

Summary:
Cancer is often seen as a disease of mutations and chromosomal abnormalities. However, some cancers, including pediatric rhabdoid tumors (RTs), lack recurrent alterations targetable by current drugs, and need alternate, informed therapeutic options. To nominate potential targets, we performed a high-throughput small-molecule screen complemented by a genome-scale CRISPR-Cas9 gene-knockout screen in a large number of RT and control cell lines. These approaches converged to reveal several receptor tyrosine kinases (RTKs) as therapeutic targets, with RTK inhibition effective in suppressing RT cell growth in vitro and against a xenograft model in vivo. RT cell lines highly express and activate (phosphorylate) different RTKs, creating dependency without mutation or amplification. Downstream of RTK signaling, we identified PTPN11, encoding the pro-growth signaling protein SHP2, as a shared dependency across all RT cell lines. This study demonstrates that large-scale perturbational screening can uncover vulnerabilities in cancers with “quiet” genomes.
Introduction:
Large-scale perturbational screening of cancer cell lines aims to provide mechanistic insight into cancer biology and identify therapeutic vulnerabilities, which may prove especially useful for cancers that lack obvious targetable genetic alterations. While direct targeting of activated oncogenes is now a proven therapeutic approach, mutations in tumor suppressor genes are more common across many cancer types and cannot be targeted directly (Lawrence et al., 2013). Here, we investigate the potential of large-scale perturbational screening to identify vulnerabilities in highly aggressive and lethal pediatric rhabdoid tumors (RT). This genomically simple cancer is driven by a single recurrent genetic event: biallelic loss of the tumor suppressor SMARCB1, or much less commonly SMARCA4, core subunits of the SWI/SNF (BAF) chromatin-remodeling complex. The average mutation burden in RT is more than twenty-fold lower than the average across cancer types; recent genomic characterization of large numbers of primary patient RT has revealed that 80% of RT have SMARCB1 loss as the sole recurrent mutation (Lawrence et al., 2013, Lee et al., 2012, Roberts et al., 2000). The cell(s) of origin have been unknown, and tumors can occur in various soft tissues including kidney, liver, or brain (these brain-localized tumors are known as AT/RT: atypical teratoid/rhabdoid tumor). Recent data reveal at least three distinct sub-classes of RT that may each arise from different progenitor cells (Torchia et al., 2015, Johann et al., 2016, Chun et al., 2016). The range of rhabdoid tumor tissue origins and sub-classes complicate treatment recommendations as dependency relationships are often unclear.
Mechanistically, we have recently demonstrated that the SWI/SNF chromatin remodeling complex is essential for the maintenance of enhancers (Mathur et al., 2016) and that inactivation of the SMARCB1 subunit of this complex, as occurs in nearly all RT, disrupts enhancer function, which impairs differentiation and thus may underlie unrestrained proliferation (Alver et al., 2017, Wang et al., 2016).
Given that the sole identified recurrent genetic event is the absence of a gene, there are no obvious therapeutic targets and mortality remains high (Wang et al., 2009). Therefore, RT constitutes a compelling model with which to investigate the potential of large-scale perturbational screening of cancer cell lines to identify therapeutic vulnerabilities.
Consequently, we collected 16 RT cell-line models (derived from tumors found in brain, kidney, muscle, and soft tissue tumors) and robustly deployed both small-molecule and genetic (CRISPR-Cas9-mediated gene knockout) perturbational screening to discover vulnerabilities in RT.
These approaches converged to reveal several receptor tyrosine kinases (RTKs) as therapeutic targets in RT, with RTK inhibition effective in suppressing RT cell growth in vitro and against a xenograft model in vivo. We find that rhabdoid cell lines from all tissue origins activate a heterogeneous range of RTKs, including PDGFRA and MET, contributing to the proliferation of RT cells and leading to dependency on these pathways. Overall, we find that RTK activation, driven in part by increased expression caused by loss of SMARCB1, rather than genomic alterations, is a hallmark of RT model systems, and that high levels of RTK expression are observed in primary RT patient samples. Both genetically and via a small-molecule inhibitor we also find that RTK-dependent RT cells are particularly sensitive to loss of the protein tyrosine phosphatase PTPN11 (encoding SHP2), a downstream effector of RTK signaling. These findings highlight the potential of large-scale perturbational screening to reveal dependencies conferred by tumor suppressor loss and suggest RTKs and SHP2 as therapeutic targets in patients with RT.
Results:
RTK inhibitors selectively target RT cell lines
Previously, we reported a small-molecule sensitivity dataset (Cancer Therapeutics Response Portal) describing the effects of a library of 481 small molecules, an informer set enriched for FDA-approved oncology drugs and clinical candidates, on the viability of 840 individual cancer cell lines (CCLs) representing 25 cancer lineages, which included four RT CCLs (Seashore-Ludlow et al., 2015, Rees et al., 2016). We tested 47 additional CCLs, including five RT CCLs, against this small-molecule collection, using area-under-concentration-response curves (AUCs) to measure sensitivity as described previously (Seashore-Ludlow et al., 2015, Rees et al., 2016). We normalized AUCs for each small molecule across all 887 CCLs by calculating a robust z-score using the median absolute deviation (ZMAD). To identify potential therapeutic vulnerabilities in these nine RT cell lines (a diverse set derived from brain, kidney, muscle, and soft tissue tumors), we focused on small molecules selective for RT CCLs relative to non-RT CCLs. Of the 15 most selective small molecules (ZMAD AUC < −3 in at least three RT CCLs), 14 were annotated receptor tyrosine kinase (RTK) inhibitors, targeting angiogenesis-related receptors such as vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), and hepatocyte growth factor receptor (HGFR/MET). Overall, RTK inhibitors as a class were highly selective for RT cell lines compared to non-RT cell lines, suggesting a remarkable dependence on these targets (Kolmogorov-Smirnov p = 7.3 × 10−11; Figure 1a, Figure S1a). The selectivity of FDA-approved RTK inhibitors for RT CCLs compared to CCLs best representing currently approved indications is presented in Figure S1b.
Figure 1).
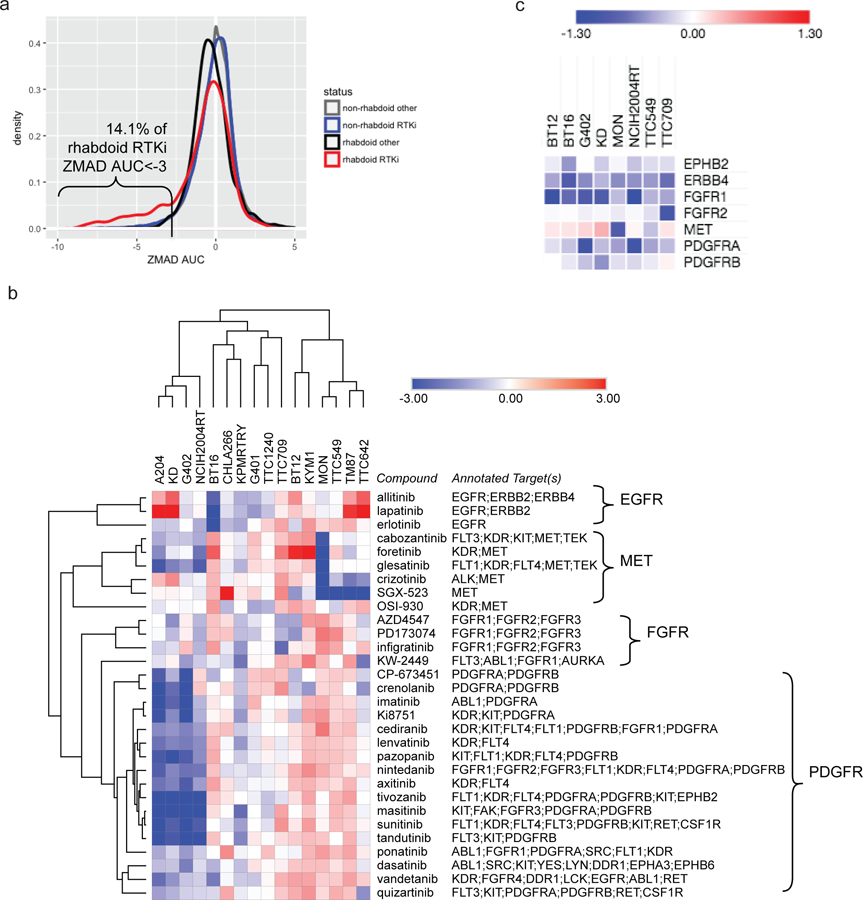
Receptor tyrosine kinases represent vulnerabilities within RT cell lines.
a) ZMAD AUC distributions for 49 annotated RTK inhibitors (and 432 non-RTK inhibitors) across 8 RT cell lines and 879 non-RT cell lines (see Figure S1a–c, Table S1). b) z-scored AUC values for 30 RTK inhibitors across 16 RT CCLs (see Table S1). c) CERES scores for 7 RTK-encoding genes with a CERES score < −0.5 and ZMAD CERES < −2 (in RT relative to 35 non-RT CCLs) in ≥1 RT CCL (see Figure S1e, Table S3). Visualization and hierarchical clustering of rows and columns (1 – Pearson correlation) were performed using Morpheus (https://software.broadinstitute.org/morpheus/).
To expand on this finding, we collected seven additional RT CCLs and tested all 16 validated SMARCB1-deficient RT CCLs against 30 RTK inhibitors, including FDA-approved therapeutics (Figure S1c). Results were consistent with high-throughput data, and further suggested distinct vulnerabilities of sets of cell lines to sets of small molecules: different RTK inhibitors potently inhibited the viability of different subsets of RT cell lines (e.g., imatinib: G402, A204, KD, NCIH2004RT, KPMRTRY; crizotinib: MON, TM87, TTC642, TTC549) while showing little efficacy against others (Figure 1b, Figure S1a, Table S1). Of note, RTK inhibitors with overlapping target profiles (Table S2, Figure S1d) clustered together in these results, suggesting on-target activity.
RTKs are preferential dependencies in RT CCLs
In parallel, we carried out genome-scale CRISPR-Cas9-mediated gene knockout screens on 8 RT CCLs with six guide RNAs targeting each gene (Aguirre et al., 2016, Meyers et al., 2017). Values were collapsed to a single dependency score per gene and adjusted per cell line using CERES to account for the confounding factor of copy number in CRISPR-Cas9 screens (Aguirre et al., 2016, Munoz et al., 2016), with normalization to pan-essential (score of −1) and non-essential (score of 0) genes. Notably, each RT cell line displayed a strong dependency on at least one RTK, although the specific dependencies again varied by cell line (Figure 1c, Figure S2e, Table S3). The strongest observed dependencies included FGFR1, MET, PDGFRA, and PDGFRB (CERES score ZMAD < −4 in at least two RT CCLs), while other RTKs were strong dependencies in a single RT CCL (e.g., PDGFRB, FGFR2, and ERBB4; Table S3).
RTKs are highly expressed and activated in RT CCLs
To nominate biomarkers for RTK dependencies, we performed RNA sequencing on RT CCLs in conjunction with the Cancer Cell Line Encyclopedia (Ghandi et al., 2019, Barretina et al., 2012). RTK-dependent RT CCLs exhibited high expression of the corresponding RTK-encoding transcript(s) relative to both non-dependent RT CCLs and non-RT CCLs (Figure 2a). Consistent with published primary tumor data (Chauvin et al., 2017), we see that rhabdoid cell lines have low promoter methylation at RTK genes. In the cell lines tested, we see that the same relevant dependent RTK genes have low promoter methylation, while RTKs not important in those cell lines have higher levels of promoter methylation (Figure S1f) (Ghandi et al., 2019). Measurement of total and phospho-protein levels using a global phospho-RTK array or individual antibodies demonstrated protein expression and phosphorylation (indicating activation) of the relevant RTKs in RT CCLs (Figure 2b, Figure S2a). Consistent with a contribution of SMARCB1 in regulation of RTK activation, retroviral restoration of SMARCB1 in the kidney RT G402 cell line reduced mRNA levels of FGFR1 and downstream RPS6KB1, but not PDGFRA or other pathway members (Figure 2c). Both RTKs showed a decrease in activated phospho- and total protein, and phospho-p70S6K also decreased, with no change in total protein (Figure 2d, Figure S2b). Of note, we did not detect SMARCB1-dependent changes in SWI/SNF binding within 100 kB of expressed RTKs (Wang et al., 2016), nor significant changes in histone acetylation (Table S4), suggesting that reduced RTK transcript levels may not be a direct consequence of SWI/SNF binding at the RTKs, although binding to a more distal enhancer cannot be excluded.
Figure 2).
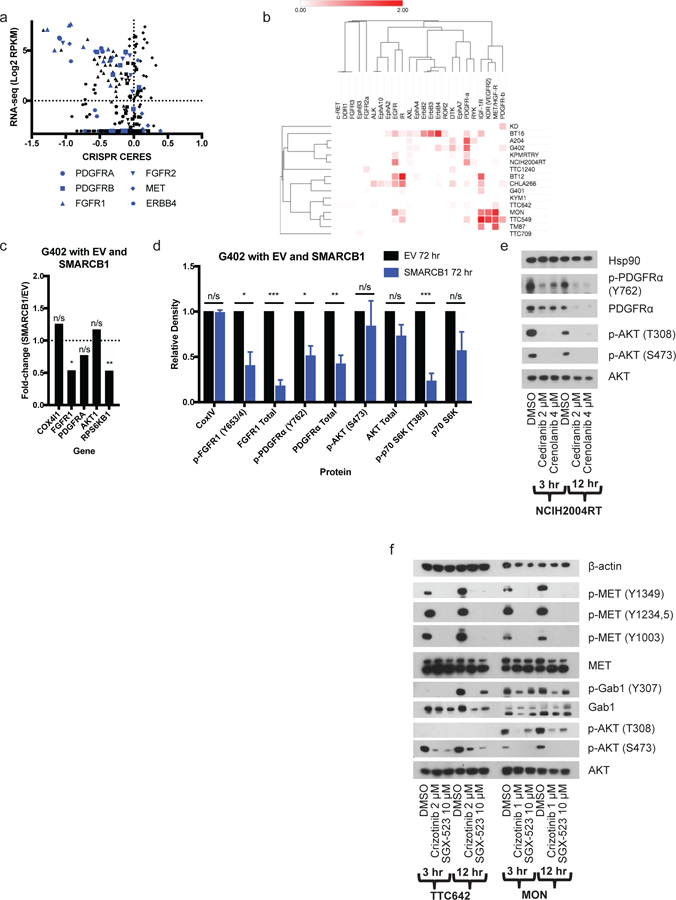
RT CCLs express activated RTKs targetable by small-molecules.
a) CRISPR CERES scores and RNA levels for 6 different RTK-encoding genes across 43 CCLs. RT CCLs are shown in blue (see Table S3). b) Heatmap of results of phospho-RTK array in 16 rhabdoid cell lines. Visualization and hierarchical clustering of rows and columns (1 – Pearson correlation) were performed using Morpheus (https://software.broadinstitute.org/morpheus) (see Figure S2a). c) Fold-change of RTK pathway genes in RNA-seq in G402 cells with empty vector (EV) vs SMARCB1 re-expression after 72 hours puromycin selection. p-values calculated using unpaired t test without assuming a consistent SD. ns, not significant; *p<0.05; **p<0.01; ***p<0.001. d) Western blots in EV and SMARCB1 G402 cells after 72 hours puromycin selection. Bar graph shows results from three biological replicates (mean + SEM) (see Figure S2b). e-f) Westerns blots on 3- and 12-hour RTK inhibitor treatment in e) PDGFRA-expressing NCIH2004RT cells and f) MET-expressing TTC642 and MON cells (see Figure S2c–e).
RTK inhibitors suppress RTK signaling alone and in combination
To relate small-molecule sensitivity data with RTK activation, we tested whether RTK inhibitors could decrease RTK phosphorylation in sensitive cell lines. As RTK inhibitors are generally ATP-competitive and have overlapping target profiles, we combined available kinase-profiling data (presented in Table S2) with our own cell-line-sensitivity data in non-RT CCLs harboring known activating mutations in RTKs to nominate the most likely relevant cellular target(s) of these molecules (Figure S1d). Combining these results, along with the clustering of inhibitors with shared targets (Figure 1b), suggested that these inhibitors were working via the targets identified by gene-knockout and genomic data (Figure 2a–b). In NCIH2004RT, A204, G401, G402, KD, and KPMRTRY cells, treatment with PDGFR-inhibiting compounds decreased phosphorylation of PDGFR and downstream AKT (Figure 2e, Figure S2c). In KYM1, TM87, TTC549, TTC642 and MON cells, treatment with MET-inhibiting compounds decreased phosphorylation of MET, GAB1, and AKT (Figure 2f, Figure S2d). In BT12 and NCIH2004RT cells, treatment with FGFR1-inhibiting compounds decreased phosphorylation of FGFR1 as well as downstream MEK1/2 or AKT (Figure S2e). In contrast to more selective inhibitors, dual-targeting small molecules such as MET/PDGFR inhibitors (cabozantinib, foretinib, glesatinib) were more broadly active in RT cell lines, including in lines that expressed and were dependent on either or both RTK (Figure 1b, Table S3). For example, treatment of MET- and PDGFRA-expressing TTC642 cells with the selective PDGFR inhibitor imatinib and the selective MET inhibitor SGX-523 alone reduced phosphorylation of only PDGFRA or MET, respectively, while treatment with the combination of imatinib and SGX-523 or the dual inhibitor cabozantinib decreased both MET and PDGFRA phosphorylation (Figure 3a–b, Figure S2d). Additionally, treatment with cabozantinib decreased p-AKT and p-MEK1/2 levels to a greater extent than PDGFR or MET inhibition alone (Figure 3b, Figure S2d). Similar patterns were observed in the TTC549 cell line (Figure S2d).
Figure 3).
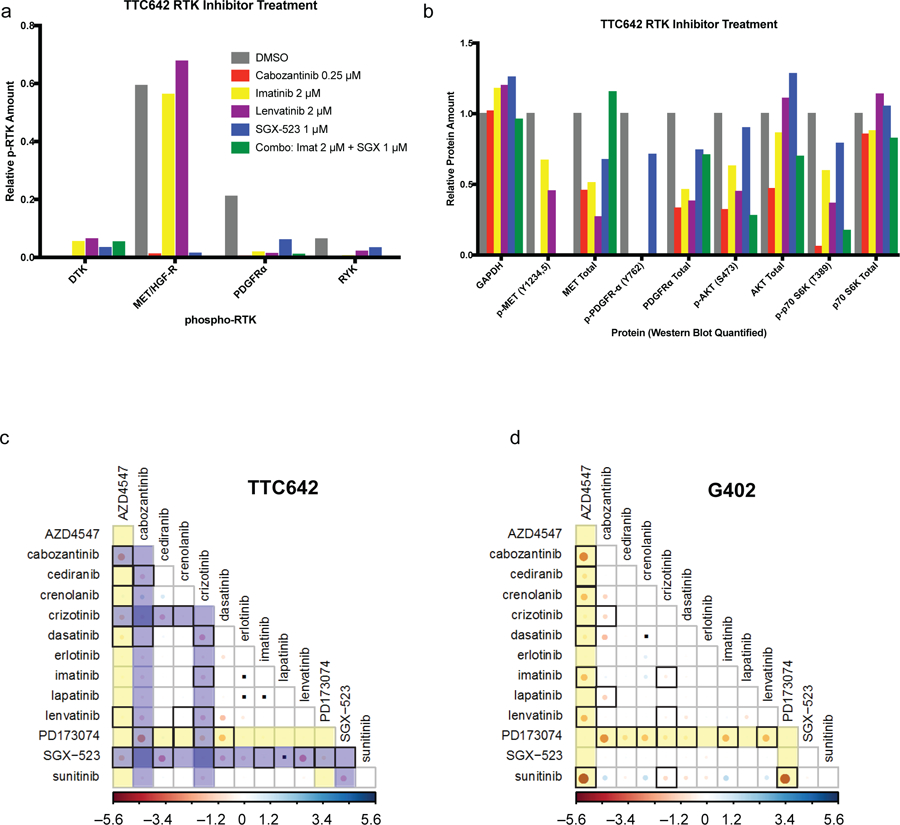
Combination of RTK inhibitors results in synergistic cell killing in RT CCLs.
a) TTC642 cells were treated with indicated inhibitors for three hours then subjected to phospho-RTK analysis with arrays. Relative phospho-RTK expression was quantified in ImageJ (see Figure S2d). b) Lysates from treated TTC642 cells were run for RTK pathway members by Western blot. Relative protein expression was quantified in ImageJ (see Figure S2d). (c-d) Pairwise combination treatments of 13 RTK inhibitors in c) TTC642 and d) G402 CCLs. FGFR inhibitors are highlighted in yellow. MET inhibitors are highlighted in blue. Red circles indicate synergistic interactions, with interactions exceeding a 99% confidence interval threshold surrounded by a black box. Inert combinations are indicated with black dots. Visualization was performed using the Corrplot package in R (see Figure S3).
As combination treatment was more effective in reducing levels of phospho-RTKs and downstream phospho-targets, we asked whether combinations of RTK inhibitors would result in synergistic cell killing. We observed that pairwise combinations of selective inhibitors of different RTKs synergized in cell lines where multiple RTKs were active, such as in TM87, TTC549 and TTC642 cells (FGFR and MET; Figure 3c, Figure S3a, b, d). Consistent with a recent report that identified synergy between FGFR and PDGFR inhibition in the G402 cell line (Wong et al., 2016), we also found that combinations of FGFR and PDGFR inhibitors were strongly synergistic in G402 cells (Figure 3d, Figure S3a), as well as in A204, KD, and NCIH2004RT cells (Figure S3c), which showed dependence on both FGFR1 and PDGFRA/B. These findings were specific to cell lines expressing multiple RTKs: for example, synergy between FGFR and PDGFR inhibitors was not observed in TM87, BT12, or BT16 cells that did not express activated PDGFRA or PDGFRB (Figure S3d).
RTK inhibitors are active against RT xenograft models
Collectively, these results provide support for use of multi-targeting inhibitors or combination treatments where multiple RTKs are active. To investigate this in vivo, we tested the multi-targeting RTK inhibitors lenvatinib, pazopanib, and cabozantinib in an orthotopic xenograft model of the luciferase-labeled TTC642 cell line. This cell line expresses PDGFRA, FGFR1, FGFR3, and MET. Responses were compared to both no chemotherapy and to a known active standard-of-care (SOC) treatment with combination doxorubicin and vincristine. Cabozantinib was also tested in combination with SOC. Mice were treated for four three-week courses (Figure 4a), and mice with a tumor burden greater than 20% of their body weight were removed from the study and scored as having progressive disease (PD). Mice with stable disease (SD), partial responses (PRs), and complete responses (CRs) were scored using bioluminescence as previously described (Stewart et al., 2014).
Figure 4).
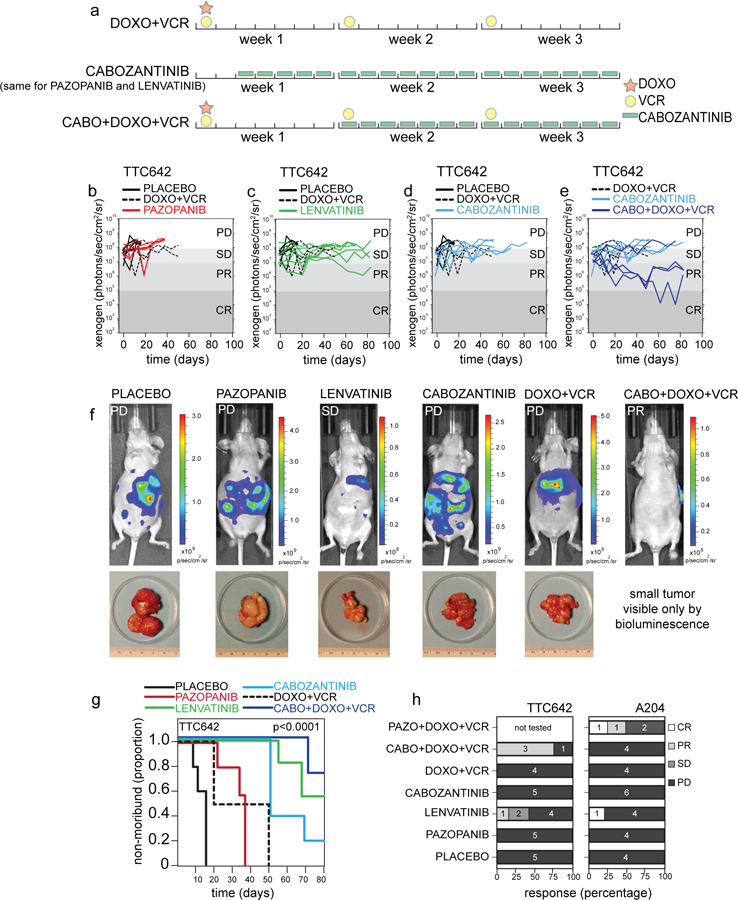
Preclinical testing of rhabdoid orthotopic xenografts.
a) Drug schedule selected for preclinical testing matched to relevant regimens used in patients. (b-e) Representative plot of tumor burden for TTC642 xenografts, as measured by bioluminescence over time, in a preclinical phase 2 study with the treatment groups listed compared to placebo control and DOXO+VCR (standard of care regimen used for rhabdoid tumor). Each line is a different mouse. f) Representative images and accompanying tumor micrographs from TTC642 xenograft mice with progressive disease in the placebo control, pazopanib, cabozantinib, and DOXO+VCR groups. Representative images of mice with stable disease in the lenvatinib group and a partial response shown for the CABO+DOXO+VCR treatment group. g) Survival curves for the TTC642 xenografts for the 6 indicated treatment groups, p-value determined by log-rank tests for each survival group. h) Histogram of the percentage of CR, PR, SD, and PD as determined by bioluminescence criteria for both TTC642 and A204 rhabdoid xenografts. The number of mice in each group is indicated. Abbreviations: CABO, cabozantinib; DOXO, doxorubicin; VCR, vincristine; PD, progressive disease; SD, stable disease; PR, partial response; CR, complete response; PAZO, pazopanib.
Mice given the standard of care treatment survived an average of 36 days, and all came off the study due to PD (Figure 4b–h). Mice treated with cabozantinib alone, which targets PDGFRA and MET, survived longer than those on SOC, staying on treatment an average of 61 days (Figure 4d). Combination of cabozantinib and SOC further increased survival; mice survived an average of 80 days and 3 of the 4 mice in this arm completed the entire treatment course with tumor shrinkage indicating PR (Figure 4e, f–h). Mice given lenvatinib, which targets PDGFRA and FGFR family members, also survived longer than SOC (average 75 days), and 3 of the 7 mice showed SD or PR (Figure 4c, f–h). Providing further evidence that targeted multi-RTK inhibition is beneficial, all the mice treated with pazopanib, which showed the weakest activity in vitro against PDGFRA- and FGFR-dependent cell lines, were removed from the study for PD by 38 days (Figure 4b, f–h).
We also tested luciferase-labeled orthotopic xenografts of the FGFR1-, FGFR3-, and PDGFRA-expressing A204 rhabdoid cell line, using the same drugs and combinations, along with a combination of pazopanib and SOC. These tumors appeared more aggressive, and most mice, including those given SOC, were removed before the end of the study for progressive disease (Figure 4h). However, three mice were able to complete the entire study, one from the lenvatinib-treatment group (CR), and two from the pazopanib and SOC group (one CR, one PR). Collectively, these results suggest that RTKi treatment in combination with SOC in vivo can provide more benefit than SOC alone, and that targeting multiple RTKs may represent a more effective treatment strategy than single inhibition in tumors with multiple active RTKs.
RTK-encoding transcripts show heterogeneous expression in RT patient samples
To better understand the heterogeneous nature of RTK activation and dependency in RT CCLs and its potential relevance to human RT, we collected public genome-wide expression data for 239 human RT samples across six studies, including two with samples collected from multiple tumor locations (Han et al., 2016, Chun et al., 2016), and analyzed expression of 58 human RTK genes. Across all studies, PDGFRA, PDGFRB, FGFR1, and FGFR2 showed the highest expression (Figure S4). However, consistent with cell-line data, we observed heterogeneity of RTK expression both within and across tissue types. For example, in the TARGET study, PDGFRA was more highly expressed in kidney relative to soft tissue tumors (padj = 0.0001). We also observed differential expression patterns for MET (highly expressed in liver samples) and ERBB4 (highly expressed in brain relative to soft tissue; padj = 0.00167; Table S5). These findings support the importance of RTK expression in RT, and are consistent with cell-line data, where CCLs with highest phospho-PDGFRA expression were from muscle (A204) and kidney (KPMRTRY, G402, and NCIH2004RT), while ERBB4 was detected predominantly in brain-derived CCLs (e.g., BT16).
PTPN11 represents a shared dependency across all RT cell lines
RTKs converge on canonical downstream effector proteins (e.g., AKT, MEK, S6K) that integrate signaling from multiple RTK pathways, and some mouse models of RT have been reported to show persistent AKT activation (Darr et al., 2013). Interestingly, however, the genes encoding these downstream effector proteins do not score as dependencies in CRISPR-Cas9 screening. While most of these downstream effector proteins have functionally homologous proteins encoded by paralogous genes, e.g., AKT1/2, MAP2K1/MAP2K2, and RPS6KB1/2, potentially making any of these genes appear non-essential when singly lost, small-molecule inhibitors tend to hit all paralogs and are also not selective for RT CCLs (Figure S1a, e).
To more systematically investigate signaling downstream of RTKs in RT cell lines, we analyzed gene dependency scores for 476 genes within the gene ontology RTK signaling network (GO_TRANSMEMBRANE_RECEPTOR_PROTEIN_TYROSINE_KINASE_SIGNALING_PATHWAY). For each gene, we calculated the probability of dependency in each RT and non-RT cell line. Of 8 genes essential across all 8 RT models (probability > 0.95), 7 were also universally essential across non-RT cell lines (minimum probability = 0.88), suggesting these dependencies were not specific to RT cell lines or RTK signaling (e.g., components of RNA polymerase II). The exception, PTPN11, encodes the tyrosine phosphatase SHP2, which acts downstream of RTKs to effect survival and proliferation and is often mutated and activated in cancer.
SHP2 has been found to be an important node in RTK signaling and essential for the growth of cancers reliant on RTK signaling (Prahallad et al., 2015, Matalkah et al., 2016, Ghandi et al., 2019). Additionally, shRNA screening (targeting 7,500 genes) in 250 cancer cell lines revealed that lines with activating mutations in RTKs were preferentially dependent upon PTPN11 (encoding SHP2), and these cell lines were sensitive to a novel allosteric SHP2 inhibitor, SHP099 (Chen et al., 2016). In our genome-scale CRISPR-Cas9 screen, RT cell lines were highly dependent on PTPN11 (dependency score range −0.96 to −1.33) (Table S6).
To determine whether rhabdoid cell lines showed preferential sensitivity to SHP2 loss, we screened the SHP099 inhibitor at eight different concentrations in 274 cancer cell lines, including 14 RT cell lines, using PRISM, a high-throughput pooled screening assay (Yu et al., 2016). CML cell lines carrying the BCR-ABL fusion were most sensitive (Gu et al., 2018), and rhabdoid cell lines showed preferential sensitivity to SHP099 compared to other lineages (Figure 5a). Of note, RT lines had lower AUC and IC50 than cell lines containing either PTPN11 activating mutations or RTK activating mutations. We confirmed these sensitivities across all 16 RT cell lines in conventional (unpooled) assays and observed that sensitivity to SHP099 also correlated with CRISPR-based sensitivity to PTPN11 loss (Figure 5b, Table S6). SHP099 treatment also decreased phospho-ERK in RT cell lines and a positive control line while having no effect on a negative control line, and no effect on upstream RTKs (Figure 5c, Figure S5a). Long-term (ten-day) treatment also inhibited colony formation in sensitive lines while having no effect in a control insensitive line (Figure 5d). We confirmed that PTPN11/SHP2 is ubiquitously expressed across rhabdoid cancer cell lines of all origins and primary human tumors (Figure S5b, c).
Figure 5).
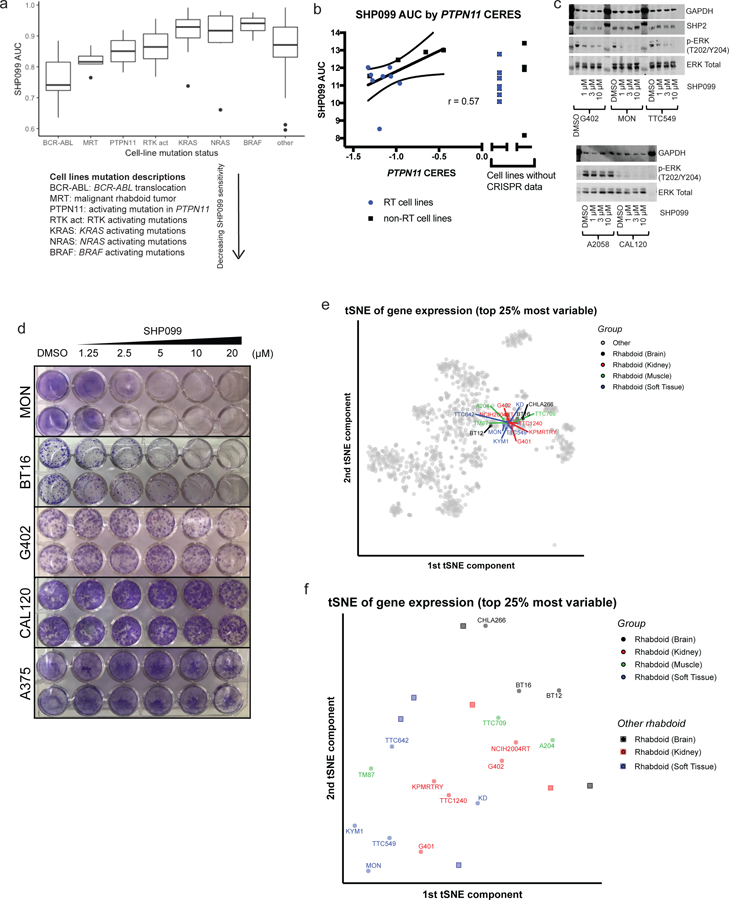
SHP2/PTPN11 inhibition is an RTK-dependent vulnerability shared across RT cell lines.
a) Pooled cell-line screening of SHP099 at 8 concentrations in 274 cancer cell lines, including 14 RT lines. Cell lines containing BCR-ABL translocation (known to sensitize to SHP2 inhibition) are the most sensitive to SHP099 (lowest AUC). RT CCLs have a lower AUC than that of CCLs containing RTK activating mutations (known to sensitize to SHP2 inhibition) or RAS/RAF mutations (known to create SHP2 independence). b) (Unpooled) validation screening SHP099 AUC (averaged across at least two replicates) correlates with CRISPR CERES scores for PTPN11 in RT cell lines and selected controls (see Figure S5a, Table S6). c) SHP099 treatment (3 hr) decreases phopho-ERK in a concentration-dependent manner in sensitive cell lines and has no effect in negative control A2058 cells (see Figure S5a). d) Crystal violet staining shows that ten-day treatment of cells with SHP099 decreases number of colonies in a concentration-dependent manner in sensitive cell lines and has no effect in negative control A375 cells. e) Gene expression based tSNE clustering of 1,165 cell lines reveals that rhabdoid cell lines cluster together. f) Clustering of 21 RT cell lines (the 16 cell lines characterized here, along with 5 more) alone does not reveal tissue-based clustering.
Gene expression and epigenetic landscape of RT cell lines
Recent studies in AT/RT and RT suggest that cell lines (and tumor samples) cluster into three to four epigenetically-defined subtypes, which also correlate with location of tumor origin (Torchia et al., 2016, Torchia et al., 2015, Johann et al., 2016). In AT/RT, for example, only one of these three defined subtypes, the spinally-located tumors, highly expressed and were dependent upon PDGFRB (Torchia et al., 2016). Therefore, we investgiated whether our RT cell lines clustered into subgroups based on RNA-seq expression as well as genome-wide methylation profiling. Our collection of 16 rhabdoid cell lines are derived from tumors found in brain (AT/RT), kidney, muscle, and soft tissue. Of note, all three of our AT/RT cell lines have been classified as AT/RT Type 2 (Torchia et al., 2016), so our findings here are limited to non-CNS and AT/RT Type 2 tumors.
We performed methylation profiling and an AT/RT-defined subgroup analysis on our rhabdoid cell lines. Four lines were classified as the “MYC” subgroup while the rest were low confidence MYC or undefined (Table S7). Of our AT/RT lines, one was defined as low confidence MYC and two were “undefined”, making it hard to draw any conclusions. Next, we clustered cancer cell lines based on gene expression using data from CCLE, and visualized results in a tSNE plot. This dataset contained 1,165 cancer cell lines, including 21 rhabdoid cell lines: the 16 rhabdoid cell lines characterized here plus five newly available. Rhabdoid tumor cell lines clustered closely together indicating conserved gene expression in RT lines regardless of tissue of origin (Figure 5e). When tSNE analysis was restricted solely to RT cell lines, tissue of origin did not appear to be a major determinant of gene expression differences between lines (Figure 5f).
Discussion:
Pediatric RT are driven by deletion or loss-of-function mutations in the SMARCB1 (or rarely SMARCA4) tumor suppressor gene. Aside from SMARCB1 loss, RT have an extremely low mutation rate and an absence of recurrent mutations in other genes. Consequently, despite being one of the most aggressive and lethal cancers, there are no obvious genetically-informed therapeutic targets. We therefore used RT as an exemplar to investigate whether large-scale, systematic high-throughput screening of cell lines could nominate new therapeutic approaches in the context of ‘quiet’ genomes driven by loss of a tumor suppressor.
In this report, high-throughput small-molecule and genetic screening across 16 RT cell lines derived from several tissue locations was used to identify specific vulnerabilities of RT cell lines relative to cell lines representing other cancer types. These approaches converged to reveal dependencies on both receptor tyrosine kinases and downstream SHP2 (PTPN11), where RT cell lines express high levels of various RTKs and are sensitive to both pharmacologic inhibition of these proteins and knockout of the genes encoding these RTKs and PTPN11.
Studies testing smaller numbers (n = 1–7) of RT and AT/RT cell lines have variably found activation and expression, and in some cases dependence upon, RTKs including EGFR (Darr et al., 2015, Kuwahara et al., 2004, Chauvin et al., 2017), FGFR (Wohrle et al., 2013, Wong et al., 2016, Chauvin et al., 2017), PDGFR (Wong et al., 2016, Torchia et al., 2016, Chauvin et al., 2017), or IGF1R (Arcaro et al., 2007), leading to uncertainty regarding the most relevant target(s). In addition to validating the known dependencies on FGFR and PDGFR, our large-scale genome-wide CRISPR-Cas9 knockout screen and follow-up screening of 30 RTK inhibitors with distinct targeting profiles enabled us to identify overexpression of and dependency on novel targets, including MET and ERBB4, in a subset of models. Our data from 16 cell lines across multiple tumor locations, combined with public tumor-expression data, support the notion that RTK expression and dependency vary both within and across tissue types. The profiling of our cell lines did not seem to reliably match the published classification schema of primary tumors, conceivably a consequence of cell culture. Notably, transcriptome analysis demonstrated that regardless of tissue of origin the gene expression profile of RT cell lines is quite similar and that the relatively limited heterogeneity between cell lines does not correlate strongly to tissue of origin, suggesting that the dependent RTKs do not constitute the greatest differential feature of the gene expression landscape.
In addition, we find that overexpression and activation of multiple RTKs is a common feature of tested RT cell lines, although it is noteworthy that the three tested AT/RT cell lines were derived from the Type 2 sub-class so we are not able to comment upon dependencies in other sub-types. RT cell lines are sensitive to appropriate single or combination treatments using existing clinically-approved RTK inhibitors, but, importantly, the diversity of RT models and patient samples suggest there is no single RTK inhibitor efficacious in all rhabdoid tumors. Immunohistochemistry (IHC) to identify the relevant activated RTK(s) may be the most proximal marker for immediate clinical investigation. In future clinical trials, the comparison of IHC with methylation classifiers is warranted in evaluation for matching patient tumors to specific RTK dependencies.
Intriguingly, we identified a shared dependency of RT cell lines on PTPN11, encoding the RTK signaling effector SHP2. Recently, an analysis of results from an shRNA knockdown screen of 7,500 genes across 250 cancer cell lines revealed that cell lines sensitive to knockdown of various RTKs were also highly sensitive to loss of PTPN11 (Chen et al., 2016). A novel allosteric inhibitor of SHP2 was developed (SHP099) and found to be efficacious in PTPN11-dependent cell lines, and an optimized compound, TNO155, has entered clinical development. While we found RT cell lines to be even more sensitive to SHP099 than cell lines with activating mutations in PTPN11 or RTKs in vitro, further studies (e.g., with TNO155) may inform whether SHP2 inhibition is an effective therapeutic strategy against a broader range of RT models and tumors in vivo.
These findings raise the question: what is the mechanistic basis by which RT are preferentially dependent upon RTKs? SWI/SNF regulates the chromatin structure at enhancers, where it facilitates transcription factor function to enable development and differentiation (Hu et al., 2011, Bossen et al., 2015, Marathe et al., 2017, Mathur et al., 2016, Wang et al., 2016, Nakayama et al., 2017). Loss of SMARCB1 in RT destabilizes the SWI/SNF complex, leading to reduced binding capacity at enhancers, which in turn results in specific reduction of the active enhancer mark H3K27ac and impaired differentiation. Restoration of SMARCB1 to RT-derived cell lines results in stabilization of the SWI/SNF complex (Wang et al., 2016) and cell cycle arrest accompanied by expression of genes associated with differentiation (Kuwahara et al., 2010, Betz et al., 2002, Mathur et al., 2016, Wang et al., 2016, Alver et al., 2017). Consequently, SMARCB1 loss may drive the growth of RT in part by preventing differentiation of highly proliferative progenitor cells in which RTKs are expressed. Here, we demonstrate that downregulation and deactivation of progenitor-active RTKs is in part dependent upon SMARCB1, as SMARCB1 re-expression results in reduced phosphorylation and expression. With respect to mechanism, this reduced activation may not be a direct consequence of altered SWI/SNF binding at the RTKs as we did not detect SMARCB1-dependent changes in SWI/SNF binding within 100 kB of expressed RTKs, although binding to a distal enhancer cannot be excluded (Wang et al., 2016).
Together, these findings seem most consistent with the following model whereby SMARCB1 loss results in impaired enhancer function, thus ‘locking in’ RT to the strongly RTK-driven progenitor state and precluding down-regulation of RTKs that would otherwise occur with differentiation. The marked dependence of RT upon RTKs is thus a result of both gene expression related to the cell of origin combined with a functional differentiation block caused by SMARCB1 loss. These lineage-related RTK dependencies may be akin to lineage dependencies upon estrogen receptor and androgen receptor in breast and prostate cancer, respectively, both of which have shown therapeutic efficacy when targeted. As cancers driven by mutation of other SWI/SNF subunits similarly have impaired ability to activate enhancers (Wang et al., 2016, Alver et al., 2017, Mathur et al., 2016), the dependency upon RTKs could extend to the myriad of other cancers in which SWI/SNF subunits are recurrently mutated, and further investigations are thus warranted.
Historically, results from pre-clinical testing have had modest predictive value in the outcomes of clinical trials. While numerous factors may contribute to predictive limitations, two have been 1) concern regarding the extent to which cell line models reflect behavior within patients, and 2) heterogeneity between the cancers of different patients that is often poorly reflected in laboratory testing due to the use of only a small number of cell lines. Collectively, our work suggests that large-scale perturbational screening of cancer cell lines uniting genetic and small-molecule approaches can be efficacious in identifying therapeutically-actionable dependencies not directly encoded by an oncogenic mutation. Our work robustly validated dependencies previously predicted from analysis of primary tumors, revealed new therapeutically targetable dependencies in RTs, and revealed the extent of heterogeneity in RTK dependence between cancers with remarkably simple genomes. Our findings further suggest that combination RTK inhibitor therapy, potentially coupled with profiling of expressed and active RTKs in patient tumors, and/or inhibition of SHP2, should be further investigated as potential therapies for these lethal pediatric cancers.
STAR METHODS TEXT
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Charles Roberts (Charles.Roberts@stjude.org).
This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human cancer cell lines
Cell lines were Mycoplasma negative, and identities were validated by SNP fingerprinting as described previously (Rees et al., 2016, Seashore-Ludlow et al., 2015, Cowley et al., 2014). A204, A375, CAL120, G401, G402, HEK293T, KYM1, LUDLU1, MeWo, MKN45, MV411, T47D, and U2OS cell lines were obtained from the Broad Biological Samples Platform. A2058 were ordered from ATCC. BT12, BT16, CHLA-266, NCIH2004RT, TM87, TTC549, TTC642, and TTC709 cells were maintained in the Roberts lab. A673 were maintained in the Stegmaier lab. KD and MON were a kind gift from Franck Bourdeaut at the Curie Institute. KP-MRT-RY was a kind gift from Yasumichi Kuwahara at the Kyoto Prefectural University of Medicine. TTC1240 was a kind gift from Timothy Triche at UCLA. Abbreviations: FBS, Fetal Bovine Serum; P/S, Penicillin Streptomycin. Cells were grown in a 37°C incubator with 5% CO2.
A204 (muscle RT), G401 (kidney RT), and G402 (kidney RT) were grown in McCoy’s 5A + 10% FBS + 1% P/S.
BT12 (AT/RT) were grown in OptiMEM + 5% FBS + 1% P/S.
BT16 (AT/RT), TM87 (muscle RT), A673 (Ewings), A2058 (melanoma), CAL120 (breast cancer), U2OS (osteosarcoma), and HEK293T (immortalized kidney) were grown in DMEM + 10% FBS + 1% P/S.
CHLA266 (AT/RT) were grown in IMDM + 20% FBS + 1% P/S + 1% ITS (Insulin, Transferrin, Selenium).
KYM1 (soft tissue RT) were grown in DMEM-F12 + 10% FBS + 1% P/S.
MV411 (AML) were grown in IMDM + 10% FBS + 1% P/S.
KD (soft tissue RT), KPMRTRY (kidney RT), MON (soft tissue RT), NCIH20004RT (kidney RT), TTC549 (extra-renal RT), TTC642 (extra-renal RT), TTC709 (muscle RT), TTC1240 (kidney RT), A375 (melanoma), LUDLU1 (lung cancer), MeWo (melanoma), MKN45 (stomach cancer), and T47D (breast cancer) were grown in RPMI + 10% FBS + 1% P/S.
Animals
Athymic nude immunodeficient female mice were purchased from Jackson Laboratories (strain code 007850). This study was carried out in strict accordance with the recommendations in the Guide to Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee at St. Jude Children’s Research Hospital. All efforts were made to minimize suffering. All mice were housed in accordance with approved IACUC protocols. Animals were housed on a 12–12 light cycle and provided food and water ad libitum.
Orthotopic Ultrasound-Guided Paranephric Xenograft Implantation
Orthotopic rhabdoid xenografts were created by injecting luciferase-labeled A204 and TTC642 cells into recipient athymic nude mice in the paranephric location. All ultrasound procedures were performed using the VEVO 2100 high-frequency ultrasound equipped with a MS-550S transducer running at 40 MHz. A204 and TTC642 cells were grown by routine culture methods. Each cell line was suspended in Matrigel (BD Worldwide, Cat#354234) at a concentration of 1×105 cells per microliter and placed on ice. Anesthetized (isoflurane 1.5% in O2 delivered at 2 liters/min) recipient nude mice were placed laterally on the imaging bed such that the left flank faced upward. In order to provide a channel for delivery of the implant, a 22 gauge catheter (BD Worldwide, Cat# 381423) was gently inserted through the skin and back muscle into the paranephric region and the hub was removed. A chilled Hamilton syringe fitted with a 27 gauge needle (1.5 inch) and loaded with 10 µl of the cell suspension was guided stereo-tactically through the catheter and positioned between the kidney and adrenal gland as visualized using ultrasound. The cells were injected into the region and the needle was left in place for 30 seconds to permit the matrigel component to set. The needle was then slowly removed, followed by gentle removal of the catheter.
Preclinical Testing
Mice with rhabdoid orthotopic xenografts generated as described above were screened weekly by Xenogen® and the bioluminescence was measured. Mice were enrolled in the study after achieving a target bioluminescence signal of 106 −107 photons/sec/cm2 to ensure tumor engraftment, and chemotherapy was started the following Monday.
A204
Mice (n = 31 total) were randomized to 7 treatment groups: Lenvatinib, Cabozantinib (CABO), Pazopanib (PAZO), Doxorubicin (DOXO) + Vincristine (VCR), DOXO + VCR + CABO, DOXO + VCR + PAZO, and placebo. The following doses were used:
| TREATMENT GROUP | DOSING |
|---|---|
| CABOZANTINIB | 30 mg/kg once daily continuous dosing days 1–21 |
| LENVATINIB | 30 mg/kg once daily continuous dosing days 1–21 |
| PAZOPANIB | 65 mg/kg twice daily continuous dosing days 1–21 |
| DOXO + VCR (standard of care) | DOXO 3.5 mg/kg day 1, VCR 0.38 mg/kg day 8 and day 15 |
| DOXO + VCR + CABOZANTINIB | DOXO 3.5 mg/kg day 1, VCR 0.38 mg/kg day 8 and day 15, CABO 30 mg/kg once daily on days 8–21 |
| DOXO + VCR + PAZOPANIB | DOXO 3.5 mg/kg day 1, VCR 0.38 mg/kg day 8 and day 15, PAZO 65 mg/kg twice daily on days 8–21 |
| PLACEBO | vehicle control (no chemotherapy) |
TTC642
Mice (n = 30 total) were randomized to 6 treatment groups: Lenvatinib, Cabozantinib, Pazopanib, DOXO + VCR, DOXO + VCR + CABO, and placebo. The same doses were used as above.
Mice received 4 courses of chemotherapy (21 days per course) and bioluminescence was monitored weekly. Disease response was classified according to bioluminescence signal. Mice with a signal of 105 photons/sec/cm2 or less (similar to background) were classified as complete response, 105-106 photons/sec/cm2 as partial response, 107 up to 108 photons/sec/cm2 (similar to enrollment signal) as stable disease, and greater than 108 photons/sec/cm2 as progressive disease, as described previously (Stewart et al., 2014). Mice with tumor burden at any time greater than 20% of body weight were also classified as progressive disease. Mice were monitored daily while receiving chemotherapy.
Xenogen® Imaging
Mice were given intraperitoneal injections of Firefly D-Luciferin (Caliper Life Sciences; 3 mg/mouse). Bioluminescent images were taken five minutes later using the IVIS® 200 imaging system. Anesthesia was administered throughout image acquisition (isoflurane 1.5% in O2 delivered at 2 liters/min).
METHOD DETAILS
CCL sensitivity profiling
Screening of 481 small molecules against 860 cancer cell lines has been described previously (Rees et al., 2016, Seashore-Ludlow et al., 2015). Briefly, CCLs were plated (500 cells/well) in their provider-recommended growth media in white, opaque tissue-culture-treated Aurora 1536-well MaKO plates (Brooks Automation) using a Multidrop Combi Reagent Dispenser (ThermoFisher). Compounds were added 24 hours after plating using an Echo 555 (Labcyte Inc.), and cellular ATP levels were assessed after 72 hours of compound treatment using CellTiterGlo (Promega) on a ViewLux Microplate Imager (PerkinElmer).
Forty-seven additional cell lines were screened and processed using the same procedure. Small molecules were tested over a 16-point concentration range (twofold dilution) in duplicate. Compounds were added 24 hours after plating and cellular ATP levels were assessed after 72 hours of treatment using CellTiterGlo (Promega). All raw and analyzed viability data, as well as cell-line and compound meta-data, are available from the NCI CTD2 Data Portal (https://ctd2.nci.nih.gov/dataPortal/).
In all, area under concentration-response curves (AUCs) for 545 compounds across 907 CCLs were included for analysis. Where duplicate CCLs were tested, AUC values were averaged, resulting in a total of 887 unique CCLs tested. For each small molecule, the AUC ZMAD was calculated in R.
Annotation of genomic features for cell lines with RTK-activating mutations were collated from the following references, and verified by the Cancer Cell Line Encyclopedia (https://portals.broadinsti-tute.org/ccle/home) where possible: EOL1 (Griffin et al., 2003); NCIH1703 (Ramos et al., 2009); SW579 (McDermott et al., 2009); KASUMI1 (Larizza et al., 2005); MOLM13 and MV411 (Quentmeier et al., 2003); EBC1 (Lutterbach et al., 2007); SNU620 (Liu et al., 2014); HS746T (Asaoka et al., 2010); IM95 (Iwai et al., 2003); DMS114 and NCIH1581 (Weiss et al., 2010); JMSU1 and UMUC1 (Tomlinson et al., 2009); NCIH716 (Mathur et al., 2014); KATOIII and SNU16 (Lorenzi et al., 1997); KMS11 and OPM2 (Ronchetti et al., 2001); RT112 and RT4 (Williams et al., 2013).
Published Kd values of binding (Davis et al., 2011) and percent inhibition at 1 μM (Uitdehaag et al., 2014) were used to determine most relevant targets and to select follow-up inhibitors for further study.
Follow-up screening
Cells were plated and screened in 384-well format as described previously (Rees et al., 2016). Briefly, cells were manually plated in white, opaque tissue-culture-treated plates (Corning) at 1000 cells/well, compounds were added (1:300 dilution) using a CyBi-Well Vario pin-transfer machine 24 hours after plating, and sensitivity was measured using CellTiterGlo 72 hours after the addition of small molecules. At least two biological replicates were done for each condition. See Key Resources Table (KRT) for small-molecule annotated targets and vendor information.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| B-actin Rabbit mAb (HRP Conjugate) | Cell Signaling | Cat#5125; RRID: AB_1903890 |
| CoxIV (3E11) | Cell Signaling | Cat#4850; RRID: AB_2085424 |
| Hsp90 | Cell Signaling | Cat#4874S; RRID: AB_2121214 |
| GAPDH (14C10) | Cell Signaling | Cat#2118; RRID: AB_561053 |
| GAPDH (D4C6R) | Cell Signaling | Cat#97166S; RRID: AB_2756824 |
| SMARCB1/SNF5 | Bethyl | Cat#A301-087; RRID: AB_2779664 |
| p-PDGFRα (Tyr754) (23B2) | Cell Signaling | Cat#2992 ; RRID: AB_390728 |
| p-PDGFRα (Tyr762) | Cell Signaling | Cat#12022; RRID: AB_2636868 |
| PDGFRα | Cell Signaling | Cat#3164; RRID: AB_2162351 |
| p-PDGFRβ (Tyr751) | Cell Signaling | Cat#3161; RRID: AB_331053 |
| PDGFRβ | Cell Signaling | Cat#3162; RRID: AB_331111 |
| VEGFR1 (clone Y103) | Abcam | Cat#ab32152; RRID: AB_778798 |
| VEGFR2 | Cell Signaling | Cat#2479; RRID: AB_2212507 |
| p-Gab1 (Tyr307) | Cell Signaling | Cat#3234; RRID: AB_2221302 |
| Gab1 | Cell Signaling | Cat#3232; RRID: AB_2304999 |
| p-Met (Tyr1003) (13D11) | Cell Signaling | Cat#3135; RRID: AB_2285325 |
| p-MET (Tyr1234/1235) (D26) XP | Cell Signaling | Cat#3077; RRID: AB_2143884 |
| p-MET (Tyr1349) (130H2) | Cell Signaling | Cat#3133; RRID: AB_2181539 |
| Met (D1C2) XP | Cell Signaling | Cat#8198; RRID: AB_10858224 |
| p-EGFR (Tyr1068) D7A5 XP | Cell Signaling | Cat#3777; RRID: AB_2096270 |
| p-EGFR (Tyr992) | Cell Signaling | Cat#2235; RRID: AB_331708 |
| p-EGFR (Tyr1045) | Cell Signaling | Cat#2237; RRID: AB_331710 |
| EGFR | Cell Signaling | Cat#4267; RRID: AB_2246311 |
| p-FGFR1 (Tyr653/654) | Cell Signaling | Cat#3471; RRID: AB_331072 |
| FGFR1 (D8E4) | Cell Signaling | Cat#9740; RRID: AB_11178519 |
| FGFR2 (Bek, C-17) | Santa Cruz | Cat#sc-122; RRID: AB_631509 |
| FGFR3 | Cell Signaling | Cat#4574; RRID: AB_2246903 |
| p-FRS2-alpha (Tyr436) | Cell Signaling | Cat#3861; RRID: AB_2231950 |
| p-AKT (Thr308) | Cell Signaling | Cat#9275; RRID: AB_329828 |
| p-AKT (Ser473) | Cell Signaling | Cat#9271S; RRID: AB_329825 |
| AKT | Cell Signaling | Cat#9272; RRID: AB_329827 |
| p-MEK1/2 (Ser217/221) | Cell Signaling | Cat#9154P; RRID: AB_2138017 |
| MEK1/2 | Cell Signaling | Cat#9122; RRID: AB_823567 |
| p-p70 S6K (Thr389) (108D2) | Cell Signaling | Cat#9234; RRID: AB_2269803 |
| p70 S6K (49D7) | Cell Signaling | Cat#2708; RRID: AB_390722 |
| SHP2 (SH_PTP2) (B-1) | Santa Cruz | Cat#sc-7384; RRID: AB_628252 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (197G2) | Cell Signaling | Cat#4377T; RRID: AB_331775 |
| p44/42 MAPK (Erk1/2) (137F5) | Cell Signaling | Cat#4695; RRID: AB_390779 |
| Anti-mouse IgG Antibody, HRP-conjugated secondary | Cell Signaling | Cat#7076; RRID: AB_330924 |
| Anti-rabbit IgG Antibody, HRP-conjugated secondary | Cell Signaling | Cat#7074; RRID: AB_2099233 |
| Goat Anti-Mouse IgG Antibody, IRDye 680RD Conjugated | LI-COR Biosciences | Cat#926-68070; RRID: AB_10956588 |
| Goat Anti-Rabbit IgG Antibody, IRDye 800CW Conjugated | LI-COR Biosciences | Cat#926-32211; RRID: AB_621843 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat#G7573 |
| Protease/Phosphatase Inhibitor Cocktail | Cell Signaling | Cat#5872 |
| Halt Protease Inhibitor Cocktail | Thermo | Cat#78425 |
| 2-mercaptoethanol | Sigma-Aldrich | Cat# M6250; CAS Number: 60-24-2 |
| TRIzol Reagent | Life Technologies | Cat#15596018 |
| cabozantinib (gene name of protein targets: FLT3;KDR;MET;RET) | SelleckChem | S1119; CAS: 849217-68-1 |
| dasatinib (EPHA2;KIT;LCK;SRC;YES1) | SelleckChem | S1021; CAS: 302962-49-8 |
| Nintedanib (FGFR1;FGFR2;FGFR3;FLT1;FLT3;KDR; PDGFRA;PDGFRB) | SelleckChem | S1363; CAS: 228559-41-9 |
| Ki8751 (KDR;KIT;PDGFRA) | SelleckChem | S1363; CAS: 228559-41-9 |
| sunitinib (FLT1;FLT3;KDR;KIT;PDGFRA;PDGFRB) | SelleckChem | S1042; CAS: 341031-54-7 |
| lenvatinib (FLT1;FLT3;KDR;KIT;PDGFRA;PDGFRB) | SelleckChem | S1164; CAS: 417716-92-8 |
| axitinib (FLT1;FLT3;KDR;KIT;PDGFRA;PDGFRB) | SelleckChem | S1005; CAS: 319460-85-0 |
| AZD4547 (FGFR1;FGFR2;FGFR3) | SelleckChem | S2801; CAS: 1035270-39-3 |
| OSI-930 (KDR;KIT) | SelleckChem | S1220; CAS: 728033-96-3 |
| KW-2449 (AURKA;FLT3) | SelleckChem | S2158; CAS: 1000669-72-6 |
| SGX-523 (MET) | SelleckChem | S1112; CAS: 1022150-57-7 |
| lapatinib (EGFR;ERBB2) | SelleckChem | S1028; CAS: 388082-77-7 |
| foretinib (KDR;MET) | SelleckChem | S1111; CAS: 849217-64-7 |
| quizartinib (FLT3) | SelleckChem | S1526; CAS: 950769-58-1 |
| imatinib (ABL1;BCR;KIT) | SelleckChem | S2475; CAS: 152459-95-5 |
| infigratinib (FGFR1;FGFR2;FGFR3) | SelleckChem | S2183; CAS: 872511-34-7 |
| tandutinib (FLT3;KIT) | SelleckChem | S1043; CAS: 387867-13-2 |
| cediranib (FLT1;FLT4;KDR) | SelleckChem | S1017; CAS: 288383-20-0 |
| crizotinib (ALK;MET) | SelleckChem | S1068; CAS: 877399-52-5 |
| pazopanib (FLT1;FLT3;KDR;KIT;PDGFRB) | SelleckChem | S1035; CAS: 635702-64-6 |
| erlotinib (EGFR;ERBB2) | SelleckChem | S1023; CAS: 183319-69-9 |
| tivozanib (FLT1;FLT3;KDR) | SelleckChem | S1207; CAS: 475108-18-0 |
| glesatinib (FLT1;FLT3;KDR;MET) | SelleckChem | S1361; CAS: 875337-44-3 |
| allitinib (EGFR;ERBB2;ERBB4) | SelleckChem | S2185; CAS: 1050500-29-2 |
| vandetanib (EGFR;KDR) | SelleckChem | S1046; CAS: 443913-73-3 |
| PD173074 (FGFR1;FGFR2;FGFR3;KDR) | SelleckChem | S1264; CAS: 219580-11-7 |
| CP-673451 (PDGFRA;PDGFRB) | SelleckChem | S1536; CAS: 343787-29-1 |
| crenolanib (PDGFRA;PDGFRB;FLT3) | SelleckChem | S2730; CAS: 670220-88-9 |
| masitinib (KIT;PDGFRA;PDGFRB) | SelleckChem | S1064; CAS: 790299-79-5 |
| ponatinib (ABL1;PDGFRA;KDR;FGFR1;SRC;KIT) | SelleckChem | S1490; CAS: 943319-70-8 |
| SHP099 (PTPN11) | MedChemExpress | HY-100388; CAS: 1801747-42-1 |
| DOXOrubicin HCl injection, USP (for mice) | Sagent Pharmaceuticals | 25021-207-05 |
| Vincristine Sulfate Injection, USP | Hospira | 61703-309-16 |
| Pazopanib free base | MedKoo Biosciences | 202161; CAS: 444731-52-6 |
| Cabozantanib free base | MedKoo Biosciences | 200595; CAS: 849217-68-1 |
| Levantinib mesylate | MedKoo Biosciences | 201080; CAS: 857890-39-2 |
| Critical Commercial Assays | ||
| Human Phospho-RTK Array | R&D Systems | Cat# ARY001B; |
| DNeasy Blood and Tissue Kit | Qiagen | Cat#69504 |
| RNeasy MinElute Cleanup Kit | Qiagen | Cat#74204 |
| TruSeq Total RNA Sample Prep Kit | Illumina | Cat# 20020597 |
| Deposited Data | ||
| Dose-response curves from 30 RTK inhibitors across 16 RT cell lines | This paper | DOI: 10.17632/2vcpns9jgw.1 |
| Density plots of AUC values of AKT and MEK inhibitors | This paper | DOI: 10.17632/2vcpns9jgw.1 |
| Association of RTK dependencies and RTK transcript expression | This paper | DOI: 10.17632/2vcpns9jgw.1 |
| Activity of RTK inhibitors in select non-RT CCLs | This paper | DOI: 10.17632/2vcpns9jgw.1 |
| Scans of phospho-RTK arrays | This paper | DOI: 10.17632/2vcpns9jgw.1 |
| Dose-response curves of pairwise combination treatments of RTK inhibitors in RT cell lines | This paper | DOI: 10.17632/2vcpns9jgw.1 |
| Experimental Models: Cell Lines | ||
| A204 (Sex: F) | Broad Biological Samples Platform (BSP) | RRID: CVCL_1058 |
| BT12 (F) | Roberts Lab | RRID: CVCL_M155 |
| BT16 (Sex: M) | Roberts Lab | RRID: CVCL_M156 |
| CHLA266 (F) |
Roberts Lab | RRID: CVCL_M149 |
| G401 (M) | BSP | RRID: CVCL_0270 |
| G402 (F) | BSP | RRID: CVCL_1221 |
| KD (F) | Franck Bourdeaut | RRID: CVCL_U757 |
| KPMRTRY (M) | Yasumichi Kuwahara | RRID: CVCL_7051 |
| KYM1 (F) | BSP | RRID: CVCL_3007 |
| MON (F) | Franck Bourdeaut | RRID: CVCL_M846 |
| NCIH2004RT (M) | Roberts Lab | RRID: CVCL_U759 |
| TM87 (M) | Roberts Lab | RRID: CVCL_8001 |
| TTC549 (F) | Roberts Lab | RRID: CVCL_8005 |
| TTC642 (M) | Roberts Lab | RRID: CVCL_8006 |
| TTC709 (M) | Roberts Lab | RRID: CVCL_8007 |
| TTC1240 (F) | Timothy Triche | RRID: CVCL_8002 |
| A375 (F) | BSP | RRID: CVCL_0132 |
| A673 (F) | Stegmaier Lab | RRID: CVCL_0080 |
| A2058 (M) | ATCC | RRID: CVCL_1059 |
| CAL120 (F) | BSP | RRID: CVCL_1104 |
| LUDLU1 (M) | BSP | RRID: CVCL_2582 |
| MeWo (M) | BSP | RRID: CVCL_0445 |
| MKN45 (F) | BSP | RRID: CVCL_0434 |
| MV411 (M) | BSP | RRID: CVCL_0064 |
| T47D (F) | BSP | RRID: CVCL_0042 |
| U2OS (F) | BSP | RRID: CVCL_0042 |
| HEK293T (F) | ATCC | RRID: CVCL_0063 |
| Experimental Models: Organisms/Strains | ||
| Athymic nude immunodeficient mice (female) | Jackson Laboratories | Strain code 007850 |
| Recombinant DNA | ||
| pBabe/FL-SMARCB1 | Robert Kingston | Sif et al., 1998 |
| pBabe/Empty Vector | Robert Kingston | Sif et al., 1998 |
| pCL-10A1 Retrovirus Packaging Vector | Wohrle et al., 2013; Novus Biologicals | Cat# NBP2-29542 |
| Software and Algorithms | ||
| ImageJ v1.50i | NIH | https://imagej.nih.gov/ij/; RRID:SCR_003070 |
| Prism 8 | GraphPad | http://www.graphpad.com/ |
| Spotfire v7.0.1 | TIBCO | http://www.tibco.com |
| MatLab R2018a | Mathworks | http://www.mathworks.com |
| CTRPv2.0 MatLab curve-fitting procedures | Rees M.G., Seashore-Ludlow B., Clemons P.A. (2019) Computational Analyses Connect Small-Molecule Sensitivity to Cellular Features Using Large Panels of Cancer Cell Lines. In: Ziegler S., Waldmann H. (eds) Systems Chemical Biology. Methods in Molecular Biology, vol 1888. Humana Press, New York, NY | https://doi.org/10.1007/978-1-4939-8891-4_14; https://github.com/remontoire-pac/ctrp-reference |
| R v3.6 | R Foundation for Statistical Computing | https://www.r-project.org/foundation/ |
| RSEM v1.3.0 | Li and Dewey, 2011 | |
| ‘Fit-SNE’ R package for t-SNE visualization | Linderman et al. 2019 | |
| ‘DESeq2’ R package v1.24.0 for differential expression | Love et al., 2010 | |
| ‘limma’ R package v3.40.2 for mRNA expression processing | Shi et al. 2010 | |
| ‘drc’ package v3.0-1 for curve fitting | Ritz and Streibig, 2005 | |
| Brain Tumor Methylation Profiler v11b4 version 2.0 | DKFZ | https://www.molecularneuropathology.org |
| Other | ||
| NuPAGE 4-12% Bis-Tris Protein Gels, 1.0 mm, 17-well | Thermo Fisher Scientific | Cat# NP0329BOX |
| Bolt 4-12% Bis-Tris Plus gels | Life Technologies | Cat# NW04120BOX |
| PVDF membrane | EMD Millipore | Cat# IPVH00010 |
| Licor Odyssey buffer TBS-T | LICOR | Cat# 927 |
| HyBlot CL film | Denville Scientific | Cat# E3012 |
Small-molecule combination screening
Viability measurements were performed in 384-well plates as described above. A 1:1 mixture of the top concentration of individual compounds, followed by serial dilution (2-fold), was used for combinations, and compounds were added using a HP D300e Digital Printer.
CRISPR screening
Genome-scale CRISPR-Cas9 mediated screening has been described previously (Aguirre et al., 2016, Meyers et al., 2017). The GeCKO 19Q1 release, which includes an additional ten cell lines screened with the GeCKO library for a total of 43, was used (data available at the DepMap Portal: https://depmap.org/portal).
Western blotting
Western blotting was performed as previously described (Rees et al., 2016). See Key Resources Table (KRT) for antibody and reagent details. Whole-cell lysates were prepared by incubating cell pellets for 30 minutes in 1% NP-40 (Sigma-Aldrich) lysis buffer containing freshly added Protease/Phosphatase Inhibitor Cocktail (Cell Signaling Technology, #5872). After incubation, lysates were spun at 4°C, 13,000 RPM for ten minutes and supernatant collected and quantitated using BCA assay (Pierce). Proteins were prepared with 4× sample buffer containing 5% 2-mercaptoethanol (Sigma-Aldrich) and heated to 95 °C for 10 min 20 μg per sample was loaded onto Novex NuPAGE or Bolt 4–12% Bis-Tris Plus gels (Life Technologies) for electrophoresis. For NuPAGE gels, proteins were then transferred to PVDF membranes (EMD Millipore) using NuPAGE transfer buffer (Life Technologies), blocked for 30 min in 5% milk (Labscientific) in PBS-T or Odyssey buffer TBS-T (LICOR, Prod# 927), and incubated overnight at 4°C with primary antibody in 1% milk in PBS-T, 5% BSA (Sigma Aldrich) in PBS-T, or Odyssey buffer with 0.2% Tween 20. All primary antibodies were diluted to 1:1000, except for B-actin HRP (1:5000), Hsp90 (1:3000), SMARCB1 (1:6000), FGFR1 (1:5000), and AKT (1:2000).
After 3×5 min washes in PBST-T or TBS-T, membranes were then incubated for one hour at room temp with either LICOR antibodies and visualization, or rabbit HRP-linked secondary antibodies (Jackson ImmunoResearch Laboraties) at 1:3000 in 1% milk in PBS-T, then washed again and visualized on HyBlot CL film (Denville Scientific) using SuperSignal West Pico or Femto chemiluminescence substrate (Thermo Scientific). For Bolt gels, dry transfer and antibody incubation (secondary antibody: 1:15000) were performed as described previously (Rees et al., 2016), with visualization using the LICOR imaging system according to the manufacturer’s instructions.
Three biological replicates of cells were prepared and run for each Western.
Phospho-RTK array
R&D Systems RTK Array (ARY001B) was used according to manufacturer’s protocol. To analyze basal phospho-RTK levels, parental rhabdoid cell lines were seeded in T75 flasks and harvested when confluent.
For analysis of phospho-RTK levels after compound treatment, rhabdoid cell lines were seeded at 2M cells/10 cM plate for 24 hr, then treated with compounds for three hours. Lysates were prepared with Halt Protease Inhibitor Cocktail (Thermo 78425), quantified using BCA assay (Pierce) and 450 μg of protein was used per array. Arrays were detected on HyBlot CL film (Denville Scientific).
Compound treatment and protein analysis
Cells were plated at 400,000 cells/well in 6-well plates. At 24 hr, plates were treated with compounds (or DMSO) for the indicated time then harvested for western blotting. Two to three biological replicates were done for each condition.
Compound treatment and colony formation assay
Rhabdoid and control cell lines were seeded at 1,500 cells/well in 300 μL volume in 24-well plates. 24 hr after plating, DMSO or SHP099 was added to wells, with each concentration in duplicate. On day 4 of treatment, media was changed and SHP099 replenished. On day 10, cells were stained with crystal violet solution. Two biological replicates were done for each condition.
SMARCB1 retroviral expression
HEK293T cells were transfected with pBabe/FL-SMARCB1 or pBabe/Empty Vector (EV) and pCL-10A1 packaging vector, and retroviral supernatants collected and infected into G402 cells as described (Wohrle et al., 2013). Media was changed at 24 hr after infection, and puromycin selection (5 μg/mL) was started at 48 hr after infection. After 48 hr selection, cells were plated at 2M cells/10 cM plate for another 24 hr, then scraped (72 hr total puromycin selection) and prepared for Western blotting to confirm SMARCB1 re-expression and to probe RTK proteins. Three biological replicates were prepared and tested each for the empty vector (EV) and SMARCB1 add-back conditions
RNA-seq (SMARCB1 add-back)
Cells were harvested and prepared for RNA-seq as described previously (Wang et al., 2016). Two biological replicates were prepared each for the empty vector (EV) and SMARCB1 add-back conditions. On average, 22 million 75bp, single-end reads were generated for each sample.
Rhabdoid cell line methylation profiling
DNA was extracted from rhabdoid cell lines using the Qiagen DNeasy Blood and Tissue Kit (Cat No. 69504) following manufacturer’s instructions, and 500ng of DNA was submitted to the St. Jude Hartwell Center for Methylation Profiling using Illumina Infinium Methylation EPIC (850K) BeadChip arrays. iDAT files were uploaded and analyzed using the Brain Tumor Methylation Profiler v11b4 version 2.0 (https://www.molecularneuropathology.org) providing methylation classifier results for each sample.
QUANTIFICATION AND STATISTICAL ANALYSIS
Xenogen® quantification and xenograft statistical analyses
The Living Image 4.3 software (Caliper Life Sciences) was used to generate a standard region of interest (ROI) encompassing the largest tumor at maximal bioluminescence signal. The identical ROI was used to determine the average radiance (photons/s/cm2/sr) for all xenografts. Survival curves of time to tumor progression were generated by the Kaplan-Meier method. Log-rank tests were used to compare survival curves across all treatment groups.
Analysis of small-molecule combination screening
Synergy calculations to assess deviation from Loewe additivity were carried out as described previously (Seashore-Ludlow et al., 2015) in R using the ‘drc’ package to fit curves and calculate EC50, AUC and IC50 values (Ritz and Streibig, 2005). Combinations were excluded from synergy analysis when the maximal effect did not reach 0.5, precluding accurate estimation of IC50 values. At least two biological replicates were done for each condition.
Western blot and phospho-RTK array quantitation
Western blots were quantified in ImageJ (v1.50i, NIH) as described: (http://lukemiller.org/index.php/2010/11/analyzing-gels-and-western-blots-with-image-j/). For phospho-RTK arrays, films were scanned, converted to grayscale, and quantified in ImageJ. A region of interest (ROI) was defined around the pair of reference dots in the upper left corner of the array. Using this ROI, Grey Mean Value was measured around the reference dots. The empty grey space directly above these dots was measured as a background control. Another ROI was defined for the pair of dots for each protein, and this ROI was also used to measure nearby empty space as a background control. The value for each pair of dots was subtracted from its corresponding background measurement. This result for each protein was then divided by the result from the reference spots, to normalize expression within each array. The resultant values were then visualized using a heat map.
RNA-seq analysis
Reads were aligned to the hg19 reference using RSEM v1.3.0 (Li and Dewey, 2011) (rsem-calculate-expression –bowtie2 –estimate-rspd –output-genome-bam). The RSEM commands, rsem-generate-data-matrix and rsem-run-ebseq, were then used to determine differentially expressed genes.
Cancer cell line RNA-seq data were obtained from the Cancer Cell Line Encyclopedia (CCLE):
Rhabdoid tumor expression samples
RNA-seq gene-level quantitation (count) data for 65 RT samples (Chun et al., 2016) were downloaded from the TARGET website (https://ocg.cancer.gov/programs/target) and analyzed for differential expression using the DESeq2 package in R (Love et al., 2014). Adjusted p-values were calculated using Benjamini-Hochberg correction.
Microarray samples GSE70678 (Johann et al., 2016) GSE35493 (Birks et al., 2013), GSE64019 (Han et al., 2016), and GSE11482 (Gadd et al., 2010) were downloaded from GEO. Differential expression in GSE64019 was analyzed using GEO2R.
Data from Torchia et al. was provided by the study authors (Torchia et al., 2015) and processed using the limma R package, with normalization and background correction using the ‘neqc’ function (Shi et al., 2010, Ritchie et al., 2015).
Rhabdoid cell line gene expression-based clustering
TPM expression data for 57,820 genes and 1,165 cell lines generated by the DepMap project were downloaded from the DepMap Portal (https://depmap.org/portal, version 19q1; CCLE_depMap_19Q1_TPM.csv) The top 25% most variable genes were identified by those with the highest standard deviations of gene expression across all 1,165 cell lines. To visualize the data, t-Distributed Stochastic Neighbor Embedding (tSNE) was performed using the FIt-SNE R package (Linderman et al., 2019). Next, the expression dataset was subsetted to only include rhabdoid tumor lines. The 25% most variable genes within rhabdoid tumors were identified as above. Next, tSNE was performed using the FIt-SNE R package (Linderman et al., 2019).
DATA AND CODE AVAILABILITY
Code available for processing small-molecule sensitivity screening data is available at https://github.com/remontoire-pac/ctrp-reference. Code available for processing genome-scale CRISPR knockout data is available at https://github.com/cancerdatasci/. Any additional scripts or code written by the authors are available upon request from the corresponding author.
Original underlying data is available online from Mendeley Data DOI: 10.17632/2vcpns9jgw.1:
Concentration-response curves from 30 RTK inhibitors across 16 RT CCLs (data underlying Figure 1b).
AUC values of AKT and MEK inhibitors in RT and control cell lines (data underlying Figure S1a).
Association of RTK dependencies and RTK transcript expression (data underlying Figure 2a, related to 1c, S1e).
Activity of RTK inhibitors in select non-RT CCLs (data underlying Figure S1d).
Scans of phospho-RTK arrays (data underlying Figure 2b, 3a, 3b)
Concentration-response curves of pairwise combination treatments of RTK inhibitors in RT cell lines (data underlying Figure 3c, d).
Supplementary Material
Acknowledgements:
This project has been supported by grants from the National Cancer Institute: the Ruth L. Kirchstein pre-doctoral fellowship F31 CA183558–03 (E.M.O.), and the Cancer Target Discovery and Development Network grants: U01 CA176058 (W.C.H.) and U01 CA176152 (S.L.S.).
E.A.S is a St. Baldrick’s Scholar with generous support from the Invictus Fund.
This work was supported by the following grants from the US National Institutes of Health: R01CA172152 (C.W.M.R.) and R01CA113794 (C.W.M.R.).
The Avalanna Fund, the Cure AT/RT Now Foundation, the Garrett B. Smith Foundation, and ALSAC/St. Jude (C.W.M.R.) provided additional support.
T.R.G. and S.L.S. are Investigators at the Howard Hughes Medical Institute.
We would like to thank members of the Roberts lab, Schreiber lab, and Broad’s Pediatric Dependencies Map Project for helpful discussion. We are also grateful to Su Wang for RNA-seq analysis guidance and Elizabeth Chun for clustering guidance and Broad’s PRISM (Profiling Relative Inhibition Simultaneously in Mixture) team for screening guidance and analysis advice.
Footnotes
Declaration of Interests:
B.A.W. is now a J&J employee.
A.T. is a consultant for Tango Therapeutics.
P.A.C. serves as on the Scientific Advisory Board for nference, Inc., and on a Scientific Advisory Panel for Pfizer, Inc.
T.R.G. is an advisor to GlaxoSmithKline, is a co-founder of Sherlock Biosciences and was a co-founder and advisor to Foundation Medicine.
K.S. has previously consulted for Novartis and Rigel Pharmaceuticals and has received research funding from Novartis.
W.C.H. is a consultant for Thermo Fisher, AjuIB, MPM Capital, and Paraxel. WCH is a founder, holds equity, and is a member of the scientific advisory board for KSQ Therapeutics.
S.L.S. serves on the Board of Directors of the Genomics Institute of the Novartis Research Foundation (“GNF”); is a shareholder and serves on the Board of Directors of Jnana Therapeutics; is a shareholder of Forma Therapeutics; is a shareholder and advises Decibel Therapeutics and Eikonizo Therapeutics; serves on the Scientific Advisory Boards of Eisai Co., Ltd., Ono Pharma Foundation, and F-Prime Capital Partners; and is a Novartis Faculty Scholar.
References:
- AGUIRRE AJ, MEYERS RM, WEIR BA, VAZQUEZ F, ZHANG CZ, BEN-DAVID U, COOK A, HA G, HARRINGTON WF, DOSHI MB, et al. 2016. Genomic copy number dictates a gene-independent cell response to CRISPR-Cas9 targeting. Cancer Discov. [DOI] [PMC free article] [PubMed]
- ALVER BH, KIM KH, LU P, WANG X, MANCHESTER HE, WANG W, HASWELL JR, PARK PJ & ROBERTS CW 2017. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat Commun, 8, 14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARCARO A, DOEPFNER KT, BOLLER D, GUERREIRO AS, SHALABY T, JACKSON SP, SCHOENWAELDER SM, DELATTRE O, GROTZER MA & FISCHER B 2007. Novel role for insulin as an autocrine growth factor for malignant brain tumour cells. Biochem J, 406, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASAOKA Y, TADA M, IKENOUE T, SETO M, IMAI M, MIYABAYASHI K, YAMAMOTO K, YAMAMOTO S, KUDO Y, MOHRI D, et al. 2010. Gastric cancer cell line Hs746T harbors a splice site mutation of c-Met causing juxtamembrane domain deletion. Biochem Biophys Res Commun, 394, 1042–6. [DOI] [PubMed] [Google Scholar]
- BARRETINA J, CAPONIGRO G, STRANSKY N, VENKATESAN K, MARGOLIN AA, KIM S, WILSON CJ, LEHAR J, KRYUKOV GV, SONKIN D, et al. 2012. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 483, 603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BETZ BL, STROBECK MW, REISMAN DN, KNUDSEN ES & WEISSMAN BE 2002. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene, 21, 5193–203. [DOI] [PubMed] [Google Scholar]
- BIRKS DK, DONSON AM, PATEL PR, SUFIT A, ALGAR EM, DUNHAM C, KLEINSCHMIDT-DEMASTERS BK, HANDLER MH, VIBHAKAR R & FOREMAN NK 2013. Pediatric rhabdoid tumors of kidney and brain show many differences in gene expression but share dysregulation of cell cycle and epigenetic effector genes. Pediatr Blood Cancer, 60, 1095–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOSSEN C, MURRE CS, CHANG AN, MANSSON R, RODEWALD HR & MURRE C 2015. The chromatin remodeler Brg1 activates enhancer repertoires to establish B cell identity and modulate cell growth. Nat Immunol, 16, 775–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAUVIN C, LERUSTE A, TAUZIEDE-ESPARIAT A, ANDRIANTERANAGNA M, SURDEZ D, LESCURE A, HAN ZY, ANTHONY E, RICHER W, BAULANDE S, et al. 2017. High-Throughput Drug Screening Identifies Pazopanib and Clofilium Tosylate as Promising Treatments for Malignant Rhabdoid Tumors. Cell Rep, 21, 1737–1745. [DOI] [PubMed] [Google Scholar]
- CHEN YN, LAMARCHE MJ, CHAN HM, FEKKES P, GARCIA-FORTANET J, ACKER MG, ANTONAKOS B, CHEN CH, CHEN Z, COOKE VG, et al. 2016. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature, 535, 148–52. [DOI] [PubMed] [Google Scholar]
- CHUN HJ, LIM EL, HERAVI-MOUSSAVI A, SABERI S, MUNGALL KL, BILENKY M, CARLES A, TSE K, SHLAFMAN I, ZHU K, et al. 2016. Genome-Wide Profiles of Extra-cranial Malignant Rhabdoid Tumors Reveal Heterogeneity and Dysregulated Developmental Pathways. Cancer Cell, 29, 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COWLEY GS, WEIR BA, VAZQUEZ F, TAMAYO P, SCOTT JA, RUSIN S, EAST-SELETSKY A, ALI LD, GERATH WF, PANTEL SE, et al. 2014. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci Data, 1, 140035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DARR J, KLOCHENDLER A, ISAAC S & EDEN A 2013. Loss of IGFBP7 expression and persistent AKT activation contribute to SMARCB1/Snf5-mediated tumorigenesis. Oncogene. [DOI] [PubMed]
- DARR J, KLOCHENDLER A, ISAAC S, GEIGER T & EDEN A 2015. Phosphoproteomic analysis reveals Smarcb1 dependent EGFR signaling in Malignant Rhabdoid tumor cells. Mol Cancer, 14, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIS MI, HUNT JP, HERRGARD S, CICERI P, WODICKA LM, PALLARES G, HOCKER M, TREIBER DK & ZARRINKAR PP 2011. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol, 29, 1046–51. [DOI] [PubMed] [Google Scholar]
- GADD S, SREDNI ST, HUANG CC, PERLMAN EJ & RENAL TUMOR COMMITTEE OF THE CHILDREN’S ONCOLOGY, G. 2010. Rhabdoid tumor: gene expression clues to pathogenesis and potential therapeutic targets. Lab Invest, 90, 724–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GHANDI M, HUANG F, JANÉ-VALBUENA J, KRYUKOV G, LO C, MCDONALD E III, BARRETINA J, GELFAND E, BIELSKI C, LI H, et al. 2019. Next generation characterization of the Cancer Cell Line Encyclopedia. Nature [DOI] [PMC free article] [PubMed]
- GRIFFIN JH, LEUNG J, BRUNER RJ, CALIGIURI MA & BRIESEWITZ R 2003. Discovery of a fusion kinase in EOL-1 cells and idiopathic hypereosinophilic syndrome. Proc Natl Acad Sci U S A, 100, 7830–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GU S, SAYAD A, CHAN G, YANG W, LU Z, VIRTANEN C, VAN ETTEN RA & NEEL BG 2018. SHP2 is required for BCR-ABL1-induced hematologic neoplasia. Leukemia, 32, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAN ZY, RICHER W, FRENEAUX P, CHAUVIN C, LUCCHESI C, GUILLEMOT D, GRISON C, LEQUIN D, PIERRON G, MASLIAH-PLANCHON J, et al. 2016. The occurrence of intracranial rhabdoid tumours in mice depends on temporal control of Smarcb1 inactivation. Nat Commun, 7, 10421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HU G, SCHONES DE, CUI K, YBARRA R, NORTHRUP D, TANG Q, GATTINONI L, RESTIFO NP, HUANG S & ZHAO K 2011. Regulation of nucleosome landscape and transcription factor targeting at tissue-specific enhancers by BRG1. Genome Res, 21, 1650–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IWAI M, MATSUDA M & IWAI Y 2003. Cloning of a cancer cell-producing hepatocyte growth factor, vascular endothelial growth factor, and interleukin-8 from gastric cancer cells. In Vitro Cell Dev Biol Anim, 39, 288–90. [DOI] [PubMed] [Google Scholar]
- JOHANN PD, ERKEK S, ZAPATKA M, KERL K, BUCHHALTER I, HOVESTADT V, JONES DT, STURM D, HERMANN C, SEGURA WANG M, et al. 2016. Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell, 29, 379–93. [DOI] [PubMed] [Google Scholar]
- KUWAHARA Y, CHARBONEAU A, KNUDSEN ES & WEISSMAN BE 2010. Reexpression of hSNF5 in malignant rhabdoid tumor cell lines causes cell cycle arrest through a p21(CIP1/WAF1)-dependent mechanism. Cancer Res, 70, 1854–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUWAHARA Y, HOSOI H, OSONE S, KITA M, IEHARA T, KURODA H & SUGIMOTO T 2004. Antitumor activity of gefitinib in malignant rhabdoid tumor cells in vitro and in vivo. Clin Cancer Res, 10, 5940–8. [DOI] [PubMed] [Google Scholar]
- LARIZZA L, MAGNANI I & BEGHINI A 2005. The Kasumi-1 cell line: a t(8;21)-kit mutant model for acute myeloid leukemia. Leuk Lymphoma, 46, 247–55. [DOI] [PubMed] [Google Scholar]
- LAWRENCE MS, STOJANOV P, POLAK P, KRYUKOV GV, CIBULSKIS K, SIVACHENKO A, CARTER SL, STEWART C, MERMEL CH, ROBERTS SA, et al. 2013. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature, 499, 214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE RS, STEWART C, CARTER SL, AMBROGIO L, CIBULSKIS K, SOUGNEZ C, LAWRENCE MS, AUCLAIR D, MORA J, GOLUB TR, et al. 2012. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest, 122, 2983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI B & DEWEY CN 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LINDERMAN GC, RACHH M, HOSKINS JG, STEINERBERGER S & KLUGER Y 2019. Fast interpolation-based t-SNE for improved visualization of single-cell RNA-seq data. Nat Methods, 16, 243–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU YJ, SHEN D, YIN X, GAVINE P, ZHANG T, SU X, ZHAN P, XU Y, LV J, QIAN J, et al. 2014. HER2, MET and FGFR2 oncogenic driver alterations define distinct molecular segments for targeted therapies in gastric carcinoma. Br J Cancer, 110, 1169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LORENZI MV, CASTAGNINO P, CHEN Q, CHEDID M & MIKI T 1997. Ligand-independent activation of fibroblast growth factor receptor-2 by carboxyl terminal alterations. Oncogene, 15, 817–26. [DOI] [PubMed] [Google Scholar]
- LOVE MI, HUBER W & ANDERS S 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUTTERBACH B, ZENG Q, DAVIS LJ, HATCH H, HANG G, KOHL NE, GIBBS JB & PAN BS 2007. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res, 67, 2081–8. [DOI] [PubMed] [Google Scholar]
- MARATHE HG, WATKINS-CHOW DE, WEIDER M, HOFFMANN A, MEHTA G, TRIVEDI A, ARAS S, BASUROY T, MEHROTRA A, BENNETT DC, et al. 2017. BRG1 interacts with SOX10 to establish the melanocyte lineage and to promote differentiation. Nucleic Acids Res. [DOI] [PMC free article] [PubMed]
- MATALKAH F, MARTIN E, ZHAO H & AGAZIE YM 2016. SHP2 acts both upstream and downstream of multiple receptor tyrosine kinases to promote basal-like and triple-negative breast cancer. Breast Cancer Res, 18, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATHUR A, WARE C, DAVIS L, GAZDAR A, PAN BS & LUTTERBACH B 2014. FGFR2 is amplified in the NCI-H716 colorectal cancer cell line and is required for growth and survival. PLoS One, 9, e98515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATHUR R, ALVER BH, SAN ROMAN AK, WILSON BG, WANG X, AGOSTON AT, PARK PJ, SHIVDASANI RA & ROBERTS CW 2016. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat Genet. [DOI] [PMC free article] [PubMed]
- MCDERMOTT U, AMES RY, IAFRATE AJ, MAHESWARAN S, STUBBS H, GRENINGER P, MCCUTCHEON K, MILANO R, TAM A, LEE DY, et al. 2009. Ligand-dependent platelet-derived growth factor receptor (PDGFR)-alpha activation sensitizes rare lung cancer and sarcoma cells to PDGFR kinase inhibitors. Cancer Res, 69, 3937–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEYERS RM, BRYAN JG, MCFARLAND JM, WEIR BA, SIZEMORE AE, XU H, DHARIA NV, MONTGOMERY PG, COWLEY GS, PANTEL S, et al. 2017. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet, 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUNOZ DM, CASSIANI PJ, LI L, BILLY E, KORN JM, JONES MD, GOLJI J, RUDDY DA, YU K, MCALLISTER G, et al. 2016. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discov, 6, 900–13. [DOI] [PubMed] [Google Scholar]
- NAKAYAMA RT, PULICE JL, VALENCIA AM, MCBRIDE MJ, MCKENZIE ZM, GILLESPIE MA, KU WL, TENG M, CUI K, WILLIAMS RT, et al. 2017. SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat Genet, 49, 1613–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRAHALLAD A, HEYNEN GJ, GERMANO G, WILLEMS SM, EVERS B, VECCHIONE L, GAMBINO V, LIEFTINK C, BEIJERSBERGEN RL, DI NICOLANTONIO F, et al. 2015. PTPN11 Is a Central Node in Intrinsic and Acquired Resistance to Targeted Cancer Drugs. Cell Rep, 12, 1978–85. [DOI] [PubMed] [Google Scholar]
- QUENTMEIER H, REINHARDT J, ZABORSKI M & DREXLER HG 2003. FLT3 mutations in acute myeloid leukemia cell lines. Leukemia, 17, 120–4. [DOI] [PubMed] [Google Scholar]
- RAMOS AH, DUTT A, MERMEL C, PERNER S, CHO J, LAFARGUE CJ, JOHNSON LA, STIEDL AC, TANAKA KE, BASS AJ, et al. 2009. Amplification of chromosomal segment 4q12 in non-small cell lung cancer. Cancer Biol Ther, 8, 2042–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REES MG, SEASHORE-LUDLOW B, CHEAH JH, ADAMS DJ, PRICE EV, GILL S, JAVAID S, COLETTI ME, JONES VL, BODYCOMBE NE, et al. 2016. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol, 12, 109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RITCHIE ME, PHIPSON B, WU D, HU Y, LAW CW, SHI W & SMYTH GK 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res, 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RITZ C & STREIBIG JC 2005. Bioassay analysis using R. Journal of Statistical Software, 12, 1–22. [Google Scholar]
- ROBERTS CW, GALUSHA SA, MCMENAMIN ME, FLETCHER CD & ORKIN SH 2000. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci U S A, 97, 13796–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RONCHETTI D, GRECO A, COMPASSO S, COLOMBO G, DELL’ERA P, OTSUKI T, LOMBARDI L & NERI A 2001. Deregulated FGFR3 mutants in multiple myeloma cell lines with t(4;14): comparative analysis of Y373C, K650E and the novel G384D mutations. Oncogene, 20, 3553–62. [DOI] [PubMed] [Google Scholar]
- SEASHORE-LUDLOW B, REES MG, CHEAH JH, COKOL M, PRICE EV, COLETTI ME, JONES V, BODYCOMBE NE, SOULE CK, GOULD J, et al. 2015. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov, 5, 1210–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHI W, OSHLACK A & SMYTH GK 2010. Optimizing the noise versus bias trade-off for Illumina whole genome expression BeadChips. Nucleic Acids Res, 38, e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEWART E, GOSHORN R, BRADLEY C, GRIFFITHS LM, BENAVENTE C, TWAROG NR, MILLER GM, CAUFIELD W, FREEMAN BB 3RD, BAHRAMI A, et al. 2014. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep, 9, 829–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOMLINSON DC, LAMONT FR, SHNYDER SD & KNOWLES MA 2009. Fibroblast growth factor receptor 1 promotes proliferation and survival via activation of the mitogen-activated protein kinase pathway in bladder cancer. Cancer Res, 69, 4613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TORCHIA J, GOLBOURN B, FENG S, HO KC, SIN-CHAN P, VASILJEVIC A, NORMAN JD, GUILHAMON P, GARZIA L, AGAMEZ NR, et al. 2016. Integrated (epi)-Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer Cell, 30, 891–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TORCHIA J, PICARD D, LAFAY-COUSIN L, HAWKINS CE, KIM SK, LETOURNEAU L, RA YS, HO KC, CHAN TS, SIN-CHAN P, et al. 2015. Molecular subgroups of atypical teratoid rhabdoid tumours in children: an integrated genomic and clinicopathological analysis. Lancet Oncol, 16, 569–82. [DOI] [PubMed] [Google Scholar]
- UITDEHAAG JC, DE ROOS JA, VAN DOORNMALEN AM, PRINSEN MB, DE MAN J, TANIZAWA Y, KAWASE Y, YOSHINO K, BUIJSMAN RC & ZAMAN GJ 2014. Comparison of the cancer gene targeting and biochemical selectivities of all targeted kinase inhibitors approved for clinical use. PLoS One, 9, e92146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG X, LEE RS, ALVER BH, HASWELL JR, WANG S, MIECZKOWSKI J, DRIER Y, GILLESPIE SM, ARCHER TC, WU JN, et al. 2016. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat Genet. [DOI] [PMC free article] [PubMed]
- WANG X, SANSAM CG, THOM CS, METZGER D, EVANS JA, NGUYEN PT & ROBERTS CW 2009. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res, 69, 8094–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEISS J, SOS ML, SEIDEL D, PEIFER M, ZANDER T, HEUCKMANN JM, ULLRICH RT, MENON R, MAIER S, SOLTERMANN A, et al. 2010. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med, 2, 62ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILLIAMS SV, HURST CD & KNOWLES MA 2013. Oncogenic FGFR3 gene fusions in bladder cancer. Hum Mol Genet, 22, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOHRLE S, WEISS A, ITO M, KAUFFMANN A, MURAKAMI M, JAGANI Z, THUERY A, BAUER-PROBST B, REIMANN F, STAMM C, et al. 2013. Fibroblast growth factor receptors as novel therapeutic targets in SNF5-deleted malignant rhabdoid tumors. PLoS One, 8, e77652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WONG JP, TODD JR, FINETTI MA, MCCARTHY F, BRONCEL M, VYSE S, LUCZYNSKI MT, CROSIER S, RYALL KA, HOLMES K, et al. 2016. Dual Targeting of PDGFRalpha and FGFR1 Displays Synergistic Efficacy in Malignant Rhabdoid Tumors. Cell Rep, 17, 1265–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YU C, MANNAN AM, YVONE GM, ROSS KN, ZHANG YL, MARTON MA, TAYLOR BR, CRENSHAW A, GOULD JZ, TAMAYO P, et al. 2016. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat Biotechnol, 34, 419–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code available for processing small-molecule sensitivity screening data is available at https://github.com/remontoire-pac/ctrp-reference. Code available for processing genome-scale CRISPR knockout data is available at https://github.com/cancerdatasci/. Any additional scripts or code written by the authors are available upon request from the corresponding author.
Original underlying data is available online from Mendeley Data DOI: 10.17632/2vcpns9jgw.1:
Concentration-response curves from 30 RTK inhibitors across 16 RT CCLs (data underlying Figure 1b).
AUC values of AKT and MEK inhibitors in RT and control cell lines (data underlying Figure S1a).
Association of RTK dependencies and RTK transcript expression (data underlying Figure 2a, related to 1c, S1e).
Activity of RTK inhibitors in select non-RT CCLs (data underlying Figure S1d).
Scans of phospho-RTK arrays (data underlying Figure 2b, 3a, 3b)
Concentration-response curves of pairwise combination treatments of RTK inhibitors in RT cell lines (data underlying Figure 3c, d).