

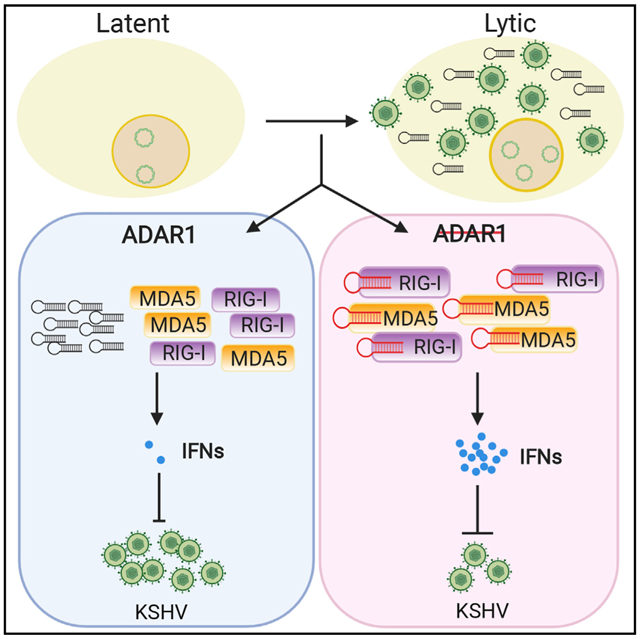
SUMMARY
Kaposi’s sarcoma-associated herpesvirus (KSHV) is a double-stranded DNA virus that exhibits two alternative life cycles: latency and lytic reactivation. During lytic reactivation, host innate immune responses are activated to restrict viral replication. Here, we report that adenosine deaminase acting on RNA 1 (ADAR1) is required for optimal KSHV lytic reactivation from latency. Knockdown of ADAR1 in KSHV latently infected cells inhibits viral gene transcription and viral replication during KSHV lytic reactivation. ADAR1 deficiency also significantly increases type I interferon production during KSHV reactivation. This increased interferon response is dependent on activation of the RIG-I-like receptor (RLR) pathway. Depletion of ADAR1 together with either RIG-I, MDA5, or MAVS reverses the increased IFNβ production and rescues KSHV lytic replication. These data suggest that ADAR1 serves as a proviral factor for KSHV lytic reactivation and facilitates DNA virus reactivation by dampening the RLR pathway-mediated innate immune response.
Graphical Abstract
In Brief
Zhang et al. report that ADAR1, a double-stranded RNA-editing enzyme, can facilitate KSHV reactivation by dampening the RIG-I/MDA5 pathway-mediated innate immune response. This study sheds light on how host cell proteins modulate the KSHV life cycle.
INTRODUCTION
Kaposi’s sarcoma-associated herpesvirus (KSHV) is the etiological agent of three human malignancies: Kaposi’s sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman’s disease (MCD) (Cesarman et al., 1995; Chang et al., 1994; Soulier et al., 1995). KSHV has a double-stranded DNA genome and can exhibit two phases of its life cycle: latency and lytic replication. During the latent phase of viral replication, the virus persists as circular episomes, and expression of viral genes is largely restricted to the latency-associated transcripts (Wong and Damania, 2017). When it switches to lytic reactivation, all viral genes are expressed, viral DNA is amplified, and progeny virions are produced. Unlike in latency, which can evade host immune surveillance to establish life-long infection, the host immune responses tend to be more pronounced during lytic infection (Ma et al., 2018).
KSHV infection has been shown to trigger innate immune responses by activating several host DNA and RNA sensors, including Toll-like receptors (TLRs), retinoic acid-inducible gene I protein (RIG-I)-like receptors (RLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), absent in melanoma 2 (AIM2)-like receptors (ALRs) and cyclic GMP-AMP synthase (cGAS) stimulator of interferon genes protein (STING) pathways (Gregory et al., 2011; Kerur et al., 2011; Lagos et al., 2008; Ma et al., 2015; West and Damania, 2008; West et al., 2014; Wu et al., 2015). Activation of these signaling pathways eventually leads to induction of type I interferons (IFNs) and proinflammatory cytokines, which establish an antiviral state and inhibit KSHV infection and replication. On the other hand, KSHV encodes several viral proteins that counteract the host immune system to facilitate its persistence (Dittmer and Damania, 2016). RIG-I and melanoma differentiation associated gene 5 (MDA5) are the two major cytosolic double-stranded RNA (dsRNA) sensors that signal through the adaptor protein mitochondrial antiviral signaling (MAVS) to activate downstream signaling and subsequent type I IFN production. Although a DNA virus, KSHV infection has been reported to also activate the RLR signaling pathway (West et al., 2014) because KSHV infection leads to production of virus- and host-derived RNAs (Zhang et al., 2018; Zhao et al., 2018) that can be recognized by MDA5 and RIG-I. Depletion of RIG-I or its adaptor MAVS has been found to increase KSHV infection, efficiency, and lytic reactivation (West et al., 2014). To counteract activation of the RLR pathway, KSHV encodes a viral deubiquitinase, ORF64, which can deubiquitinate RIG-I to prevent RIG-I-mediated IFN induction (Inn et al., 2011).
dsRNA usually arises from evading pathogens, but some cellular RNAs do contain RNA duplexes, such as the Alu hairpins in 3′ UTRs of mature mRNAs in the cytoplasm (Capshew et al., 2012). Aberrant recognition of these RNA structures leads to chronic inflammation, which is hazardous to the host. Therefore, host cells have developed efficient mechanisms, such as RNA base modification, to prevent these endogenous RNA duplexes from being detected by host pattern recognition receptors (PRRs). Several recent studies have demonstrated that adenosine deaminase acting on RNA 1 (ADAR1), a dsRNA adenosine (A)-to-inosine (I) editing enzyme, plays a critical role in avoiding autoinflammation by preventing PRRs from sensing host-derived RNAs that contain RNA duplexes (Mannion et al., 2014). Murine Adar1−/− cells showed reduced A-to-I editing of IFN-inducible RNA species, which led to dsRNA ligand sensing by dsRNA-activated protein kinase R (PKR) and MDA5 (Ishizuka et al., 2019). Moreover, ADAR1 knockout in human neural progenitor cells results in IFN production and cell death (Chung et al., 2018). ADAR1 mutations lead to Aicardi-Goutieres syndrome (AGS), an autoimmune disease associated with spontaneous IFN production (Rice et al., 2012). There are three ADARs (ADAR1–ADR3) in mammalian cells, and ADAR1 is the active A-to-I editor of Alu elements in human cells (Nishikura, 2010). A-to-I editing events have also been observed in several different viruses, and they play important roles during viral infection (Samuel, 2011). In the case of KSHV, ADAR1-mediated RNA editing of the KSHV Kaposin transcript has been reported, which eliminates its transforming activity (Gandy et al., 2007). However, whether ADAR1 plays a role in the innate immune response to KSHV is currently not known.
In this study, we found that depletion of ADAR1 in KSHV latently infected cells inhibited viral gene transcription and viral replication during KSHV reactivation from latency. The inhibited viral replication is due to increased type I IFN and proinflammatory cytokine production, which is dependent on the RIG-I/MDA5-MAVS signaling pathway. Our findings collectively suggest that ADAR1 plays a proviral role in facilitating KSHV lytic reactivation by preventing viral induced host RNA duplexes from activating the RLR signaling pathway. Our findings therefore indicate that ADAR1 facilitates DNA virus replication by dampening the RLR pathway-mediated innate immune response.
RESULTS
Suppression of ADAR1 Expression Inhibits KSHV Reactivation
We measured the editing activity of ADAR in KSHV-infected cells, iSLK.219, and found that ADAR activity was gradually increased during KSHV reactivation from latency (Figure S1). To further determine whether ADAR1 is involved in regulating KSHV lytic reactivation, we used small interfering RNA (siRNA) to suppress ADAR1 expression in iSLK.219 cells, which contain a latent KSHV genome and a doxycycline (Dox)-inducible RTA (replication and transcription activator) protein to enable entry into the lytic cycle. The cells also express a constitutive GFP marker and an RFP marker driven by a lytic cycle-specific promoter, which can be used as an indicator for lytic reactivated cells (Myoung and Ganem, 2011). We found that there were markedly fewer RFP-positive cells after Dox treatment in ADAR1-depleted cells than in control cells (Figure 1A), indicating that suppression of ADAR1 inhibited KSHV lytic reactivation. The RFP (red fluorescent protein) and GFP intensities in the cells were further quantified by a plate reader and presented as fold induction of the RFP/GFP ratio (Figure 1B). Consistent with these data, we found that KSHV genome copies in the supernatant were also significantly reduced in ADAR1-silenced cells (Figure 1C). The knockdown efficiency of ADAR1 was confirmed by real-time PCR (Figure 1D). We next examined the mRNA expression level of several KSHV genes to identify the effect of ADAR1 on viral gene transcription. Expression of the KSHV genes viral interleukin-6 (vIL-6) (latent and lytic gene), ORF39 (early gene), and K8.1 (late gene) was successfully induced upon reactivation, whereas ADAR1-depleted cells showed an impaired ability to induce transcription of these lytic genes compared with control cells, especially 72 h after reactivation (Figures 1E-1G). Consistent with these results, expression of the KSHV proteins vIL-6, ORF45, and K8α was also reduced in ADAR1-deficient cells (Figure 1H). Taken together, these data indicate ADAR1 is required for optimal KSHV lytic reactivation from latency.
Figure 1. Suppression of ADAR1 Inhibits KSHV Lytic Reactivation.

iSLK.219 cells were transfected with control non-specific (NS) siRNA or ADAR1 siRNA for 48 h and then treated with doxycycline (Dox; 0.2 μg/mL).
(A) GFP- and RFP-positive cells were imaged 0, 48, and 72 h post-Dox treatment.
(B) GFP and RFP intensities were measured by a Clariostar plate reader, and the RFP/GFP ratio was calculated at the indicated times.
(C) KSHV genome copy numbers in the supernatants were measured by real-time PCR.
(D–G) RNA was extracted from the cells, and the knockdown efficiency of ADAR1 (D) and the mRNA expression levels of the KSHV vIL-6 (E), ORF39 (F), and K8.1
(G) genes were measured by real-time PCR. Target mRNA expression was normalized to β-actin mRNA and presented as fold induction.
(H) Cell lysates were collected at the indicated time points, and western blots were performed with the indicated antibodies.
The data shown are representative of three independent experiments. Data from (B)–(G) are presented as mean ± SD. *, p < 0.05 by Student’s t test. Scale bar, 500 μm.
See also Figure S1.
Knockdown of ADAR1 Increases IFNβ Production and Induces a Large Array of Antiviral Genes during KSHV Lytic Reactivation
Because ADAR1 has been reported to prevent dsRNA from triggering innate immune signaling (Mannion et al., 2014), we next checked the induction of IFNβ in ADAR1-deficient cells during KSHV lytic reactivation. As shown in Figure 2A, IFNβ mRNA expression following KSHV reactivation was significantly enhanced in ADAR1 depleted cells 48 h and 72 h after Dox treatment. We also measured IFNβ production in the supernatant and found that IFNβ protein was readily detected in ADAR1-depleted cells 72 h after Dox treatment, but not in any of the samples from control cells (Figure 2B). Similar results were observed with three individual ADAR1 siRNAs, confirming the specificity of the ADAR1 siRNAs we used (Figures S2A-S2D). We also checked the mRNA expression profile of several IFN-related genes, including IFNα1, IFNγ, signal transducer and activator of transcription 1 (STAT1), and IL-8. We found that mRNA induction of these genes was also significantly increased in ADAR1-suppressed cells 72 h post-reactivation (Figures 2C-2F). IFNβ is known to bind IFNα/β receptor (IFNAR) and induce IFN-stimulated genes (ISGs) for effective antiviral responses. We further performed a Human Interferon & Receptors PCR Array (QIAGEN) analysis of 84 genes whose expression is controlled by or that are involved in cell signaling mediated by IFN ligands and receptors. After Dox treatment for 72 h, KSHV reactivation led to a more than 2-fold induction in multiple genes. Among these genes, 30 showed a much higher induction (>2-fold) in ADAR1 siRNA-transfected cells than in non-specific (NS) siRNA-transfected control cells. The overall gene expression profile of all 84 genes tested is shown as a heatmap in Figure 2G, and raw data are listed in Table S1. In summary, ADAR1 deficiency leads to a markedly higher induction of type I IFN signaling during KSHV lytic reactivation.
Figure 2. Knockdown of ADAR1 Increases Type I IFN and Cytokine Production during KSHV Reactivation.

iSLK.219 cells were transfected with NS or ADAR1 siRNA for 48 h and then treated with Dox (0.2 μg/mL) for 0, 48, and 72 h.
(A) The fold induction of IFNβ mRNA expression level was measured by real-time PCR.
(B) IFNβ production in the supernatant was measured by ELISA.
(C–F) The mRNA expression of the IFNα1 (C), IFNγ (D), STAT1 (E), and IL-8 (F) genes was measured by real-time PCR, and the fold change was normalized to β-actin mRNA.
(G) Heatmap of the Human Interferons & Receptors PCR array. RNA was extracted from the indicated samples, and the mRNA levels of the indicated genes were analyzed using the Human Interferons & Receptors RT2 Profiler PCR Array. The mRNA levels of genes were normalized to β-actin. The heatmap was generated using the web-based tool Morpheus (https://software.broadinstitute.org/morpheus/).
The data shown are representative of three independent experiments, except for (G), which is representative of two experiments. Data from (A)–(F) are presented as mean ± SD. *, p < 0.05 by Student’s t test. A1, ADAR1.
Knockdown of ADAR1, but Not ADAR2, Results in Loss of A-to-I Editing of KSHV Transcripts and Increases IFNβ Production during Lytic Reactivation
Considering that both ADAR1 and ADAR2 have been shown to possess A-to-I editing activity, we further examined whether ADAR2 also plays a role in KSHV lytic reactivation. Kaposin RNA is the only transcript in KSHV that has been reported to undergo A-to-I editing, and the amount of edited Kaposin transcript increases dramatically as the lytic cycle progresses, leading to highly penetrant A-to-G transition at 72 h (Arias et al., 2014; Gandy et al., 2007). To evaluate the A-to-I editing activity of ADAR1 and ADAR2 in KSHV transcripts, we depleted ADAR1 or ADAR2 in iSLK.219 cells using siRNA and confirmed the knockdown efficiency with real-time PCR (Figures 3A and 3B). We next sequenced the editing region of Kaposin mRNA from cells transfected with each siRNA after Dox treatment for 72 h. A large portion of cDNAs derived from Kaposin RNA transcripts have G at the previously reported rKSHV.219 genome coordinate site 117,809 in control and ADAR2-deficient cells, whereas only the original unedited nucleotide (A) was seen at this site in the Kaposin transcript from ADAR1-suppressed cells (Figure 3C). This indicates that A-to-I editing of the KSHV Kaposin transcript is dependent on ADAR1 but not ADAR2. In contrast to ADAR1, knockdown of ADAR2 did not enhance IFNβ production during KSHV lytic reactivation compared with control cells (Figures 3D and 3E). We also compared the mRNA expression level of ADAR1 and ADAR2 because one previous study reported that ADAR2 mRNA is much lower than ADAR1 in KSHV-infected PEL cells (Gandy et al., 2007). Consistent with that study, we found that ADAR1 mRNA is about 50-fold more abundant than ADAR2 mRNA in iSLK.219 cells (Figure S3). Although we do not exclude the possibility that ADAR2 may also be involved in RNA editing during KSHV reactivation, because of its extremely low abundance, its contribution is likely to be minor. Taken together, these data indicate that ADAR1, but not ADAR2, is responsible for dsRNA editing during KSHV lytic reactivation.
Figure 3. ADAR1, but Not ADAR2, Is Involved in KSHV Lytic Reactivation-Induced IFN.

iSLK.219 cells were transfected with NS, ADAR1, or ADAR2 siRNA, and then lytic reactivation was induced with 0.2 μg/mL Dox for the indicated times.
(A and B) The knockdown efficiency of ADAR1 (A) and ADAR2 (B) was confirmed with real-time PCR.
(C) A-to-I editing in KSHV Kaposin transcripts. Sanger sequencing of Kaposin cDNA 72 h after Dox treatment from iSLK.219 cells. *indicates the edited position at rKSHV.219 (NCBI:txid651668) genome coordinate 117809.
(D) Transcription of the IFNβ gene was quantified by real-time PCR. The mRNA expression fold change was normalized to β-actin mRNA.
(E) IFNβ production in the supernatant was measured by ELISA.
The data shown are representative of three independent experiments. Data from (A), (B), (D), and (E) are presented as mean ± SD. *, p < 0.05 by Student’s t test.
See also Figures S3-S5.
The ADAR1 p110 and p150 Isoforms Are Responsible for Preventing Innate Immune Activation during KSHV Lytic Reactivation
The ADAR1 gene encodes two protein isoforms, referred to as p110 and p150, both of which possess A-to-I editing activity (Samuel, 2011). To further dissect the role of each isoform in innate immune response activation during KSHV lytic reactivation, we first designed several siRNAs that are supposed to specifically target p110 or p150. However, all of these siRNAs turned out to target both isoforms (data not shown). We next attempted to generate ADAR1, p110, or p150 knockout (KO) iSLK.219 cell lines using CRISPR technology. A previous study successfully generated ADAR1 KO and p150 KO 293T cell lines and also found that p110 KO was not feasible (Chung et al., 2018). We employed the same short guide RNA (sgRNA) sequences used in that study. However, although we screened more than 100 clones for each gene, we were not able to get a clean KO cell clone. Nevertheless, we did generate several cell clones in which ADAR1 or p150 expression was significantly reduced. For convenience, we still named these clones KO cells. Robust IFNβ production was detected in supernatant from ADAR1 KO cells 72 h after KSHV reactivation (Figure S4A), which confirmed the results from the siRNA experiments. On the other hand, although the amount of IFNβ produced from p150 KO cells is significantly more than that from WT cells, it is much less than that from ADAR1 KO cells. This indicates that p150 plays a partial role in preventing KSHV reactivation-induced innate immune responses, and p110 likely also contributes to some extent. Consistently, IFNβ mRNA and mRNA of two IFNβ-inducible genes, RIG-I and MDA5, were markedly induced in ADAR1 KO and p150 KO cells, with lower induction in p150 KO cells (Figures S4B-S4D). Phosphorylation of TBK1 and IRF3, which indicates the activation status of several canonical innate immune pathways, is also enhanced in ADAR1 KO and p150 KO cells compared with WT iSLK.219 cells after KSHV lytic reactivation (Figure S4E). Taken together, these data suggest that the ADAR1 isoforms p110 and p150 contribute to preventing innate immune activation during KSHV lytic reactivation.
A-to-I Editing of the Kaposin Transcript Does Not Induce IFNβ Production, Nor Does It Affect KSHV Lytic Reactivation
Because we confirmed that Kaposin transcript undergoes A-to-I editing by ADAR1, we next evaluated whether this editing affects innate immune activation and KSHV lytic reactivation. We constructed plasmids that expressed the wild-type or edited Kaposin A transcripts. The RNA secondary structures of both transcripts were subjected to the RNA Mfold program and are depicted in Figure S5A. Each plasmid was co-transfected with an IFNβ promoter luciferase reporter plasmid into 293T cells, and the expression of both transcripts was readily detected (Figure S5B). However, neither transcript was able to activate IFNβ promoter activity (Figure S5C). We also suppressed Kaposin transcript expression in iSLK.219 cells using three individual siRNAs (Figure S5D). Depletion of Kaposin did not seem to affect KSHV virion production and release into the medium after Dox-induced lytic reactivation (Figure S5E). Moreover, knockdown of Kaposin did not affect KSHV viral gene expression either (Figure S5F). Hence, depletion of Kaposin does not appear to affect KSHV lytic reactivation.
Suppression of ADAR1 Enhances the RLR Signaling Pathway during KSHV Reactivation
Previous studies have shown that dsRNA was induced in iSLK.219 cells (West et al., 2014) and BC-3 cells (Zhao et al., 2018) during KSHV lytic reactivation. RIG-I and MDA5 are two major dsRNA sensors, and both are IFN-inducible genes. Because we have shown that KSHV reactivation led to IFNβ induction and that suppression of ADAR1 further enhanced this induction, we next monitored the transcription of the MDA5 and RIG-I genes. KSHV reactivation lead to limited induction of MDA5 and RIG-I mRNA in cells transfected with control siRNA, whereas knockdown of ADAR1 significantly increased induction of both genes 72 h post-reactivation (Figures 4A and 4B). Correspondingly, the protein expression of both MDA5 and RIG-I was markedly increased only in ADAR1-suppressed cells treated with Dox for 72 h (Figure 4C). We also checked activation of the RLR signaling pathway by looking at TBK1 and IRF3 phosphorylation. KSHV reactivation induced phosphorylation of TBK1 and IRF3 in control cells, whereas knockdown of ADAR1 significantly increased the phosphorylation level of both proteins, especially 72 h after reactivation (Figure 4C).
Figure 4. Depletion of ADAR1 Enhances Activation of the RLR Signaling Pathway during KSHV Reactivation.

iSLK.219 cells were transfected with NS or ADAR1 siRNAs, and then lytic reactivation was induced with 0.2 μg/mL Dox for the indicated times.
(A and B) Transcription of the MDA5 (A) and RIG-I (B) genes was quantified by real-time PCR. The mRNA fold change was normalized to β-actin mRNA.
(C) Cell lysates were collected 0, 48, and 72 h after Dox treatment, and western blots were performed with the indicated antibodies.
(D) Cell lysates were harvest 0 and 48 h after Dox treatment, and RIP-qPCR was performed with the J2 antibody or immunoglobulin G (IgG) as a negative control.
The data shown are representative of three independent experiments. Data from (A) and (B) are presented as mean ± SD. *, p < 0.05 by Student’s t test.
See also Figure S6.
Many cellular RNAs have been reported to specifically interact with MDA5 or RIG-I during KSHV lytic reactivation, including GINS1, NOP14, and several vault RNAs (vtRNAs) (Zhao et al., 2018), and thus can serve as ligands that activate the RIG-I/MAD5 pathway. Moreover, GINS1 and NOP14 RNAs have already been reported to harbor adenosine sites that can be edited by ADAR1 (Brümmer et al., 2017; Peng et al., 2017). To evaluate whether ADAR1 can prevent potential recognition of these cellular RNAs by MDA5 or RIG-I, we performed RNA immunoprecipitation quantitative PCR (RIP-qPCR) using the J2 antibody, which specifically interacts with dsRNA, in iSLK.219 cells transfected with control or ADAR1 siRNA.
GINS1 and NOP14 RNAs were significantly enriched by the J2 antibody in ADAR1 knockdown cells compared with control cells after KSHV reactivation (Figure 4D). Although the J2 antibody may not share the exact same structural/sequence preferences as MDA5 and RIG-I, these data suggest that, in the absence of ADAR1, it is possible that unedited cellular RNAs such as GINS1 and NOP14 RNA are more easily recognized by MDA5 or RIG-I and therefore induce robust innate immune responses. The three vtRNAs and Kaposin RNA were not further enriched in ADAR1 knockdown cells compared with control cells.
To rule out the possibility that Dox itself can trigger IFNβ production, we transfected NS or ADAR1 siRNA into iSLK.RTA cells, which harbor a Dox-inducible RTA gene but lack the KSHV genome, and then treated with Dox. After ADAR1 was depleted in iSLK.RTA cells (Figure S6A), IFNβ mRNA was increased in ADAR1-depleted cells even without Dox (0 h), but Dox treatment did not affect this trend, suggesting that Dox by itself (or RTA expression alone) does not induce IFNβ production (Figure S6B). Moreover, Dox treatment or RTA expression alone did not induce IFNβ mRNA expression in cells transfected with control siRNA (Figure S6B). Finally, Dox treatment or RTA expression alone also did not affect or only moderately increased the mRNA level of MDA5 or RIG-I (Figures S6C and S6D).
Enhanced IFNβ Production by Depletion of ADAR1 Is Dependent on the RLR Signaling Pathway
To confirm whether enhanced IFNβ induction in ADAR1-depleted cells during KSHV reactivation is dependent on the RLR pathway, we further knocked down MDA5 or RIG-I in ADAR1-deficient iSLK.219 cells and then induced lytic reactivation. As described above, IFNβ induction was increased in ADAR1-depleted cells. However, this increase was almost completely abolished in cells transfected with MDA5 and ADAR1 siRNA (Figure 5A). IFNβ mRNA induction was also significantly decreased in cells with RIG-I and ADAR1 siRNAs compared with cells with ADAR1 siRNA alone, but to a much lesser extent compared with cells with MDA5 depletion (Figure 5A). The knockdown efficiency of ADAR1, MDA5, and RIG-I was confirmed by real-time PCR (Figures 5B-5D). Additional silencing of MDA5 or RIG-I in ADAR1-depleted cells almost completely abolished KSHV reactivation-induced phosphorylation of TBK1 and IRF3 compared with cells transfected with ADAR1 siRNA alone (Figure 5E). Consistent with these data, IFNβ in the supernatant from cells transfected with MDA5 and ADAR1 siRNA together or RIG-I and ADAR1 siRNA together was significantly reduced compared with that from cells transfected with only ADAR1 siRNA (Figure 5F). Finally, depletion of MDA5 together with ADAR1 rescued KSHV viral gene transcription, which was inhibited by knockdown of ADAR1 alone, whereas depletion of RIG-I and ADAR1 together only partially rescued viral gene transcription (Figures 5G and 5H), probably because depletion of RIG-I only partially reduced IFNβ production (Figures 5A and 5F). Taken together, these data suggest that enhanced innate immune responses during KSHV reactivation in ADAR1-deficient cells is dependent on activation of the RLR signaling pathway.
Figure 5. Enhanced IFNβ Production by Depletion of ADAR1 Is Dependent on the RLR Signaling Pathway.
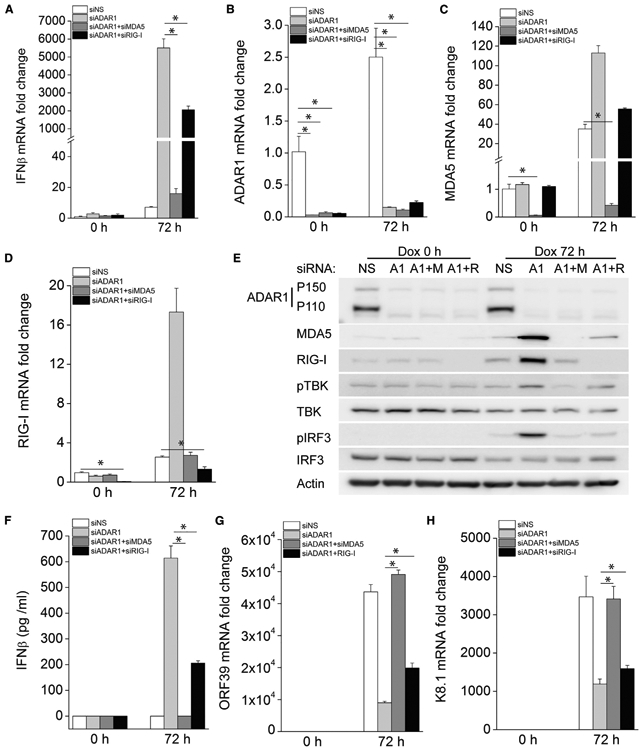
iSLK.219 cells were transfected with the indicated siRNAs for 48 h, and then KSHV lytic reactivation was induced with Dox (1 μg/mL) for 72 h.
(A) The fold induction of IFNβ mRNA expression levels was measured by real-time PCR.
(B–D) The knockdown efficiency of ADAR1 (B), MDA5 (C), and RIG-I (D) was confirmed by real-time PCR.
(E) Cells were harvested 72 h post-Dox treatment and immunoblotted with the indicated antibodies.
(F) IFNβ production in the supernatant was measured by ELISA.
(G and H) The mRNA expression levels of KSHV ORF39 (G) and K8.1 (H) were measured by real-time PCR. Target mRNA expression levels were normalized to β-actin mRNA and presented as fold induction.
The data shown are representative of three independent experiments. Data from (A)–(D) and (F)–(H) are presented as mean ± SD. *, p < 0.05 by Student’s t test. M, MDA5; R, RIG-I.
Depletion of the RLR Adaptor Protein MAVS in ADAR1-Deficient Cells Rescues KSHV Lytic Reactivation
It is known that dsRNA sensing by RIG-I or MDA5 leads to activation of the common adaptor protein MAVS, which then mediates activation of downstream signaling and subsequent type I IFN induction. To further confirm that ADAR1 facilitates KSHV lytic reactivation by inhibiting RLR-dependent IFNβ production, we transfected iSLK.219 cells with ADAR1 siRNA alone or together with MAVS siRNA and then induced lytic reactivation with Dox. As we showed previously, depletion of ADAR1 significantly reduced RFP-positive cells, an indicator of KSHV lytic reactivation, compared with control cells (Figure 1A). However, additional knockdown of MAVS in ADAR1-deficient cells showed a similar level of RFP-positive cells compared with control cells (Figures 6A and 6B). We also confirmed the knockdown efficiency of ADAR1 and MAVS by real-time PCR (Figures 6C and 6D). IFNβ mRNA induction in ADAR1-deficient cells induced by KSHV reactivation was completely abolished in the absence of MAVS (Figure 6E). Accordingly, transcription of viral genes was significantly inhibited in ADAR1-depleted cells but rescued by depletion of ADAR1 and MAVS together (Figures 6F and 6G). Furthermore, immunoblot analysis demonstrated that expression of the viral proteins K8α and ORF45 was decreased in ADAR1-deficient cells, especially at the 72-h time point, and rescued when MAVS was also depleted along with ADAR1 (Figure 6H). Finally, the increased IRF3 phosphorylation in ADAR1-depleted cells was not observed when MAVS and ADAR1 were depleted together (Figure 6H).
Figure 6. Concurrent Depletion of MAVS in ADAR1-Deficient Cells Rescues KSHV Reactivation.

iSLK.219 cells were transfected with ADAR1 siRNA alone or together with MAVS siRNA for 48 h and then reactivated with Dox (0.2 μg/mL) for 0, 48 and 72 h.
(A) A representative image of a field of cells expressing GFP and RFP with or without Dox treatment for 72 h.
(B) GFP and RFP intensities were measured by a Clariostar plate reader, and the ratio of RFP/GFP was calculated for the indicated times.
(C–G) The mRNA expression of the ADAR1 (C), MAVS (D), IFNβ (E), KSHV ORF39 (F), and K8.1 (G) genes was measured by real-time PCR, and the fold change was normalized to β-actin mRNA.
(H) Cells were harvested at different time points, and western blots were performed with the indicated antibodies.
The data shown are representative of three independent experiments. Data from (B)–(G) are presented as mean ± SD. *, p < 0.05 by Student’s t test. Scale bar, 500 μm. MA, MAVS.
Knockdown of ADAR1 in PEL Cells Increases IFNβ Expression and Inhibits KSHV Reactivation
To further confirm the role of ADAR1 in modulating KSHV lytic reactivation, we also performed similar experiments in TREx BCBL1-RTA cells, which is a KSHV-infected PEL-derived B cell line. Similar to iSLK.219 cells, TREx BCBL1-RTA cells carry an RTA gene under the control of a Dox-inducible promoter but lack the GFP and RFP markers. We knocked down ADAR1 in TREx BCBL1-RTA cells (Figure 7A) and discovered that suppression of ADAR1 resulted in significantly increased IFNβ, IFNα1, and IFNγ mRNA expression (Figures 7B-7D). We next performed the same Human Interferons & Receptors PCR array analysis in TREx BCBL1-RTA cells. After Dox treatment for 48 h, KSHV reactivation lead to a more than 2-fold induction of multiple genes in cells transfected with NS siRNA or ADAR1 siRNA. Of these genes, 15 showed much higher induction (>2-fold) in ADAR1 siRNA-transfected cells than in control siRNA-transfected cells (Figure 7E; Table S2). In summary, ADAR1 deficiency leads to markedly higher induction of type I IFNs signaling in PEL cells during lytic replication.
Figure 7. ADAR1 Deficiency Increases Type I IFN Production and Inhibits KSHV Reactivation in PEL by Activating the RLR Pathway.

TREx BCBL1-RTA cells were transfected with NS or ADAR1 siRNA for 48 h and then reactivated with Dox (0.2 μg/mL) for 48 h.
(A–D) The mRNA expression of ADAR1 (A), IFNβ (B), IFNα1 (C), and IFNγ (D) was measured by real-time PCR and normalized to β-actin mRNA.
(E) Heatmap of the Human Interferons & Receptors PCR array. RNA was extracted from the indicated samples, and the mRNA levels of the indicated genes were analyzed using the Human Interferons & Receptors RT2 Profiler PCR Array. The mRNA levels of genes were normalized to β-actin. The heatmap was generated using the web-based tool Morpheus (https://software.broadinstitute.org/morpheus/).
TREx BCBL1-RTA cells were transfected with ADAR1 siRNA alone or together with MAVS siRNA for 48 h and then reactivated with Dox (0.2 μg/mL) for 48 h.
(F–J) The mRNA expression of ADAR1 (F), MAVS (G), IFNβ (H), KSHV ORF39 (I), and K8.1 (J) was measured by real-time PCR and normalized to β-actin mRNA.
(K) Cells were harvested 48 h post-Dox treatment, and immunoblots were performed with the indicated antibodies.
The data shown are representative of three independent experiments, except for (E), which is representative oftwo experiments. Data from (A)–(D) and (F)–(J) are presented as mean ± SD. *, p < 0.05 by Student’s t test.
See also Table S2.
Enhanced IFNβ induction upon ADAR1 depletion in TREx BCBL1-RTA cells is also dependent on the RLR pathway because depletion of MAVS together with ADAR1 reversed IFN induction (Figures 7F-7H). Accordingly, KSHV viral gene transcription and viral protein expression were significantly reduced in ADAR1-suppressed cells and rescued to a similar level as control cells when MAVS was silenced together with ADAR1 (Figures 7I-7K). In conclusion, we demonstrated that suppression of ADAR1 in KSHV-positive PEL inhibits KSHV lytic reactivation because of increased type I IFN production, which is dependent on activation of the RLR signaling pathway.
DISCUSSION
KSHV lytic reactivation activates innate immune pathways, which leads to production of type I IFNs and subsequent antiviral responses. KSHV has evolved several mechanisms to curb these responses for successful viral infection and replication. For example, KSHV vIRF1, a viral homolog of cellular IRFs (interferon regulatory factors), can inhibit activation of the TLR3 and cGAS-STING pathways, facilitating KSHV replication (Jacobs et al., 2013; Ma et al., 2015). In addition, utilization of host proteins to restrict IFNβ responses during KSHV lytic reactivation has also been reported. NLRX1 has been identified to facilitate KSHV lytic replication by suppressing the type I IFN response (Ma et al., 2017). In this study, we demonstrated that the host dsRNA-editing enzyme ADAR1 plays a pivotal role in facilitating KSHV reactivation. Suppression of ADAR1 in iSLK.219 and TREx BCBL1-RTA cells led to markedly increased type I IFN production during KSHV lytic reactivation, which subsequently activates expression of many ISGs and proinflammatory cytokines, significantly impairing KSHV viral gene transcription and virion production. Although some studies have shown that ADAR1 plays an antiviral role during infection with certain RNA viruses (Samuel, 2011), our data suggest that ADAR1 acts as a proviral factor during KSHV lytic replication.
Previous studies have reported that ADAR1 controls innate immune responses to RNA through the RLR signaling pathway (Mannion et al., 2014). RIG-I and MDA5 are the major cytosolic sensors of dsRNA, and both of them have been reported to restrict KSHV lytic reactivation (West et al., 2014). We also investigated whether induction of type I IFN in ADAR1-deficient cells during KSHV reactivation is dependent on the RLR pathway. Our data showed that additional depletion of MDA5/RIG-I or MAVS together with ADAR1 greatly diminished induction of IFNβ and rescued KSHV reactivation in ADAR1-deficient cells, suggesting that the enhanced KSHV reactivation-induced IFN responses in ADAR1-suppressed cells are mainly dependent on activation of RLR-MAVS signaling. We also investigated the potential triggers for the RLR pathway. Our data show that GINS1 and NOP14 RNAs, which have been reported previously to be enriched by MDA5 pull-down during KSHV reactivation (Zhao et al., 2018), were significantly enriched by the J2 antibody in ADAR1-depleted cells, suggesting that these cellular RNAs could be targets of ADAR1. These data suggest that, in the absence of ADAR1, it is possible that these unedited cellular RNAs can be more easily recognized by MDA5 or RIG-I to trigger activation of the RLR pathway. The three vtRNAs, which have been reported previously to be enriched by RIG-I pull-down during KSHV reactivation (Zhao et al., 2018), were not further enriched by the J2 antibody in ADAR1-depleted cells. This may be because they were not edited by ADAR1.
Several studies have shown that ADAR1 displays antiviral activities against a subset of RNA viruses by directly modifying viral RNAs and accumulating deleterious mutations in the viral genomes (Samuel, 2011). ADAR1 also edits Epstein-Barr virus (EBV) BART6 microRNA, miR-BART6-5p, to antagonize its activity regarding EBNA2 expression in EBV-infected cells (Iizasa et al., 2010). In the case of KSHV, there is only one study that has reported that ADAR1 performs A-to-I editing in the KSHV Kaposin (K12) transcript, which eliminates its transforming activity (Gandy et al., 2007). Notably, this editing level is much higher during lytic replication than in latency, probably because of increased ADAR1 activity during reactivation, as our data suggest (Figure S1). Our data confirmed that ADAR1 indeed carries out A-to-I editing in the Kaposin transcript. However, neither wild-type nor the edited Kaposin transcripts could induce IFNβ promoter-driven luciferase activity. Kaposin does not seem to be essential for KSHV lytic replication either. Therefore, our data suggest that loss of A-to-I editing in the Kaposin transcript by ADAR1 is not responsible for activation of innate immune responses or defective KSHV lytic replication in ADAR1-deficient cells. However, although a previous study showed that Kaposin is the only KSHV transcript that undergoes A-to-I editing (Arias et al., 2014), it is still possible that other KSHV transcripts are also edited by ADAR1, which might be important for immune activation or KSHV lytic replication. For example, several circular RNAs (circRNAs) have been identified in KSHV-infected cells as well as in KSHV-positive patients. The KSHV circRNAs are located within open reading frames (ORFs) of viral lytic genes whose expression is activated upon induction of the lytic cycle (Tagawa et al., 2018; Toptan et al., 2018). Cellular circRNAs tend to form 16- to 26-bp imperfect RNA duplexes and act as inhibitors of PKR to regulate innate immunity (Liu et al., 2019). circRNAs have also been shown to be highly enriched in RNA editing events, and knockdown of ADAR1 significantly and specifically upregulates circRNA expression (Ivanov et al., 2015). Therefore, future studies will focus on determining whether ADAR1 is involved in KSHV circRNA editing and whether this editing affects KSHV lytic reactivation and host innate immune responses. To date, no previous studies have reported that ADAR1 is involved in regulating RLR-mediated innate immune responses against DNA viruses. Our data clearly show that ADAR1 prevents activation of the RLR-mediated innate immune response, which, in turn, facilitates KSHV lytic reactivation.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Blossom Damania (damania@med.unc.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate any unique datasets or code. The published article includes all datasets generated or analyzed during this study.
EXPERIMENT MODEL AND SUBJECT DETAIL
Cell culture
iSLK.219 cells (a kind gift from Dr. Don Ganem) were maintained in DMEM medium (Corning) containing 10% tetracycline (Tet)-free FBS (Sigma), 1% Pen-Strep (Corning), 10 μg/ml puromycin (Corning), 250 μg/ml Geneticin (Corning), and 400 μg/ml hygromycin B (Corning). BCBL1-TREx-RTA cells (a kind gift from Dr. Jae Jung) were cultured in RPMI 1640 (Corning) medium supplemented with 10% tetracycline (Tet)-free FBS, 1% Pen-Strep, 1% L-glutamine, 1% sodium bicarbonate, 0.05 mM β-mercaptoethanol, and 20 μg/ml hygromycin. 293FT (Thermo Fisher, R7007) were maintained in DMEM medium containing 10% FBS (Millipore) and 1% Pen-Strep. All cells were maintained at 37°C in a 5% CO2 laboratory incubator subject to routine cleaning and decontamination.
iSLK.219 knockout cell lines
ADAR1 KO and ADAR1p150 KO iSLK.219 cell lines were generated via CRISPR-Cas9 genome editing. All gRNA sequences are the same as those used in a previous study (Chung et al., 2018) and are listed in Table S3. Briefly, gRNAs were cloned into LentiCRISPRv2 blast plasmid (Addgene) and transfected into 293FT cells along with ViraPower Lentiviral Packaging Mix (Invitrogen) using Lipofectamine 2000 (Invitrogen). Two days post transfection, the supernatant was collected, centrifuged and then transferred to iSLK.219 cells. Two days after infection, the iSLK.219 cells were selected with 10 μg/ml blasticidin (Invivogen) for 5 days and single cell colonies were isolated in 96-well plates and validated by immunoblotting using ADAR1 antibody.
METHOD DETAILS
RNA interference, KSHV reactivation and fluorescence microscopy
iSLK.219 cells were transfected with 80 nM siRNA (Dharmacon, siRNA sequences are listed in Table S3) or control non-targeting (NS) pool (Dharmacon, D-001810-10-50) using Lipofectamine RNAiMax (Invitrogen). At 48 hours post-transfection, cells were replenished with complete DMEM medium containing doxycycline at the indicated concentration for KSHV reactivation. BCBL1-TREx-RTA cells were transfected with siRNA using Lonza nucleofector V kit according to the manufacturer’s recommendations. At 48 hours post-transfection, cells were replenished with complete RPMI 1640 medium containing 0.1 μg/ml doxycycline for KSHV reactivation. Cells were collected at the indicated times for later analysis. GFP and RFP images of the cells were taken by a fluorescence microscope at the indicated times using the same exposure time setting. One representative image of each sample is shown in the manuscript. The RFP/GFP fluorescence intensity in each well (triple wells for each sample) was then measured by a microplate reader right after the microscopy. The fold induction was calculated and normalized to NS treated cells at Dox 0h.
Quantitative Real-time PCR and PCR arrays
Total RNA was extracted with RNeasy Plus Mini Kit (QIAGEN) and cDNA synthesis was performed using SensiFAST cDNA Synthesis Kit (Bioline). Real-time qPCR was performed with Sensi-Fast SYBR (Bioline). The housekeeping gene ACTB was used for normalization. All primer sequences are listed in Table S4.
Total RNA was extracted from iSLK.219 or BCBL1-TREx-RTA cells and cDNA synthesis was performed as described above. Human Interferons and Receptors RT2 Profiler PCR Array was purchased from QIAGEN (Cat #: PAHS-064Z), which contains 84 primer sets against human interferon signaling related genes and 5 housekeeping gene primer sets. Real-time PCR was then performed according to the manufacturer’s protocol. Fold-change is the normalized gene expression (2^(- Delta Ct)) in the Test Sample divided by the normalized gene expression (2^(- Delta Ct)) in the Control Sample. The heatmap was generated using the web-based tool Morpheus (https://software.broadinstitute.org/morpheus/).
Kaposin mRNA A-to-I editing by Sanger sequencing
Total RNA was extracted from iSLK.219 cells transfected with NS, ADAR1 or ADAR2 siRNA followed by Dox treatment for 72h, and cDNA synthesis was performed as described above. Gene specific amplification was done with Q5 High-Fidelity DNA polymerase (NEB) following the manufacturer’s guidelines. The primer sequences used for amplifying Kaposin are listed in Table S4. PCR products were purified with EZNA Cycle Pure Kit (OMEGA) and sequenced by Eurofins Genomics.
Plasmid construction and luciferase assays
Total RNA was extracted from iSLK.219 cells and cDNA was synthesized as described above. Kaposin A transcript was amplified by PCR and then cloned into pcDNA3.1 plasmid (Thermo Fisher) using NEBbuilder HiFi DNA Assembly Cloning Kit (NEB). The edited Kaposin plasmid was generated based on unedited Kaposin plasmid using the Q5 Site-Directed Mutagenesisi Kit (NEB). All primer sequences are listed in Table S4. 293T cells were co-transfected with empty vector or plasmids encoding wild-type or edited kaposin transcript along with the IFNβ promoter luciferase reporter plasmid. Luciferase activity was measured 24 hours post-transfection using the Dual-Luciferase Reporter Assay System (Promega).
ADA activity assay
iSLK.219 were induced with 0.2 μg/ml doxycycline for 0, 24, 48 and 72 hours, and then lysed in ADA assay buffer containing protease inhibitor cocktail. ADA activity was measured and quantified using Adenosine Deaminase (ADA) Activity Assay Kit (Fluorometric) (Abcam, ab204695) according to the manufacturer’s protocols.
Immunoblotting and ELISA
Whole cell lysates were prepared in SDS-sample buffer (60mM Tris.HCl pH6.8, 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.01% bromophenol blue) and separated by SDS-PAGE and then transferred to nitrocellulose membranes (GE Healthcare). The membrane was blocked in PBS-T (PBS with 0.1% Tween20) with 5% nonfat milk (Apex) and then incubated with the indicated primary antibody at 4°C overnight. Please refer to the KEY RESOURCES TABLE for antibodies used in this study. The vIL-6 antibody was purified from the supernatant of v6m 12.1.1 hybridomas (ATCC) using magnetic Protein A/G beads (Thermo Fisher). IFNβ protein in the supernatant were quantified by human IFN-beta DuoSet ELISA kit (R&D Systems) according to the manufacturer’s protocol. IFNβ protein levels (pg/ml) were calculated based on the standard curve generated in the assay.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ADAR1 | Santa Cruz | Cat# sc-271854; RRID: AB_10708553 |
| Phospho-IRF3 | Abcam | Cat# ab76493; RRID: AB_1523836 |
| IRF3 | Cell signaling | Cat# 4302; RRID: AB_1904036 |
| Phospho-TBK1 | Cell signaling | Cat# 5483; RRID: AB_10693472 |
| TBK1 | Cell signaling | Cat# 3504; RRID: AB_2255663 |
| MDA5 | Cell signaling | Cat# 5321; RRID: AB_10694490 |
| RIG-I | Cell signaling | Cat# 3743; RRID: AB_2269233 |
| MAVS | Cell signaling | Cat# 3993; RRID: AB_823565 |
| KSHV ORF45 | Thermo Fisher | Cat# MA5-14769; RRID: AB_10999794 |
| KSHV K8alpha | Santa Cruz | Cat# sc-57889; RRID: AB_783765 |
| KSHV vIL-6 | This laboratory | N/A |
| J2 mAb | Scicons | Cat# 10010200; RRID: AB_2651015 |
| Mouse IgG | Santa Cruz | Cat# sc-2025; RRID: AB_737182 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine RNAiMAX | Invitrogen | Cat# 13778150 |
| Lipofectamine 2000 | Invitrogen | Cat# 11668019 |
| Doxycycline Hydrochloride | Fisher Scientific | Cat# BP26535 |
| Opti-MEM | GIBCO | REF# 31985-070 |
| 2-Mercaptoethanol | GIBCO | REF# 21985-023 |
| Tetracycline-free FBS | Clontech | Cat# 631106 |
| Puromycin | Corning | REF# 61-385-PA |
| Hygromycin B | Corning | REF# 30-240-CR |
| Geneticin | GIBCO | REF# 10131-035 |
| Protease inhibitor cocktail | Sigma-Aldrich | Cat# 11697498001 |
| 5X siRNA buffer | Fisher Scientific | REF# B-002000-UB-100 |
| Q5 High-Fidelity DNA Polymerase | NEB | Cat# M0491S |
| DMEM | Corning | REF# 17-205-CV |
| RPMI 1640 | Corning | REF# 10-040-CV |
| Poly (I:C) HMW | Invivogen | Cat# tlrl-pic |
| Dynabeads Protein G | Invitrogen | Cat# 10004D |
| ViraPower Lentiviral Packaging Mix | Invitrogen | Cat# K497500 |
| Critical Commercial Assays | ||
| RNeasy Plus Mini Kit | QIAGEN | Cat# 74136 |
| DNeasy Mini Kit | QIAGEN | Cat# 69506 |
| ADA Activity Assay Kit-Fluorometric | Abcam | Cat# Ab204695 |
| EZNA Cycle Pure Kit | OMEGA | Cat# D6492-01 |
| SensiFAST cDNA Synthesis Kit | Bioline | Cat# BIO-65054 |
| SensiFAST SYBR Lo-ROX Kit | Bioline | Cat# BIO-94050 |
| IFN-beta DupSet ELISA Kit | R&D Systems | Cat# DY8234-05 |
| Cell Line Nucleofector Kit V | Lonza | Cat# VCA-1003 |
| Human Interferons and Receptors RT2 Profiler PCR Array | QIAGEN | Cat# PAHS-064Z |
| RNeasy MinElute Cleanup Kit | QIAGEN | REF# 74204 |
| NEBbuilder HiFi DNA Assembly Cloning Kit | NEB | Cat# E5520S |
| Q5 Site-Directed Mutagenesis Kit | NEB | Cat# E0554S |
| Dual-Luciferase Reporter Assay System | Promega | Cat# E1980 |
| Experimental Models: Cell Lines | ||
| Human: iSLK.219 | Don Ganem laboratory | Myoung and Ganem, 2011 |
| Human: TREx BCBL1-RTA | Jae Jung laboratory | Nakamura et al., 2003 |
| Human: 293FT | Thermo Fisher | Cat# R70007 |
| Oligonucleotides | ||
| See Tables S3 and S4 for oligonucleotide information | N/A | |
| siRNA and sgRNA sequences (Listed in Table S3) | Dharmacon | N/A |
| Primers sequences (Listed in Table S4) | This paper | N/A |
| Recombinant DNA | ||
| LentiCRISPRv2 blast | Addgene | Plasmid# 98293 |
| pcDNA3.1 | Thermo Fisher | Cat# V79020 |
| IFNβ reporter luciferase reporter | Zhijian Chen laboratory | N/A |
| Software and Algorithms | ||
| Morpheus | Morpheus | https://software.broadinstitute.org/morpheus |
| NIS Elements AR 4.60 | Nikon | N/A |
| Origin 9 | Origin Lab | N/A |
| Illustrator 2019 | Adobe | N/A |
| RNA Mfold | The RNA institute | http://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form |
RNA Immunoprecipitation quantitative PCR
iSLK.219 cells were transfected with NS and ADAR1 siRNA followed by Dox treatment for 0 and 48 hours. The cells were harvested in RNA immunoprecipitation (RIP) buffer (25 mM HEPES pH7.2, 75 mM NaCl, 5 mM MgCl2, 0.1% NP-40, 1 U/μl RNase inhibitor), sonicated and then centrifuged at 15,000 rpm for 15 min. Supernatants were incubated with 25 μl of ProtG Dynabeads (Thermo Fisher) pre-coupled to 2 μg J2 mAb (Scicons) or mouse-IgG (Santa Crus) for 3 hours at 4°C. The beads were washed fourtimes with 1M urea RIP buffer and bound RNA was isolated with RNeasy MinElute Cleanup Kit (QIAGEN). Eluted RNA was incubated at 70°C for 3 min and then reverse transcribed with the SensiFAST cDNA Synthesis Kit (Bioline). Real-time qPCR was performed as described above, and enrichments were calculated using 10% of input material as a reference for each immunoprecipitation.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all figures, error bars show mean ± SD. Statistical significance of differences in mRNA expression level, IFNβ concentration and viral genome copies were determined using Student’s t test (two-tailed). For all tests, a p value of < 0.05 was considered statistically significant. * indicates p < 0.05.
Supplementary Material
Highlights.
Suppression of ADAR1 inhibits KSHV lytic reactivation from latency
ADAR1 deficiency increases type I interferon production during KSHV lytic reactivation
ADAR1 facilitates DNA virus reactivation
Reactivation results from dampening of RLR pathway-mediated innate immune response
ACKNOWLEDGMENTS
This work was supported by the U.S. National Institutes of Health grants CA019014, DE028211, CA096500, CA239583, and CA163217. B.D. is a Leukemia and Lymphoma Society Scholar and a Burroughs Wellcome Fund Investigator in Infectious Disease. The graphical abstract was made using the BioRender program.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107564.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Arias C, Weisburd B, Stern-Ginossar N, Mercier A, Madrid AS, Bellare P, Holdorf M, Weissman JS, and Ganem D (2014). KSHV 2.0: a comprehensive annotation of the Kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 10, e1003847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brümmer A, Yang Y, Chan TW, and Xiao X (2017). Structure-mediated modulation of mRNA abundance by A-to-I editing. Nat. Commun 8, 1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capshew CR, Dusenbury KL, and Hundley HA (2012). Inverted Alu dsRNA structures do not affect localization but can alter translation efficiency of human mRNAs independent of RNA editing. Nucleic Acids Res. 40, 8637–8645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesarman E, Chang Y, Moore PS, Said JW, and Knowles DM (1995). Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med 332, 1186–1191. [DOI] [PubMed] [Google Scholar]
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, and Moore PS (1994). Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266, 1865–1869. [DOI] [PubMed] [Google Scholar]
- Chung H, Calis JJ, Wu X, Sun T, Yu Y, Sarbanes SL, Thi VLD, Shilvock AR, Hoffmann H-H, and Rosenberg BR (2018). Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172, 811–824.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer DP, and Damania B (2016). Kaposi sarcoma-associated herpesvirus: immunobiology, oncogenesis, and therapy. J. Clin. Invest 126, 3165–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandy SZ, Linnstaedt SD, Muralidhar S, Cashman KA, Rosenthal LJ, and Casey JL (2007). RNA editing of the human herpesvirus 8 kaposin transcript eliminates its transforming activity and is induced during lytic replication. J. Virol 81, 13544–13551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory SM, Davis BK, West JA, Taxman DJ, Matsuzawa S, Reed JC, Ting JP, and Damania B (2011). Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331, 330–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizasa H, Wulff B-E, Alla NR, Maragkakis M, Megraw M, Hatzigeorgiou A, Iwakiri D, Takada K, Wiedmer A, Showe L, et al. (2010). Editing of Epstein-Barr virus-encoded BART6 microRNAs controls their dicer targeting and consequently affects viral latency. J. Biol. Chem 285, 33358–33370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inn K-S, Lee S-H, Rathbun JY, Wong L-Y, Toth Z, Machida K, Ou J-HJ, and Jung JU (2011). Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol 85, 10899–10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, Miller BC, Du PP, Yates KB, Dubrot J, et al. (2019). Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, Piechotta M, Levanon EY, Landthaler M, Dieterich C, and Rajewsky N (2015). Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 10, 170–177. [DOI] [PubMed] [Google Scholar]
- Jacobs SR, Gregory SM, West JA, Wollish AC, Bennett CL, Blackbourn DJ, Heise MT, and Damania B (2013). The viral interferon regulatory factors of kaposi’s sarcoma-associated herpesvirus differ in their inhibition of interferon activation mediated by toll-like receptor 3. J. Virol 87, 798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, and Chandran B (2011). IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos D, Vart RJ, Gratrix F, Westrop SJ, Emuss V, Wong P-P, Robey R, Imami N, Bower M, Gotch F, and Boshoff C (2008). Toll-like receptor4 mediates innate immunity to Kaposi sarcoma herpesvirus. Cell Host Microbe 4, 470–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CX, Li X, Nan F, Jiang S, Gao X, Guo SK, Xue W, Cui Y, Dong K, Ding H, et al. (2019). Structure and Degradation of Circular RNAs Regulate PKR Activation in Innate Immunity. Cell 177, 865–880.e21. [DOI] [PubMed] [Google Scholar]
- Ma Z, Jacobs SR, West JA, Stopford C, Zhang Z, Davis Z, Barber GN, Glaunsinger BA, Dittmer DP, and Damania B (2015). Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA 112, E4306–E4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Hopcraft SE, Yang F, Petrucelli A, Guo H, Ting JPY, Dittmer DP, and Damania B (2017). NLRX1 negatively modulates type I IFN to facilitate KSHV reactivation from latency. PLoS Pathog. 13, e1006350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Ni G, and Damania B (2018). Innate Sensing of DNA Virus Genomes. Annu. Rev. Virol 5, 341–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellåker C, Vesely C, Ponting CP, McLaughlin PJ, et al. (2014). The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 9, 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myoung J, and Ganem D (2011). Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J. Virol. Methods 174, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, and Jung JU (2003). Global changes in Kaposi’s sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. Journal of virology 77, 4205–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K (2010). Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem 79, 321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng L, Lee LJ, Xiong H, Su H, Rao J, Xiao D, He J, Wu K, and Liu D (2017). Characterization of RNA editome in primary and metastatic lung adenocarcinomas. Oncotarget 8, 11517–11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. (2012). Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet 44, 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE (2011). Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 411, 180–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d’Agay MF, Clauvel J-P, Raphael M, Degos L, et al. (1995). Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 86, 1276–1280. [PubMed] [Google Scholar]
- Tagawa T, Gao S, Koparde VN, Gonzalez M, Spouge JL, Serquiña AP, Lurain K, Ramaswami R, Uldrick TS, Yarchoan R, and Ziegelbauer JM (2018). Discovery of Kaposi’s sarcoma herpesvirus-encoded circular RNAs and a human antiviral circular RNA. Proc. Natl. Acad. Sci. USA 115, 12805–12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toptan T, Abere B, Nalesnik MA, Swerdlow SH, Ranganathan S, Lee N, Shair KH, Moore PS, and Chang Y (2018). Circular DNA tumor viruses make circular RNAs. Proc. Natl. Acad. Sci. USA 115, E8737–E8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West J, and Damania B (2008). Upregulation of the TLR3 pathway by Kaposi’s sarcoma-associated herpesvirus during primary infection. J. Virol 82, 5440–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JA, Wicks M, Gregory SM, Chugh P, Jacobs SR, Zhang Z, Host KM, Dittmer DP, and Damania B (2014). An important role for mitochondrial antiviral signaling protein in the Kaposi’s sarcoma-associated herpesvirus life cycle. J. Virol 88, 5778–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JP, and Damania B (2017). Modulation of oncogenic signaling networks by Kaposi’s sarcoma-associated herpesvirus. Biol. Chem 398, 911–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JJ, Li W, Shao Y, Avey D, Fu B, Gillen J, Hand T, Ma S, Liu X, Miley W, et al. (2015). Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell Host Microbe 18, 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Dittmer DP, Mieczkowski PA, Host KM, Fusco WG, Duncan JA, and Damania B (2018). RIG-I Detects Kaposi’s Sarcoma-Associated Herpesvirus Transcripts in a RNA Polymerase III-Independent Manner. MBio 9, e00823–e00818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ye X, Dunker W, Song Y, and Karijolich J (2018). RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat. Commun 9, 4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code. The published article includes all datasets generated or analyzed during this study.