acidic (pH 4.4) to neutral (pH 7.4), the emission intensity increased by 8-fold (Fig. 2C). The pKa value for fluorescence change was determined as 5.7, which corresponds well to the reported pKa value of quinoline (pKa = 5–6) (Fig. 2D). These data supported that the pH-dependent responses in fluorescence intensity of EGFP-66Qui was due to Qui, although the pH profile of EGFP-66Qui remained similar to that of common FPs.
Interestingly, we observed that EGFP-66Qui turned from colorless to light red after exposure to ambient light for a while, which prompted us to check whether it was photoactivable. Indeed, illuminating an EGFP-66Qui protein freshly prepared in dark with 365 nm light for a few minutes turned it in red (Fig. 3A). After illumination, the absorption spectrum changed correspondingly, with absorption peak shifted from 385 to 523 nm in neutral phosphate buffer (pH = 7.4) (Fig. 3B).
Fig. 3.
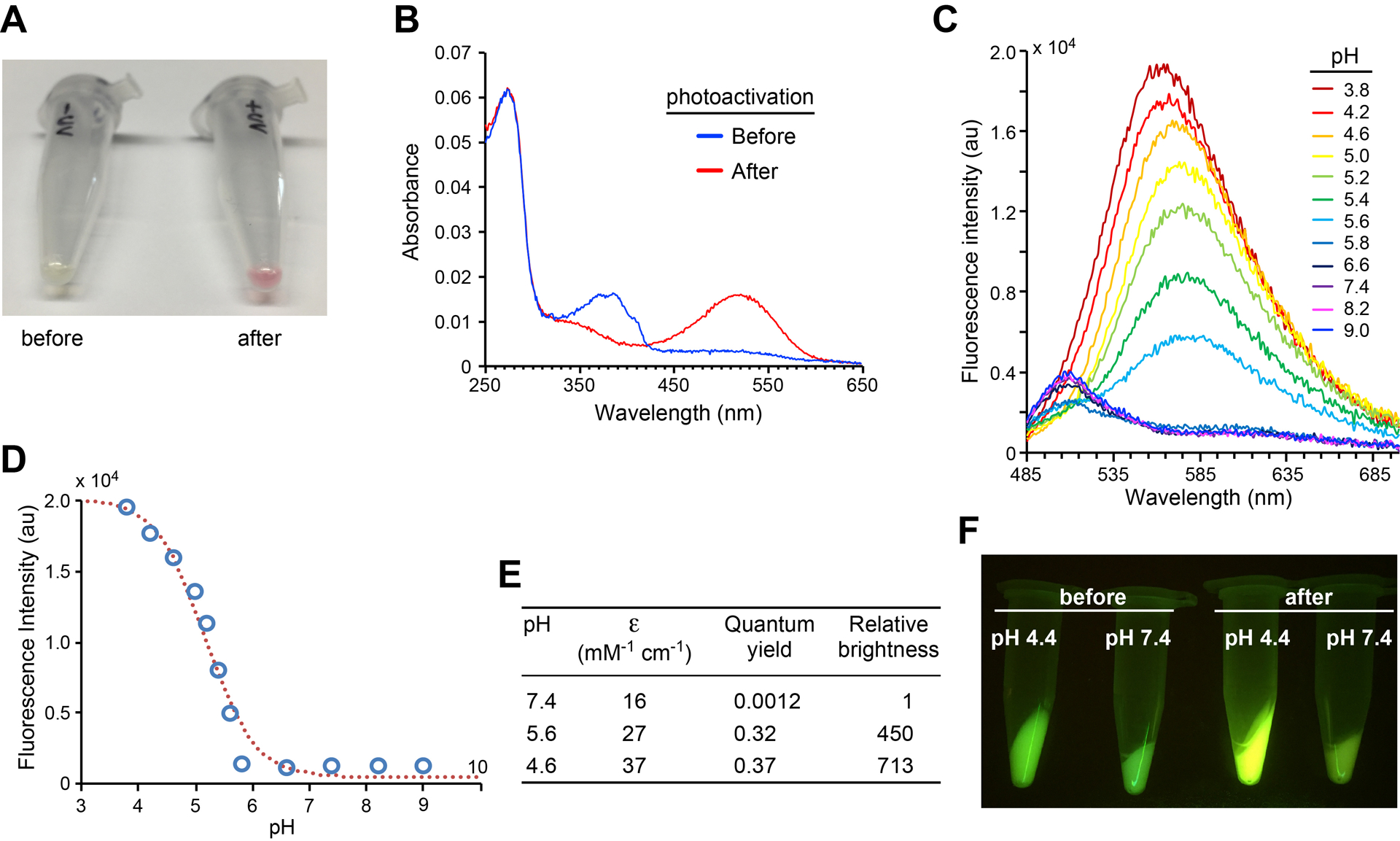
Photoactivation of EGFP-66Qui results in abFP that fluoresces at acidic pH only. (A) A photograph of EGFP-66Qui protein samples before and after illumination with a handhold UV lamp (365 nm). (B) Absorption spectral change of EGFP-66Qui upon photoactivation in phosphate buffer (100 mM, pH 7.4). (C) Fluorescence emission spectra of photoactivated EGFP-66Qui in buffers with indicated pH values. (D) pH profile of photoactivated EGFP-66Qui. Fluorescence emission intensity at 560 nm measured in c was plotted against pH. (E) Extinction coefficient (ε, measured at 523 nm) and quantum yield of photoactivated EGFP-66Qui at different pH. Relative brightness is the product of these two values normalized to that at pH 7.4. (F) Fluorescence images of bacterial pellets expressing EGFP-66Qui before and after photoactivation in different pH aqueous media. Adapted with permission from Fu, C. et al. (2018). Genetically Encoding Quinoline Reverses Chromophore Charge and Enables Fluorescent Protein Brightening in Acidic Vesicles. Journal of the American Chemical Society, 140, 11058–11066. Copyright (2018) American Chemical Society.
When excited at 475 nm, the photoactivated EGFP-66Qui showed ratiometric spectral changes (Fig. 3C). From pH 5.8 to pH 9.0, the main fluorescence emission maxima were at ~510 nm; when pH was tuned to more acidic values (pH 3.8 to pH 5.6), the main fluorescence emission maxima red-shifted to 566–582 nm. Excitingly, the pH profile of photoactivated EGFP-66Qui changed dramatically: when pH value dropped from neutral (pH 7.4) to acidic (pH 3.8), the fluorescence intensity of the peak around 570 nm increased (Fig. 3C). The pKa value for fluorescence change was determined as 5.2, which corresponded well to that of quinoline, suggesting that Qui still functioned as the pH-sensitive motif after photoactivation (Fig. 3D).
The photoactivated EGFP-66Qui possessed several unusual characteristics. First, it was able to fluoresce strongly at extreme acidic pH 3.8, which hasn’t been reported with other FPs. Second, it was almost not fluorescent at physiological pH 7.4 at 570 nm, which will be invaluable for reducing the background fluorescent noises. Meanwhile, its weak fluorescence at 510 nm at pH 7.4 will help localize the target proteins. The extinction coefficient and quantum yield of the photoactivated EGFP-66Qui were measured at pH values 7.4, 5.6, and 4.6 (Fig. 3E). When pH shifted from neutral value (pH 7.4) to acidic values (pH 5.6 and 4.6), its brightness increased by 450-fold and 713-fold respectively. Taken together, the photoactivated EGFP-66Qui presented a desired pH profile of fluorescence that is inverse to conventional FPs and successfully covered a whole acidic pH range that one can possible encounter in live cells. These features will allow photoactivated EGFP-66Qui to specify sharp boundaries between acidic organelles and vesicles with surrounding environment. Therefore, we named EGFP-66Qui as abFP based on its unique features.
Upon illumination wild type GFP can photoconvert from 398 nm absorbing state to 475 nm absorbing state with a decarboxylation at Glu222 (van Thor, Gensch, Hellingwerf, & Johnson, 2002). Glu222 locates close to the chromophore and takes part in its hydrogen bonding network. To examine whether photoactivation of abFP is also associated with decarboxylation, we analyzed abFP after UV illumination with mass spectrometry. ESI-TOF-MS of the intact protein indicates loss of 44 Da (MW of CO2) after photoactivation, suggesting decarboxylation. To map the exact position where decarboxylation happened, photoactivated abFP was digested with trypsin and analyzed with tandem MS, which unambiguously indicate that the decarboxylation occurred at Glu222 (Fu et al., 2018).
We examined abFP’s ability to sense pH in live E. coli cells. E. coli cells expressing abFP were collected and treated with or without photoactivation (365 nm illumination). Then the cells were exchanged into aqueous buffers of pH 7.4 or 4.4. The cell pellets were excited at 470 ± 20nm and photographed through a 530 nm filter. Only the cells photoactivated and in the pH 4.4 buffer showed strong yellow fluorescence (Fig. 3F), suggesting that abFP can work as a photoactivatable indicator for acidic pH in live bacterial cells.
3. Expression of abFP in mammalian cells to sense acidic pH
3.1. Genetic incorporation of Qui into proteins in mammalian cells
To test and optimize Qui incorporation in mammalian cells, a HeLa-EGFP-182TAG cell line, which contains EGFP gene with a TAG stop codon at position 182 stably integrated into its genome, was used as a reporter (W. Wang et al., 2007). Only when the Uaa is incorporated at the 182TAG site will full length EGFP be produced to generate green fluorescence signal for detection. We transfected the reporter cell line with plasmid pMP-QuiRS-1xtRNA or pMP-QuiRS-3xtRNA, which contains one copy of MmQuiRS and a single or three copies of tRNAPyl. After transfection, the cells were incubated with 0, 0.1, 0.5, and 1 mM of Qui at 37 °C for 24 h or 48 h followed by flow cytometric analysis. In the presence of Qui, strong fluorescence was detected, suggesting efficient incorporation of Qui in mammalian cells. Along with the increase of tRNA copy number, Qui concentration, and incubation time, the total fluorescence intensity of reporter cells increased (Fu et al., 2018). Therefore, plasmid pMP-QuiRS-3xtRNA, 1 mM of Qui, and 48 h of incubation time were chosen for Qui incorporation in mammalian cell experiments unless indicated otherwise.
The cytotoxicity of Uaa or contaminants in the Uaa preparation can significantly impair Uaa incorporation and application in mammalian cells. To evaluate the potential cytotoxicity of Qui, we performed cell viability assay using alamarBlue. Qui did not cause obvious toxicity to HeLa-EGFP-182TAG reporter cells or HeLa cells under a broad range of Qui concentrations (0, 0.1, 0.25, 0.5, 0.75, and 1 mM), indicating that Qui is suitable for applications in mammalian cells (Fu et al., 2018).
3.2. Expression of abFP in mammalian cells to sense extracellular acidic pH
To quickly adjust extracellular pH through buffer exchange, we displayed abFP or wild type EGFP on mammalian cell surface by fusing them to the N-terminus of human interlueukin-2 receptor α-subunit Tac, which directs fused proteins to the extracellular side of the plasma membrane (Tan, Waites, Liu, Krantz, & Edwards, 1998). Plasmids pMP-QuiRS-3xtRNA and pcDNA3.1-EGFP66TAG-Tac were co-transfected into HeLa cells. After transfection, the cells were incubated with 1 mM Qui for 48 h. After being photoactivated with UV illumination at 385 nm, the cells were exposed to buffers with various pH and then imaged using wildfield fluorescence microscope. Cells expressing abFP showed weak fluorescence at pH 7.2. When pH reduced to more acidic values of 5.6, 4.6, and 3.8, the fluorescence intensity increased drastically and peaked at pH 3.8 (Fig. 4A). In contrast, the control cells expressing wild type EGFP (expressed by plasmid pcDNA3.1-EGFP-Tac) showed strong fluorescence at pH 7.2, and the fluorescence intensity decreased precipitously when pH shifted to more acidic values of 5.6, 4.6, and became almost non-detectable at pH 3.8 (Fig. 4B). Immunoblot results showed that the expression level of abFP-Tac and EGFP-Tac remained relatively stable after pH changes, indicating that the variations of fluorescence intensity were not due to changes in protein amount but due to pH sensitivity.
Fig. 4.
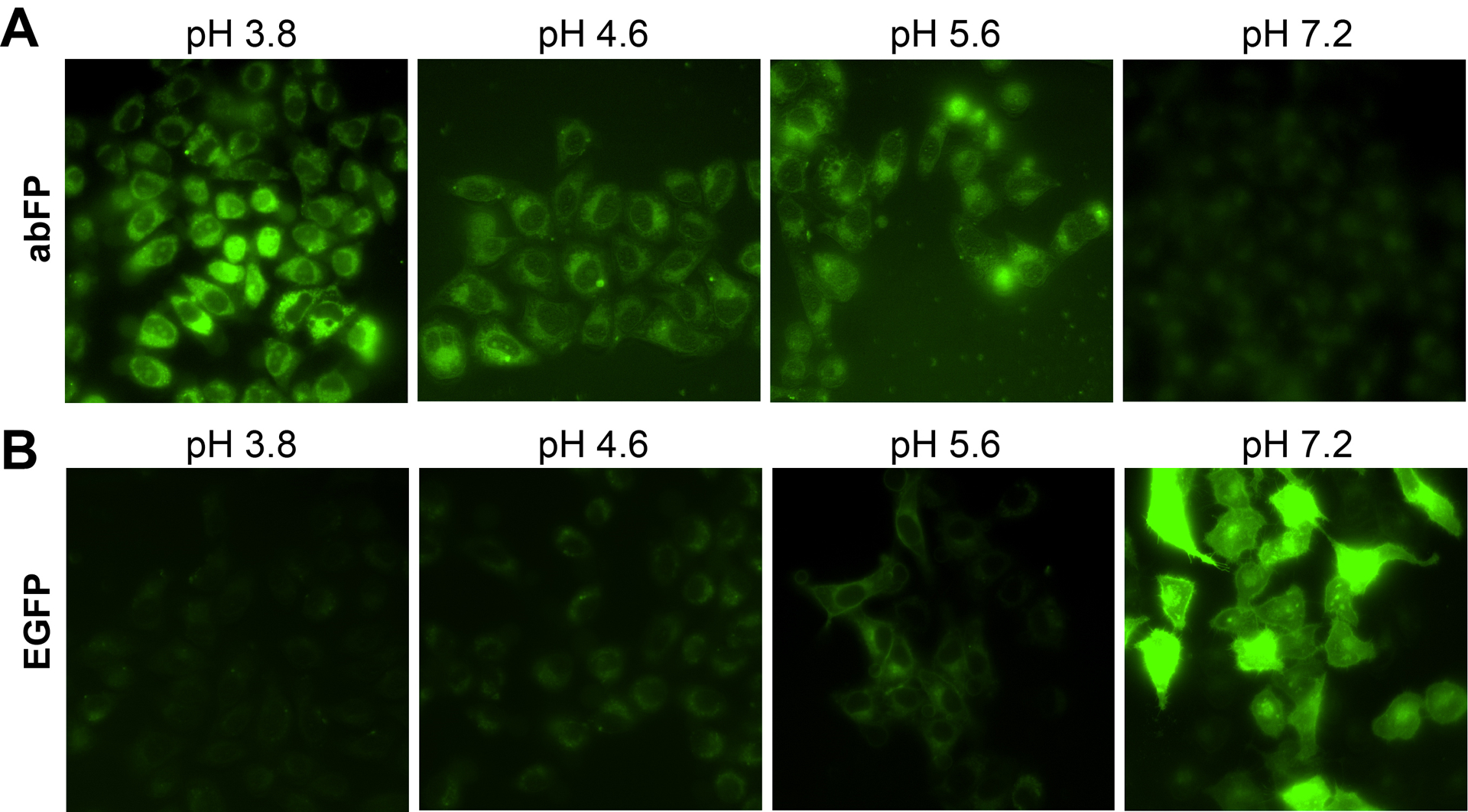
Fluorescence images of HeLa cells expressing abFP (A) or EGFP (B) exposed to buffers of indicated pH values. abFP and EGFP were expressed at the extracellular side of the cell surface. Adapted with permission from Fu, C. et al. (2018). Genetically Encoding Quinoline Reverses Chromophore Charge and Enables Fluorescent Protein Brightening in Acidic Vesicles. Journal of the American Chemical Society, 140, 11058–11066. Copyright (2018) American Chemical Society.
4. Using abFP to fluorescently image receptor endocytosis in mammalian cells
Endocytosis internalizes extracellular materials and is involved in nutrient uptake, pathogen entry, cell adhesion and migration, cell polarity, and receptor-mediated signaling (Sorkin & von Zastrow, 2009). Recently, it has been reported that many receptors initiate signaling from not only cell surfaces but also other subcellular locations, such as endosomes and Golgi apparatus (Calebiro et al., 2009; Ferrandon et al., 2009; Irannejad et al., 2017; Irannejad et al., 2013; Mullershausen et al., 2009; Tsvetanova & von Zastrow, 2014). Following activation, signaling receptors can be rapidly endocytosed and then moved through a series of endosomal compartments for downstream events. After internalization, some receptors are sorted to lysosomes for degradation, while many receptors remain active in endosomes and continue signaling by interacting with downstream effectors (Sorkin & von Zastrow, 2009).
The ability to track the internalization and re-localization of targeting receptors to the acidic subcellular compartments in real time would facilitate the understanding of endocytic trafficking of receptors and the emerging “location bias” of receptor functional selectivity. To demonstrate abFP’s unique ability in this regard, we fused abFP or wild type EGFP to the δ opioid receptor (DOR) for fluorescent imaging in mammalian cells (Fig. 5A). DOR belongs to the G-protein coupled receptor family and can be activated by endogenous peptide ligands and opiate drugs. Activation of DOR is usually accompanied by rapid agonist-induced internalization of receptor, followed by receptor trafficking to lysosomes via endosomes.
Fig. 5.
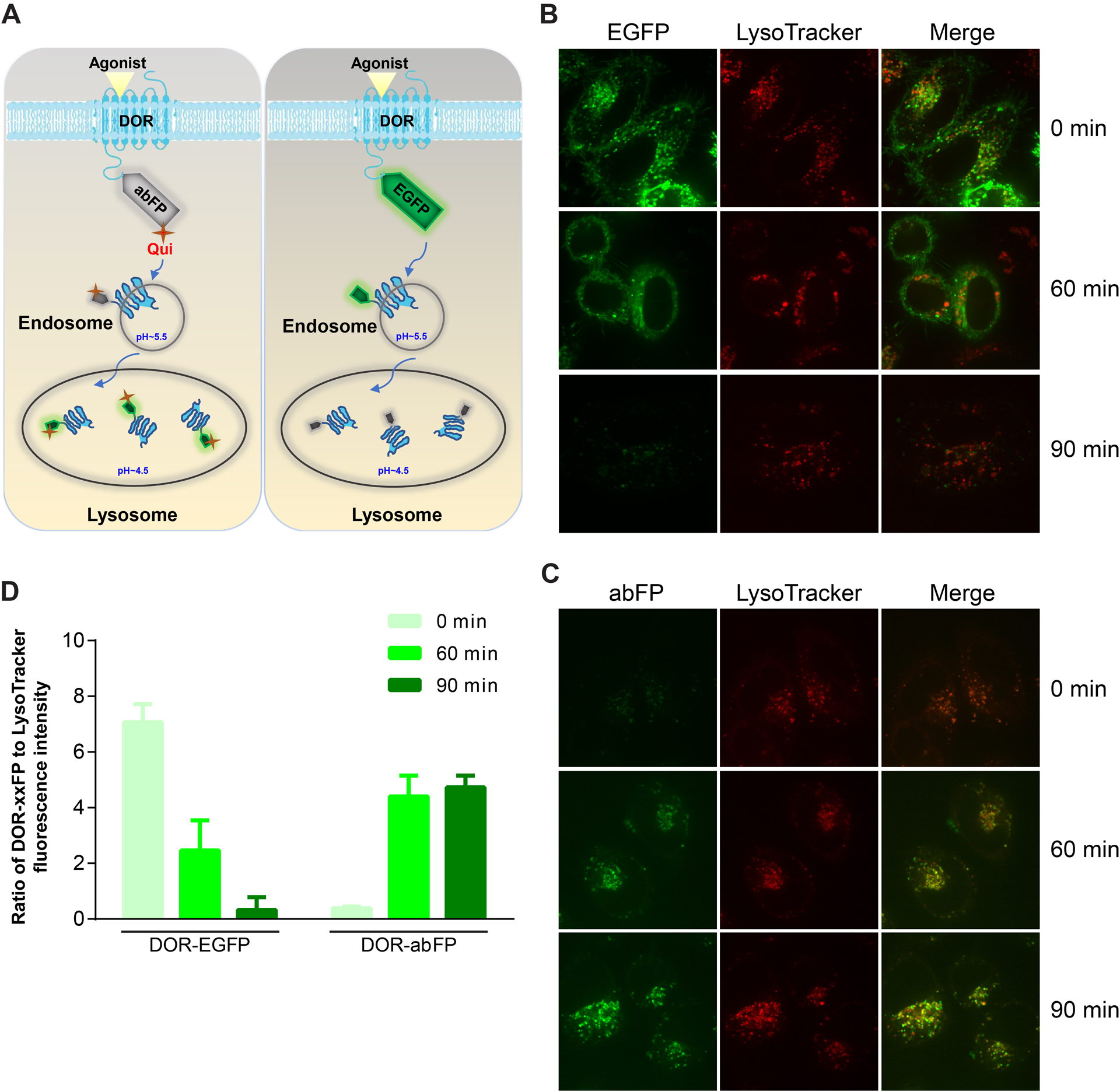
abFP enables fluorescence imaging of endocytosed opioid receptor DOR in mammalian cells. (A) Scheme showing the endocytosis of abFP or EGFP tagged DORs to lysosome upon agonist induction. (B) Confocal fluorescence images of HeLa cells expressing EGFP-tagged DOR. Cells were imaged before (t = 0) and after (t = 60, 90 min) agonist induction. (C) Confocal fluorescence images of HeLa cells expressing abFP-tagged DOR. (D) Ratio of the DOR-xxFP to LysoTracker Red fluorescence intensity measured from confocal fluorescence images representatively shown in B and C. Adapted with permission from Fu, C. et al. (2018). Genetically Encoding Quinoline Reverses Chromophore Charge and Enables Fluorescent Protein Brightening in Acidic Vesicles. Journal of the American Chemical Society, 140, 11058–11066. Copyright (2018) American Chemical Society.
We fused abFP or wild type EGFP to the C-terminus of DOR to generate DOR-abFP and DOR-EGFP plasmids. HeLa cells was co-transfected with the DOR-abFP and pMP-QuiRS-3xtRNA plasmids or transfected with the DOR-EGFP plasmid. Expressed DOR-abFP was photoactivated at 385 nm for 10 min and then imaged with fluorescence confocal microscopy. For comparison, LysoTracker Red, a cell-permeable small molecule probe that selectively accumulates in acidic organelles, were applied to each sample. Before agonist stimulation (t = 0 min), cells expressing DOR-EGFP showed strong green fluorescence. The zones of fluorescence did not overlap with that of LysoTracker Red, indicating that DOR-EGFP did not locate at acidic environment. Contrary to DOR-EGFP, the fluorescence of DOR-abFP was negligible under the same condition, consistent with abFP’s low fluorescence in nonacidic environment. The cells were then treated with DADLE, a synthetic polypeptide DOR agonist, to initiate the receptor internalization. During 60 min of agonist stimulation, the fluorescence intensity of cells expressing DOR-EGFP decreased gradually and the fluorescent zones still did not overlap with that of LysoTracker Red. After 90 min, the fluorescence of DOR-EGFP became almost non-detectable, suggesting that DOR-EGFP had trafficked to acidic environment (Fig. 5B). In contrast, the green fluorescence of cells expressing DOR-abFP emerged after agonist stimulation and accumulated with extended stimulation time (Fig. 5C). Moreover, the fluorescence signal of DOR-abFP overlapped extensively with the red fluorescence of LysoTracker Red in puncta, suggesting that the DOR-abFP receptors had trafficked to lysosomes.
As demonstrated, in neutral environment DOR-abFP was almost nonfluorescent, while at acidic locations it fluoresced strongly, enabling us to image DOR receptors within acidic vesicles with minimum background fluorescence and a sharp contrast. We normalized the fluorescence intensity of DOR-abFP and DOR-EGFP by calculating the ratio of their fluorescence intensity over that of LysoTracker Red (Fig. 5D). Obviously, DOR-abFP presented an increasing trend in fluorescence during endocytosis, which was opposite to DOR-EGFP. Specifically, the normalized fluorescence intensity of DOR-abFP increased by more than 4-fold after the DOR was endocytosed from cell surface into the lysosomes, providing satisfying signal over noise ratio. Immunoblot results showed that the expression level of DOR-EGFP or DOR-abFP did not change significantly during experiments, suggesting that the observed change in fluorescence intensity was not due to the change of FP protein amount but because of pH sensing.
5. Protocol
5.1. Equipment
Fiber optic illuminator and filter
Shaker/incubator
UV–visible spectrophotometer
Analytical balance
Sonicator
Centrifuge
Nanodrop (Thermo Scientific)
Spectrophotometer (UVIKON XS Spectrophotometer)
Plate reader (Tecan the infinite M1000 Pro)
FluoroLog spectrophotometer (HORIBA)
FACS (BD LSRFortessa™ cell analyzer)
Confocal microscope (Yokagawa CSU22 spinning disk confocal microscope)
5.2. Chemicals
2xYT media (Sigma)
Chloramphenicol (Sigma)
Ni-NTA agarose (Macherey Nagel)
Kanamycin (Thermo Fisher)
Qui (Chemical synthesis of Qui is described in the Supporting Information of our recent publication (Fu et al., 2018))
Tetracycline (Sigma)
Dimethyl sulfoxide (DMSO) (Sigma)
NaOH (0.1 N)
Isopropyl β-D-1-thiogalactopyranoside (IPTG) (Invitrogen)
Tris–HCl buffer (pH 8) (Sigma)
NaCl (Sigma)
Imidazole (Sigma)
Tween 20 (Sigma)
Glycerol (Sigma)
Lysozyme (Sigma)
Methanol (Sigma)
Dulbecco’s PBS (Sigma)
Trypsin-EDTA(Sigma)
LysoTracker Red DND-99 (Life Technologies)
Agonist DADLE (Sigma)
Lipofectamine2000 (Invitrogen)
Sodium acetate/acetic acid buffer (100 mM) (pH 3.8 to 5.2) (Sigma)
Phosphate buffer (100 mM) (pH 5.6 to 9.0) (Sigma)
5.3. Expression of abFP in E. coli and purification
Prepare LB broth media, 2xYT media and LB-agar plate supplemented with kanamycin sulfate 50 μg/mL and chloramphenicol 40 μg/mL (LB-agar-Kan50Cm40).
Co-transform plasmids pTak-EGFP66TAG and pBK-MmQuiRS into NiCo21(DE3) chemical competent cells (New England Biolabs). The cells were plated on an LB-agar-Kan50Cm40 plate and cultured overnight at 37 °C.
A single colony was inoculated into 4 mL LB-Kan50Cm40 to make a start culture. The culture was grown overnight at 37 °C with shaking.
The start culture was subcultured in 400 mL 2xYT-Kan50Cm40 and grown at 37 °C with shaking.
When OD600 reached 0.4~0.5, the culture was induced with Qui (120 mg/mL, 1 mL) and IPTG (0.5 M, 400 μL). The culture was grown at 30 °C with shaking for another 6 h, and cells were pelleted (5,000 g, 10 min, 4 °C) and stored at −80 °C until purification.
-
The cell pellet was resuspended in 30 mL lysis buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole, 1% v/v Tween20, 10% v/v glycerol, lysozyme 1 mg/mL, DNase 0.1 mg/mL, Complete™ Mini protease inhibitor, EDTA-free 1 tablet) and incubated at 4 °C for 1 h. The lysed solution was sonicated with a Sonic Dismembrator (Fisher Scientific, 30% amplitude, 2 min, 1 sec on, 1 sec off) and centrifuged at 15,000 rpm for 30 min at 4 °C. The supernatant was collected, mixed with 400 μL Protino® Ni-NTA agarose, and then incubated at 4 °C for 1 h. The slurry was loaded onto a Poly-Prep® Chromatography Column. The column was washed with 1 mL washing buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole, 10% v/v glycerol) for 4 times, and the abFP protein was eluted with 200 μL elution buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 250 mM imidazole, 10%v/v glycerol) for 5 times. The eluted fractions were combined and passed through a chitin resin column (2 mL slurry, New England Biolabs) to eliminate E. coli protein contaminations, and the flow through was collected. The resin was washed with 1 mL washing buffer for 4 times, and the flow through was collected. All flow-through fractions were combined and concentrated into ~50 μL with an Amicon® Ultra 10K centrifugal filters (EMD Millipore). Then the concentrated abFP was buffer-exchanged into 1x phosphate buffered saline (1xPBS, pH 7.4) using the same centrifugal filter.
Note: If your protein of interest is difficult to purify, such as membrane proteins and insoluble proteins, we suggest to optimize the purification procedures with a small initial volume (25 to 50 mL) of culture media before proceeding to large scale purification.
5.4. Characterization of the optical properties of purified abFP
-
AbFP was photoactivated by 365 nm light illumination for 10 min using a handhold UV lamp.
Note: Before testing on live cells, we suggest to optimize the illumination time and the distance of the light source to your purified protein samples for the best photoactivation results. Photoactivation status can be monitored by measuring the absorption spectrum as shown in Fig. 3B.
The absorption spectra of AbFP before and after photoactivation was measured on UVIKON XS Spectrophotometer in a 100 μL quartz microcuvette with black side walls (Hellma 105–251-QS, Hellma Ltd). The measurements were carried out in 100 mM PBS buffer (pH = 7.4).
The fluorescence emission spectra of AbFP were measured on a Fluorolog-3 (Horiba Jobin Yvon) in a 100 μL quartz microcuvette with black side walls (Hellma 105–251-QS, Hellma Ltd) under different pH. For pH ranging from 3.8 to 5.2, sodium acetate/acetic acid buffer (100 mM) was used; for pH ranging from 5.6 to 9.0, phosphate buffer (100 mM) was used.
To detect the molar extinction coefficient of abFP, the absorbance and concentration of abFP were measured. The absorbance at 523 nm was measured using a UV-vis, which is the peak absorption wavelength of photoactivated abFP. The protein concentration was determined by comparison of the intensity of Coomassie blue-stained band of abFP to that of BSA standards with known concentrations. The molar extinction coefficient ε was calculated using the following equation: ε = A/cl, where A is the absorbance, c is the molar concentration, and l is the path length.
The quantum yields of abFP at different pH were determined by comparison of the absorbance and integrated fluorescence emission intensity relative to an orange fluorescent protein mKO2 using the following equation: QYsample = (Emisample/EmimKO2) × (AbsmKO2/Abssample) × QYmKO2, where QY represents quantum yield, Emi represents integrated emission intensity, and Abs represents absorbance.
5.5. Genetic incorporation of Qui in HeLa-EGFP-182TAG reporter cells
The HeLa-EGFP-182TAG cells were cultured in DMEM medium (Gibco) supplemented with 10% (v/v) FBS (Corning) without antibiotics at 37 °C with 5% CO2.
One day before transfection, 4.5×104 of HeLa-EGFP-182TAG reporter cells were seeded in a Greiner Bio-One 24 well-cell culture dish containing 500 μL/well of DMEM media with 10% FBS and incubated at 37 °C in a CO2 incubator. Plasmid pMP-QuiRS-1xtRNA (500 ng) or pMP-QuiRS-3xtRNA (500 ng) was transfected into target cells using 2.5 μL of lipofectamine 2000 following manufacturer’s instructions. Six hours post transfection, the media containing transfection complex were replaced with fresh DMEM media with 10% FBS in the presence of 0, 0.1, 0.5, and 1 mM of Qui.
After incubation at 37 °C for 24 h or 48 h, transfected cells were detached using a trypsin–EDTA solution (Sigma) and collected by centrifugation (1500 rpm, 5 min, r.t.). The cells were resuspended in 300 μL of FACS buffer (1×PBS, 2% FBS, 1 mM EDTA, 0.1% sodium azide, 0.28 μM DAPI) and analyzed with the BD LSRFortessa™ cell analyzer.
5.6. Cell viability assay to test Uaa toxicity
When HeLa or HeLa-EGFP-182TAG cells reach 60–80% confluency, the cells were detached using a trypsin–EDTA solution (Sigma) followed by neutralization with DMEM containing 10% FBS. The detached cells were pelleted by centrifugation (1200 rpm, 3 min, r.t.). After removing supernatants, cell pellets were re-suspended in DMEM with 10% FBS for cell counting using a hemocytometer.
9×103 of HeLa or HeLa-EGFP-182TAG cells were then seeded in 96-well plates containing 100 μL/well of DMEM with 10% FBS and incubated overnight to allow for attachment and recovery. Qui was added at 0, 0.1, 0.25, 0.5, 0.75, and 1 mM of final concentration into the culture media, and cells were incubated for another 24 to 48 h.
-
The cell viability was assayed using the alamarBlue cell viability reagent (Invitrogen) following manufacturer’s instructions. Briefly, alamarBlue reagent (20 μL) was added to each well, and cells were incubated for 4 h. Fluorescence (Excitation 560 nm; Emission 590 nm) was then recorded using a Labsystems Fluoroskan Ascent plate reader (Infinite M1000 Pro).
Note: If you obtain a custom-synthesized Qui, we suggest to test the cytotoxicity of the compound to mammalian cells prior to incorporation, because impurities in the Uaa preparation could impair mammalian cell health and incorporation efficiency.
5.7. Widefield fluorescence microscopy
One day before transfection, 4.5×104 of HeLa cells were seeded into each compartment of 35 mm tissue culture dishes (four compartments, Greiner Bio-One) and incubated overnight to allow for attachment and recovery.
Each compartment was transfected with pcDNA3.1-EGFP-Tac (500 ng), or co-transfected with pMP-QuiRS-3×tRNA (500 ng) together with pcDNA3.1-EGFP66TAG-Tac (500 ng) using Lipofectamine 2000 (2.5 μL) following manufacturer’s instructions. Six hours post transfection, the transfection complex was removed and replaced with fresh DMEM containing 10% FBS. Qui (1 mM) was added to the cells containing pMP-QuiRS-3×tRNA and pcDNA3.1-EGFP66TAG-Tac.
After incubation for additional 48 h, the DMEM media was changed to citric acid-Na2HPO4 buffer (for pH values of 3.8, 4.6, 5.6) or phosphate buffer (for pH 7.2). abFP-expressing cells were then photoactivated using a LED light source (385 nm, 40 mW/cm2, Prizmatix) for 10 min.
After waiting for 5 min, cells were imaged using a Nikon inverted microscope equipped with an environmental chamber controlling temperature and CO2. Excitation: 480/40 nm; Emission: 525/50 nm.
5.8. Confocal fluorescence microscopy
One day before transfection, 4.5×104 of HeLa cells were seeded into each compartment of 35 mm tissue culture dishes (four compartments, Greiner Bio-One) and incubated overnight to allow for attachment and recovery.
Each compartment was transfected with pcDNA3.1-EGFP-Tac (500 ng), or co-transfected with pMP-QuiRS-3×tRNA (500 ng) together with pcDNA3.1-EGFP66TAG-Tac (500 ng) using Lipofectamine 2000 (2.5 μL) following manufacturer’s instructions. Six hours post transfection, the transfection complex was removed and replaced with fresh DMEM containing 10% FBS. Qui (1 mM) was added to the cells containing pMP-QuiRS-3×tRNA and pcDNA3.1-EGFP66TAG-Tac.
After incubation for additional 48 h, the culture media was replaced with DMEM (without phenol red, Gibco) containing 10% FBS and 30 mM HEPES (Gibco). The cells were then photoactivated at 385 nm using a LED light source (Prizmatix, 40 mW/cm2) for 10 min.
LysoTracker Red DND-99 (50 nM, Life Technologies) was added into each compartment and incubate for 10 min. Cells were then imaged for t = 0 min.
-
Agonist DADLE (10 μM, Sigma) was added to stimulate the DORs. Cells were imaged at 60 and 90 mins after agonist addition (t = 60 min, and t = 90 min).
Note: Imaging can be similarly performed at more time points.
A Yokagawa CSU22 spinning disk confocal microscope equipped with an environmental chamber controlling temperature and CO2 and a 100x magnification oil objective (NA 1.4) was used. For EGFP and abFP signals, cells were excited at 491 nm and emission detected at 525/60 nm; for LysoTracker signal, cells were excited at 561 nm and emission detected at 645/65 nm. Quantitative image analysis was carried out on raw images using ImageJ software (http://rsb.info.nih.gov/ij).
6. Summary
We describe here an abFP that fluoresces in acidic pH but becomes almost nonfluorescent at or above physiological pH, possessing a pH profile opposite to that of common FPs. The unique features of abFP make it suitable for real-time imaging of acidic organelles and vesicles in live cells. We showed that abFP fluoresced strongly inside acidic vesicles but remained virtually nonfluorescent at other subcellular structures, reducing the interfering background fluorescence and making the acidic vesicles distinct. In addition, abFP is photoactivatible, which may be useful for pulse-chase labeling of subpopulations of cells, organelles, or proteins and for super-resolution imaging of single molecules (Lippincott-Schwartz & Patterson, 2009). abFP is made possible by installing a positive charge onto the chromophore through replacing a chromophore-building tyrosine with the Uaa Qui via genetic code expansion. This strategy may be similarly applied to other FPs to enable them for acidic imaging. Moreover, we expect that Qui can be readily incorporated into abFP and other proteins of interest in mammalian cells (W. Wang et al., 2007), neurons (W. Wang et al., 2007), stem cells (B. Shen et al., 2011), C. elegans (Parrish et al., 2012), zebrafish (Chen et al., 2017), and mouse (Chen et al., 2017; Kang et al., 2013), in which the QuiRS/tRNAPyl pair should be compatible for use. Therefore, abFP has the potential to facilitate the study of molecules residing in acidic organelles and vesicles in different cells and organisms.
Acknowledgment
This work was supported by NIH RF1MH114079.
References
- Adie EJ, Francis MJ, Davies J, Smith L, Marenghi A, Hather C, … Game S (2003). CypHer 5: a generic approach for measuring the activation and trafficking of G protein-coupled receptors in live cells. Assay Drug Dev Technol, 1(2), 251–259. doi: 10.1089/15406580360545062 [DOI] [PubMed] [Google Scholar]
- Asanuma D, Takaoka Y, Namiki S, Takikawa K, Kamiya M, Nagano T, … Hirose K (2014). Acidic-pH-activatable fluorescence probes for visualizing exocytosis dynamics. Angew Chem Int Ed Engl, 53(24), 6085–6089. doi: 10.1002/anie.201402030 [DOI] [PubMed] [Google Scholar]
- Bonam SR, Wang FJ, & Muller S (2019). Lysosomes as a therapeutic target. Nature Reviews Drug Discovery, 18(12), 923–948. doi: 10.1038/s41573-019-0036-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, … Lohse MJ (2009). Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol, 7(8), e1000172. doi: 10.1371/journal.pbio.1000172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey JR, Grinstein S, & Orlowski J (2010). Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol, 11(1), 50–61. doi: 10.1038/nrm2820 [DOI] [PubMed] [Google Scholar]
- Chen Y, Ma J, Lu W, Tian M, Thauvin M, Yuan C, … Li D (2017). Heritable expansion of the genetic code in mouse and zebrafish. Cell Res, 27(2), 294–297. doi: 10.1038/cr.2016.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudakov DM, Matz MV, Lukyanov S, & Lukyanov KA (2010). Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev, 90(3), 1103–1163. doi: 10.1152/physrev.00038.2009 [DOI] [PubMed] [Google Scholar]
- Cossart P, & Helenius A (2014). Endocytosis of viruses and bacteria. Cold Spring Harb Perspect Biol, 6(8). doi: 10.1101/cshperspect.a016972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT, … Vilardaga JP (2009). Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol, 5(10), 734–742. doi: 10.1038/nchembio.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C, Kobayashi T, Wang N, Hoppmann C, Yang B, Irannejad R, & Wang L (2018). Genetically Encoding Quinoline Reverses Chromophore Charge and Enables Fluorescent Protein Brightening in Acidic Vesicles. J Am Chem Soc, 140(35), 11058–11066. doi: 10.1021/jacs.8b05814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppmann C, Lacey VK, Louie GV, Wei J, Noel JP, & Wang L (2014). Genetically encoding photoswitchable click amino acids in Escherichia coli and mammalian cells. Angew Chem Int Ed Engl, 53(15), 3932–3936. doi: 10.1002/anie.201400001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irannejad R, Pessino V, Mika D, Huang B, Wedegaertner PB, Conti M, & von Zastrow M (2017). Functional selectivity of GPCR-directed drug action through location bias. Nat Chem Biol, 13(7), 799–806. doi: 10.1038/nchembio.2389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, … von Zastrow M (2013). Conformational biosensors reveal GPCR signalling from endosomes. Nature, 495(7442), 534–538. doi: 10.1038/nature12000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JY, Kawaguchi D, Coin I, Xiang Z, O’Leary DD, Slesinger PA, & Wang L (2013). In vivo expression of a light-activatable potassium channel using unnatural amino acids. Neuron, 80(2), 358–370. doi: 10.1016/j.neuron.2013.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneen M, Farinas J, Li Y, & Verkman AS (1998). Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys J, 74(3), 1591–1599. doi: 10.1016/s0006-3495(98)77870-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey VK, Louie GV, Noel JP, & Wang L (2013). Expanding the library and substrate diversity of the pyrrolysyl-tRNA synthetase to incorporate unnatural amino acids containing conjugated rings. Chembiochem, 14(16), 2100–2105. doi: 10.1002/cbic.201300400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MZ, & Schnitzer MJ (2016). Genetically encoded indicators of neuronal activity. Nat Neurosci, 19(9), 1142–1153. doi: 10.1038/nn.4359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, & Patterson GH (2009). Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol, 19(11), 555–565. doi: 10.1016/j.tcb.2009.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield FR, & McGraw TE (2004). Endocytic recycling. Nat Rev Mol Cell Biol, 5(2), 121–132. doi: 10.1038/nrm1315 [DOI] [PubMed] [Google Scholar]
- Miesenbock G, De Angelis DA, & Rothman JE (1998). Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature, 394(6689), 192–195. doi: 10.1038/28190 [DOI] [PubMed] [Google Scholar]
- Mullershausen F, Zecri F, Cetin C, Billich A, Guerini D, & Seuwen K (2009). Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors. Nat Chem Biol, 5(6), 428–434. doi: 10.1038/nchembio.173 [DOI] [PubMed] [Google Scholar]
- Parrish AR, She X, Xiang Z, Coin I, Shen Z, Briggs SP, … Wang L (2012). Expanding the genetic code of Caenorhabditis elegans using bacterial aminoacyl-tRNA synthetase/tRNA pairs. ACS Chem Biol, 7(7), 1292–1302. doi: 10.1021/cb200542j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Knolker HJ, & Simons K (2010). Subcellular targeting strategies for drug design and delivery. Nat Rev Drug Discov, 9(1), 29–42. doi: 10.1038/nrd2897 [DOI] [PubMed] [Google Scholar]
- Saftig P, & Klumperman J (2009). Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol, 10(9), 623–635. doi: 10.1038/nrm2745 [DOI] [PubMed] [Google Scholar]
- Sakhrani NM, & Padh H (2013). Organelle targeting: third level of drug targeting. Drug Des Devel Ther, 7, 585–599. doi: 10.2147/DDDT.S45614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, & Ballabio A (2013). Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol, 14(5), 283–296. doi: 10.1038/nrm3565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Xiang Z, Miller B, Louie G, Wang W, Noel JP, … Wang L (2011). Genetically encoding unnatural amino acids in neural stem cells and optically reporting voltage-sensitive domain changes in differentiated neurons. Stem Cells, 29(8), 1231–1240. doi: 10.1002/stem.679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Rosendale M, Campbell RE, & Perrais D (2014). pHuji, a pH-sensitive red fluorescent protein for imaging of exo- and endocytosis. J Cell Biol, 207(3), 419–432. doi: 10.1083/jcb.201404107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinning A, & Hubner CA (2013). Minireview: pH and synaptic transmission. FEBS Lett, 587(13), 1923–1928. doi: 10.1016/j.febslet.2013.04.045 [DOI] [PubMed] [Google Scholar]
- Sorkin A, & von Zastrow M (2009). Endocytosis and signalling: intertwining molecular networks. Nature Reviews Molecular Cell Biology, 10(9), 609–622. doi: 10.1038/nrm2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto JK, Dellas N, Noel JP, & Wang L (2011). Stereochemical basis for engineered pyrrolysyl-tRNA synthetase and the efficient in vivo incorporation of structurally divergent non-native amino acids. ACS Chem Biol, 6(7), 733–743. doi: 10.1021/cb200057a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan PK, Waites C, Liu Y, Krantz DE, & Edwards RH (1998). A leucine-based motif mediates the endocytosis of vesicular monoamine and acetylcholine transporters. J Biol Chem, 273(28), 17351–17360. doi: 10.1074/jbc.273.28.17351 [DOI] [PubMed] [Google Scholar]
- Tsien RY (2009). Constructing and exploiting the fluorescent protein paintbox (Nobel Lecture). Angew Chem Int Ed Engl, 48(31), 5612–5626. doi: 10.1002/anie.200901916 [DOI] [PubMed] [Google Scholar]
- Tsvetanova NG, & von Zastrow M (2014). Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol, 10(12), 1061–1065. doi: 10.1038/nchembio.1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Thor JJ, Gensch T, Hellingwerf KJ, & Johnson LN (2002). Phototransformation of green fluorescent protein with UV and visible light leads to decarboxylation of glutamate 222. Nat Struct Biol, 9(1), 37–41. doi: 10.1038/nsb739 [DOI] [PubMed] [Google Scholar]
- Wang L (2017). Engineering the Genetic Code in Cells and Animals: Biological Considerations and Impacts. Acc Chem Res, 50(11), 2767–2775. doi: 10.1021/acs.accounts.7b00376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Brock A, Herberich B, & Schultz PG (2001). Expanding the genetic code of Escherichia coli. Science, 292(5516), 498–500. doi: 10.1126/science.1060077 [DOI] [PubMed] [Google Scholar]
- Wang L, & Schultz PG (2004). Expanding the genetic code. Angew Chem Int Ed Engl, 44(1), 34–66. doi: 10.1002/anie.200460627 [DOI] [PubMed] [Google Scholar]
- Wang L, Xie J, Deniz AA, & Schultz PG (2003). Unnatural amino acid mutagenesis of green fluorescent protein. J Org Chem, 68(1), 174–176. doi: 10.1021/jo026570u [DOI] [PubMed] [Google Scholar]
- Wang N, Yang B, Fu C, Zhu H, Zheng F, Kobayashi T, … Wang L (2018). Genetically Encoding Fluorosulfate-l-tyrosine To React with Lysine, Histidine, and Tyrosine via SuFEx in Proteins in Vivo. J Am Chem Soc, 140(15), 4995–4999. doi: 10.1021/jacs.8b01087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Takimoto JK, Louie GV, Baiga TJ, Noel JP, Lee KF, … Wang L (2007). Genetically encoding unnatural amino acids for cellular and neuronal studies. Nat Neurosci, 10(8), 1063–1072. doi: 10.1038/nn1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Lacey VK, Ren H, Xu J, Burban DJ, Jennings PA, & Wang L (2014). Proximity-enabled protein crosslinking through genetically encoding haloalkane unnatural amino acids. Angew Chem Int Ed Engl, 53(8), 2190–2193. doi: 10.1002/anie.201308794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitomirsky B, & Assaraf YG (2016). Lysosomes as mediators of drug resistance in cancer. Drug Resistance Updates, 24, 23–33. doi: 10.1016/j.drup.2015.11.004 [DOI] [PubMed] [Google Scholar]