

Abstract
Aims
Retinoic acid‐related orphan receptor γ (RORγ), a master regulator of T‐helper 17 (Th17) cell function and differentiation, is an attractive target for treatment of Th17‐driven diseases. This first‐in‐human study aimed to investigate the pharmacokinetics, pharmacodynamics, safety and tolerability of the inverse RORγ agonist AZD0284.
Methods
We conducted a phase I, randomized, single‐blind, placebo‐controlled, two‐part, first‐in‐human study with healthy subjects receiving single (4–238 mg) or multiple (12–100 mg) oral doses of AZD0284 or placebo after overnight fasting. Subjects in the one single dose cohort additionally received a single dose of AZD0284 after a high‐calorie meal. AZD0284 plasma concentrations, as well as inhibition of ex vivo‐stimulated interleukin (IL)‐17A release in whole blood, were frequently measured after both single and multiple dosing.
Results
Eighty‐three men participated in the study. AZD0284 was absorbed rapidly into plasma after oral dosing and exhibited a terminal half‐life of 13–16 hours. Both the area under the concentration‐time curve (AUC) and maximum concentration (C max) increased subproportionally with increasing dose (95% confidence intervals of slope parameter were 0.71–0.84 and 0.72–0.88 for AUC and C max, respectively). Food intake delayed the absorption of AZD0284 but did not affect the overall exposure or half‐life. AZD0284 showed dose‐dependent reduction of ex vivo‐stimulated IL‐17A release after both single and multiple doses. No significant safety concerns were identified in the study.
Conclusions
AZD0284 was well tolerated, rapidly and dose‐dependently absorbed, and reduced stimulated IL‐17A release after single and multiple dosing. The results of this study support further clinical development of AZD0284.
Keywords: autoimmune disease, phase I clinical trial, psoriasis, RORγ, Th17 cells
What is already known about this subject
The nuclear retinoic acid‐related orphan receptor γ (RORγ) is a master transcriptional factor of Th17 immune cells.
RORγ is an interesting therapeutic target for the treatment of Th17‐driven disease as current treatment is restricted to injected biologics.
The inverse RORγ agonist AZD0284 have shown to be a potent inhibitor of RORγ in a primary human Th17 cell assay.
What this study adds
Single and multiple oral doses (4–238 mg) of the inverse RORγ agonist, AZD0284, showed rapid, dose‐dependent systemic absorption in healthy male subjects.
A dose‐dependent reduction of ex vivo‐stimulated IL‐17A release in whole blood was observed after both single and multiple doses.
AZD0284 was safe and well tolerated in the dose range studied, with stable and predictable pharmacokinetics, and offers the potential to treat Th17‐driven disease with an orally active small‐molecule drug.
1. INTRODUCTION
Psoriasis is an inflammatory and autoimmune skin disease affecting 2–3% of the adult population. T cells have recently been identified to play an important role in the pathogenesis of psoriasis, driving inflammation by interacting with keratinocytes and endothelial cells.1, 2, 3 Specifically, several studies point to an important role of the interleukin (IL)‐23/T‐helper 17 (Th17)/IL‐17 axis in the development of psoriasis.4 Monoclonal antibodies directed towards IL‐12/23, IL‐17 and the IL‐17 receptor have shown excellent efficacy in patients with psoriasis, with up to 80% achieving a 75% improvement in the Psoriasis Area Severity Index (PASI) after 12–16 weeks of treatment.5
Retinoic acid‐related orphan receptor γ (RORγ) is a member of the nuclear receptor family of transcription factors and is essential for cytotoxic and T‐helper cell development in the thymus. It acts as the master transcription factor of Th17 cell differentiation and is induced by IL‐23 signalling. RORγ is therefore an interesting pharmacological target for the treatment of Th17‐mediated disease, and orally bioavailable inhibitors of RORγ have been presented as an alternative to antibody therapy.6, 7, 8, 9 Positive data in psoriasis have been reported following 4 weeks oral administration of the inverse RORγ agonist VTP‐43742, resulting in a 30% reduction in the PASI score relative to placebo.10
Besides its role in immune cells, RORγ is also known to be expressed in the liver, where it has been shown to affect several genes involved in the metabolism of steroids, bile acids, lipids, glucose and xenobiotics.11, 12 Thus, inhibition of RORγ could potentially interfere with the regulation of such metabolic pathways and monitoring of, for example, bile acids, triglycerides and glucose levels is warranted.
AZD0284 is an inverse RORγ agonist with high potency as measured by its ability to inhibit IL‐17A production in a primary human Th17 cell assay.7 Inhibition of IL‐17A production by AZD0284 has also been demonstrated in human whole‐blood samples following stimulation of T cells with Cytostim (betadex sulfobutyl ether sodium).13 Furthermore, in a preclinical mouse model of skin inflammation, AZD0284 significantly reduced the number of IL‐17A producing cells in the ear (unpublished data on file, Research and Early Development, Respiratory, Inflammation and Autoimmune, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden).
From a drug substance perspective, AZD0284 has low solubility and moderate permeability, which implies potential absorption limitations. Fraction absorbed and oral bioavailability (F) of the compound may thus vary with dose and formulation. Different options to increase solubility are available, including using nanosuspensions, amorphous formulations and addition of solubility enhancers. A solution including Captisol has previously been shown to increase the exposure of AZD0284 in rats (manuscript in progress, Department of Early Product Development, Pharmaceutical Sciences, R&D, AstraZeneca, Gothenburg, Sweden).
In this paper, we report data from the first‐in‐human study of AZD0284, evaluating the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of AZD0284 following single and multiple oral doses in solution with Captisol. We show that AZD0284 is well tolerated, rapidly absorbed into plasma, exhibits consistent PK across doses and is capable of systemic RORγ inhibition within the dose range studied.
2. METHODS
2.1. Study design
A two‐part, randomized, single‐blind, placebo‐controlled, dose escalation study was designed to evaluate the safety, tolerability, PK, and PD of AZD0284 following single and multiple oral doses (ClinicalTrials.gov identifier: NCT02976831). Healthy adult male and female subjects aged 18–50 years, with a body mass index of 18–30 kg m−2 were eligible for the study. All subjects were required to be of good health, and have normal laboratory parameters and electrocardiograms (ECGs) at enrolment. The use of prescription or over‐the‐counter medications was not allowed, except for acetaminophen. Nonchildbearing females were enrolled if they had a negative pregnancy test, were postmenopausal or had documentation of sterilization. Subjects were screened up to 28 days prior to dosing.
In Part 1, subjects were first randomized (3:1) to receive a single dose of either AZD0284 (4, 12, 36, 50, 108 or 238 mg, n = 6 per cohort) or placebo (n = 2 per cohort) under fasting conditions. The 238 mg dose was given both as a single dose and as a fractionated dose (two 119 mg doses administered 1 hour apart). Subjects were discharged on day 3 after all assessments had been completed. In the 50 mg dose cohort, subjects returned to the clinic after a washout period of at least 7 days and received an additional 50 mg dose following a standard high‐fat breakfast, as recommended by the US Food and Drug Administration (FDA). This was done to provide an exposure comparison between fed and fasted conditions. Sentinel dosing was employed in all cohorts. In the 108 mg dose cohort, urine was collected from subjects up to 48 hours after dosing to investigate the renal excretion of AZD0284.
In Part 2, subjects were dosed with a single dose of AZD0284 (12, 36 or 100 mg, n = 6 per cohort) or placebo (n = 3 per cohort) on day 1. After a washout period of 3 days, each subject received repeated twice‐daily administration of drug on days 4 to 9, followed by a single dose in the morning of day 10. This design allowed estimation of time‐dependent PK following multiple dosing. Subjects were discharged 3 days after the last dose when all assessments were completed. In both parts of the study, there was a final follow‐up visit 5–7 days after the final dose. All morning doses were given after overnight fasting and subjects were fasted for 4 hours prior to evening doses.
The study took place between December 2016 and May 2017 at the PAREXEL Early Phase Unit in London. It was conducted in accordance with the applicable regulatory and International Conference on Harmonization – Good Clinical Practice requirements. A Local Research Ethics Committee provided approval of the study and written informed consent was obtained from potential subjects prior to screening.
2.2. Treatments
A solution of AZD0284 or placebo containing Captisol (1:2 concentration ratio) was administered orally at a total volume of 240 mL. While the volume was kept constant, the concentrations of AZD0284 and Captisol were increased with increasing dose.
2.3. Assessments
To assess the primary endpoint of safety and tolerability of AZD0284, clinical laboratory tests, ECGs, physical examinations and telemetry were conducted, and adverse events were monitored. In Part 2, the potential of AZD0284 to interfere with gene expression of detoxifying enzymes or lipid and glucose metabolic genes was investigated by measuring bile acids, triglycerides and glucose levels (both post prandial and in the fasted state). Furthermore, the risk for CYP3A4‐mediated drug–drug interactions was assessed by the biomarker 4β‐hydroxy‐cholesterol (4β‐OH‐cholesterol).14
To characterize the PK of AZD0284 (secondary endpoint), plasma samples were collected and assayed using a validated bioanalytical method based on protein precipitation, followed by liquid chromatography with tandem mass spectrometric detection (LC–MS/MS). The method was validated over the calibration curve range of 5–5000 nmol L−1 with weighted linear regression with weighting factor 1x −2. Urine samples were analysed for AZD0284 using the LC–MS/MS method following dilution. The lower limit of quantification was 50 nmol L−1 with higher limits of quantification of 50000 nmol L−1.
Quality control (QC) samples were prepared at four different analyte concentrations and analysed with each batch of samples. The analysis was acceptable if at least one‐half of the QC samples at each concentration and two‐thirds of all QC samples in the curve range were within the range of ±15% of the nominal concentration. Furthermore, the overall accuracy and precision (relative standard deviation (%RSD)) of all QC samples should be within ±15% of the nominal concentration and ≤15%, respectively.
To assess pharmacodynamic effects of AZD0284, IL‐17A levels were measured in whole‐blood samples following ex vivo stimulation with Cytostim according to the method described by Russell et al.13 Whole‐blood samples were diluted 2‐fold with buffer prior to transfer to assay plates. In Part 1, two baseline samples were taken, one of which was spiked with 10 μM exogenous AZD0284 prior to Cytostim stimulation. The spiked samples served as a positive control to determine the maximum achievable inhibition with AZD0284 in the assay. In Part 2, positive control samples were collected and analysed also at all post‐dose timepoints. This was done to allow for monitoring and adjustment of between‐experiment variability over the 10‐day dosing period.
2.4. Statistical methods
Due to the exploratory nature of the study, the sample size was not based on formal statistical considerations, but rather experience from previous similar phase I studies with other compounds. Safety data were summarized using the population of subjects who received at least one dose of the study drug or placebo. PK data were analysed from those subjects who received the study drug and had evaluable PK data. PD data were analysed from subjects receiving at least one dose of study drug or placebo, and who had at least the baseline and one post‐dose measurement of IL‐17A.
PK parameters including maximum concentration (C max), time to reach maximum concentration (T max), area under the concentration–time curve extrapolated to time of last measurable concentration last time point (AUClast) or extrapolated to infinity following a single dose (AUC), or over a 24‐hour dosing interval at steady state (AUCτ) and terminal half‐life (t ½λz) were analysed using noncompartmental methods with Phoenix WinNonlin v. 6.3 (Certara, St Louis, MO, USA). Dose‐proportionality was analysed by using the power model approach.
Change from baseline in stimulated whole‐blood IL‐17A levels was analysed using a linear mixed model with treatment, time and a treatment by time interaction as fixed effects and subject as a random effect. IL‐17A levels were log‐transformed prior to analysis. In Part 2, IL‐17A levels were also corrected for between‐experiment variability by adjusting the value of each sample with the change from baseline of the corresponding positive control sample, eg if the positive control value had changed by +30% from baseline, the value of IL‐17A was divided by 1.3 prior to log‐transformation and analysis.
2.5. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY,15 and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20.16
3. RESULTS
A total of 83 subjects were randomized in the study. None of the randomized subjects withdrew or were withdrawn from the study. Treatments and subject demographics for Part 1 and 2 are summarized in Table 1.
Table 1.
Demographics
| Part 1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo (n = 14) | 4 mg AZD0284 (n = 6) | 12 mg AZD0284 (n = 6) | 36 mg AZD0284 (n = 6) | 50 mg AZD0284 (n = 6) | 108 mg AZD0284 (n = 6) | 238 mg AZD0284 (n = 6) | 238 mg AZD0284 fractionated (n = 6) | Total AZD0284 (n = 42) | |
| Male, n (%) | 14 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 42 (100) |
| Mean age, years (SD) | 32.3 (7.7) | 32.3 (7.1) | 34.7 (7.6) | 28.5 (3.8) | 27.7 (6.6) | 36.7 (7.1) | 29.8 (5.9) | 33.5 (7.3) | 31.9 (6.8) |
| White, n (%) | 11 (78.6) | 4 (66. 7) | 5 (83.3) | 6 (100) | 4 (66. 7) | 5 (83.3) | 5 (83.3) | 4 (66. 7) | 33 (78.6) |
| Black, n (%) | 1 (7.1) | 2 (33.3) | 1 (16.7) | 0 (0.0) | 1 (16.7) | 1 (16.7) | 1 (16.7) | 1 (16.7) | 8 (19.0) |
| Other, n (%) | 2 (14.3) | 1 (16.7) | 1 (16.7) | 1 (2.4) | |||||
| Mean BMI, kg m −2 (SD) | 24.4 (2.5) | 24.8 (3.3) | 24.4 (2.4) | 23.07 (2.4) | 22.5 (1.3) | 23.9 (1.6) | 24.7 (2.3) | 24.1 (4.3) | 23.9 (2.6) |
| Part 2 | |||||
|---|---|---|---|---|---|
| Placebo (n = 9) | 12 mg AZD0284 (n = 6) | 36 mg AZD0284 (n = 6) | 100 mg AZD0284 (n = 6) | Total AZD0284 (n = 18) | |
| Male, n (%) | 9 (100) | 6 (100) | 6 (100) | 6 (100) | 18 (100) |
| Mean age, years (SD) | 32.7 (7.8) | 34.5 (5.8) | 36.3 (5.2) | 37.3 (6.5) | 36.1 (5.6) |
| White, n (%) | 9 (100) | 3 (50) | 6 (100) | 6 (100) | 15 (83.3) |
| Asian, n (%) | 0 | 1 (16.7) | 0 | 0 | 1 (5. 6) |
| Other, n (%) | 0 | 2 (33.3) | 0 | 0 | 2 (11.1) |
| Mean BMI, kg m −2 (SD) | 24.7 (2.2) | 25.2 (2.4) | 26.0 (2.8) | 24.0 (1.6) | 25.1 (2.3) |
Abbreviations: BMI, body mass index; SD, standard deviation.
3.1. Safety and tolerability
Overall, single and multiple doses of AZD0284 were well tolerated. There were no serious adverse events or adverse events (AEs) that led to discontinuation. All other AEs reported were of mild to moderate intensity (Supporting Information Tables S1 and S2). In Part 1, 12 subjects receiving AZD0284 reported a total of 13 AEs. No AEs were reported under fed conditions. In Part 2, nine subjects receiving AZD0284 reported a total of 13 AEs. For placebo, 21 AEs were reported in six subjects (two and four subjects in Parts 1 and 2, respectively). The most commonly reported AE following single dosing was nasopharyngitis (three events reported by three subjects receiving AZD0284), whereas headache was the most commonly reported AE following multiple dosing (reported by one subject receiving placebo and four subjects receiving AZD0284). No trends were seen in bile acids, triglycerides or glucose levels, and plasma levels of 4β‐OH‐cholesterol were unchanged after treatment.
3.2. Pharmacokinetics
Plasma concentrations of AZD0284 indicated rapid absorption following single and multiple administration (Figure 1A,B). PK parameters estimated for Part 1 are summarized in Table 2. The plasma exposure increased in a subproportional manner in the 4–238 mg dose range. In a power model, the 95% CI of the slope parameter did not include 1 for either AUC (0.71–0.84) or C max (0.72–0.88). Dose‐normalized AUC and C max values are shown in Figure 2. Fractionating the top dose of 238 mg dose into two 119 mg doses given 1 hour apart resulted in an approximately 2‐fold increase in C max and AUC compared to the nonfractionated 238 mg dose (Figure 1A and Table 2) and dose‐normalized AUC and C max comparable to the 108 mg dose (Figure 2). Low intersubject variability was observed with a coefficient of variation (CV) less than 20% for most of the estimated PK parameters. The mean terminal half‐life was consistent following both single and multiple dosing (range 13–16 hours). Approximately 19% of an oral AZD0284 dose was excreted unchanged in urine, resulting in a mean renal clearance of 1.4 L h−1. Intake of food prior to dosing delayed the absorption of AZD0284 and decreased C max by approximately 3.6 hours and 33%, respectively, but no change was observed in AUC (Figure 3). Following twice‐daily dosing of AZD0284, steady‐state plasma concentrations were generally achieved after 4 days (Figure 1B). Accumulation ratios were about 2‐fold for C max and AUC, and data did not indicate any time‐dependent pharmacokinetics (Table 3).
Figure 1.
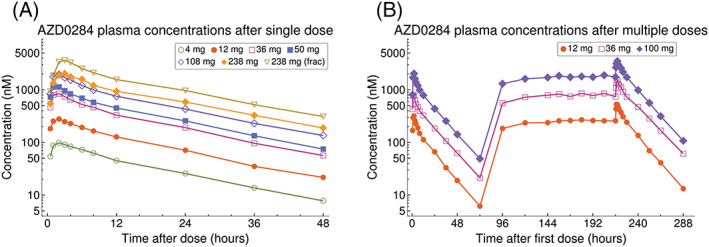
(A) Observed mean AZD0284 plasma concentrations after single doses. (B) Observed mean AZD0284 plasma concentrations after single doses on day 1, followed by twice‐daily dosing on days 4–9 and single doses on day 10
Table 2.
Summary of PK parameters following a single dose of AZD0284 (Part 1)
| PK parameter | Dose (mg) | n | Geometric mean (CV%) |
|---|---|---|---|
| AUC (h*nmol L −1 ) | 4 | 6 | 1751 (3.6) |
| 12 | 6 | 4808 (9.8) | |
| 36 | 6 | 12960 (23) | |
| 50 | 6 | 17410 (20) | |
| 50a | 6 | 17100 (20) | |
| 108 | 6 | 30200 (15) | |
| 238 | 6 | 36230 (36) | |
| 238b | 6 | 63000 (13) | |
| C max (nmol L −1 ) | 4 | 6 | 97.9 (10) |
| 12 | 6 | 278 (12) | |
| 36 | 6 | 823 (27) | |
| 50 | 6 | 1141 (20) | |
| 50a | 6 | 765.9 (12.4) | |
| 108 | 6 | 2079 (17) | |
| 238 | 6 | 2067 (43) | |
| 238b | 6 | 3885 (9.7) |
| Arithmetic mean (SD) | |||
|---|---|---|---|
| t ½λz (h) | 4 | 6 | 13.6 (1.7) |
| 12 | 6 | 13.6 (1.6) | |
| 36 | 6 | 13.4 (1.2) | |
| 50 | 6 | 13.6 (0.90) | |
| 50a | 6 | 13.9 (1.4) | |
| 108 | 6 | 14.4 (2.8) | |
| 238 | 6 | 15.1 (3.1) | |
| 238b | 6 | 14.5 (1.3) |
| Median (range) | |||
|---|---|---|---|
| t max (h) | 4 | 6 | 2.0 (2.0, 2.0) |
| 12 | 6 | 2.0 (2.0, 2.1) | |
| 36 | 6 | 2.0 (1.0, 2.0) | |
| 50 | 6 | 1.5 (1.0, 2.0) | |
| 50a | 6 | 5.1 (4.0, 6.0) | |
| 108 | 6 | 1.5 (1.0, 4.0) | |
| 238 | 6 | 2.5 (2.0, 3.0) | |
| 238b | 6 | 2.5 (2.0, 4.0) |
Abbreviations: AUC, area under plasma concentration–time curve from time zero extrapolated to infinity; C max, observed maximum plasma concentration; %CV, coefficient of variation; SD, standard deviation; t ½λz, terminal half‐life; t max, time to reach observed maximum concentration.
50 mg AZD0284 given 30 min after a standard high‐fat FDA breakfast.
238 mg given as two 119 mg doses separated by 1 hour.
Figure 2.
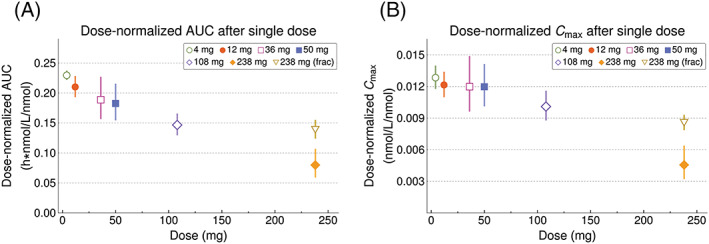
Geometric means (95% CI) of dose‐normalized AUC (A) and C max (B) after single doses of AZD0284
Figure 3.
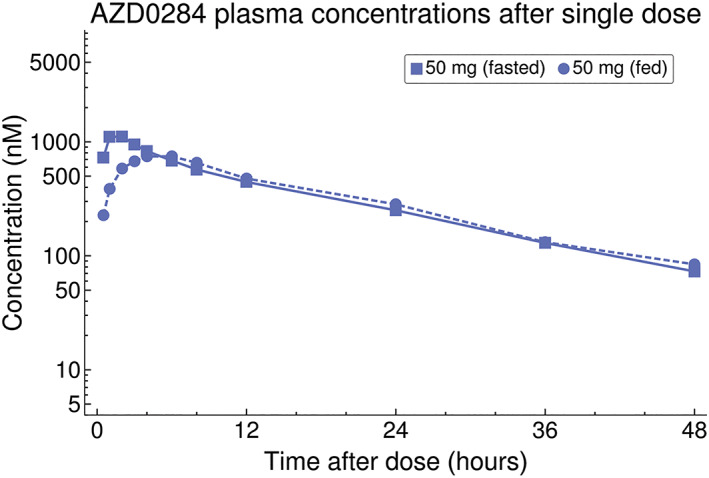
Observed mean AZD0284 plasma concentrations after a single dose of 50 mg taken either after overnight fasting or a standard high‐fat breakfast
Table 3.
Summary of PK parameters following multiple dosing of AZD0284 (Part 2)
| PK parameter | Dose (mg) | Day | n | Geometric mean (CV%) |
|---|---|---|---|---|
| AUC (nmol*h L –1 ) | 12 | 1 | 6 | 4545 (8.6) |
| 36 | 1 | 6 | 12730 (18) | |
| 100 | 1 | 6 | 29520 (39) | |
| AUC last (nmol*h L –1 ) | 12 | 10 | 6 | 9003 (18) |
| 36 | 10 | 6 | 29200 (19) | |
| 100 | 10 | 6 | 63660 (19) | |
| AUC τ (nmol*h L –1 ) | 12 | 1 | 6 | 2261 (10) |
| 12 | 10 | 6 | 4389 (14) | |
| 36 | 1 | 6 | 5725 (19) | |
| 36 | 10 | 6 | 12710 (16) | |
| 100 | 1 | 6 | 13400 (47) | |
| 100 | 10 | 6 | 29470 (20) | |
| C max (nmol L –1 ) | 12 | 1 | 6 | 334.7 (10) |
| 12 | 10 | 6 | 539.6 (13) | |
| 36 | 1 | 6 | 810.9 (20) | |
| 36 | 10 | 6 | 1514 (15) | |
| 100 | 1 | 6 | 1903 (54) | |
| 100 | 10 | 6 | 3535 (18) |
| Arithmetic mean (SD) | ||||
|---|---|---|---|---|
| t ½λz (h) | 12 | 1 | 6 | 13.3 (1.1) |
| 12 | 10 | 6 | 13.8 (1.2) | |
| 36 | 1 | 6 | 15.2 (1.5) | |
| 36 | 10 | 6 | 16.4 (2.0) | |
| 100 | 1 | 6 | 15.2 (2.3) | |
| 100 | 10 | 6 | 14.6 (1.2) |
| Median (range) | ||||
|---|---|---|---|---|
| t max (h) | 12 | 1 | 6 | 1.0 (1.0, 2.0) |
| 12 | 10 | 6 | 1.5 (1.0, 2.0) | |
| 36 | 1 | 6 | 1.5 (1.0, 2.0) | |
| 36 | 10 | 6 | 2.0 (1.0, 2.0) | |
| 100 | 1 | 6 | 2.0 (1.0, 2.0) | |
| 100 | 10 | 6 | 2.0 (1.0, 2.0) |
| Geometric mean (CV%) | ||||
|---|---|---|---|---|
| RA AUCτ | 12 | 10 | 6 | 1.94 (13) |
| 36 | 10 | 6 | 2.22 (11) | |
| 100 | 10 | 6 | 2.20 (27) | |
| RA Cmax | 12 | 10 | 6 | 1.61 (14) |
| 36 | 10 | 6 | 1.87 (9.8) | |
| 100 | 10 | 6 | 1.86 (34) | |
| TCP | 12 | 10 | 6 | 0.966 (8.7) |
| 36 | 10 | 6 | 0.999 (4.0) | |
| 100 | 10 | 6 | 0.998 (20) |
Abbreviations: AUC, area under plasma concentration–time curve from time zero extrapolated to infinity; AUClast, area under the plasma concentration–curve from time zero to time of last measurable concentration; AUCτ, area under the plasma concentration–time curve over a dosing interval; C max, observed maximum plasma concentration; %CV, coefficient of variation; RAAUCτ, accumulation ratio AUCτ,D10/AUCτ,D1; RACmax, accumulation ratio C max,D10/C max,D1; SD, standard deviation; t ½λz, terminal half‐life; t max, time to reach observed maximum concentration; TCP, temporal change parameter AUCτ,D10/AUCD10.
3.3. Pharmacodynamics
AZD0284 demonstrated dose‐dependent inhibition of stimulated IL‐17A release after both single and multiple dosing (Figure 4). Although variable, the inhibition profiles appeared similar to the PK profiles. Maximum achievable inhibition (mean value 67–69% in positive control samples) was observed at early timepoints (near C max) following single doses of >50 mg and at steady state following a twice‐daily dose of 100 mg.
Figure 4.
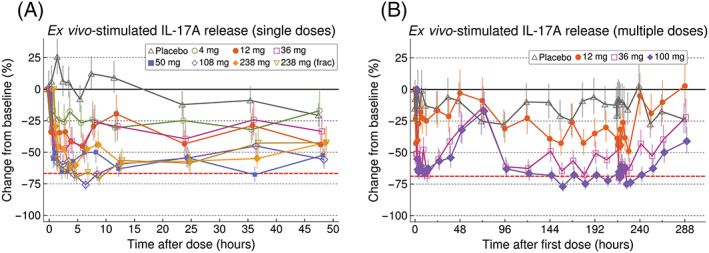
Estimated percentage change from baseline (±SE) of ex vivo‐stimulated IL‐17A in whole blood following single (A) and multiple (B) doses of AZD0284 or placebo. The red dashed lines show the mean reduction observed in positive control samples taken at baseline following addition of a maximal concentration of exogenous AZD0284
4. DISCUSSION
Considerable progress has been made to understand the complex pathogenesis of inflammatory diseases such as psoriasis. This has facilitated the development of more efficacious and targeted treatment alternatives, such as therapeutic antibodies targeting IL‐23 and IL‐17.17 However, not all patients have access to these efficacious but expensive treatments. Oral treatment options are currently based on other mechanisms, such as the phosphodiesterase inhibitor apremilast, which has significantly lower efficacy compared to injectable biologics along with well‐characterized tolerability issues. Hence, there is still a significant unmet need in the oral treatment of psoriasis, especially for patients with mild to moderate disease and children.18, 19, 20
The inverse RORγ agonist AZD0284, with a mode of action that is anticipated to result in an effect similar to neutralization of IL‐17 and IL‐23, has the potential to become a new oral therapy for patients with psoriasis. Data from this first‐in‐human study support that orally dosed AZD0284 can achieve a high level of systemic RORγ inhibition, while being well tolerated. No safety concerns were raised throughout the investigated dose range and bile acid, triglyceride and glucose data suggest that AZD0284 does not significantly affect the regulation of genes involved in lipid and glucose metabolism. Furthermore, AZD0284 is not likely to cause drug–drug interactions mediated by CYP3A4 induction as the plasma levels of 4β‐OH‐cholesterol remained unchanged throughout dosing.
From a PK perspective, AZD0284 exhibited low intersubject variability in exposure levels, with a consistent half‐life across doses and a modest 2‐fold accumulation on multiple dosing, as expected from the single dose PK. Of note, however, is that the observed plasma exposure of AZD0284 increased in a subproportional manner following both single and multiple dosing, most likely caused by a dose‐dependent decrease in oral bioavailability. AZD0284 was given as a solution containing Captisol to increase solubility, and a possible explanation to the decreasing bioavailability is binding of AZD0284 to Captisol. The ratio between AZD0284 and Captisol concentrations was kept constant with increasing dose, which meant higher concentrations of Captisol were used for higher doses. This may have limited the relative amount of AZD0284 available for absorption.21 To overcome absorption limitations and enable the study of higher plasma exposures, the highest single dose (238 mg) was fractionated into two 119 mg doses given 1 hour apart. By fractionating the dose, oral bioavailability was increased and a similar dose‐normalized AUC as the 108 mg single dose was observed. This allowed for higher doses to be tested in the multiple dosing part of the study.
Food intake resulted in a 33% decrease in C max and a later t max due to delayed absorption but did not alter AUC. These results suggest that AZD0284 could be given together with food without any meaningful alterations in exposure. The results may not be translatable to other formulations of AZD0284, however, and further food interaction studies may be needed as the development of AZD0284 continues.
As anticipated from in vitro experiments, AZD0284 could inhibit ex vivo‐stimulated release of IL‐17A in whole blood. Maximum inhibition was limited to approximately 70% in both positive control samples taken at baseline and samples taken after AZD0284 dosing. The reason for the incomplete inhibition in this assay is not known but may be due to preformed IL‐17A protein or RORγ‐independent mechanisms of IL‐17A synthesis induced by Cytostim.
It is also not known how much inhibition of RORγ is required to translate into clinical efficacy. The two doses of the inverse RORγ agonist VTP‐43742 tested in psoriasis patients were reported to achieve a very high level of inhibition in an ex vivo IL‐17A release assay similar to the one used for AZD0284.22 Both these doses also resulted in a significant reduction in PASI score after 4 weeks of dosing.10 The 100 mg dose of AZD0284 studied in the multiple dosing part of this study is expected to achieve comparable RORγ inhibition as VTP‐43742, but it remains to be tested if this translates to a reduction in PASI score and if also lower levels of inhibition can be enough to achieve clinically relevant effects.
In conclusion, this first‐in‐human study has demonstrated that oral administration of AZD0284 in a Captisol solution results in rapid and dose‐dependent absorption, with low interindividual variability in estimated PK parameters and consistent exposure over 10 days. A high degree of systemic RORγ inhibition was achieved within the investigated dose range and AZD0284 was well tolerated with no safety concerns. These findings support further clinical development of AZD0284 as a treatment of Th17‐mediated disease.
COMPETING INTERESTS
S. Asimus, R. Palmér, H. Forsman, C. Lundin, M. Olsson, R. Pehrson, J. Mo, M. Russell, S. Carlert , D. Close and D. Keeling are employees of AstraZeneca and may own stock or stock options. M. Albayaty is an employee of Parexel. AstraZeneca provided funding to Parexel for the conduct of this study. Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca's data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
CONTRIBUTORS
S.A., R.P., M.O., S.C., and D.K. were involved in design of the study and data analysis. M.A. was the principal investigator for the study and was involved in study design, protocol development, obtaining ethics approval, clinical study conduct, safety evaluations and dose escalation decisions. H.F., C.L., and D.C. contributed to study design and study conduct. R.P., J.M., and M.R. contributed to set up of the IL‐17A assay and data analysis. S.A. and R.P. wrote the first draft of the manuscript. All authors contributed to interpretation of the data and writing or critical review of the manuscript.
Supporting information
Supporting Information Table S1. Adverse events by MedDRA Preferred Term following single doses of AZD0284 or placebo
Supporting Information Table S2. Adverse events by MedDRA Preferred Term following multiple doses of AZD0284 or placebo
ACKNOWLEDGEMENTS
We thank the volunteers and site staff who participated in this study. The first two authors contributed equally to this work.
AstraZeneca funded this study and participated in the study design, data collection, data analysis, data interpretation and the writing of the study report. AstraZeneca reviewed the publication, without influencing the opinions of the authors, to ensure medical and scientific accuracy, and the protection of intellectual property. The corresponding author had access to all data in the study and had the final responsibility for the decision to submit the manuscript for publication.
Asimus S, Palmér R, Albayaty M, et al. Pharmacokinetics, pharmacodynamics and safety of the inverse retinoic acid‐related orphan receptor γ agonist AZD0284. Br J Clin Pharmacol. 2020;86:1398–1405. 10.1111/bcp.14253
PI Statement: The authors confirm that the Principal Investigator for this paper is Muna Albayaty and that she had direct clinical responsibility for the subjects included in the study.
REFERENCES
- 1. Boehncke WH, Schon MP. Psoriasis. Lancet. 2015;386(9997):983‐994. [DOI] [PubMed] [Google Scholar]
- 2. Eberle FC, Brück J, Holstein J, Hirahara K, Ghoreschi K. Recent advances in understanding psoriasis. F1000Res. 2016;5:1‐9. 770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lowes MA, Suarez‐Farinas M, Krueger JG. Immunology of psoriasis. Annual Review of Immunology. 2014;32:227‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cai Y, Fleming C, Yan J. New insights of T cells in the pathogenesis of psoriasis. Cellular & Molecular Immunology. 2012;9(4):302‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gaspari AA, Tyring S. New and emerging biologic therapies for moderate‐to‐severe plaque psoriasis: mechanistic rationales and recent clinical data for IL‐17 and IL‐23 inhibitors. Dermatologic Therapy. 2015;28(4):179‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Olsson RI, Xue Y, von Berg S, et al. Benzoxazepines achieve potent suppression of IL‐17 release in human T‐helper 17 (TH 17) cells through an induced‐fit binding mode to the nuclear receptor RORgamma. ChemMedChem. 2016;11(2):207‐216. [DOI] [PubMed] [Google Scholar]
- 7. Narjes F, Xue Y, von Berg S, et al. Potent and orally bioavailable inverse agonists of RORgammat resulting from structure‐based design. Journal of Medicinal Chemistry. 2018;61(17):7796‐7813. [DOI] [PubMed] [Google Scholar]
- 8. Lin H, Song P, Zhao Y, Xue LJ, Liu Y, Chu CQ. Targeting Th17 cells with small molecules and small interference RNA. Mediators of Inflammation. 2015;2015:1‐11. 290657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kojetin DJ, Burris TP. REV‐ERB and ROR nuclear receptors as drug targets. Nature Reviews. Drug Discovery. 2014;13(3):197‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vitae Pharmaceuticals Achieves Proof‐of‐Concept with First‐in‐Class RORyt Inhibitor in Moderate to Severe Psoriasis. Press release for Vitae Pharmaceuticals, March 16, 2016: https://www.globenewswire.com/news-release/2016/03/16/820582/0/en/Vitae-Pharmaceuticals-Achieves-Proof-of-Concept-with-First-in-Class-RORyt-Inhibitor-in-Moderate-to-Severe-Psoriasis.html.
- 11. Kang HS, Angers M, Beak JY, et al. Gene expression profiling reveals a regulatory role for ROR alpha and ROR gamma in phase I and phase II metabolism. Physiological Genomics. 2007;31(2):281‐294. [DOI] [PubMed] [Google Scholar]
- 12. Chen Y, Coulter S, Jetten AM, Goldstein. Identification of human CYP2C8 as a retinoid‐related orphan nuclear receptor target gene. The Journal of Pharmacology and Experimental Therapeutics. 2009;329(1):192‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Russell M, Mellkvist‐Roos A, Mo J, Hidi R. Simple and robust two‐step ex vivo whole blood stimulation assay suitable for investigating IL‐17 pathway in a clinical laboratory setting. Journal of Immunological Methods. 2018;454:71‐75. [DOI] [PubMed] [Google Scholar]
- 14. Diczfalusy U, Kanebratt KP, Bredberg E, Andersson TB, Böttiger Y, Bertilsson L. 4beta‐hydroxycholesterol as an endogenous marker for CYP3A4/5 activity. Stability and half‐life of elimination after induction with rifampicin. British Journal of Clinical Pharmacology. 2009;67(1):38‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018;46(D1):D1091‐D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alexander SPH, Kelly E, Mathie A, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Introduction and Other Protein Targets. Br J Pharmacol. 2019;176(Suppl 1):S1‐S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Woo YR, Cho DH, Park HJ. Molecular mechanisms and management of a cutaneous inflammatory disorder: psoriasis. International Journal of Molecular Sciences. 2017;18(12):1‐26. 2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sticherling M, Augustin M, Boehncke WH, et al. Therapy of psoriasis in childhood and adolescence ‐ a German expert consensus. Journal der Deutschen Dermatologischen Gesellschaft. 2011;9(10):815‐823. [DOI] [PubMed] [Google Scholar]
- 19. Raut AS, Prabhu RH, Patravale VB. Psoriasis clinical implications and treatment: a review. Critical Reviews in Therapeutic Drug Carrier Systems. 2013;30(3):183‐216. [DOI] [PubMed] [Google Scholar]
- 20. Fotiadou C, Lazaridou E, Ioannides D. Management of psoriasis in adolescence. Adolescent Health, Medicine and Therapeutics. 2014;5:25‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fine‐Shamir N, Beig A, Zur M, Lindley D, Miller JM, Dahan A. Toward successful cyclodextrin based solubility‐enabling formulations for oral delivery of lipophilic drugs: solubility‐permeability trade‐off, biorelevant dissolution, and the unstirred water layer. Molecular Pharmaceutics. 2017;14(6):2138‐2146. [DOI] [PubMed] [Google Scholar]
- 22. McGeehan GM, Palmer SA, Bryson CC, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of VTP‐43742, a RORγt inhibitor, in normal healthy volunteers. Journal of Immunology. 2016;196(1 Supplement):71.4. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Table S1. Adverse events by MedDRA Preferred Term following single doses of AZD0284 or placebo
Supporting Information Table S2. Adverse events by MedDRA Preferred Term following multiple doses of AZD0284 or placebo