

Abstract
Aims
To characterize the pharmacokinetics (PK) of moxetumomab pasudotox, an anti‐CD22 recombinant immunotoxin, in adults with relapsed or refractory hairy cell leukaemia, we examined data from a phase 1 study (Study 1001; n = 49) and from the pivotal clinical study (Study 1053; n = 74).
Methods
Data from both studies were pooled (n = 123) to develop a population PK model. Covariates included demographics, disease state, liver and kidney function, prior treatment, and antidrug antibodies (ADAs). Exposure–response and exposure–safety were analysed separately by study. A 1‐compartment model with linear elimination from the central compartment and 2 clearance (CL) rates was developed.
Results
Moxetumomab pasudotox was cleared more rapidly after cycle 1, day 1 (CL1 = 24.7 L/h) than subsequently (CL2 = 3.76 L/h), with high interindividual variability (116 and 109%, respectively). In Study 1053, patients with ADA titres >10 240 showed ~4‐fold increase in CL. Higher exposures (≥median) were related to higher response rates, capillary leak syndrome and increased creatinine (Study 1053 only), or grade ≥3 adverse events (Study 1001 only). Clinical benefits were still observed in patients with lower exposure or high ADA titres.
Conclusion
Despite a high incidence of immunogenicity with increased clearance, moxetumomab pasudotox demonstrated efficacy in hairy cell leukaemia.
Keywords: cancer, modelling and simulation, pharmacokinetic–pharmacodynamic, pharmacokinetics
What is already known about this subject
Moxetumomab pasudotox, an anti‐CD22 immunotoxin, showed encouraging clinical activity in patients with relapsed or refractory hairy cell leukaemia in 2 prior clinical studies.
We developed a population pharmacokinetic model to characterize the pharmacokinetics of moxetumomab pasudotox in adults with relapsed or refractory hairy cell leukaemia based on the 2 prior clinical studies.
What this study adds
Higher response rates, higher capillary leak syndrome rates and increased creatinine occurred with increased PK exposure.
Patients with higher baseline CD19+ B‐cell counts had elevated moxetumomab pasudotox clearance and numerically lower response rates.
The relationship between moxetumomab pasudotox exposure and safety/efficacy may help optimize dosage and guide future study design.
1. INTRODUCTION
Hairy cell leukaemia (HCL) is a rare haematological malignancy characterized by pancytopenia and increased susceptibility to infection1 that was first described as a distinctive malignancy in 1958.2 Although purine nucleoside analogue therapy (cladribine or pentostatin) provides 73– to 98% complete response (CR) rates in the first‐line treatment of patients with HCL,3, 4 relapse is common. Approximately 50% of patients reportedly relapse within 16 years,5 with 1 study of patients aged ≤40 years at first diagnosis with HCL reporting a 51% relapse rate among complete responders at a median of 54 months after initially responding to cladribine.3 Subsequent treatments following relapse are associated with lower CR rates, shorter periods of remission4, 5 and increased treatment‐related toxicities, including immunosuppression.6 Thus, improved treatment options for patients with relapsed or refractory HCL are needed.
Immunotoxins, which consist of an antibody domain fused to a bacterial toxin payload for efficient killing of targeted cells, have been investigated7, 8, 9 and shown to induce response in patients with HCL.10, 11, 12 Moxetumomab pasudotox (formerly HA22 and CAT‐8015; AstraZeneca Pharmaceuticals, LP) is a recombinant immunotoxin targeting CD22 that consists of an immunoglobulin light chain variable domain and a heavy chain variable domain fused to a truncated form of Pseudomonas exotoxin A.13 It was approved by the US Food and Drug Administration in September 2018 for adults with relapsed or refractory HCL who received at least 2 prior systemic therapies, including treatment with a purine nucleoside analogue.14 In a phase 1 study (Study 1001 [CAT‐8015‐1001]) of patients with relapsed or refractory HCL (median of 2 prior treatments), those in the highest dosing group (50 μg/kg every other day × 3, for 1–16 cycles) achieved 64% CR and 88% objective response (OR) rates per investigator assessment. Bone marrow aspirate flow cytometry conducted at local study sites to assess minimal residual disease (MRD) showed that 11/32 (34%) evaluable complete responders had MRD negativity.15 A pivotal study of moxetumomab pasudotox (Study 1053 [CAT‐8015‐1053]) was conducted in patients with relapsed or refractory HCL who had at least 2 prior systemic therapies (at least 1 of which included a purine analogue). The primary end point, durable CR, was defined as CR with a >180‐day haematological remission (resolution of cytopenias), as assessed by blinded independent review and was achieved in 30% of the patients.15 The CR rate was 41% (OR, 75%), with 85% of complete responders achieving MRD negativity on bone marrow biopsy immunohistochemistry. The incidence of treatment‐related serious adverse events (AEs) was low (haemolytic uraemic syndrome [HUS], 7.5%; capillary leak syndrome [CLS], 5%), reversible and manageable.
To characterize the pharmacokinetics (PK) of moxetumomab pasudotox in adults with relapsed or refractory HCL, we examined data from the phase 1 study and the pivotal clinical study and developed a population PK model. The influence of covariates on PK, including demographics and immunogenicity, among others, was examined. In addition, potential relationships between moxetumomab pasudotox exposure and efficacy end points, as well as the occurrence of AEs of special interest, were analysed for each of the studies.
Three processes (Processes 1, 2, and 3) were developed for the manufacture of moxetumomab pasudotox, and clinical trial materials from all 3 processes were administered to patients with HCL (see Methods). Because the formulation of moxetumomab pasudotox used in Study 1053 (which was manufactured by Process 3) had higher bioactivity than the compound used in Study 1001 (manufactured by Process 1 or Process 2), the influence of process material differences on moxetumomab pasudotox PK was also evaluated.
2. METHODS
2.1. Studies
Data from 2 studies evaluating moxetumomab pasudotox in patients with HCL were analysed. Study 1001 (NCT00586924) was a phase 1, multicentre, dose‐escalation trial of moxetumomab pasudotox in adults with HCL who had received at least 2 prior systemic therapies, including at least 2 purine analogues, unless the response to the first course lasted less than 2 years.15, 16 Study 1053 (NCT01829711) was a pivotal, international, single‐arm study of moxetumomab pasudotox in adults with HCL who had received at least 2 prior therapies, including 1 course of a purine nucleoside analogue or 1 course of a BRAF inhibitor after a single purine nucleoside analogue course.17 Both studies were conducted in accordance with the principles of the Declaration of Helsinki, the International Conference on Harmonisation Good Clinical Practice guidelines, and local regulatory requirements. The study protocols were approved by the institutional review board or independent ethics committee at each study site. All patients provided written informed consent prior to study participation.
Details regarding the design of both studies have been published.15, 16, 17 Briefly, in Study 1001, moxetumomab pasudotox was administered via 30‐minute intravenous infusion at doses of 5, 10, 20, 30, 40 or 50 μg/kg on days 1, 3 and 5 of each 28‐day cycle.16 In Study 1053, moxetumomab pasudotox was administered intravenously over 30 minutes at a dose of 40 μg/kg on days 1, 3 and 5 of each 28‐day cycle.17
Process 2 (lyophilized formulation) was developed due to a limited supply of the investigational product manufactured under Process 1 (frozen formulation), and to allow for a more scalable product. Because Process 2 was not optimal for routine commercial production due to low yield and manufacturing variability, Process 3 was developed as the commercial process. Process 3 (lyophilized formulation) was designed to provide significant improvement in process control and consistency, manufacturing facility fit, and product quality. As a result, Process 3 material has higher bioactivity relative to Process 2 material.
Due to the higher bioactivity and reduction in deamidation of Process 3 material compared with Process 1 or Process 2 material, a dose adjustment was implemented to ensure that a similar amount of biologically equipotent active product would be delivered to patients receiving Process 3 material compared with patients receiving Process 1 or Process 2 material. The recommended dose from the phase 1 study (Study 1001) of 50 μg/kg of Process 2 material was determined to be equivalent to the phase 3 dose (Study 1053) of 40 μg/kg of Process 3 material with respect to bioactivity. Because of the differences in the bioactivity of the Process 1, Process 2 and Process 3 materials, exposure–response and exposure–safety analyses were conducted separately for Study 1001 and Study 1053.
Patients with at least 1 evaluable PK sample after the first dose were included in both the population PK model and the exposure–response analysis. The PK sampling schedules for each study are shown in Table S1.
2.2. Bioanalytical methods
Moxetumomab pasudotox plasma concentrations in both studies were determined using a validated enzyme‐linked immunosorbent assay developed by MedImmune. A bioassay developed by the National Cancer Institute (NCI) that measured moxetumomab pasudotox in plasma by assaying cytotoxicity of diluted plasma toward CD22+ cells was also used in Study 1001. Twenty‐one patients had PK samples analysed using both MedImmune and NCI PK assays. For these 21 patients, only the MedImmune PK assay measurements were retained in the analysis data set. The lower limit of quantification of the MedImmune and NCI assays were 40 and 16 ng/mL, respectively.
In Study 1053, results from 3 assays (screening, neutralizing antibody and specificity) and titres were reported. Immunogenicity testing utilized a tiered strategy to screen and characterize antidrug antibody (ADA) responses. Samples were first tested with an ADA screening assay. Samples determined to be ADA positive by the screening assay were subsequently evaluated in a cell‐based neutralization assay. For neutralizing antibody (Nab)‐positive samples, ADA titres were also reported. In Study 1001, immunogenicity was tested by MedImmune and the NCI by an ADA screening assay and neutralizing antibody assay, respectively; all samples were tested using the screening and/or neutralizing antibody enzyme‐linked immunosorbent assay, regardless of positivity.
2.3. PK model
A population PK model was developed using the pooled PK data from Study 1001 and Study 1053. A 1‐compartment model, with linear elimination from the central compartment and 2 clearance (CL) rates, was developed (Figure 1) and is described by the following equation:
Figure 1.
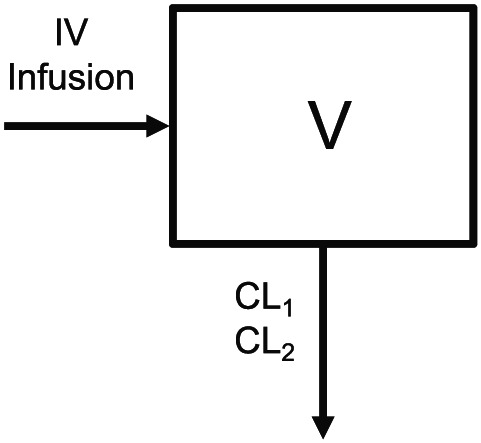
Schematic of moxetumomab pasudotox population pharmacokinetic model structure. V indicates the central volume of distribution, CL1 is clearance from the central compartment after first dose of moxetumomab pasudotox (cycle 1, day 1), and CL2 is clearance from the central compartment for any subsequent doses. IV, intravenous
C is the moxetumomab pasudotox concentrations in the central compartment. CL1 represents the linear clearance from the central compartment after the first dose of moxetumomab pasudotox in cycle 1. CL2 represents the linear clearance from the central compartment for all subsequent doses. V is the volume of distribution of the central compartment. The 2‐clearance model was chosen because moxetumomab pasudotox exhibited dose‐proportional PK and was rapidly cleared from systemic circulation with faster clearance of the initial dose than of subsequent doses (see Supplementary information 1 for nonlinear mixed‐effects modelling (NONMEM) code).
The population PK analysis evaluated the effect of demographics (age, sex, body weight and race), disease state (spleen size and percentage of bone marrow infiltration), liver function (aspartate aminotransferase, alanine aminotransferase, total bilirubin and serum albumin), kidney function (creatinine clearance), prior treatment (previous rituximab use), process material, and ADAs (immunogenicity screen positive and neutralizing ADA positive) on moxetumomab pasudotox PK. PK parameter–covariate relationships were also examined graphically to evaluate potential association with the variability observed in the PK of moxetumomab pasudotox. If initial graphical exploration indicated a relationship between a covariate and a PK parameter, suggesting an influence on the interindividual variability of the PK of moxetumomab pasudotox, the covariate–PK relationship was then assessed in the nonlinear mixed‐effects model framework. Covariates that were missing or not evaluated for >15% of the patients in the pooled analysis were excluded from further covariate analysis.
The stepwise covariate model (SCM) proceeded in 2 stepwise phases – a forward inclusion phase and a backward elimination phase. In each step of the forward phase, covariate–parameter relations were tested 1 at a time. The most significant relation, if it was statistically significant, was retained in the next step. In Step 2, each remaining covariate–parameter relation was again added 1 at a time to the model and the most significant covariate was retained. This proceeded until no more relations were significant with P < .01 and, thus, the forward SCM model had been established.
The SCM model was subsequently subjected to a stepwise backward elimination of relations. This is essentially the reverse of the forward search. Each covariate–parameter relation was omitted from the model 1 at a time. The least significant relationship, given that it was not statistically significant with P < .001, was removed from the model. This was repeated until no more relations could be removed, establishing the final SCM model. The P‐values were derived from the change in the objective function value provided by NONMEM based on likelihood ratio test for nested models. The final population PK model was developed by including the covariates that remained in the final backward elimination model of the SCM process to the base model.
Individual concentrations of moxetumomab pasudotox were pooled, along with dosing information and selected covariates, to form the NONMEM data set. NONMEM software v7.3 or higher (ICON Development Solutions, Ellicott City, MD), using a first‐order conditional estimation method with an interaction maximum‐likelihood estimation method, was used to derive the PK model parameters. Assessments of goodness‐of‐fit plots and visual predictive checks were performed for model evaluation. The precision of the model parameters was evaluated during model building. Multiple covariates were tested for their ability to explain interindividual variability in PK parameters.
Since ADA titre data were only available for Study 1053, it could not be evaluated as a covariate in the pooled population PK analysis. A separate subgroup analysis of only Study 1053 PK data evaluated ADA titre as a covariate in the population PK model. ADA titre was evaluated as a time‐varying covariate on moxetumomab pasudotox clearance (CL2). For ADA titre, an initial NONMEM run was carried out to identify the ADA titre cut‐off (based on ADA titre steps, i.e. 1280, 2560, 5120, 10 240), above which there is an ADA impact on CL2. Based on this analysis, ADA titre >10 240 was found to significantly increase moxetumomab pasudotox clearance. Based on an ADA titre cut‐off of 10 240, a categorical variable was derived to classify the ADA titre as ≤10 240 or > 10 240. The titre variable was set to 1 if the ADA titre was >10 240 (0 otherwise) at each time point for the individual (See Supplementary information 2 for NONMEM code). The relationship between baseline CD19+ B cells and PK was assessed graphically.
2.4. Exposure–efficacy and exposure–safety analyses
Because of the known difference in the bioactivity of moxetumomab pasudotox used to manufacture clinical trial material for the 2 studies, exposure–response analyses were done separately for each study. The Cmax and AUC predicted by the model were derived and examined as PK exposure parameters in the Study 1001 and Study 1053 populations. For Study 1001, the efficacy end points were CR and OR based on investigator response assessment. For Study 1053, the efficacy end points included rates of durable CR, CR and OR, all based on blinded independent central review. The safety categories that were evaluated for both clinical studies were HUS, CLS, increased creatinine (≥2‐grade worsening from baseline) and grade ≥ 3 treatment‐emergent AEs (TEAEs).
2.5. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY,18 and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20.19
3. RESULTS
For the population PK analyses, data were pooled from Study 1001 and Study 1053; for the analysis of exposure–efficacy and exposure–safety, data were analysed separately because of process material differences.
3.1. Population PK
Data from 123 patients (49 from Study 1001; 74 from Study 1053) treated with moxetumomab pasudotox at doses of 5 (n = 3), 10 (n = 3), 20 (n = 3), 30 (n = 3), 40 (n = 4 Study 1001; n = 74 Study 1053) and 50 (n = 33) μg/kg were used to develop the population PK model. As shown in Table 1, patient covariates were generally similar between the 2 studies. Additional patient and disease characteristics for each study were previously reported.15, 16 Prior use of rituximab was more common in Study 1053 than in Study 1001 (71.6 vs 44.9%, respectively), probably because of prevailing treatment patterns at the time of the later study.
Table 1.
Summary statistics of patient covariates by study
| Parameter | Study 1001 (n = 49) | Study 1053 (n = 74) | Pooled population (n = 123) |
|---|---|---|---|
| Median age, y (range) | 57.0 (40.0–77.0) | 60.0 (36.0–84.0) | 58.0 (36.0–84.0) |
| Race, n (%) | |||
| White | 46 (94) | 66 (89) | 112 (91) |
| Nonwhite | 3 (6) | 5 (7) | 8 (7) |
| Missing* | 0 (0) | 3 (4) | 3 (2) |
| Male, n (%) | 41 (84) | 59 (80) | 100 (81) |
| Median weight, kg (range) | 86.0 (52.1–152.0) | 80.0 (42.4–123.1) | 82.8 (42.4–152.0) |
| Median ALT, U/L (range) | 26.0 (13.0–77.0) | 17.5 (6.0–53.0) | 20.0 (6.0–77.0) |
| Median AST, U/L (range) | 20.0 (7.0–41.0) | 20.0 (0.5–57.0) | 20.0 (6.0–77.0) |
| Median albumin, g/dL (range) | 3.6 (2.9–4.8) | 4.1 (2.7–4.9) | 3.9 (2.7–4.9) |
| Median blood urea nitrogen, mg/dL (range) | 14.0 (8.0–27.0) | 15.1 (7.1–50.0) | 15.1 (7.1–50.0) |
| Missing, n (%)* | 0 (0) | 19 (26) | 19 (15) |
| Median creatinine clearance, mL/min (range) | 107.6 (62.0–187.9) | 106.1 (42.0–238.8) | 107.3 (42.0–238.8) |
| Median spleen size, cm (range)† | 14.0 (0.01–22.5) | 13.1 (0.01–20.9) | 13.3 (0.01–22.5) |
| Missing % (n/total)* | 1 (2) | 1 (1) | 2 (2) |
| BM infiltration by leukaemia | |||
| Median BM infiltration by leukaemia, % (range) | 80.0 (5.0–99.0) | 70.0 (0.0–95.0) | 75.0 (0.0–99.0) |
| Missing, n (%)* | 3 (6) | 1 (1) | 4 (3) |
| Process material, n (%) | |||
| 1 | 32 (65) | 0 | 32 (26) |
| 2 | 17 (35) | 0 | 17 (14) |
| 3 | 0 | 74 (100) | 74 (60) |
| Prior rituximab use, n (%) | 22 (45) | 53 (72) | 75 (61) |
| Median baseline CD19+ B‐cell count, cells/μL (range) | N/A | 80.5 (1.0–17,487) | 80.5 (1.0–17,487) |
| Missing, n (%)* | 49 (100) | 16 (22) | 65 (53) |
| Median baseline lymphocyte count, 109 cells/L (range) | N/A | 0.8 (0.2–29.9) | 0.8 (0.2–29.9) |
| Missing, n (%)* | 49 (100) | 16 (22) | 65 (53) |
| Median baseline lymph node SPD, mm (range) | N/A | 988 (144–23,930) | 988 (144–23,930) |
| Missing, n (%)* | 49 (100) | 57 (77) | 106 (86) |
| Positive baseline ADA, % (n/N) | 4 (1/28) | 61 (43/71) | 44 (44/99) |
| Missing/NE, n (%)* | 43 (21/49) | 4 (3/74) | 20 (24/123) |
| ADA positive anytime, % (n/N) | 68 (19/28) | 87 (64/74) | 81 (83/102) |
| NE, % (n/total)* | 43 (21/49) | 0 (0/74) | 17 (21/123) |
| Positive NAb at baseline, % (n/N) | 0 (0/44) | 55 (39/71) | 34 (39/115) |
| Missing, % (n/N)* | 10 (5/49) | 4 (3/74) | 7 (8/123) |
| NAb‐positive anytime, % (n/N) | 74 (36/47) | 82 (61/74) | 80 (97/121) |
| Missing, % (n/N)* | 4 (2/49) | 0 (0/74) | 2 (2/123) |
Total number of patients including those with missing covariates.
For patients with splenectomies, baseline spleen size was imputed to a very small value (0.01 cm).
ADA, antidrug antibody; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BM, bone marrow; BUN, blood urea nitrogen; N/A, not available; NAb, neutralizing antibody; NE, not evaluated; SPD, sum of product of diameters.
Moxetumomab pasudotox exhibited dose‐proportional PK and was rapidly cleared from systemic circulation with faster clearance of the initial dose than of subsequent doses (Figure 2). The typical estimates of CL1, CL2 and central volume were 24.7 L/h, 3.76 L/h and 6.51 L, respectively, with high interindividual variability for both CL1 (116%) and CL2 (109%; Table 2). Interindividual variability for volume was moderate (37.4%). These clearance estimates were consistent with the observed short half‐life (elimination half‐life 1.2 h for later cycles) and rapid clearance of moxetumomab pasudotox from systemic circulation. Goodness‐of‐fit plots (Figure S1) and the visual predictive checks (Figure S2) indicated that the model adequately described data from both studies. Model fit to representative individual patient PK profiles is shown in Figure S3. None of the covariates examined in the pooled population PK analysis significantly affected the PK of moxetumomab pasudotox.
Figure 2.
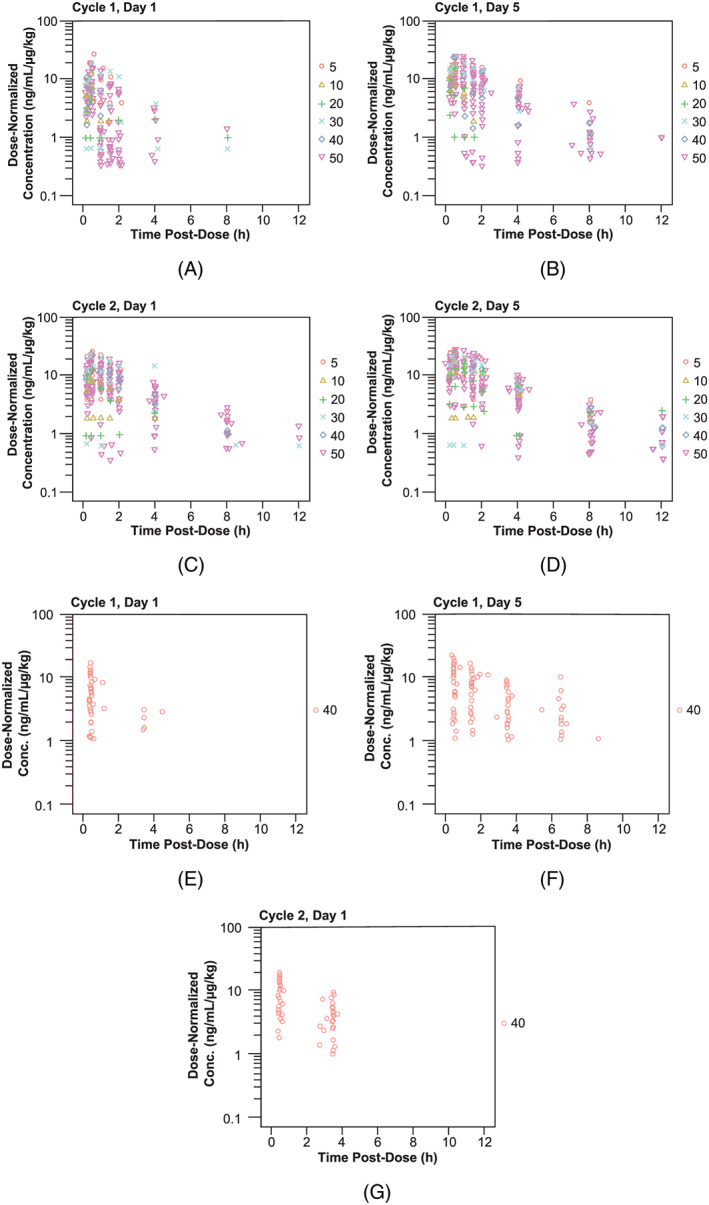
Dose‐normalized moxetumomab concentration–time profiles following intravenous administration. Doses were (A–D) 5–50 μg/kg in study 1001 and (E–G) 40 μg/kg in study 1053
Table 2.
Summary of pharmacokinetic parameters from the base pharmacokinetic model
| Parameter | Point estimate | Standard error | % RSE |
|---|---|---|---|
| CL1, L/h | 24.7 | 3.71 | 15.0 |
| CL2, L/h | 3.76 | 0.601 | 16.0 |
| Vd, L | 6.51 | 0.357 | 5.48 |
| Variance (CL1) | 1.34 (CV = 116%) | 0.228 | 17.0% |
| Variance (CL2) | 1.19 (CV = 109%) | 0.189 | 15.9% |
| Variance (Vd) | 0.140 (CV = 37.4%) | 0.0207 | 14.8% |
| Covariance (CL1 and CL2) | 0.910 (R = .721) | 0.202 | 22.2% |
| Covariance (CL1 and Vd) | 0.294 (R = .679) | 0.0652 | 22.2% |
| Covariance (CL2 and Vd) | 0.355 (R = .870) | 0.0566 | 15.9% |
| Additive error (MEDI assay), ng/mL | 20.5 | 0.461 | 2.25% |
| Additive error (NCI assay), ng/mL | 8.95 | 0.359 | 4.01% |
| Proportional error | 0.414 | 0.00297 | 0.717% |
% RSE, percent relative standard error (100% × SE/EST); CL1, clearance for first dose (cycle 1, day 1); CL2, clearance for all subsequent doses; CV, coefficient of variation; MEDI, MedImmune; NCI, National Cancer Institute; R, correlation coefficient; SD, standard deviation; Vd, volume of distribution.
Posthoc analysis revealed an approximately 4‐fold increase in moxetumomab pasudotox clearance (CL2) among NAb‐positive patients with high (>10 240) ADA titres in Study 1053. In these NAb‐positive patients with high ADA titres, the median cycle to develop an ADA titre >10 240 was cycle 5 (range, 2–5). The estimated median model‐predicted area under the concentration–time curve (AUC) following the 40‐μg/kg moxetumomab pasudotox dose was 186 h ng/mL with high ADA titres compared with 752 h ng/mL in those with low ADA titres (Figure S4 ). ADA titre (stratified by ≤10 240 or >10 240) was included as a covariate in the final subgroup analysis PK model. Additionally, this posthoc analysis showed that patients with high baseline CD19+ B‐cell counts in Study 1053 had more rapid moxetumomab pasudotox clearance (CL1; Figure S5) compared with patients with low baseline CD19+ B‐cell counts.
3.2. Exposure–efficacy analysis
The median maximum moxetumomab pasudotox concentration (Cmax) and AUC values for Studies 1001 and 1053 are shown in Tables S2 and S3, respectively. In both studies, OR rates were higher for patients with moxetumomab pasudotox Cmax and AUC values greater than or equal to the median values relative to those with lower median values (using PK parameters estimated at cycle 2 day 1), although ORs were also observed in patients with lower PK exposure (Figure 3). Similar results were obtained at cycle 1, day 1 (data not shown). Similar trends were observed for CR in the 2 studies and for durable CR in Study 1053, with each of these efficacy outcomes occurring more frequently (but not exclusively) in patients with above‐median Cmax and AUC than in patients with lower exposure.
Figure 3.
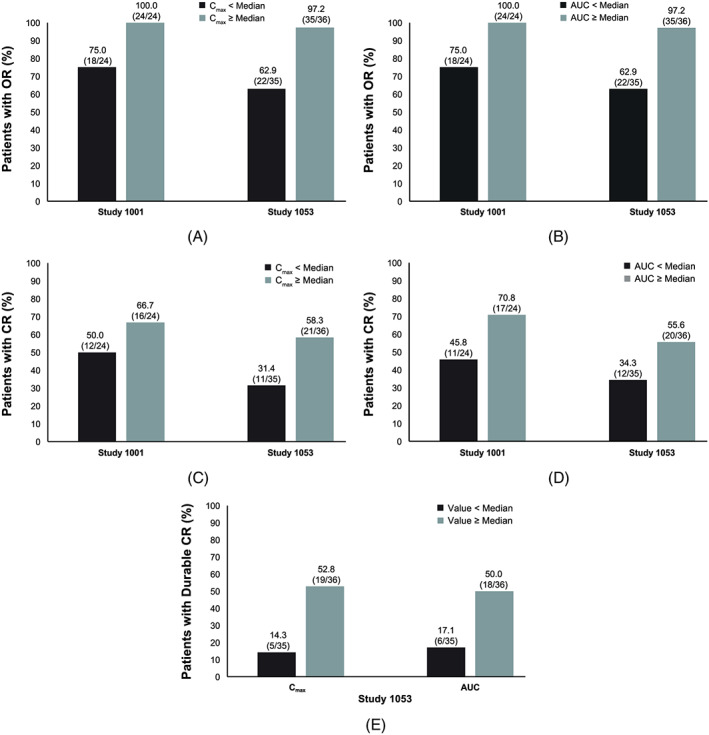
Efficacy outcomes stratified by model‐predicted PK parameters at cycle 2 day 1, including: (a) objective response rates stratified by predicted Cmax; (B) objective response rates stratified by predicted AUC; (C) complete response rates stratified by predicted Cmax; (D) complete response rates stratified by predicted AUC, and (E) durable complete response rates stratified by predicted Cmax and predicted AUC in study 1053. Model‐predicted medians for cycle 2 day 1: For study 1001, Cmax = 660.64 ng/mL; AUC = 2002 h ng/mL. For study 1053, Cmax = 423.1 ng/mL; AUC = 652.8 h ng/mL. AUC, area under the moxetumomab pasudotox concentration–time curve; Cmax, maximum moxetumomab pasudotox concentration; CR, complete response; dCR, durable complete response; OR, objective response
Consistent with previous data showing higher moxetumomab pasudotox clearance in patients with high baseline CD19+ B‐cell counts, lower rates of OR (65.5 vs 89.7%), CR (34.5 vs 48.3%), and durable CR (20.7 vs 34.5%) were observed in patients with CD19+ B‐cell counts above the median (Figure S6), although the number of patients with CD19+ B‐cell data available was limited (n = 58, all from Study 1053). Because a post hoc analysis found that ADA and NAb‐positive patients with a high ADA titre (>10 240) had an approximately 4‐fold increase in moxetumomab pasudotox clearance, the efficacy end points were analysed by ADA titre cut‐off ≤10 240 or >10 240. Results indicate that patients with a high ADA titre had a lower rate of durable CR (20.0 vs 38.8%) and, to a lesser extent, CR (36.0 vs 46.9%), but that OR rates were comparable between the 2 ADA titre categories (76.0 vs 77.6%; Figure S7). Notably, although 25/74 patients had an ADA titre exceeding 10 240, 5 NAb‐positive patients still achieved durable CR.
3.3. Exposure–safety analysis
Among patients experiencing CLS in Study 1053, the median time to onset was 37 days. Although no relationship was observed between moxetumomab pasudotox PK exposure and the incidence of CLS in Study 1001, patients with higher PK exposure in Study 1053 had a numerically higher incidence of CLS relative to those with lower PK exposure (Figure 4). However, the overall incidence of CLS was relatively low in that study. The incidence rates of HUS were 4.1% (2/49) and 7.5% (6/80) in Study 1001 and Study 1053, respectively; because of this, exposure–safety relationships for HUS were not assessed. No incidence of increased creatinine (at least 2‐grade worsening from baseline) was reported in Study 1001; however, 31.3% of patients in Study 1053 experienced such an increase (Figure 4). A higher incidence of increased creatinine was observed in patients with greater than median PK exposure vs patients with median or lower PK exposure.
Figure 4.
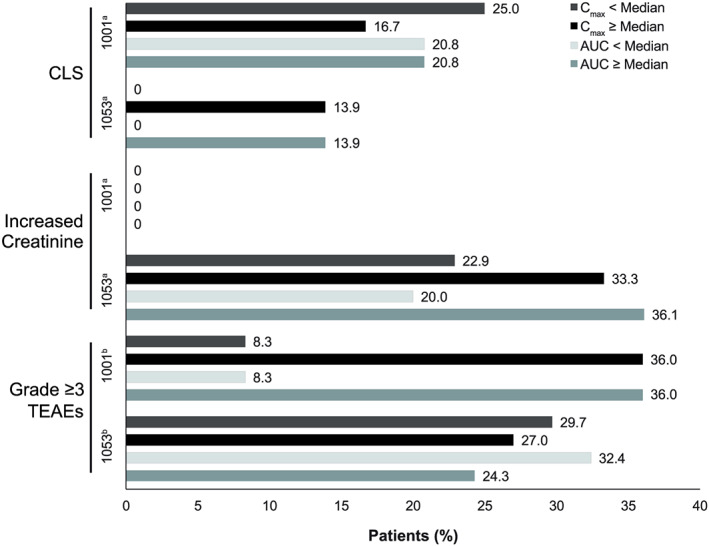
Incidence of selected adverse events, with events stratified by model‐predicted median Cmax and AUC in study 1001 and study 1053. aMedians for cycle 2 day 1: Study 1001, Cmax = 660.64 ng/mL; AUC = 2002 h ng/mL. For study 1053, Cmax = 423.1 ng/mL; AUC = 652.8 h ng/mL. bMedians for cycle 1 day 1: For Study 1001, Cmax = 286.74 ng/mL; AUC = 169.53 h ng/mL. For Study1053, Cmax = 188.89 ng/mL; AUC = 122.03 h ng/mL.
AUC, area under the moxetumomab pasudotox concentration–time curve; CLS, capillary leak syndrome; Cmax, maximum plasma drug concentration; Gr, grade; TEAE, treatment‐emergent adverse event
In Study 1001, grade 3 or 4 TEAEs occurred more frequently in patients with higher PK exposure at cycle 1 day 1 (Figure 4). The overall incidence of grade 3 or 4 TEAEs was 30.3% (10/33) in patients receiving the highest moxetumomab pasudotox dose (50 μg/kg). In Study 1053, no trends for grade 3 or 4 TEAEs by cycle 1 day 1 PK exposure were observed. The incidence rates in patients with greater than or equal to the median PK exposure or with less than the median PK exposure, as determined by Cmax and AUC, were similar to the overall incidence rate of TEAEs in the Study 1001 50‐μg/kg dose group.
4. DISCUSSION
The PK profile of moxetumomab pasudotox was previously described in the published clinical studies.15, 16, 20 Using data from patients enrolled in Studies 1001 and 1053, this analysis produced a population PK model to further assess the PK properties of moxetumomab pasudotox. The PK is characterized by a 1‐compartment model with 2 different linear clearance parameters: CL1 (24.7 L/h) and CL2 (3.76 L/h). CL1 captures the rapid systemic clearance following the first dose of moxetumomab pasudotox, while CL2 reflects the slower clearance observed with subsequent doses. The difference in clearance levels probably reflects rapid post‐treatment depletion of B cells expressing CD22, the target antigen, resulting in a reduced sink effect. This may also be observed when comparing the Cmax values across different doses. The Cmax for the first dose was lower than that for later doses owing to the faster clearance linked to the B‐cell depletion. However, after the first dose, the observed Cmax for all subsequent doses was similar, indicating that moxetumomab pasudotox clearance for subsequent cycles is similar. Similar cases of altered PK attributable to pharmacodynamic effects (target cell elimination resulting in reduced target antigen‐mediated clearance) have been documented for monoclonal antibodies.21, 22
Exposure–efficacy analyses of both studies revealed that patients with higher moxetumomab pasudotox exposure had higher OR, CR and durable CR rates compared with patients with lower exposure levels. None of the covariates examined had significant effects on PK in this pooled analysis of patients in Studies 1001 and 1053. In a subsequent subgroup analysis of Study 1053, however, patients with ADA titres exceeding 10 240 showed an approximately 4‐fold increase in moxetumomab pasudotox clearance relative to those with lower ADA titres. Rates of durable CR and, to some extent, CR were also lower in these patients however, OR rates were similar. Notably, the median cycle at which patients developed an ADA titre exceeding 10 240 was cycle 5, and 20% of these high‐titre patients still achieved a durable CR.
A possible relationship between CD19+ B cells and PK exposure also was explored in a limited subset of patients in Study 1053 for whom these values were available (n = 58). Patients with higher baseline CD19+ B‐cell values, which is also a measure of disease burden, tended to have elevated moxetumomab pasudotox clearance, lower PK exposure and numerically lower response rates.
The lower exposure to moxetumomab pasudotox in patients with elevated baseline B‐cell counts suggests that possibly a higher dose could achieve improved efficacy in this patient subset. However, the reduced efficacy in the patients with elevated B‐cell counts and lower exposure could reflect more severe disease that is less responsive to treatment. Evaluation of dose‐adjustments based on baseline patient characteristics would need to be carefully evaluated within a dose escalation study, given the potentially dose‐limiting toxicities of the immunotoxin.
Possible exposure–safety relationships were explored using model‐predicted median Cmax and AUC. In previous studies, CLS, HUS and increased creatinine (≥2‐grade worsening from baseline) were identified as moxetumomab pasudotox‐related AEs and were treated as AEs of special interest in both studies.16 In Study 1053, the incidence of CLS stratified by median model‐predicted Cmax and AUC was greater in patients with higher moxetumomab pasudotox exposure (13.9% [5/36] vs 0% [0/35] for each). However, in Study 1001, patients with model‐predicted Cmax at median (661 ng/mL) or less had a CLS incidence of 25.0% (6/24) vs 16.7% (2/24) in those with Cmax greater than median; patients with model‐predicted AUC less than or equal to the median and greater than the median (2002 h ng/mL) both had a CLS incidence of 20.8% (5/24). Overall, a numerically greater incidence of CLS was observed in patients with high moxetumomab pasudotox exposure compared with patients with low exposure in Study 1053; however, this trend was not observed in Study 1001.
In an analysis of Study 1053, increased creatinine occurred in 31.3% of patients who had quantifiable PK and safety data, with greater frequency observed in those with higher moxetumomab pasudotox exposure. Given the limited sample sizes, the rates of HUS in both studies were too low (<10%) to draw any meaningful conclusions regarding exposure–safety relationships, and no such analysis was performed.
In Study 1001, grade 3 or 4 AEs were more common in patients with greater moxetumomab pasudotox exposure, while no such relationship was observed in Study 1053. All but 1 of these events (10/11) in the dose‐ranging Study 1001, however, occurred in the highest dose group (moxetumomab pasudotox 50 μg/kg, using Process 1 or 2).
The incidence of grade 3 or 4 AEs in both PK strata from Study 1053 40‐μg/kg group were similar to that of the 50‐μg/kg group. All patients in Study 1053 received moxetumomab pasudotox 40 μg/kg, prepared using Process 3, which had comparable bioactivity to the 50 μg/kg dose in Study 1001, prepared using Process 1 or 2.
In conclusion, moxetumomab pasudotox has demonstrated efficacy in HCL, despite a high incidence of immunogenicity. However, the effect of immunogenicity of immunotoxins on efficacy may depend on the baseline immunocompetence of the patient population and the speed of response to treatment. In HCL, the response to treatment is generally rapid relative to the development of high‐titre ADA responses. Thus, the relationship of ADA responses to clinical outcomes may not be readily extrapolated to different diseases.
COMPETING INTERESTS
This study was funded by AstraZeneca. D. Kuruvilla, Y.L. Chia, K. Balic, N.S. Yao, X. Li, N. Standifer, M. Liang, C.‐M. Tseng, R. Faggioni and L. Roskos are employees of AstraZeneca and may own stock/options in the company. R.J. Kreitman and I. Pastan are co‐inventors on immunotoxin patents that are assigned to the government of the USA, as represented by the Secretary of the Department of Health and Human Services on behalf of the National Institutes of Health, and are licensed by AstraZeneca. No author received an honorarium or other form of financial support related to the development of this manuscript.
CONTRIBUTORS
R.F., L.R., and N.S.Y. contributed to the study design. N.S.Y. participated in the collection and assembly of data. D.K., K.B., X.L., N.S., N. S. Y. participated in data analysis. All authors participated in data interpretation and collaborated in the preparation of the manuscript, supported by a professional medical writer funded by MedImmune (AstraZeneca), and critically reviewed and provided revisions to the manuscript. All authors provided final approval of the manuscript.
DATA AVAILABILITY STATEMENT
No data have been shared.
Supporting information
TABLE S1 Blood sampling schedules for pharmacokinetic analyses in Studies 1001 and 1053
TABLE S2 Summary statistics of model‐predicted pharmacokinetic exposure in the exposure–efficacy population for Study 1001, by dose group
TABLE S3 Summary statistics of model‐predicted pharmacokinetic exposure in the exposure–efficacy population for Study 1053
FIGURE S1 Standard goodness‐of‐fit plots of base model
FIGURE S2 Visual predictive check of the final PK model vs time after previous dose on for cycle 1, day 5. LLOQ, lower limit of quantification; MEDI, MedImmune; Moxe, moxetumomab pasudotox; NCI, National Cancer Institute; PK, pharmacokinetic. Visual predictive check was obtained through n = 2000 simulations from the final PK model. Black dashed line represents the LLOQ for MEDI assay (LLOQ = 40 ng/mL). Red dashed line represents the LLOQ for NCI assay (LLOQ = 16 ng/mL). The grey shaded area represents the area between the 5th and 95th percentiles from the simulations. The solid white line represents the 50th percentile of the simulations.
FIGURE S3 Population PK model fit to individual PK profiles (representative subjects)
IPRED, individual predictions; PK, pharmacokinetic; PRED, population prediction.
FIGURE S4 Effect of high ADA titre (>10 240) on moxetumomab pasudotox PK exposure on cycle 5, day 1 in Study 1053. Solid blue line presents the simulated moxetumomab pasudotox PK profile for ADA titre (≤10 240), dashed line represents PK profile in patients with ADA titre >10 240, and black dashed line denotes the LLOQ of the assay (40 ng/mL)
ADA, antidrug antibody; LLOQ, lower limit of quantification; PK, pharmacokinetic.
FIGURE S5 Relationship between model‐predicted moxetumomab pasudotox clearance from first dose (CL1) and baseline CD19+ cell count in Study 1053. In the left panel, the solid line represents Loess smoothing line, and the grey shaded area represents 95% confidence intervals.
FIGURE S6 Relationship between baseline CD19+ B cells and efficacy end points in Study 1053. Median value was 80.5. Durable complete response (CR) was CR with >180‐day haematological remission, as assessed by blinded independent review. OR, objective response.
FIGURE S7 Relationship between antidrug antibody (ADA) titre and efficacy end points in Study 1053. The ADA cut‐off value was 10 240. Durable complete response (CR) was CR with >180‐day haematological remission as assessed by blinded independent review
ACKNOWLEDGEMENTS
This study was sponsored by AstraZeneca. Medical writing and editorial support were provided by Peloton Advantage, LLC, an OPEN Health company, and funded by AstraZeneca. The authors acknowledge Inna Vainshtein, PhD, of AstraZeneca for analysing and providing input on pharmacodynamic and antidrug antibody data.
Kuruvilla D, Chia YL, Balic K, et al. Population pharmacokinetics, efficacy, and safety of moxetumomab pasudotox in patients with relapsed or refractory hairy cell leukaemia. Br J Clin Pharmacol. 2020;86:1367–1376. 10.1111/bcp.14250
The authors confirm that the Principal Investigator for this paper is Robert J. Kreitman and that he had direct clinical responsibility for patients.
REFERENCES
- 1. Grever MR, Abdel‐Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with classic hairy cell leukemia. Blood. 2017;129(5):553‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bouroncle BA, Wiseman BK, Doan CA. Leukemic reticuloendotheliosis. Blood. 1958;13(7):609‐630. [DOI] [PubMed] [Google Scholar]
- 3. Rosenberg JD, Burian C, Waalen J, Saven A. Clinical characteristics and long‐term outcome of young hairy cell leukemia patients treated with cladribine: a single‐institution series. Blood. 2014;123(2):177‐183. [DOI] [PubMed] [Google Scholar]
- 4. Zinzani PL, Pellegrini C, Stefoni V, et al. Hairy cell leukemia: evaluation of the long‐term outcome in 121 patients. Cancer. 2010;116(20):4788‐4792. [DOI] [PubMed] [Google Scholar]
- 5. Else M, Dearden CE, Matutes E, et al. Long‐term follow‐up of 233 patients with hairy cell leukaemia, treated initially with pentostatin or cladribine, at a median of 16 years from diagnosis. Br J Haematol. 2009;145(6):733‐740. [DOI] [PubMed] [Google Scholar]
- 6. Thompson PA, Ravandi F. How I manage patients with hairy cell leukaemia. Br J Haematol. 2017;177(4):543‐556. [DOI] [PubMed] [Google Scholar]
- 7. Alewine C, Hassan R, Pastan I. Advances in anticancer immunotoxin therapy. Oncologist. 2015;20(2):176‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kreitman RJ. Immunoconjugates and new molecular targets in hairy cell leukemia. Hematology am Soc Hematol Educ Program. 2012;2012:660‐666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kreitman RJ, Pastan I. Immunoconjugates in the management of hairy cell leukemia. Best Pract Res Clin Haematol. 2015;28(4):236‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kreitman RJ, Stetler‐Stevenson M, Margulies I, et al. Phase II trial of recombinant immunotoxin RFB4(dsFv)‐PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol. 2009;27(18):2983‐2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kreitman RJ, Wilson WH, Bergeron K, et al. Efficacy of the anti‐CD22 recombinant immunotoxin BL22 in chemotherapy‐resistant hairy‐cell leukemia. N Engl J Med. 2001;345(4):241‐247. [DOI] [PubMed] [Google Scholar]
- 12. Kreitman RJ, Wilson WH, White JD, et al. Phase I trial of recombinant immunotoxin anti‐tac (Fv)‐PE38 (LMB‐2) in patients with hematologic malignancies. J Clin Oncol. 2000;18(8):1622‐1636. [DOI] [PubMed] [Google Scholar]
- 13. Kreitman RJ, Pastan I. Antibody fusion proteins: anti‐CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res. 2011;17(20):6398‐6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lumoxiti [package insert]. Wilmington, DE: AstraZeneca pharmaceuticals. 2018.
- 15. Kreitman RJ, Tallman MS, Robak T, et al. Minimal residual hairy cell leukemia eradication with moxetumomab pasudotox: phase 1 results and long‐term follow‐up. Blood. 2018;131(21):2331‐2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kreitman RJ, Tallman MS, Robak T, et al. Phase I trial of anti‐CD22 recombinant immunotoxin moxetumomab pasudotox (CAT‐8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol. 2012;30(15):1822‐1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kreitman RJ, Dearden C, Zinzani PL, et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia. 2018;32(8):1768‐1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A.J. , Irelan, S. , … Bryant, C. (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic acids research, 46(D1): D1091‐D106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alexander SPH, Kelly E, Mathie A, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: transporters. Br J Pharmacol. 2019;176(Suppl 1):S397‐S493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wayne AS, Shah NN, Bhojwani D, et al. Phase 1 study of the anti‐CD22 immunotoxin moxetumomab pasudotox for childhood acute lymphoblastic leukemia. Blood. 2017;130(14):1620‐1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Glassman PM, Balthasar JP. Mechanistic considerations for the use of monoclonal antibodies for cancer therapy. Cancer Biol Med. 2014;11(1):20‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meijer RT, Koopmans RP, ten Berge IJ, Schellekens PT. Pharmacokinetics of murine anti‐human CD3 antibodies in man are determined by the disappearance of target antigen. J Pharmacol Exp Ther. 2002;300(1):346‐353. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Blood sampling schedules for pharmacokinetic analyses in Studies 1001 and 1053
TABLE S2 Summary statistics of model‐predicted pharmacokinetic exposure in the exposure–efficacy population for Study 1001, by dose group
TABLE S3 Summary statistics of model‐predicted pharmacokinetic exposure in the exposure–efficacy population for Study 1053
FIGURE S1 Standard goodness‐of‐fit plots of base model
FIGURE S2 Visual predictive check of the final PK model vs time after previous dose on for cycle 1, day 5. LLOQ, lower limit of quantification; MEDI, MedImmune; Moxe, moxetumomab pasudotox; NCI, National Cancer Institute; PK, pharmacokinetic. Visual predictive check was obtained through n = 2000 simulations from the final PK model. Black dashed line represents the LLOQ for MEDI assay (LLOQ = 40 ng/mL). Red dashed line represents the LLOQ for NCI assay (LLOQ = 16 ng/mL). The grey shaded area represents the area between the 5th and 95th percentiles from the simulations. The solid white line represents the 50th percentile of the simulations.
FIGURE S3 Population PK model fit to individual PK profiles (representative subjects)
IPRED, individual predictions; PK, pharmacokinetic; PRED, population prediction.
FIGURE S4 Effect of high ADA titre (>10 240) on moxetumomab pasudotox PK exposure on cycle 5, day 1 in Study 1053. Solid blue line presents the simulated moxetumomab pasudotox PK profile for ADA titre (≤10 240), dashed line represents PK profile in patients with ADA titre >10 240, and black dashed line denotes the LLOQ of the assay (40 ng/mL)
ADA, antidrug antibody; LLOQ, lower limit of quantification; PK, pharmacokinetic.
FIGURE S5 Relationship between model‐predicted moxetumomab pasudotox clearance from first dose (CL1) and baseline CD19+ cell count in Study 1053. In the left panel, the solid line represents Loess smoothing line, and the grey shaded area represents 95% confidence intervals.
FIGURE S6 Relationship between baseline CD19+ B cells and efficacy end points in Study 1053. Median value was 80.5. Durable complete response (CR) was CR with >180‐day haematological remission, as assessed by blinded independent review. OR, objective response.
FIGURE S7 Relationship between antidrug antibody (ADA) titre and efficacy end points in Study 1053. The ADA cut‐off value was 10 240. Durable complete response (CR) was CR with >180‐day haematological remission as assessed by blinded independent review
Data Availability Statement
No data have been shared.