

Abstract
Aims
There is a trend for more flexibility in timing of evidence generation in relation to marketing authorization, including the option to complete phase III trials after authorization or not at all. This paper investigated the relation between phase II and III clinical trial efficacy in oncology.
Methods
All oncology drugs approved by the European Medicines Agency (2007–2016) were included. Phase II and phase III trials were matched based on indication and treatment and patient characteristics. Reported objective response rates (ORR), median progression‐free survival (PFS) and median overall survival (OS) were analysed through weighted mixed‐effects regression with previous treatment, treatment regimen, blinding, randomization, marketing authorization type and cancer type as covariates.
Results
A total of 81 phase II‐III matches were identified including 252 trials. Mean (standard deviation) weighted difference (phase III minus II) was −4.2% (17.4) for ORR, 2.1 (6.7) months for PFS and −0.3 (5.1) months for OS, indicating very small average differences between phases. Differences varied substantially between individual indications: from −46.6% to 47.3% for ORR, from −5.3 to 35.9 months for PFS and from −13.3 to 10.8 months for OS. All covariates except blinding were associated with differences in effect sizes for at least 1 outcome.
Conclusions
The lack of marked average differences between phases may encourage decision‐makers to regard the quality of design and total body of evidence instead of differentiating between phases of clinical development. The large variability emphasizes that replication of study findings remains essential to confirm efficacy of oncology drugs and discern variables associated with demonstrated effects.
Keywords: clinical trials, conditional marketing authorization, efficacy, oncology, phase, regulation
What is already known about this subject
Drugs are increasingly being evaluated based on a single pivotal trial, sometimes with only phase II data.
Objective response rates reported in phase II trials are higher than in phase III trials in solid malignancies.
What this study adds
There are no marked average differences in effect estimates between phase II and phase III trials in oncology, but differences are subject to large variations between indications.
Differences in efficacy estimates measured in trials are associated with multiple covariates.
Replication of oncology trials remains essential to confirm efficacy of oncology drugs and discern variables associated with demonstrated effects.
1. INTRODUCTION
In drug regulation, historically, the standard requirement for approval of a marketing authorization (MA) was at least 2 confirmatory high‐quality phase III randomized controlled trials.1, 2 More recently, an increased flexibility towards these evidentiary standards has been exhibited by regulators. Between 2005–2012, 37% of indications approved by the Food and Drug Administration were based on a single pivotal trial.3 MA based on a single pivotal trial was granted in 45% of the European Medicines Agency (EMA) approvals during 2012–2016.4 These percentages are vastly different throughout therapeutic areas, with antineoplastic and immunomodulatory agents at the very top with 74% of approvals based on a single pivotal trial. Approval based on more limited evidence is especially apparent in special marketing authorization schemes such as accelerated approval in the USA and conditional MA in Europe, with 75% and 85% of MAs based on a single pivotal trial.4, 5 Additionally, a significant portion of drugs are approved based on uncontrolled trials or phase II trials only.4, 5, 6, 7, 8, 9
Even though the benefit/risk balance assessed by regulators may be positive, a concern voiced by other stakeholders is that deciding on appropriate use of drugs approved based on relatively more limited data will be impossible, either because relevant clinical endpoints are not available or because of the uncertainty considering the reported effect sizes.10, 12, 13 This concern is illustrated by recent studies that show that evidence for clinical effectiveness on survival or quality of life is often delayed or even lacking years after approval of conditional MA products.14, 15 Health technology assessment bodies and other stakeholders have repeatedly emphasized that determining the value of drugs is extremely complicated if evidence is too limited to reliably estimate long‐term clinically relevant effects.10, 12
Uncertainties in established effects may be exacerbated through the presence of a gap between clinical trials' efficacy and effectiveness in practice, which appears to be present for some therapeutic areas but not for others.16, 17, 18, 19, 20, 21 With increasing amounts of drugs being approved before phase III data are available, it is vital to know whether such a gap also exists between phases II and III. It is expected that in general, phase III trials will only be available if phase II results are positive. Even if there is no true underlying difference in effects between the phases, corrected for other factors, this selection would lead on average to a smaller effect in phase III due to regression to the mean.
Indeed, previous research for phase II and phase III trials within advanced solid malignancies found that of 43 phase III trials, 81% had lower objective response rates (ORR) than their preceding phase II trials for the new drug arm. The mean difference was 12.9%. No predictors for this difference were found.22 The lack of explanatory factors endorses regression to the mean as a relevant factor as this cannot be predicted. So far, no studies have evaluated whether differences between phase II and phase III trials also apply for survival.
The goal of this study is to investigate differences in estimated effect sizes between phase II and phase III clinical trials for oncology drugs. Outcome measures included are objective response rate, median progression‐free survival and median overall survival. Additionally, several factors are explored for associations with efficacy differences.
2. METHODS
2.1. Drug selection
We searched the publicly available EMA website to identify all the new active substances approved, refused or withdrawn by the EMA for the treatment of cancer (ATC codes L + V23) between 1 January 2007 and 31 December 2016. Drugs targeted at side effects of cancer treatment and cancer pain, supportive treatments, generics and biosimilars were excluded. For each product, all indications were assessed separately. For each indication, all efficacy trials listed in the EMA Committee for Medicinal Products for Human Use European public assessment report were retrieved. Additionally, phase III studies not included in the EMA evaluation (e.g. published afterwards) were located through searches in PubMed following a strategy that included the international nonproprietary name, approved indication and the keywords clinical trial and phase III. Only indications with both phase II and phase III clinical trials were selected for further analysis. Trials for indications with only phase II results were included only for descriptive analysis, to assess whether such trials had different characteristics from phase II trials within indications for which phase III trials were available.
2.2. Data collection
From the trials, quantitative data on objective response rate (ORR), median progression‐free survival (PFS) and/or median overall survival (OS) were collected. In trials with multiple arms, these efficacy outcomes were extracted with respect to the group of patients receiving the investigational drug for the indication of interest. If available, relative effect measures (e.g. hazard ratios) were also included if the comparator treatment was equal between phase II and III studies. If ORR outcomes were reported for the number of assessable patients, these results were recalculated according to the intent‐to‐treat principle. Furthermore, the following other features were collected: number of participants, randomization (yes/no), blinding (yes/no; including single‐, double‐ and triple‐blinding in yes), treatment regimen (dosing, regimen, combination treatments), previous treatment (yes/no), marketing authorization type (standard or conditional), cancer type (haematological or solid), and mean age of the patient population (or median if mean was not reported).
2.3. Data categorization and analysis
Each combination of phase II and III trials was reviewed, and compatibility was assessed based on an algorithm that defined whether the drug regimen and target population were identical. Only the studies involving identical patients (in terms of mean age [within 10%] and disease severity) and using the same agents following the same treatment schedule and administered with a dosing at least 85% identical were deemed valid for comparison. This algorithm was based on previous research.22 When an outcome (ORR, PFS, OS) was reported within 1 or more trials for 1 of the phases, but not for the other phase, this set was excluded from the analysis for this outcome but could still be included for any of the other 2 outcomes. If median survival (PFS or OS) was not reached, these trials were excluded for this specific outcome. The exclusion of studies in which median survival was not reached may lead to exclusion of entire sets but also to biased (lower) estimates when other studies within that phase for the same drug–indication combination do report median survival. How often this occurred was recorded.
A separate analysis was carried out for each of the 3 endpoints as dependent variables within a linear mixed‐effects model with clinical development phase (II/III) as a fixed effect. Each trial entered the model individually but was grouped according to its matched drug–indication combination by including a random effect for drug–indication combination. The outcomes in each phase were weighted according to the trial size (number of patients in the applicable arm) within each drug–indication phase II or phase III set of trials. Subsequently, each drug–indication combination had an equal weight in the analysis. Six covariates were tested for fixed effects: randomization, blinding, being a combination treatment, having received previous treatment, marketing authorization type, and cancer type. Starting with a full model, covariates and their interactions were estimated and tested. We assessed the homoscedasticity and normality of residuals graphically and through Kolmogorov–Smirnov tests. Data analyses were performed in R software version 3.5.2. using the package lme4 designed for (non)linear mixed‐effects models.
3. RESULTS
3.1. Inclusion of drug–indication combinations
In total, 81 phase II‐phase III drug–indication combinations (called sets) were identified for which the age of the patient population, dosing regimen and disease‐related characteristics were appraised as identical between development phases. There were 136 phase II and 116 phase III trials available for these sets. Often, within a set, multiple phase II studies corresponded with a single phase III study or vice versa. Within 9 sets, median survival was reported for at least 1 trial in each phase, but not for all trials. One trial did not reach median PFS and 8 did not reach median OS. Figure 1 shows the flowchart for the selection of phase II‐phase III matching sets for the 3 outcomes ORR, median PFS and median OS. Table 1 shows the characteristics of the included indications and trials.
Figure 1.
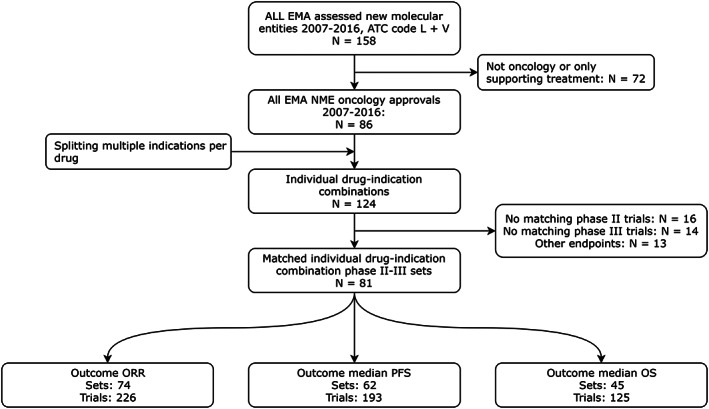
Flowchart for the inclusion of phase II‐phase III matched sets and trials. EMA: European Medicines Agency; NME: new molecular entity; ORR: objective response rate; PFS: progression‐free survival; OS: overall survival
Table 1.
Characteristics of included trials. ORR = objective response rate; PFS = progression‐free survival; OS = overall survival; CMA = conditional marketing authorization; SD = standard deviation
| Indications | All | ORR | Median PFS | Median OS | ||||
|---|---|---|---|---|---|---|---|---|
| 81 | 74 | 62 | 45 | |||||
| Haematological (%) | 20 (25) | 17 (23) | 12 (19) | 7 (16) | ||||
| CMA (%) | 22 (27) | 20 (27) | 16 (26) | 8 (18) | ||||
| Phase II | Phase III | Phase II | Phase III | Phase II | Phase III | Phase II | Phase III | |
| Trials | 136 | 116 | 121 | 105 | 108 | 85 | 66 | 59 |
| Randomized (%) | 44 (32) | 116 (100) | 39 (32) | 105 (100) | 34 (31) | 85 (100) | 17 (26) | 59 (100) |
| Blinded (%) | 10 (7) | 43 (37) | 8 (7) | 37 (35) | 5 (5) | 28 (33) | 4 (6) | 17 (29) |
| Combination (%) | 54 (40) | 53 (46) | 48 (40) | 49 (47) | 41 (38) | 35 (41) | 19 (29) | 18 (31) |
| Previous treatment (%) | 93 (68) | 78 (67) | 82 (68) | 70 (67) | 76 (70) | 59 (69) | 45 (68) | 40 (68) |
| Mean participants (SD) | 85 (101) | 319 (199) | 86 (105) | 314 (197) | 88 (112) | 319 (187) | 88 (134) | 335 (207) |
3.2. Mean effect sizes in phase II and phase III
Mean (standard deviation [SD]) ORR, weighted for the size of the trials, was 41.2% (23.0) for phase II and 37.0% (24.8) for phase III. For median PFS, the weighted means (SD) were 8.2 (4.2) and 10.3 (8.6) months for phase II and phase III respectively, whereas these were 14.7 (6.3) and 14.4 (6.7) months for median OS. Unweighted mean (SD) ORR was 41.2% (22.9) for phase II and 37.1% (24.4) for phase III. Unweighted median PFS and OS for phase II and phase III were 8.4 (4.6), 10.3 (8.5) for PFS and 14.8 (6.3) and 14.4 (6.7) for OS, indicating almost no effect of weighting. Scatterplots of outcomes in phase II vs phase III for all drug–indication combinations are provided in Figure 2.
Figure 2.
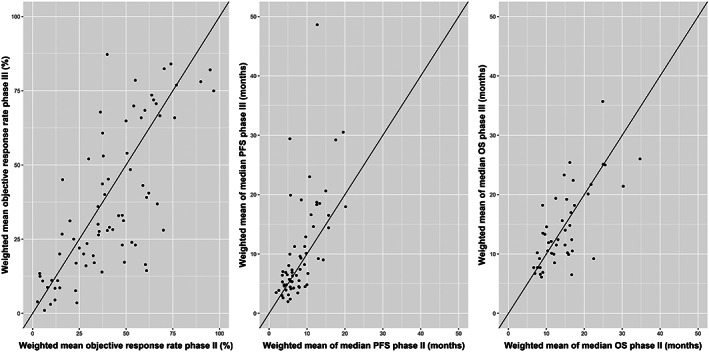
Scatterplots of phase II vs phase III results for objective response rate (ORR), median progression‐free survival (PFS) and median overall survival (OS). Line: Y = x
3.3. Mean differences between effect sizes in phase II and phase III
Unadjusted mean differences in results between phases were small. Mean (SD) weighted difference (phase III minus phase II) for ORR was −4.2% (17.4), indicating a slightly higher weighted average objective response rate in phase II trials. For median PFS, mean weighted difference was 2.1 (6.7) months, whereas the mean difference was even smaller for median OS: −0.25 (5.1) months. Comparative effects were available for only 8 drug–indication combinations (6 of them reporting a hazard ratio for median PFS and 5 reporting a hazard ratio for median OS), rendering it impossible to perform statistical analyses.
3.4. Differences in effect sizes between phase II and phase III in individual indications
The large standard deviation for mean differences indicates that the differences between effect sizes in phase II and in phase III in individual indications varied substantially. For individual drug–indication sets the ORR varied between phase II and phase III from −46.6% to 47.3%. For PFS, this range was −5.3 to 35.9 months and for OS the range was −13.3 to 10.8 months. Differences in effects between phases in individual drug–indication sets can also be expressed as a percentage of the size of the measured effect in phase II (e.g. an ORR of 30% in phase III and 20% in phase II means a 50% higher ORR in phase III relative to phase II). The mean of these percentages was 43.9% for ORR and 50.8% and 24.9% for median PFS and OS, respectively.
3.5. Factors associated with effect sizes
In the multivariable linear mixed effects model, there was no association between phase and effect estimates for any of the efficacy measures (see Table 2). All 6 covariates except blinding were associated with the effect size in at least 1 of the analyses of efficacy outcomes, but only previous treatment had a clear association with all 3 efficacy outcomes. Interaction effects were also evaluated. The only consistent interaction found was between haematological indication and phase for ORR and PFS (both P < .01). This interaction indicated that the mean difference for haematological vs nonhaematological products was 22.4% in phase II and 33.5% in phase III for ORR and 1.4 and 7.5 months for median PFS. The interaction was not observed for median OS.
Table 2.
Summarizing multivariable regression analyses for the objective response rate (ORR), median progression‐free survival (PFS) and median overall survival (OS). MA = marketing authorization. * P < .05
| Reference | Coef. | 95% confidence interval | ||
|---|---|---|---|---|
| Objective response rate (%) | ||||
| Intercept | 37.8% | 30.6% | 44.9% | |
| Phase III | Phase II | −5.0% | −10.2% | 0.2% |
| Randomization | Nonrandomized | 1.7% | −4.4% | 7.9% |
| Blinding | No blinding | 1.3% | −4.5% | 7.1% |
| Conditional marketing authorization* | Standard MA | 9.6% | 0.8% | 18.4% |
| Haematological indication* | Solid tumours | 27.7% | 18.2% | 37.1% |
| Previous treatment* | No previous treatment | −17.5% | −23.0% | −11.9% |
| Combination therapy* | Monotherapy | 10.7% | 4.5% | 17.0% |
| Median progression‐free survival (mo) | ||||
| Intercept | 7.1 | 4.8 | 9.6 | |
| Phase III | Phase II | 1.3 | −0.6 | 3.3 |
| Randomization | Nonrandomized | 0.2 | −2.0 | 2.2 |
| Blinding* | No blinding | 2.7 | 0.5 | 4.8 |
| Conditional marketing authorization* | Standard MA | 5.4 | 2.6 | 8.4 |
| Haematological indication* | Solid tumours | 4.3 | 0.7 | 7.6 |
| Previous treatment* | No previous treatment | −3.2 | −5.1 | −1.4 |
| Combination therapy | Monotherapy | 2.1 | 0.0 | 4.1 |
| Median overall survival (mo) | ||||
| Intercept | 16.5 | 13.2 | 19.5 | |
| Phase III | Phase II | −1.6 | −3.7 | 0.7 |
| Randomization | Nonrandomized | 1.7 | −0.6 | 4.4 |
| Blinding | No blinding | 0.2 | −2.2 | 2.3 |
| Conditional marketing authorization | Standard MA | 5.0 | 0.2 | 9.7 |
| Haematological indication | Solid tumours | −4.0 | −9.4 | 1.5 |
| Previous treatment* | No previous treatment | −3.7 | −6.2 | −1.2 |
| Combination therapy | Monotherapy | 0.2 | −2.4 | 2.9 |
3.6. Drug–indication combinations with only phase II trials
There were 14 drug–indication combinations that only had phase II results, together including 25 trials. The mean ORR (n = 24) for these trials was 47.1% (range 10.5–90.5%), the average median PFS (n = 18) was 9.2 months (range 1.2–24.7) and the average median OS (n = 9) was 21.9 months (range 3.0–45.8). The average difference in measured ORR between trials for the drugs that had multiple phase II trials (n = 8), was 14.1% (SD: 7.3%), unweighted and uncorrected for covariates. These findings indicate that results between phase II trials are similarly variable as results between phase II and phase III trials.
4. DISCUSSION
Our results show that there are no consistent differences in objective response rate, median PFS or median OS between development phases for oncology drugs, when trial results are available for both phases. Although the mean difference in effect sizes between phases was small, there were large variations in differences between phase II and III through all individual drug–indication combinations, which was only partly explained by covariates describing the type of drug and design of the studies.
The associations of covariates with effect sizes were mostly as could be expected. Larger effect sizes were noted in untreated populations, combination regimens, haematological indications and conditionally approved products. The higher effect sizes within conditionally approved products somewhat mitigate the uncertainty associated with more preliminary evidence related to the benefit/risk balance. For OS, not all these effects were significant and the effect for haematological drugs reversed, which is probably explained by a combination of the small sample size and the fact that correlation between surrogate endpoints and OS is not consistent throughout oncology indications.24 Based on this study, we cannot conclude or dismiss an association between effect size and blinding or randomization.
4.1. Implications of the study findings
Historically, in evaluations of evidence by stakeholders such as regulators, health technology assessment bodies and clinical guideline developers, more emphasis is placed on randomized controlled phase III trials rather than on phase II trials.1, 2 However, approvals based on only nonrandomized, uncontrolled phase II trials are becoming more common.4, 5, 6, 7 Whether the effects measured in such trials translate to clinically relevant effects in practice remains unclear.
The lack of a mean difference between phases in our dataset indicates that the lack of a phase III study in itself may not necessarily mean that the effects from phase II are problematically biased. Our results also showed that trials for drugs approved based on solely phase II results showed on average greater effect sizes than phase II trials for drugs for which phase III trials were available. This suggests that phase III trials may not always be necessary if adequate phase II trials demonstrate convincing effects. However, although mean differences were small, there were large variations in differences between phases in the dataset. The gap between effect sizes in phase II and in phase III can represent a big proportion of the measured effect size in phase II. A similarly large difference was found between trials for drugs with multiple phase II trials without a phase III trial. These similarities indicate that a large effect size measured in a phase II trial is no guarantee for an equally impressive effect in a replication phase II trial or a phase III trial.
Many factors are associated with positive development pathways such as the type and size of the effect measured, the number of institutions included in phase II trials, trial characteristic, the inclusion of biomarker‐driven objectives, funding sources, and interactions with regulators.25, 26, 27, 28, 29, 30, 31, 32, 33 Our study confirms that factors related to the indication, the study population and the quality of the trial can be associated with efficacy differences between studies, irrespective of phase. More importantly, our study highlights that significant unexplained variation remains despite included covariates. Thus, when oncology approvals are based on a single trial, there is a risk that the estimated efficacy may be greatly reduced or even absent in practice. Such unpredictable variability in effect sizes makes it very difficult for stakeholders to determine the appropriate treatment line and reimbursement level. Replicating studies with slightly modified population and trial characteristics can help to confirm efficacy and to discern variables associated with demonstrated effects. This is at odds with the recent trend of increased flexibility of regulators in accepting single phase II or single phase III trials for marketing authorization.4 Our finding that multiple other variables are associated with differences in effect sizes between trials suggests that we could have introduced additional variables to the regression model. For example, the inclusion of biomarker‐driven objectives has been shown to be associated with a higher probability of achieving development milestones.33 Differences in effect sizes due to the covariates we included had no interaction with the differences between development phases except for having a haematological indication. We expect that the inclusion of more covariates would not lead to more interactions with phase but it might have provided us with additional information regarding the reasons for the big variations in effect sizes. Finding all covariates that may be associated with differences in effect sizes in similar populations was not an objective of this study. This emphasizes that replication of trials remains essential. In conclusion, based on our results, replication of oncology trials and subsequent consideration of the quality of design and total body of available evidence should be preferred above demanding phase III trials in all situations.
4.2. Previous research
As far as we know, no previous studies have quantitatively assessed the efficacy gap between development phases for survival outcomes. One previous study has highlighted the efficacy gap in objective response rate between phase II and phase III studies for solid malignancies, finding a 12.9% higher ORR reported in phase II as opposed to phase III studies.22 Our results show a smaller difference (8.5% when restricted to solid malignancies). Because the matching mechanisms were comparable, this difference is probably explained by their shorter inclusion period, their inclusion of products that were never submitted to regulators and their lack of correction for covariates such as previous treatment and treatment regimen.
4.3. Limitations
We applied a selection mechanism to phase II–phase III sets. This was necessary to produce valid comparisons but also means that we excluded many trials for which no match existed in the corresponding phase. Additionally, it was impossible to match trials based on all known patient characteristics (for example genetic scores), which may explain some of the observed variation not attributable to the included covariates. The risk of such effects strengthens our suggestion that replication of clinical trials in slightly different populations can provide increased confidence in effect sizes. Although the variability between phase II trials for drugs that only had phase II trials seemed similar to the variability between phase II and phase III in our cohort, the higher effect sizes for drugs that do not have phase III results suggests important differences between the 2 types of drugs. Our conclusions should be interpreted accordingly. Selection bias is also introduced through the exclusion of trials for which median survival was not reached. However, the effect on PFS results is minor, since it affects only 1 set. The effect on OS is harder to estimate, as median survival not being reached can have multiple reasons, including a shorter trial duration or increased efficacy. Overall, we find consistent results throughout all 3 efficacy outcomes which strengthens our confidence in the conclusions. Nonrandomized studies are never blinded, which means that there may be confounding between the 2 covariates. However, we included both blinding as well as randomization in the multivariable model to mitigate this issue. Regulatory evaluations are often based on comparative efficacy (to active control or placebo). Thus, another approach could be to look at differences in comparative effect sizes instead of differences in absolute effect sizes between phase II and phase III. We have found that it is very rare for phase II and phase III trials to include the same comparator within the same matched population (only 8 drug–indication combinations in our dataset). This indicates that, in reality, comparisons between results in phase II and results in phase III must also be done on absolute rather than comparative effects, which asserts our approach and conclusions.
4.4. Conclusions
Overall, effect estimates in this dataset of trials measuring objective response rate and median progression free and/or overall survival did not show marked average differences between phase II and phase III trials for oncology drugs. These results may encourage decision‐makers such as clinicians, regulators or health technology assessment bodies to regard the quality of design and total body of evidence, including patient population and trial characteristics, instead of differentiating between phases of clinical development. The large variability in effect size differences throughout individual indications emphasizes that proper replication of study findings remains essential to confirm efficacy of oncology drugs and discern variables associated with demonstrated effects.
ACKNOWLEDGEMENTS
R.A.V.'s work is partly financed by the governmental Dutch National Health Care Institute. None of the authors have been paid to write this article.
COMPETING INTERESTS
R.A.V. reports funding from the Governmental Dutch National Health Care Institute for the submitted work. All other authors declare no support from any organization for the submitted work; A.M.H. reports unrestricted grants from GlaxoSmithKline, outside the submitted work. H.G.M.L. reports that he is past‐chairman of the Dutch Medicines Evaluation Board and past‐member of the EMA Committee for Medicinal Products for Human Use. He also reports that he is a member of the Lygature Leadership Team. The other authors declare no other relationships or activities that could appear to have influenced the submitted work.
CONTRIBUTORS
R.A.V., H.G.L. and A.K.M.‐T. designed the research. R.A.V., A.T.M.M. and S.V.B. performed the data extraction and preparation. R.A.V., A.T.M.M., S.V.B., K.C.B.R. and A.K.M.‐T. analysed the data. R.A.V., A.T.M.M., A.M.H., W.G.G., K.C.B.R., H.G.L. and A.K.M.‐T. interpreted the data. All authors contributed to writing the manuscript. All authors approved the final manuscript.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in the European Medicines Agency document repository, at https://www.ema.europa.eu/en/medicines, and from published clinical trials for oncology indications available in the public domain through PubMed: https://www.ncbi.nlm.nih.gov/pubmed/.
Vreman RA, Belitser SV, Mota ATM, et al. Efficacy gap between phase II and subsequent phase III studies in oncology. Br J Clin Pharmacol. 2020;86:1306–1313. 10.1111/bcp.14237
This study did not involve human subjects.
The views expressed in this article are the personal views of the authors and must not be understood or quoted as being made on behalf of or reflecting the position of the Dutch National Health Care Institute.
REFERENCES
- 1. International Council for Harmonisation (ICH) . E8 General Considerations for Clinical Trials. Switserland. 1997. [Internet]. [cited 2018 Sep 7]. Available from: http://www.ich.org/products/guidelines/efficacy/efficacy-single/article/general-considerations-for-clinical-trials.html
- 2. European Medicines Agency . CPMP. E8 (CPMP/ICH/291/95) General Considerations for Clinical Trials. London. 1997. [Internet]. [cited 2018. Sep 7]. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_001255.jsp&mid=WC0b01ac0580032ec4
- 3. Downing NS, Aminawung JA, Shah ND, Krumholz HM, Ross JS. Clinical trial evidence supporting FDA approval of novel therapeutic agents, 2005‐2012. JAMA. 2014;311(4):368‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morant AV, Vestergaard HT. European marketing authorizations granted based on a single pivotal clinical trial: the rule or the exception? Clinical Pharmacology & Therapeutics. 2018;104(1):169‐177. [DOI] [PubMed] [Google Scholar]
- 5. Naci H, Smalley KR, Kesselheim AS. Characteristics of preapproval and Postapproval studies for drugs granted accelerated approval by the US Food and Drug Administration. JAMA. 2017;318(7):626‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vreman RA, Bouvy JC, Bloem LT, et al. Weighing of evidence by health technology assessment bodies: retrospective study of reimbursement recommendations for conditionally approved drugs. Clin Pharmacol Ther. 2019;105(3):684‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pregelj L, Hwang TJ, Hine DC, et al. Precision medicines have faster approvals based on fewer and smaller trials than other medicines. Health Aff. 2018;37(5):724‐731. [DOI] [PubMed] [Google Scholar]
- 8. Darrow JJ, Avorn J, Kesselheim AS. New FDA breakthrough‐drug category — implications for patients. N Engl J Med. 2014;370(13):1252‐1258. [DOI] [PubMed] [Google Scholar]
- 9. Luijn JCFV, Gribnau FWJ, Leufkens HGM. Availability of comparative trials for the assessment of new medicines in the European Union at the moment of market authorization. Br J Clin Pharmacol. 2007;63(2):159‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ermisch M, Bucsics A, Vella Bonanno P, et al. Payers' views of the changes arising through the possible adoption of adaptive pathways. Front Pharmacol. 2016;7 10.3389/fphar.2016.00305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vella Bonanno P, Ermisch M, Godman B, et al. Adaptive pathways: possible next steps for payers in preparation for their potential implementation. Front Pharmacol. 2017;8 10.3389/fphar.2017.00497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gellad WF, Kesselheim AS. Accelerated approval and expensive drugs — a challenging combination. N Engl J Med. 2017;376:2001‐2004. [DOI] [PubMed] [Google Scholar]
- 13. Darrow JJ, Avorn J, Kesselheim AS. The FDA breakthrough‐drug designation — four years of experience. N Engl J Med. 2018;378(15):1444‐1453. [DOI] [PubMed] [Google Scholar]
- 14. Davis C, Naci H, Gurpinar E, Poplavska E, Pinto A, Aggarwal A. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European medicines agency: retrospective cohort study of drug approvals 2009‐13. BMJ. 2017;359:j4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bloem LT, Mantel‐Teeuwisse AK, Leufkens HGM, De Bruin ML, Klungel OH, Hoekman J. Postauthorization changes to specific obligations of conditionally authorized medicines in the European Union: a retrospective cohort study. Clin Pharmacol Ther. 2019;105(2):426‐435. [DOI] [PubMed] [Google Scholar]
- 16. Eichler H‐G, Abadie E, Breckenridge A, et al. Bridging the efficacy–effectiveness gap: a regulator's perspective on addressing variability of drug response. Nat Rev Drug Discov. 2011;10(7):495‐506. [DOI] [PubMed] [Google Scholar]
- 17. Laupacis A, Mamdani M. Observational studies of treatment effectiveness: some cautions. Ann Intern Med. 2004;140(11):923‐924. [DOI] [PubMed] [Google Scholar]
- 18. Anglemyer A, Horvath HT, Bero L. Healthcare outcomes assessed with observational study designs compared with those assessed in randomized trials. Cochrane Database Syst Rev. 2014;(4):MR000034 10.1002/14651858.MR000034.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ankarfeldt MZ, Adalsteinsson E, Groenwold RH, Ali MS, Klungel OH. A systematic literature review on the efficacy–effectiveness gap: comparison of randomized controlled trials and observational studies of glucose‐lowering drugs. Clin Epidemiol. 2017;9:41‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martin K, Bégaud B, Latry P, Miremont‐Salamé G, Fourrier A, Moore N. Differences between clinical trials and postmarketing use. Br J Clin Pharmacol. 2004;57(1):86‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wallerstedt SM, Hoffmann M. Evaluating beneficial drug effects in a non‐interventional setting: a review of effectiveness studies based on Swedish prescribed drug register data. Br J Clin Pharmacol. 2016;83:1309‐1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zia MI, Siu LL, Pond GR, Chen EX. Comparison of outcomes of phase II studies and subsequent randomized control studies using identical chemotherapeutic regimens. JCO. 2005;23:6982‐6991. [DOI] [PubMed] [Google Scholar]
- 23. WHO Collaborating Centre for Drug Statistics Methodology , Guidelines for ATC classification and DDD assignment 2013. Oslo, 2012.
- 24. Davis S, Tappenden P, Cantrell A. A Review of Studies Examining the Relationship between Progression‐Free Survival and Overall Survival in Advanced or Metastatic Cancer. Sheffield: School of Health and Related Research, University of Sheffield; 2012. [PubMed] [Google Scholar]
- 25. Balk EM, Bonis PAL, Moskowitz H, et al. Correlation of quality measures with estimates of treatment effect in meta‐analyses of randomized controlled trials. JAMA. 2002;287(22):2973‐2982. [DOI] [PubMed] [Google Scholar]
- 26. El‐Maraghi RH, Eisenhauer EA. Review of phase II trial designs used in studies of molecular targeted agents: outcomes and predictors of success in phase III. JCO. 2008;26(8):1346‐1354. [DOI] [PubMed] [Google Scholar]
- 27. Putzeist M, Mantel‐Teeuwisse AK, Aronsson B, et al. Factors influencing non‐approval of new drugs in Europe. Nat Rev Drug Discov. 2012;11(12):903‐904. [DOI] [PubMed] [Google Scholar]
- 28. Liberti L, Stolk P, McAuslane JN, Schellens J, Breckenridge AM, Leufkens H. Observations on three endpoint properties and their relationship to regulatory outcomes of European oncology marketing applications. Oncologist. 2015;20(6):683‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chan JK, Ueda SM, Sugiyama VE, et al. Analysis of phase II studies on targeted agents and subsequent phase III trials: what are the predictors for success? JCO. 2008;26(9):1511‐1518. [DOI] [PubMed] [Google Scholar]
- 30. Wood L, Egger M, Gluud LL, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ. 2008;336(7644):601‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. US Food and Drug Administration . 22 Case Studies Where Phase 2 and Phase 3 Trials Had Divergent Results. FDA 2017.
- 32. Dumbrava EI, Meric‐Bernstam F, Yap TA. Challenges with biomarkers in cancer drug discovery and development. Expert Opin Drug Discovery. 2018;13:685‐690. [DOI] [PubMed] [Google Scholar]
- 33. Paller CJ, Huang EP, Luechtefeld T, et al. Factors Affecting Combination Trial Success (FACTS): Investigator Survey Results on Early‐Phase Combination Trials. Front Med [Internet]. 2019 [cited 2019 Nov 18];6. Available from: https://www.frontiersin.org/articles/10.3389/fmed.2019.00122/full [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available in the European Medicines Agency document repository, at https://www.ema.europa.eu/en/medicines, and from published clinical trials for oncology indications available in the public domain through PubMed: https://www.ncbi.nlm.nih.gov/pubmed/.