

Abstract
Aims
The orexin system is involved in anxiety behaviour and corresponding physiological reactions and constitutes a target for treatment of anxiety disorders. ACT‐539313 is a potent, selective orexin‐1 receptor antagonist being developed for the treatment of anxiety disorders. This first‐in‐human study investigated its single‐dose pharmacokinetics (PK) including food effect, pharmacodynamics (PD), safety and tolerability.
Methods
This double‐blind, placebo‐controlled, randomized study included 40 healthy male subjects. Ascending oral doses of 10–400 mg ACT‐539313 were investigated in 5 dose groups of 8 subjects (of whom 2 received placebo per dose group). At 100 mg, subjects received ACT‐539313 in fasted and fed conditions in a fixed sequential design. PK, PD (objective and subjective measures of sedation and effects on central nervous system), safety and tolerability were assessed.
Results
In fasted conditions, ACT‐539313 was rapidly absorbed (median time to maximum plasma concentration [Cmax] 0.7–3.5 h) and cleared from plasma with a mean terminal half‐life of 3.3–5.7 h across dose levels. A 1.63‐fold (90% confidence interval: 1.26–2.11) increase in Cmax and no change in area under the concentration–time curve extrapolated to infinity was observed under fed compared to fasted conditions. No relevant PD signals were detected except for a trend of reduced saccadic peak velocity around time to Cmax. The most commonly reported adverse events were somnolence and headache. All adverse events were transient and of mild or moderate intensity. No treatment‐related effects on vital signs, clinical laboratory or 12‐lead electrocardiogram were observed.
Conclusions
ACT‐539313 exhibits good safety and tolerability at single doses of up to and including 400 mg that warrant further investigations.
Keywords: first‐in‐human, pharmacodynamics, pharmacokinetics, safety and tolerability, selective orexin‐1 receptor antagonist
What is already known about this subject
Current anxiolytic treatments are often associated with a safety burden and lack of efficacy in a relevant number of patients.
The orexin system is involved in anxiety behaviour and corresponding physiological reactions and constitutes a target for treatment of anxiety disorders.
Limited clinical data of selective orexin‐1 receptor antagonists are available.
What this study adds
The terminal half‐life of ACT‐539313 was 3.3–5.7 h across dose levels, while area under the concentration–time curve extrapolated to infinity increased approximately dose‐proportionally and maximum plasma concentration less than dose‐proportionally.
No relevant treatment‐related effects on sedation or activation of the central nervous system were observed.
ACT‐539313 exhibits good safety and tolerability after single‐dose administration in healthy male subjects up to 400 mg.
1. INTRODUCTION
The orexin peptides (orexin‐A and orexin‐B) were discovered at the end of the last century and act as endogenous ligands on the orexin‐1 and ‐2 receptors (OX1 and OX2 receptors).1, 2 The nomenclature of the peptides and receptors conforms with the IUPHAR/BPS guidelines classification.3 The orexin‐containing neurons are located in the perifornical area of the lateral hypothalamus, which mediates survival‐related processes, such as circadian rhythm, arousal, fight‐flight responses or feeding behaviour.4
While dual orexin receptor antagonists (DORAs) have been shown to promote sleep by inhibiting the output of wake active neurons,5, 6, 7, 8, 9, 10 antagonism of the OX2 receptor alone is sufficient to prolong sleep in rodents11, 12 and humans.13, 14 In addition, evidence has emerged that demonstrates involvement of the orexin signalling via OX1 receptors in anxiety‐like responses in rodents.11, 15, 16, 17 Orexin neurons have strong projections to anatomical sites such as the amygdala, the bed nucleus of the stria terminalis and the periaqueductal grey, which are well known centres of stress and anxiety signal integration.18, 19
Consistent with these anatomical connections, stimulation of orexin peptide expression resulted in increased anxiety‐related behaviour20 and associated physiological reactions in rodents.21 Johnson et al.15 observed increased orexin concentrations in cerebrospinal fluid in subjects with panic disorder compared to controls. Subsequently, first encouraging results in rats showed that selective blockage of the OX1 receptor resulted in attenuated panic‐ and anxiety‐associated responses in CO2‐induced panic models17, 22 as well as a decrease in conditioned fear reactions.23
Taken together, this evidence indicates that the orexin system represents a novel target for treatment of anxiety disorders. Although a broad spectrum of anxiolytic drugs with various modes of action is available, several drawbacks remain. Antidepressants, such as selective serotonin/serotonin‐noradrenaline reuptake inhibitors, which are widely used to treat anxiety disorders, initially cause increases in anxiety before showing efficacy after several weeks of treatment and are associated with a range of adverse effects such as weight gain, sleep disturbances or sexual dysfunction24 as well as withdrawal syndromes.25 Tricyclic antidepressants or monoamine oxidase inhibitors, shown to have similar efficacy against anxiety as selective serotonin reuptake inhibitors, both come with a significant safety burden.25 While benzodiazepines are immediately effective, they can cause sedation, cognitive impairment, and abuse and dependence liabilities.24 Overall, still up to 50% of patients remain unresponsive to treatment.25 Therefore, there is an unmet medical need for anxiolytic drugs, especially with an advantageous safety and tolerability profile.
ACT‐539313 ((4‐methyl‐2‐[1,2,3]triazol‐2‐yl‐phenyl)‐[(R)‐3‐(3‐[1,2,3]triazol‐2‐yl‐benzyl)‐morpholin‐4‐yl]‐methanone) is an orally active, reversible, selective OX1 receptor antagonist (1‐SORA) that readily crosses the blood–brain barrier. It shows antagonism at the human OX1 receptor in a Ca2+‐release assay, with an apparent equilibrium dissociation constant (Kb) of 0.69 nM and has similar potency at human and different animal orexin receptors. The apparent Kb at the human OX2 receptor is 42 nM, thus the potency is approximately 60‐fold lower.
The compound has anxiolytic effects in animal models of fear and stress‐related disorders. In a social interaction stress rat model, ACT‐539313 dose‐dependently reduced behavioural and physiological stress responses, such as locomotion, heart rate, body temperature, and mean arterial pressure. CYP3A4 is the major cytochrome P450 enzyme involved in the in vitro metabolism of ACT‐539313 in man. So far, limited clinical data on 1‐SORA in humans are available.26, 27, 28 Therefore, exploration of ACT‐539313 will contribute to the understanding of the role of the orexin system in subjects with anxiety disorders.
Here, we report the results of the first administration of ACT‐539313 to healthy male subjects in a single‐ascending dose study including tolerability and safety assessments, PK in plasma, and assessment of potential sedative and other central nervous system (CNS) effects using a PD test battery.
2. METHODS
2.1. Subjects
Healthy male subjects, aged between 18 and 55 years and with a body mass index (BMI) between 18 and 29.9 kg m−2, were recruited in this study. Eligibility of the study participants was assessed at a screening examination, which took place between 3 and 21 days prior to first drug administration. The subjects were in good health, as assessed by medical history, physical examination, 12‐lead electrocardiogram (ECG) and clinical laboratory tests (including haematology, clinical chemistry, urinalysis, virus serology and drug screening). Vital signs had to be within the normal range, defined as 90–140 mmHg for systolic blood pressure, 50–90 mmHg for diastolic blood pressure, and 50–90 beats min−1 for pulse rate. History of narcolepsy and cataplexy were among the exclusion criteria.
Prior to any study procedure, written informed consent was obtained from each participant after adequate explanation of the objectives, methods and potential hazards of the study. The ethics committee (Ethik‐Kommission des Landes Berlin, Germany) and the German Health Authorities (BfArM) approved the protocol (EudraCT number: 2015–003059‐23). The study adhered to the Declaration of Helsinki and was conducted according to good clinical practice.
2.2. Study conduct
This was a single‐centre, double‐blind, placebo‐controlled, single‐ascending oral dose study including a nested cross‐over food‐effect part. Each dose level was investigated in a separate group of 8 subjects (6 on active drug, 2 on placebo). ACT‐539313 doses were 10, 30, 100, 200 and 400 mg, and effects were compared to placebo. The study treatment was administered as capsules of 10 and 100 mg. A starting dose of 10 mg was selected based on the PD properties in animals and by comparing the plasma exposure in animals with predicted human exposure derived from a physiologically based PK model. The predicted area under the plasma concentration–time curve (AUC) and the maximum plasma concentration (Cmax) were 987 ng h/mL and 198 ng/mL, while the AUC and Cmax at the lowest‐observed‐adverse‐effect level in rats were 28 100 ng h/mL and 5250 ng/mL, respectively. The resulting safety margins of AUC and Cmax expressed as the ratio of observed levels in the rat and predicted values in human are 28 and 27.
For the monkey, the other toxicological species, the safety margins were larger. The study treatment was administered orally to subjects under fasted conditions except for the 100‐mg cohort. At this dose level, the first treatment period under fasted conditions ended after the observation period of 96 hours on Day 5. Following a wash‐out period of approximately 6 weeks, and review of tolerability and safety data from the next higher dose level (200 mg), a second treatment period under fed conditions was performed after intake of a high‐fat and high‐calorie standardized breakfast. Dosing was done in sitting position.
Safety and tolerability were evaluated by monitoring of adverse events (AEs), clinical laboratory variables, vital signs, 12‐lead ECG, body weight, body temperature and physical examination. Blood pressure, pulse rate and ECGs were recorded from subjects in the supine position after having rested for 5 minutes. Venous blood samples were taken at regular time points up to 96 hours after treatment administration for measuring ACT‐539313 plasma levels and plasma protein binding. Subjects were in the clinic until 48 hours after dosing.
The decision to proceed with the next higher dose level was taken after review of the safety, tolerability, PD and PK data of the previous dose level(s).
2.3. Bioanalysis
Concentrations of ACT‐539313 were measured using a validated liquid chromatography coupled to tandem mass spectrometry (LC–MS/MS) method. The internal standard, a stable isotope‐labelled analogue of ACT‐539313, was dissolved in acetonitrile/dimethyl‐sulfoxide (1:1 volume:volume) and added to the plasma sample. After vortex‐mixing (Genie 2, VWR, Dietikon, Switzerland), the samples were centrifuged (Eppendorf 5810R, Vaudaux‐Eppendorf, Schönenbuch, Switzerland) for 20 minutes at 3250 g and 4°C. The supernatant was injected into the high‐performance LC column.
The chromatographic system consisted of a pump (LC‐20 AD, Shimadzu Schweiz GmbH, Reinach, Switzerland), an analytical column (Symmetry C18, Waters Corporation, Milford, MA, USA; particle size 3.5 μm, 2.1 mm × 50 mm), and a gradient elution with mobile phases consisting of water containing 0.1% formic acid and methanol.
ACT‐539313 concentrations were determined with a mass spectrometer (API 4000, SCIEX, Framingham, MA, USA) operating in electrospray ionization positive‐ion mode.
The method was linear in the concentration range 1.0–3000 ng mL−1. The limit of quantification was 1.0 ng mL−1. The performance of the method was monitored using quality control samples. Inter‐batch precision (coefficient of variation in % [CV%]) was ≤8.0%, whereas accuracy ranged from −1.4 to +0.7%.
Plasma protein binding was determined using a rapid equilibrium dialysis device (Thermo Fisher Scientific, Waltham, MA, USA). Plasma aliquots were subjected to equilibrium dialysis against 100 mmol/L phosphate‐buffered saline for 3 h at 37°C. Concentration analysis was performed by LC–MS/MS.
2.4. PK analysis
PK parameters were determined by model‐independent methods (WinNonlin Version 6.4; Pharsight, Mountain View, CA, USA) whereby AUC was calculated using the linear trapezoidal method.29
2.5. PD assessments
A battery of objective and subjective PD tests (saccadic eye movements, unstable tracking, simple reaction time, critical flicker fusion [CFF], visual analogue scale [VAS] Bond and Lader, VAS Bowdle, Karolinska sleepiness scale [KSS]) was performed pre‐dose and at regular time points up to 12 hours after drug administration under fasted conditions to assess potential sedative and CNS effects of ACT‐539313. A PD training session was included on Day −1. All tests were presented on a computer screen using the Berisoft software (Berisoft US LLC, Redwood City, CA, USA), with exception of the saccadic peak velocity assessment for which the light stimulus was projected on a screen with the Compumedics STIM 2 systems (Compumedics Ltd, Abbotsford, Victoria, Australia), and the CFF for which the Vienna Test System label FLIM (Schuhfried GmbH, Mödling, Austria) was used.
Saccadic peak velocity is one of the most sensitive variables for measuring sedation. The effects of 1 night of sleep deprivation are detectable by a decrease in peak velocity of 9–10%.30, 31, 32, 33 In a quiet room with dimmed illumination, saccadic eye movements were recorded using surface electrodes placed on the skin lateral of the eyes and connected to NuAmps amplifiers and Compumedics acquisition and analysis software as described by Richard et al.33 A stimulus was projected at variable intervals at 15–20° of the eyeball rotation. Peak velocity (°/s), the highest velocity reached during the saccade, was measured.
The unstable tracking test was performed using customized equipment and software as described previously.33, 34, 35 In this adaptive test, subjects were instructed to keep an unstable bar in the middle of a horizontal plane by counteracting or reverse its movements with the aid of a computer control stick. If the unstable bar hit the edges of the plane, the subject had to start again. If performed well, the signal instability increased. The average signal instability (mean λ) mastered by the subject was the primary outcome of the test.
The simple reaction time test was performed to assess the psychomotor function. For 5 minutes, subjects were required to press a key when a red square appeared on screen and the mean reaction time in ms was recorded.33, 36
The CFF test is a measure of the CNS activation.37 Subjects are required to discriminate flickering from constant (i.e. fusion) light, and vice versa, in a set of light‐emitting diodes flashing with in‐ or decreasing frequency. The frequency at which the impression changes from flicker to fusion (ascending frequency starting at 20 Hz) or from fusion to flicker (descending frequency starting from 60 Hz) is known as the flicker fusion threshold. Decreasing values of flicker fusion frequency indicate an increase in sedation.
The VAS Bond and Lader38 has previously been applied to assess sedative effects of drugs.7, 30, 39 Three factors corresponding to alertness, mood and calmness can be derived.
Psychedelic effects, such as internal and external perception, feeling high and feeling drowsy, were monitored by an adapted version of the VAS originally described by Bowdle et al.40 KSS was used to assess subjects' subjective sleepiness using a 9‐point scale.41
2.6. Statistical analysis
Safety and tolerability variables were summarized descriptively by dose group. Subjects treated with placebo were pooled for the analyses.
PK parameters were summarized with geometric mean and 95% confidence interval (CI) or, for time to Cmax (tmax), with median and range. Dose proportionality was explored as previously described.42 Food effect was explored by a mixed‐effects model using log‐transformed values of the PK parameters as dependent variable, treatment period (fed/fasted) as fixed effect, and subject as random effect. Geometric mean ratios (test/reference) and 90% CI are calculated from the corresponding back‐transformed least‐square means for treatment period of the mixed‐effects models.
Exploratory analyses were performed to compare the effects of different treatments vs placebo on PD variables. Mean changes from baseline (saccadic eye movements, unstable tracking, simple reaction time, CFF, VAS, KSS) were explored using mixed models based on least square means of absolute values with dose, time, and dose by time as fixed effects, subject as random effect, and the average baseline value as covariate. Treatments effects were analyzed as the contrasts between placebo and the different ACT‐539313 doses.
2.7. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY,43 and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20.44, 45
3. RESULTS
Forty healthy male subjects with a mean age of 41.8 years (range: 23–55 years) and a mean BMI of 25.0 kg m−2 (range: 20.3–29.2 kg m−2) participated in this study (Supplementary Table S1). The first subject was screened on 02 February 2016 and the study duration was 4 months. The different dose groups were well balanced for all the demographic variables and no subject withdrew prematurely from the study.
3.1. PK
ACT‐539313 concentration–time profiles across the studied doses are presented in Figure 1 (arithmetic mean ± standard deviation). PK parameters from the noncompartmental analysis are summarized in Table 1. Under fasted conditions, ACT‐539313 was absorbed with a median tmax ranging from 0.7 to 1.0 hours at low doses (i.e. 10–30 mg) and 2.0 to 3.5 hours at doses of ≥100 mg. The elimination phase was characterized by an apparent mean terminal half‐life (t1/2) of 3.3– 5.7 hours independent of dose. The observed increase in AUC extrapolated to infinity (AUC0‐inf) was approximately dose‐proportional, whereas Cmax increased dose‐proportionally from 10 to 30 mg and less than proportionally thereafter (Figure 3). At all dose levels, > 99% of the compound was bound to plasma protein at 2 hours postdose. The fraction unbound increased from 0.26% at the lowest dose to 0.76% at the highest dose.
Figure 1.
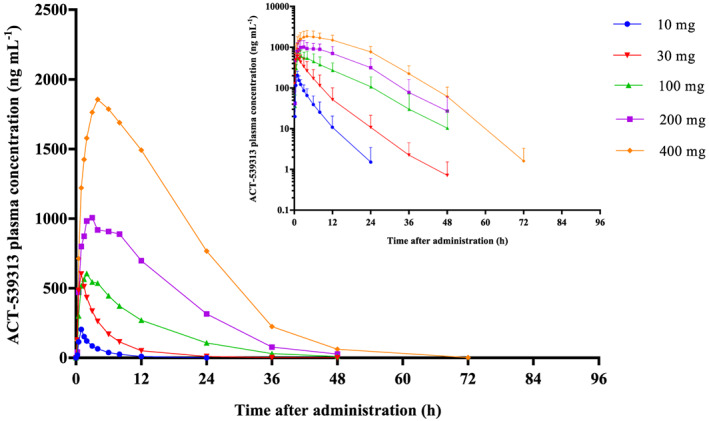
Arithmetic mean plasma concentration–time profiles of ACT‐539313 in healthy male subjects (n = 6) after single‐dose administration of 10, 30, 100, 200 and 400 mg (linear scale) under fasted conditions. The inset shows the profiles on a semilogarithmic scale. For clarity, standard deviation is only displayed for curves on a semilogarithmic scale
Table 1.
Pharmacokinetic parameters of ACT‐539313 in healthy male subjects after administration of a single dose of 10, 30, 100 (fasted and fed), 200 and 400 mg
| Dose (mg) | n | Cmax (ng/mL) | tmax (h) | AUC0‐inf (ng h/mL) | t1/2 (h) | Fraction unbound (%) |
|---|---|---|---|---|---|---|
| 10 | 6 | 198.3 (149.2, 263.5) | 1.0 (1.0, 1.0) | 677.0 (392.3, 1168.4) | 3.3 (2.3, 4.9) | 0.26 (0.21, 0.32) |
| 30 | 6 | 580.6 (368.7, 914.2) | 0.7 (0.5, 1.0) | 2621.4 (1375.2, 4997.0) | 5.6 (4.1, 7.7) | 0.29 (0.21, 0.38) |
| 100 (fasted) | 6a | 651.8 (439.6, 966.3) | 2.0 (1.5, 12.0) | 7710.2 (4715.4, 12607.1) | 5.0 (3.7, 7.0) | 0.4 (0.32, 0.51) |
| 100 (fed) | 6a | 1064.8 (730.1, 1553.1) | 3.0 (2.0, 4.0) | 7987.3 (4448.7, 14340.6) | 4.0 (3.0, 5.5) | ND |
| 200 | 6 | 1142.9 (739.9, 1765.3) | 2.5 (1.0, 8.0) | 17980.9 (11354.5, 28474.4) | 5.7 (4.6, 7.1) | 0.54 (0.39, 0.77) |
| 400 | 6 | 2071.5 (1545.3, 2776.9) | 3.5 (2.0, 11.8) | 39407.8 (28957.1, 53630.2) | 5.2 (4.1, 6.6) | 0.76 (0.65, 0.87) |
AUC0‐inf, area under the concentration‐time curve from zero to infinity; CI, confidence interval; Cmax, maximum plasma concentration; ND, not done; tmax, time to maximum concentration; t1/2, terminal half‐life.
Data are geometric means (and 95% CI) or, for tmax, the median (and range).
The food effect was evaluated in a cross‐over design, i.e., the same subjects that received 100 mg ACT‐539313 under fasted conditions also received the treatment under fed conditions (after a wash‐out period of at least 6 weeks).
Figure 3.
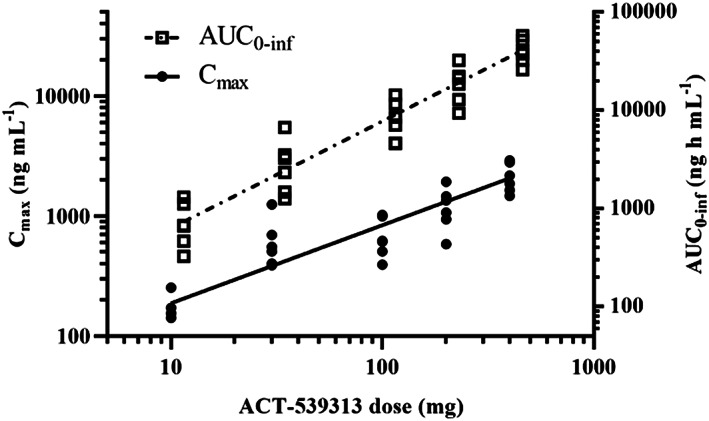
Graphical representation of dose proportionality of area under the concentration–time curve extrapolated to infinity (AUC0‐inf) and the less than dose‐proportional increase of maximum plasma concentration (Cmax) for ACT‐539313
A 63% increase in Cmax was observed in the presence of food (ratio of geometric means fed/fasted [90% CI], 1.63 [1.26–2.11]), whereas AUC0‐inf remained unchanged after a high‐fat and high‐calorie meal (ratio of geometric means fed/fasted [90% CI], 1.04 [0.89–1.21]). Absorption of ACT‐539313 was slightly delayed by food as indicated by a value for tmax of 3 hours when compared to 2 hours under fasted conditions (median difference [90% CI], 0.5 hours [−4.49–2.45]), and elimination rate was marginally increased with a t1/2 of 4.0 hours under fed and 5.0 hours (ratio of geometric means fed/fasted [90% CI], 0.80 [0.60–1.06]) under fasted conditions (Figure 2).
Figure 2.
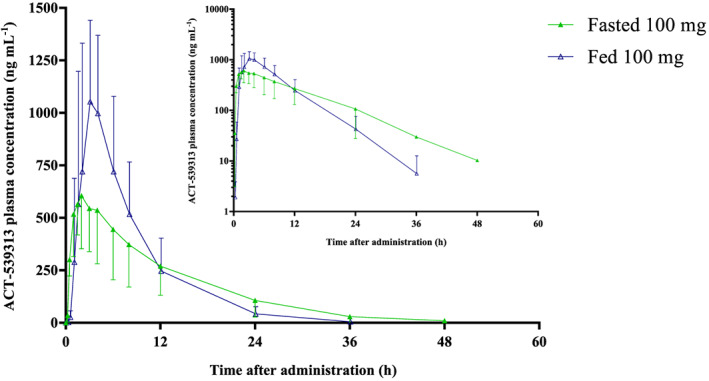
Arithmetic mean (± standard deviation) plasma concentration–time profiles of ACT‐539313 in healthy male subjects (n = 6) after single‐dose administration of 100 mg (linear scale) under fasted and fed conditions. The inset shows the profiles on a semilogarithmic scale
3.2. PD
Figure 4 shows the time course for the mean change from baseline after ACT‐539313 and placebo on saccadic peak velocity, unstable tracking, simple reaction time, and subjective alertness. The estimated treatment effect assessed during the 12‐hour period after dosing as compared to placebo revealed that treatment with 200 and 400 mg ACT‐539313 resulted in a statistically significant decrease of saccadic peak velocity with a maximum effect 3 hours postdose (−47 and −43°/s or −9.2 and −8.3% for 200 and 400 mg, P = .0190 and .0243, respectively). The effects returned to baseline within 12 hours after drug intake. Overall, no relevant changes in unstable tracking results were detected (Table 2). The maximum mean decrease in tracking performance was 0.11 observed 3 hours postdose in the 400 mg‐dose group, corresponding to a decrease of <5% (P = .3158). No consistent changes were seen for the simple reaction time and the alertness score.
Figure 4.
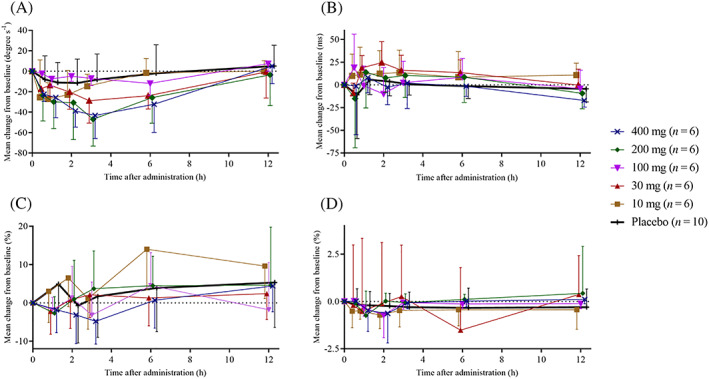
Time course of the mean (± standard deviation) change from baseline for (A) saccadic peak velocity, (B) simple reaction time, (C) unstable tracking performance and (D) alertness after administration of single doses of ACT‐539313 under fasted conditions (10, 30, 100, 200, 400 mg) or placebo
Table 2.
Maximum mean changes from baseline (CFB) in pharmacodynamic tests
| Dose (mg) | n | Saccadic peak velocity (°/s) | Unstable tracking, mean λ | Simple reaction time (ms) | VAS alertness (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Baseline | CFB | Baseline | CFB | Baseline | CFB | Baseline | CFB | ||
| 10 | 6 | 522.0 (93.1) | −27.1 (16.0) | 2.15 (0.37) | 0.14 (0.16) | 334.3 (28.8) | 13.5 (24.7) | 50.62 (0.81) | −0.72 (0.72) |
| 30 | 6a | 491.3 (52.8) | −28.9 (22.0) | 2.19 (0.29) | 0.05 (0.16) | 297.5 (13.1) | 24.5 (23.1) | 50.41 (2.59) | −1.52 (3.32) |
| 100 (fasted) | 6 | 500.4 (48.2) | −11.9 (14.4) | 2.27 (0.54) | 0.10 (0.19) | 339.3 (21.3) | 19.0 (36.7) | 50.07 (0.12) | −0.72 (1.20) |
| 200 | 6 | 509.6 (76.6) | −46.9 (26.4) | 1.95 (0.14) | 0.09 (0.33) | 330.1 (36.5) | −15.2 (54.1) | 50.06 (0.30) | −0.74 (1.28) |
| 400 | 6 | 518.2 (74.9) | −43.2 (22.6) | 2.55 (0.67) | 0.11 (0.14) | 315.6 (31.1) | −17.1 (7.4) | 50.02 (0.11) | −0.63 (1.56) |
| Placebo | 10 | 502.6 (53.9) | −11.5 (23.5) | 2.33 (0.36) | 0.12 (0.25) | 335.1 (34.5) | −10.9 (48.3) | 50.36 (0.83) | −0.34 (1.05) |
Data are presented as mean (standard deviation) for baseline and maximum CFB.
n = 4 for simple reaction time due to missing baseline assessments.
Moreover, ACT‐539313 had no relevant effects on CFF, mood, calmness, subjective internal and external perception, feeling drowsy, feeling high, and KSS (data not shown).
3.3. Safety and tolerability
No serious or severe AE occurred, and all AEs resolved without sequelae. ACT‐539313 was well tolerated and there was no increase in number and intensity of AEs with increasing doses.
In total, 13 AEs were reported by 11 subjects (27.5%). Under fasted conditions, of the 30 subjects treated with ACT‐539313, 9 (30%) subjects reported a total of 11 AEs. None of the 10 subjects treated with placebo under fasted conditions reported AEs.
After administration of 100 mg ACT‐539313 under fed conditions, 1 subject (16.7%) reported 1 AE, while 3 (50%) subjects reported a total of 4 AEs under fasted conditions. The only AEs reported by more than 1 subject were somnolence and headache reported by 5 (16.7%) and 3 (10.0%) subjects, respectively, on ACT‐539313 (Table 3). While all 3 cases of headache were reported at least 7 hours after study treatment administration, all 5 cases of somnolence had an onset 30 minutes after ACT‐539313 administration. Out of 13 AEs, 10 were related to study treatment, of which 8 were mild and 2 were moderate in intensity. All of them resolved within 13 hours after onset. Both moderate AEs were headache events, had the longest duration (of 5 and 13 h), and were reported for the 10 and 100 mg doses under fed conditions, respectively. Overall, no apparent dose relationship in number, intensity or duration of AEs was observed.
Table 3.
Summary of adverse events (AEs) by treatment reported after single‐dose administration of ACT‐539313 (including adverse events judged to be unrelated to study drug)
| Treatment | ACT‐539313 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 10 mg | 30 mg | 100 mg | 200 mg | 400 mg | Placebo | Total | |||
| (n = 6) | (n = 6) | (n = 6) | (n = 6) | (n = 6) | (n = 10) | (n = 40) | |||
| Condition | Fasted | Fed | Fasted | Fed | |||||
| Total subjects with AEs | 2 | 1 | 3 | 1 | 1 | 2 | ‐ | 1 | 11 |
| Total number of AEs | 2 | 1 | 4 | 1 | 1 | 3 | ‐ | 1 | 13 |
| Number of subjects reporting the AEs | |||||||||
| Somnolence | ‐ | ‐ | 2 | ‐ | 1 | 2 | ‐ | ‐ | 5 |
| Headache | 1 | 1 | ‐ | 1 | ‐ | ‐ | ‐ | ‐ | 3 |
| Hypervigilance | ‐ | ‐ | 1 | ‐ | ‐ | ‐ | ‐ | ‐ | 1 |
| Restlessness | ‐ | ‐ | ‐ | ‐ | ‐ | 1 | ‐ | ‐ | 1 |
| Toothache | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1 | 1 |
| Medical device site irritation | 1 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 1 |
| Rhinitis | ‐ | ‐ | 1 | ‐ | ‐ | ‐ | ‐ | ‐ | 1 |
There were no clinically relevant effects of ACT‐539313 on vital signs, ECG recordings, body weight, body temperature, clinical laboratory variables or physical examination.
4. DISCUSSION
This is the first manuscript reporting on administration of a 1‐SORA compound to human subjects. In this study, ACT‐539313 was administered for the first time to healthy subjects, exploring safety, tolerability, PK, and PD properties.
The PK profile of ACT‐539313 was characterized by a fast absorption with a median tmax of 0.7 to 3.5 hours. Afterwards, plasma levels decreased with a t1/2 of 3.3–5.7 hours across dose levels. Peak exposure Cmax increased in a dose‐proportional manner from 10 to 30 mg only. Thereafter, the increase was less than dose‐proportional, which might be explained by the limited solubility of ACT‐539313 in aqueous solutions (practically insoluble at pH 6.5). Overall AUC0‐inf, however, was approximately dose‐proportional.
The effect of food on the PK of ACT‐539313 was assessed according to recommendations of the FDA.46 In the presence of food, the absorption of ACT‐539313 was delayed as indicated by a median tmax that shifted from 2 hours under fasted to 3 hours under fed conditions and a t1/2 decreased from 5 hours under fasted to 4 hours under fed conditions. When compared to the fasted state, exposure as based on AUC0‐inf was not affected by food intake whereas Cmax was increased by 63%. Taken together, these observations could indicate an apparent increase in the rate but not the extent of absorption of ACT‐539313 in the presence of food. When compared to the bioequivalence limit of a geometric mean ratio (test/reference) of exposure of 0.8–1.25, the geometric mean ratio and its 90% CI for AUC remained within the limits, confirming the absence of a food effect on exposure.46 For Cmax and t1/2, the geometric mean ratios and their 90% CI were not within bioequivalence limits. The presence of a food effect on tmax of ACT‐539313 was not confirmed by the analysis of the median difference between fed and fasted state due to considerable variability. A faster solubilization of lipophilic compounds after a high‐fat meal could have caused the enhanced rate of absorption and, subsequently, an increase in Cmax.47
The ACT‐539313 fraction unbound to plasma protein was <1% in all dose groups. The increase of dose was associated with an increase of the free fraction from 0.26 to 0.76%. Based on in vitro data, concentration‐dependent plasma protein binding was observed across several species and was, therefore, expected.
A series of PD tests were performed in order to explore the effects of ACT‐539313 on various CNS functions. The VAS Bond and Lader and the VAS Bowdle were implemented to assess potential changes in subjective feelings of sedation or psychedelic effects.38, 40 No remarkable changes were observed for any score except for the changes in saccadic eye movements. Saccadic peak velocity, as an objective measure of sedation,32 was significantly decreased at the highest 2 dose levels compared to placebo, with the lowest values observed at 3 hours postdose, which is in line with the observed median tmax of the compound at higher doses. However, the largest decrease from baseline was, with 47°/s, still smaller than decreases up to approximately 87 or 60°/s observed for the DORAs daridorexant and almorexant at clinically relevant doses after single‐dose administration.7, 48 In line with the frequent reporting of somnolence, a significant feature of DORAs is the distinct decrease in alertness‐assessing PD variables. Besides saccadic peak velocity, simple reaction time was observed to increase dose‐dependently after single‐dose administration of the DORA suvorexant,49 while no dose dependency could be established for ACT‐539313, as reaction times even decreased at the highest dose level. Moreover, for DORAs daridorexant and almorexant, decreases of 15.5% in changes from baseline and 6.1% in least square means compared to placebo after single‐dose administration, respectively, were reported for adaptive tracking,7, 48 whereas the largest corresponding decrease from baseline for ACT‐539313 was <5%. Regarding subjective assessments, subjects felt up to 20% less alert, based on the VAS alertness score, following single‐dose administration of the DORAs daridorexant and almorexant7, 48 or following the first dose of the selective OX2 receptor antagonist (2‐SORA) seltorexant,14 but the score remained unchanged upon ACT‐539313 administration (Figure 4).
Overall, these effects confirm CNS activity of the 1‐SORA ACT‐539313 without profound effects on sedation, vigilance or visuo‐motor coordination, which have been described for DORAs or 2‐SORAs. Furthermore, saccadic peak velocity has also been considered as a biomarker for the anxiolytic component of benzodiazepines and newly developed compounds with potential anxiolytic effects.50
Single‐dose administrations of ACT‐539313 were well tolerated. The maximum tolerated dose was not reached. No severe or serious AEs were reported and no subject was prematurely withdrawn from the study. The intensity of the AEs was mostly mild with only 2 moderate AEs reported. All AEs resolved without sequelae. Somnolence and headache were reported most frequently. Similarly, in a study with JNJ‐61393215 (a high affinity and potent 1‐SORA) the most common treatment‐emergent AEs in all treatment groups were somnolence and headache. The overall incidence of somnolence following single‐dose administrations was approximately 15% for subjects on active drug which is comparable to the 17% observed for ACT‐539313. However, the proportions of subjects who manifested somnolence appeared comparable between the placebo and JNJ‐61393215 treatment arms.51
As the orexin system is known to promote wakefulness,52 similar AEs have been reported for DORAs7, 10, 48, 53, 54 or 2‐SORAs.13, 14 However, the contribution of the OX2 receptor in maintaining the sleep–wake cycle is larger than of the OX1 receptor,55 which explains the low number of sleep‐associated AEs for the 1‐SORAs ACT‐539313 and JNJ‐61393215 compared to DORAs and 2‐SORAs: while somnolence was reported by 17% of subjects after a single dose of ACT‐539313 or 18% after single and multiple doses of JNJ‐61393215,51 33 and 53% of subjects reported somnolence after ascending single oral dose administrations of the DORAs almorexant7 and daridorexant,48 whereas following multiple‐dose administration of suvorexant56 57% of subjects reported somnolence. Furthermore, 87% of subjects reported somnolence after the first administration of the 2‐SORA seltorexant in a multiple‐dose trial.14 Based on the apparent potencies of ACT‐539313 at OX1 and OX2 receptors, a contribution of OX2 receptor antagonism, however, to the observed somnolence cannot be completely excluded.
Neither the number of subjects reporting any AEs, the total number, the intensity nor the duration of AEs indicated a dose‐dependency. The tolerability and safety profile in the presence of food showed a decrease of the number of AEs compared to fasted conditions, however, the good tolerability profile at higher doses indicates that this finding may be linked to the small sample size. No treatment‐related pattern was detected that suggested an effect of single doses of ACT‐539313 on vital signs, clinical chemistry, haematology, urinalysis, body weight, body temperature, ECG variables or morphology.
The PK profile of ACT‐539313 is compatible with a twice daily dosing regimen regardless of food intake. As such, multiple‐dose studies have in the meantime been conducted, of which the results are to be reported separately. Together with the good safety and tolerability profile and PD properties, further investigations are warranted to assess ACT‐539313 as the first 1‐SORA for the treatment of anxiety disorders.
COMPETING INTERESTS
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author). Actelion Pharmaceuticals Ltd, the predecessor of Idorsia Pharmaceuticals Ltd, provided funding for this clinical trial. At the time of the study, P.K. and J.D. were employees of Actelion Pharmaceuticals Ltd and own stocks and stock options of Idorsia Pharmaceuticals Ltd. M.O. is an employee of Idorsia Pharmaceuticals Ltd. R.K. and G.G. were employees of Parexel International GmbH, respectively. There are no other relationships or activities that could appear to have influenced the submitted work. Parexel International GmbH received financial compensation for the clinical conduct.
CONTRIBUTORS
P.K. and J.D. contributed to the study design and protocol. G.G. and R.K. conducted the study. P.K. directed the study at Actelion/Idorsia. P.K., M.O., R.K. and J.D. analysed/interpreted the data, critically revised the manuscript, agreed on the content, and approved the final version for publication.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supporting information
TABLE S1 Demographic characteristics of subjects per cohort and overall
ACKNOWLEDGEMENTS
We thank Anne‐Helene Clugery for project management Zsofia Molnar for monitoring, Susanne Globig and Stéphane Delahaye for bioanalytical assessments, and Mariya Antonova, Aprova, the predecessor of Aixial, Brno, Czech Republic, for the statistical analysis.
Kaufmann P, Ort M, Golor G, Kornberger R, Dingemanse J. First‐in‐human study with ACT‐539313, a novel selective orexin‐1 receptor antagonist. Br J Clin Pharmacol. 2020;86:1377–1386. 10.1111/bcp.14251
The authors confirm that the Principal Investigator for this study is Georg Golor and that he had direct clinical responsibility for subjects.
REFERENCES
- 1. de Lecea L, Kilduff TS, Peyron C, et al. The hypocretins: hypothalamus‐specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95(1):322‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sakurai T, Amemiya A, Ishii M, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein‐coupled receptors that regulate feeding behavior. Cell. 1998;92(4):573‐585. [DOI] [PubMed] [Google Scholar]
- 3. Alexander SP, Christopoulos A, Davenport AP, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol. 2017;174(Suppl 1):S17‐S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chase MH. A unified survival theory of the functioning of the hypocretinergic system. J Appl Physiol. 2013;115(7):954‐971. [DOI] [PubMed] [Google Scholar]
- 5. Black J, Pillar G, Hedner J, et al. Efficacy and safety of almorexant in adult chronic insomnia: a randomized placebo‐controlled trial with an active reference. Sleep Med. 2017;36:86‐94. [DOI] [PubMed] [Google Scholar]
- 6. Hoch M, van Gorsel H, van Gerven J, Dingemanse J. Entry‐into‐humans study with ACT‐462206, a novel dual orexin receptor antagonist, comparing its pharmacodynamics with almorexant. J Clin Pharmacol. 2014;54(9):979‐986. [DOI] [PubMed] [Google Scholar]
- 7. Hoever P, de Haas S, Winkler J, et al. Orexin receptor antagonism, a new sleep‐promoting paradigm: an ascending single‐dose study with almorexant. Clin Pharmacol Ther. 2010;87:593‐600. [DOI] [PubMed] [Google Scholar]
- 8. Parks GS, Warrier DR, Dittrich L, et al. The dual Hypocretin receptor antagonist Almorexant is permissive for activation of wake‐promoting systems. Neuropsychopharmacology. 2016;41(4):1144‐1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuriyama A, Tabata H. Suvorexant for the treatment of primary insomnia: a systematic review and meta‐analysis. Sleep Med Rev. 2017;35:1‐7. [DOI] [PubMed] [Google Scholar]
- 10. Hoever P, Dorffner G, Benes H, et al. Orexin receptor antagonism, a new sleep‐enabling paradigm: a proof‐of‐concept clinical trial. Clin Pharmacol Ther. 2012;91(6):975‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dugovic C, Shelton JE, Aluisio LE, et al. Blockade of orexin‐1 receptors attenuates orexin‐2 receptor antagonism‐induced sleep promotion in the rat. J Pharmacol Exp Ther. 2009;330(1):142‐151. [DOI] [PubMed] [Google Scholar]
- 12. Mang GM, Dürst T, Bürki H, et al. The dual orexin receptor antagonist Almorexant induces sleep and decreases orexin‐induced locomotion by blocking orexin 2 receptors. Sleep. 2012;35(12):1625‐1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brooks S, Jacobs GE, de Boer P, et al. The selective orexin‐2 receptor antagonist seltorexant improves sleep: an exploratory double‐blind, placebo controlled, crossover study in antidepressant‐treated major depressive disorder patients with persistent insomnia. J Psychopharmacol. 2019;33:202‐209. [DOI] [PubMed] [Google Scholar]
- 14. van der Ark PD, Golor G, van Nueten L, Nandy P, de Boer P. Multiple daytime administration of the selective orexin‐2 receptor antagonist JNJ‐42847922 induces somnolence in healthy subjects without residual central effects. J Psychopharmacol. 2018;32(12):1330‐1340. [DOI] [PubMed] [Google Scholar]
- 15. Johnson PL, Truitt W, Fitz SD, et al. A key role for orexin in panic anxiety. Nat Med. 2010;16(1):111‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson PL, Samuels BC, Fitz SD, et al. Orexin 1 receptors are a novel target to modulate panic responses and the panic brain network. Physiol Behav. 2012;107(5):733‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnson PL, Samuels BC, Fitz SD, Lightman SL, Lowry CA, Shekhar A. Activation of the orexin 1 receptor is a critical component of CO2‐mediated anxiety and hypertension but not bradycardia. Neuropsychopharmacology. 2012;37(8):1911‐1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marcus JN, Aschkenasi CJ, Lee CE, et al. Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol. 2001;435(1):6‐25. [DOI] [PubMed] [Google Scholar]
- 19. Trivedi P, Yu H, MacNeil DJ, Van der Ploeg LH, Guan XM. Distribution of orexin receptor mRNA in the rat brain. FEBS Lett. 1998;438(1‐2):71‐75. [DOI] [PubMed] [Google Scholar]
- 20. Heydendael W, Sengupta A, Beck S, Bhatnagar S. Optogenetic examination identifies a context‐specific role for orexins/hypocretins in anxiety‐related behavior. Physiol Behav. 2014;130:182‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bonnavion P, Jackson AC, Carter ME, de Lecea L. Antagonistic interplay between hypocretin and leptin in the lateral hypothalamus regulates stress responses. Nat Commun. 2015;6:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bonaventure P, Dugovic C, Shireman B, et al. Evaluation of JNJ‐54717793 a novel brain penetrant selective orexin 1 receptor antagonist in two rat models of panic attack provocation. Front Pharmacol. 2017;8:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Steiner MA, Gatfield J, Brisbare‐Roch C, et al. Discovery and characterization of ACT‐335827, an orally available, brain penetrant orexin receptor type 1 selective antagonist. ChemMedChem. 2013;8(6):898‐903. [DOI] [PubMed] [Google Scholar]
- 24. Perna G, Schruers K, Alciati A, Caldirola D. Novel investigational therapeutics for panic disorder. Expert Opin Investig Drugs. 2015;24(4):491‐505. [DOI] [PubMed] [Google Scholar]
- 25. Farach FJ, Pruitt LD, Jun JJ, Jerud AB, Zoellner LA, Roy‐Byrne PP. Pharmacological treatment of anxiety disorders: current treatments and future directions. J Anxiety Disord. 2012;26(8):833‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boss C, Roch C. Recent trends in orexin research—2010 to 2015. Bioorg Med Chem Lett. 2015;25(15):2875‐2887. [DOI] [PubMed] [Google Scholar]
- 27. Gottschalk MG, Richter J, Ziegler C, et al. Orexin in the anxiety spectrum: association of a HCRTR1 polymorphism with panic disorder/agoraphobia, CBT treatment response and fear‐related intermediate phenotypes. Transl Psychiatry. 2019;(9):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zarrabian S, Riahic E, Karimi S, Razavi Y, Haghparast A. The potential role of the orexin reward system in future treatments for opioid drug abuse. Brain Res. 2018. 10.1016/j.brainres.2018.11.023 [DOI] [PubMed] [Google Scholar]
- 29. Gibaldi M, Perrier D. Pharmacokinetics. New York: Marcel Dekker; 1982. [Google Scholar]
- 30. van Steveninck AL, Schoemaker HC, Pieters MS, Kroon R, Breimer DD, Cohen AF. A comparison of the sensitivities of adaptive tracking, eye movement analysis and visual analog lines to the effects of incremental doses of temazepam in healthy volunteers. Clin Pharmacol Ther. 1991;50:172‐180. [DOI] [PubMed] [Google Scholar]
- 31. van Steveninck AL, Schoemaker HC, den Hartigh J, et al. Effects of intravenous temazepam. I. Saccadic eye movements and electroencephalogram after fast and slow infusion to pseudo steady state. Clin Pharmacol Ther. 1994;55:535‐545. [DOI] [PubMed] [Google Scholar]
- 32. van Steveninck AL, van Berckel BNM, Schoemaker RC, Breimer DD, van Gerven JMA, Cohen AF. The sensitivity of pharmacodynamic tests for the central nervous system effects of drugs on the effects of sleep deprivation. J Psychopharmacol. 1999;13(1):10‐17. [DOI] [PubMed] [Google Scholar]
- 33. Richard M, Kaufmann P, Kornberger R, Dingemanse J. First‐in‐man study of ACT‐709478, a novel selective triple T‐type calcium channel blocker. Epilepsia. 2019;60(5):968‐978. [DOI] [PubMed] [Google Scholar]
- 34. Verster JC, Roth T. Predicting psychopharmacological drug effects on actual driving performance (SDLP) from psychometric tests measuring driving‐related skills. Psychopharmacology (Berl). 2012;220(2):293‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jex HR, McDonnell JD, Phatak AV. A “critical” tracking task for manual control research. IEEE Transac Humn Fact Electro. 1966;HFE‐7:138‐145. [Google Scholar]
- 36. Dinges DF, Powell JW. Microcomputer analyses of performance on a sustained operations. Behav Res Methods Instrum Comput. 1985;17:652‐655. [Google Scholar]
- 37. Smith JM, Misiak H. Critical flicker frequency (CFF) and psychotropic drugs in normal human subjects – a review. Psychopharmacology (Berl). 1976;47:175‐182. [DOI] [PubMed] [Google Scholar]
- 38. Bond A, Lader M. The use of analogue scales in rating subjective feelings. Br J Med Psychol. 1974;47:211‐218. [Google Scholar]
- 39. Van Steveninck AL, Mandema JW, Tuk B, et al. A comparison of the concentration‐effect relationships of midazolam for EEG‐derived parameters and saccadic peak velocity. Br J Clin Pharmacol. 1993;36(2):109‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bowdle TA, Radant AD, Cowley DS, Kharasch ED, Strassman RJ, Roy‐Byrne PP. Psychedelic effects of ketamine in healthy volunteers: relationship to steady‐state plasma concentrations. Anesthesiology. 1998;88(1):82‐88. [DOI] [PubMed] [Google Scholar]
- 41. Akerstedt T, Gillberg M. Subjective and objective sleepiness in the active individual. Int J Neurosci. 1990;52(1‐2):29‐37. [DOI] [PubMed] [Google Scholar]
- 42. Gough K, Hutchison M, Keene O, et al. Assessment of dose proportionality: report from the statisticians in the pharmaceutical industry/pharmacokinetics UK joint working party. Drug Inf J. 1995;29:1039‐1048. [Google Scholar]
- 43. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018;46(D1):D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alexander SPH, Christopoulos A, Davenport AP, et al. The Concise Guide to PHARMACOLOGY 2019/20: G protein‐coupled receptors. Br J Pharmacol. 2019;176(S1):S21‐S141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. Br J Pharmacol. 2019;176(S1):S297‐S396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. FDA . Guidance for industry: food effect bioavailability and, fed bioequivalence studies. In, 2002.
- 47. Bruderer S, Hurst N, de Kanter R, et al. First‐in‐humans study of the safety, tolerability, and pharmacokinetics of ACT‐451840, a new chemical entity with antimalarial activity. Antimicrob Agents Chemother. 2015;59(2):935‐942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muehlan C, Heuberger J, Juif PE, Croft M, Van Gerven J, Dingemanse J. Accelerated development of the dual orexin receptor antagonist ACT‐541468: integration of a microtracer in a first‐in‐human study. Clinicl Pharmaco Therapeu. 2018;104(5):1022‐1029. [DOI] [PubMed] [Google Scholar]
- 49. Sun H, Kennedy WP, Wilbraham D, et al. Effects of Suvorexant, an orexin receptor antagonist, on sleep parameters as measured by polysomnography in healthy men. Sleep Med. 2013;36:259‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen X, Broeyer F, de Kam M, Baas J, Cohen A, van Gerven J. Pharmacodynamic response profiles of anxiolytic and sedative drugs. Br J Clin Pharmacol. 2017;83(5):1028‐1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Salvadore G, Brooks S, Cathy B, et al. F13. Safety, tolerability, pharmacokinetic and Pharmacodynamic properties of the selective Orexin‐1 receptor antagonist JNJ‐61393215: results from the first‐in‐human and multiple ascending dose studies. Biol Psychiatry. 2019;85:S217‐S218. [Google Scholar]
- 52. Mahler S, Moorman D, Smith R, James M, Aston‐Jones G. Motivational activation: a unifying hypothesis of orexin/hypocretin function. Nat Neurosci. 2014;17(10):1298‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Citrome L. Suvorexant for insomnia: a systematic review of the efficacy and safety profile for this newly approved hypnotic – what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2014;68(12):1429‐1441. [DOI] [PubMed] [Google Scholar]
- 54. Hoever P, De Haas SL, Dorffner G, Chiossi E, Van Gerven JM, Dingemanse J. Orexin receptor antagonism: an ascending multiple‐dose study with almorexant. J Psychopharmacol. 2012;26:1071‐1080. [DOI] [PubMed] [Google Scholar]
- 55. de Lecea L, Huerta R. Hypocretin (orexin) regulation of sleep‐to‐wake transitions. Front Pharmacol. 2014;5:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yee KL, McCrea J, Panebianco D, et al. Safety, tolerability, and pharmacokinetics of Suvorexant: a randomized rising‐dose trial in healthy men. Clin Drug Investig. 2018;38(7):631‐638. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Demographic characteristics of subjects per cohort and overall
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.