

Abstract
Periodontitis is a complex disease: (a) various causative factors play a role simultaneously and interact with each other; and (b) the disease is episodic in nature, and bursts of disease activity can be recognized, ie, the disease develops and cycles in a nonlinear fashion. We recognize that various causative factors determine the immune blueprint and, consequently, the immune fitness of a subject. Normally, the host lives in a state of homeostasis or symbiosis with the oral microbiome; however, disturbances in homeostatic balance can occur, because of an aberrant host response (inherited and/or acquired during life). This imbalance results from hyper‐ or hyporesponsiveness and/or lack of sufficient resolution of inflammation, which in turn is responsible for much of the disease destruction seen in periodontitis. The control of this destruction by anti‐inflammatory processes and proresolution processes limits the destruction to the tissues surrounding the teeth. The local inflammatory processes can also become systemic, which in turn affect organs such as the heart. Gingival inflammation also elicits changes in the ecology of the subgingival environment providing optimal conditions for the outgrowth of gram‐negative, anaerobic species, which become pathobionts and can propagate periodontal inflammation and can further negatively impact immune fitness. The factors that determine immune fitness are often the same factors that determine the response to the resident biofilm, and are clustered as follows: (a) genetic and epigenetic factors; (b) lifestyle factors, such as smoking, diet, and psychosocial conditions; (c) comorbidities, such as diabetes; and (d) local and dental factors, as well as randomly determined factors (stochasticity). Of critical importance are the pathobionts in a dysbiotic biofilm that drive the viscious cycle. Focusing on genetic factors, currently variants in at least 65 genes have been suggested as being associated with periodontitis based on genome‐wide association studies and candidate gene case control studies. These studies have found pleiotropy between periodontitis and cardiovascular diseases. Most of these studies point to potential pathways in the pathogenesis of periodontal disease. Also, most contribute to a small portion of the total risk profile of periodontitis, often limited to specific racial and ethnic groups. To date, 4 genetic loci are shared between atherosclerotic cardiovascular diseases and periodontitis, ie, CDKN2B‐AS1(ANRIL), a conserved noncoding element within CAMTA1 upstream of VAMP3, PLG, and a haplotype block at the VAMP 8 locus. The shared genes suggest that periodontitis is not causally related to atherosclerotic diseases, but rather both conditions are sequelae of similar (the same?) aberrant inflammatory pathways. In addition to variations in genomic sequences, epigenetic modifications of DNA can affect the genetic blueprint of the host responses. This emerging field will yield new valuable information about susceptibility to periodontitis and subsequent persisting inflammatory reactions in periodontitis. Further studies are required to verify and expand our knowledge base before final cause and effect conclusions about the role of inflammation and genetic factors in periodontitis can be made.
Keywords: periodontitis, inflammation, genetics, cardiovascular disease, pleiotropy, microbial ecology
1. INTRODUCTION
Patients with periodontitis show inflammatory destruction of the supporting tissues around the teeth. Loss of connective tissue and collagen in the gingiva is characteristic, along with loss of periodontal ligament and resorption of alveolar bone. Thus the tooth roots become exposed to the oral environment, and the root and root cementum are colonized with a bacterial biofilm, which can calcify to form dental calculus. The chronicity and mostly slow progression of this disease results in tooth mobility, loss of chewing function, esthetic disturbances and, ultimately, if left untreated, tooth exfoliation. Moreover, periodontal inflammation has systemic effects; it can induce low grade systemic inflammation, which has negative effects on other organs.
Traditionally, the most common forms of periodontitis have been separated into 2 types: aggressive periodontitis and chronic periodontitis.1 However, it has recently been acknowledged that the scientific basis for this classification is weak and based on a variable clinical presentation.2, 3, 4, 5 In particular, in light of the complexity of the causative factors for periodontitis that will be discussed in this paper, the distinction between various clinical presentations has been removed.5, 6 The shortcomings of clinical diagnoses included substantial overlap and lack of clear pathobiology‐based distinctions between the stipulated categories, diagnostic imprecision, and treatment implementation difficulties. Although the 1999 classification1 provided a workable framework that has been used extensively in both clinical practice and scientific investigations in periodontology during the past 17 years, the shortcomings were deemed too great for further utility,5, 6 and a new classification was introduced.7
Epidemiological research has shown that severe periodontitis occurs in about 7%‐14% of the population in western Europe and North America, depending on the definitions used for severe periodontitis, and depending on the specific study population evaluated.8, 9, 10 In populations and countries with low availability of dental care with limited dental health awareness, and when limited preventive measures are available, the prevalence of severe periodontitis may be 10%‐15%.9 It may well be that the genetic susceptibility factors are more pronounced in certain racial/ethnic populations; the prevalence of severe periodontal disease in central and east sub‐Saharan Africa, and within some racial/ethnic groups in the USA, was found to be up to 20%.8, 9
2. CHRONIC INFLAMMATORY DISEASES
Humans can suffer from a range of chronic immune‐mediated inflammatory conditions, collectively called chronic inflammatory diseases. These include autoimmune diseases, allergies, immune deficiencies, and perhaps some psychiatric disorders such as depression.11 More than 10% of people in all populations may suffer from 1 or more chronic inflammatory diseases. For many chronic inflammatory diseases, the question remains as to which factors contribute to their onset and progression, while, on the other hand, which mechanisms keep the immune system tolerant for self as well as for the daily challenges of trillions of antigens from bacterial, fungal, and viral microorganisms. In essence, uncontrolled and unresolved inflammation is the basis of chronic inflammatory diseases. Chronic inflammatory diseases share common causal factors, including genetic factors (ie, pleiotropy) and immune pathways.11, 12, 13, 14, 15, 16 This knowledge has helped in our general understanding of individual chronic inflammatory diseases, including periodontal disease.17, 18
Chronic inflammatory diseases are complex conditions because they often involve multiple causal components that play a role simultaneously, and they interact with each other, often in an unpredictable way. In other words, the disease is the result of complex interactions between genetics and environment, such as microbial communities (biofilms) and the host response, which is hard to explain by a few individual factors.14, 15, 19, 20 Thus it should be noted that each of the separate causal components vary in relative importance, which varies in individual cases, yielding a tremendous number of combinations of causal factors that impact upon how each case may develop and progress.14, 15, 19, 21 Nevertheless, clinically, many cases of a given chronic inflammatory disease share causal components, which of course is helpful for determining diagnosis, prognosis, and treatment.
Complex chronic inflammatory diseases are recognized as nonlinear systems.20, 22, 23, 24 Nonlinearity in complex systems means that cause and effect are disproportional so that various causal pathways may account for mechanically different effects, which vary in individuals. For example, in one individual a causal pathway may contribute little to the disease, while in another individual the same pathway has a major effect. Disease progression rate fluctuates, or rather can move (‘cycle’) from one state (homeostasis, ie, a stable state with high resilience) to another (disease activity, ie, relapse) and back again, in often unpredictable ways.20, 23 For many chronic inflammatory diseases, it has been established that the disease can relapse (occurrence of bursts of activity) after a period of remission or quiescence.20, 24 Many clinicians have noted heterogeneity in the clinical course of patients with chronic inflammatory disease, attesting to the nonlinearity of the condition. For patients suffering from rheumatoid arthritis, there are large variations reported on the frequency and time periods of disease remission,25 and this nonlinearity can be related back to stability or changes in the patients’ immune fitness.11, 22 Such periods of stability and disease progression appear also to be characteristic of periodontitis.
The immune function of any individual can be equated with immune fitness; the way the host deals with the challenges and perturbations encountered during life, including normal inflammation‐resolving mechanisms.11, 14, 26 There is a wide variety of determinants (ie, causal factors) that regulate the immune system, and these factors are both intrinsic and acquired. The intrinsic causal factors are the inherited risk factors, ie, genetic susceptibility. Common to chronic inflammatory diseases is the fact that they are associated with 100s of disease‐associated genetic variants (single nucleotide polymorphisms).13, 15, 27 Chronic inflammatory diseases are understood to be polygenic, and the various chronic inflammatory diseases often share particular single nucleotide polymorphisms that are considered to play a role in immune fitness. This is the concept of pleiotropy and demonstrates the existence of common pathogenic pathways for different chronic inflammatory diseases based on shared single nucleotide polymorphisms.12, 16, 27
In addition to variations in genomic sequences that have been associated with chronic inflammatory diseases, there are epigenetic modifications of DNA that are in part acquired during life, but can also be inherited and thus can be intrinsic.28, 29, 30 It has been established that aging, microbial exposure, dietary factors, systemic conditions (eg, obesity, diabetes, osteoporosis, and depression), environmental factors (eg, pollution), as well as modifiable risk and lifestyle factors, such as smoking, stress, and alcohol consumption, have the capacity to induce epigenetic changes.29 Interestingly, microRNA sequences have also been implicated in DNA methylation abnormalities.31 The major epigenetic modifications are DNA methylation, as well as histone modification involving acetyl, methyl, and phosphate groups, leading to molecular alterations of the normal molecular structure of DNA, and to remodeling of chromatin. For example, acetylation of histones alters accessibility of chromatin and allows DNA binding proteins (transcription factors) to interact with exposed sites to transcribe the gene, or vice versa, DNA accessibility for functional transcription may be inhibited. Thus epigenetic mechanisms determine gene expression levels; epigenetic changes, especially in promoters, enhancer sequences, and DNA sequences for long noncoding RNA, can contribute to altered on/off mechanisms of gene transcription, or cause genes to behave in an aberrant way. In chronic inflammatory diseases, epigenetic mechanisms can, like single nucleotide polymorphisms, contribute to gene expression and function, altered immune function, and the magnitude of the inflammatory responses.29, 30, 32, 33 Interestingly, for epigenetic patterns, the DNA modifications can be shared by various chronic inflammatory diseases, especially via microinhibitory RNA sequences that may play an important role in intracellular regulation and feedback loops.33, 34
Further, the microbiomes that are in and on all the various ecologic niches of the body are of importance for determining normal immune function and may induce an aberrant host response in various chronic inflammatory diseases. For example, the mucous surfaces of the complete gastrointestinal tract, starting with the mouth, are lined with biofilm, normally without eliciting any pathological immune reaction.35 A series of studies36, 37, 38, 39, 40 support an important role for local (peripheral) Treg cells in the maintenance of mucosal or oral tolerance that avoids damaging immune reactions to microorganisms colonizing the mucosal surfaces of the human body. In fact, earlier research indicated that Treg cells were induced by microbial‐produced metabolites, in particular short‐chain fatty acids, thus suggesting that the intestinal biofilm induced its own tolerance.36, 39, 40 More recent research showed that there is a reciprocal and functional metabolite‐driven loop, where the gut bacteria control the local Treg cells numbers via their metabolites, and the local Treg cells alter the gut bacteria composition and determine which bacteria are tolerated.37, 38 Interestingly, the importance of Treg cells for immune tolerance and biofilm control has recently been recognized for periodontitis.41 This mucosal or oral tolerance is critically important in controlling destructive immune reactions to the commensal flora of the gastrointestinal tract.
Lifestyle factors constitute an important group of risk factors that can have a significant impact on immune function and immune fitness. For many chronic inflammatory diseases, lifestyle factors such as smoking, poor diet, and nutrition are considered important extrinsic and shared causal factors.19, 42, 43, 44 Smoking is one of the major sources of toxic chemical exposure to humans (there are at least 4,500 components in cigarette smoke) and can induce epigenetic alterations, which in turn leads to heightened inflammation.45, 46 Also, in general, oxidative stress is increased, with an excess of free oxygen radicals leading to cell damage. Apoptosis, programmed cell death, is pathologically both decreased and increased in smokers in different cellular compartments. Smoking impacts the immune‐inflammatory system and has consequences for inflammatory cellular activation. The nuclear factor‐kappa B pathway, which when activated results in the production of proinflammatory mediators, has autocrine, paracrine, and endocrine consequences. In smokers, low grade systemic inflammation is often present and macrophage elastase (matrix metalloproteinase‐12) is elevated. The immune system in general shows signs of dysfunction, with an increased tendency to produce autoantibodies, and reduced polymorphonuclear neutrophil chemotaxis and phagocytic capabilities.43 Also, smoking has an impact on the microbiome of the gastrointestinal tract; smokers with inflammatory bowel disease have a dysbiotic intestinal microbiome.45 However, the mechanisms by which smoking may induce a dysbiotic gut microbiota are not yet clear. There are indications that there is a vicious cycle, in that the inflammatory process of inflammatory bowel diseases may induce the dysbiosis, but the dysbiosis itself perpetuates and worsens inflammatory reactions in Crohn's disease and ulcerative colitis. Interestingly, there have been some reports that indicate that smoking may be protective for ulcerative colitis; however, there seems to be little data to explain these observations.45
Similar to smoking, the central aspect of poor diet and nutrition is the induction or increase in low grade systemic inflammation. The result of the high intake of animal fats (saturated fatty acids), red meat, salt, refined carbohydrates, fried foods, and low intake of fruit, vegetables, fiber, vitamin C, and other antioxidants, and shortage of vitamin D, results in increased inflammation. This can be through direct actions on the immune system or through epigenetic modifications that lead to nuclear factor‐kappa B or activator protein‐1 activation.19 Conversely, exercise, and low‐calorie diets (fruit, vegetables, and fish), perhaps probiotics and prebiotics, can act on the nuclear receptors and enzymes that upregulate oxidative metabolism and reduce the production of proinflammatory molecules. For example, the enzymatic conversion of polyunsaturated fatty acids from fish is essential for the generation of specialized proresolving mediators such as lipoxins, resolvins, protectins, and maresins.47, 48
Also, the effects of nutrition on the commensal gut microbiota are becoming increasingly understood as important.42, 44, 49 For example, good diet may help to maintain eubiosis, while poor dietary habits tend to induce a gut dysbiosis. The interaction of the gut microbiome and systemic health has now been established.50 A dysbiotic intestinal microbiome (with a reduced diversity) through poor diet leads to inflammation of the mucosal lining, and in turn, inflammation (“leaky gut”) and dysregulated mucosal immunity (altered Treg cells function). Together, this will induce or further propagate a poor quality gut microbiome, with reduced production of useful metabolites.19
Other aspects within the cluster of lifestyle factors are psychological stress and other types of psychosocial elements.11 Psychosocial aspects, including cognitive behavior (ie, coping with stress), have been identified as being part of the shared, causal factors deterministic to immune fitness. Over the years there has been consistent evidence for psychosocial factors having the capability to suppress immune functions at various levels.51, 52, 53, 54 In fact, it has been established that there is a bidirectional loop between the immune and nervous systems.52, 55, 56 Basically, neurons produce various immune mediators and cytokines that have effects on immune cells, and vice versa; immune cells produce neuropeptides and neurotransmitters, as well as express receptors for neurological mediators. Interestingly, susceptibility for depression is increased in situations of chronic inflammatory diseases.52, 55, 56 Between the various chronic diseases (morbidities), bi‐ or triple comorbidity is not uncommon.57 This is not surprising as the various chronic inflammatory diseases share genetic and epigenetic risk factors (pleiotropy is outlined above), immune pathways, and have shared lifestyle and psychosocial risk factors.11 For example, heart failure is associated with various comorbidities,57 and there is evidence of an association of psoriasis with rheumatoid arthritis, depression, inflammatory bowel disease, and cardiovascular disease.58 Also, a meta‐analysis showed a 48% increased risk of incident cardiovascular disease in patients with rheumatoid arthritis.59 Recently, the importance of the risk for cardiovascular comorbidity in patients with rheumatoid arthritis has been further stressed.60 In fact, one chronic inflammatory disease can negatively affect the immune fitness for another disorder; for example, this has been suggested for rheumatoid arthritis. The systemic inflammation in rheumatoid arthritis, as evidenced by elevated levels of C‐reactive protein and the consistent elevated erythrocyte sedimentation rate, may contribute to a heightened inflammatory burden, which is atherogenic.21, 61 Interestingly, a dysbiotic subgingival microbiome in periodontitis may affect the pathobiology and immune reactions of several autoimmune diseases such as rheumatoid arthritis, Sjogren's syndrome, systemic lupus erythematosus, and Crohn's disease.62 For rheumatoid arthritis, it is speculated that an altered oral microbiome with the outgrowth of Porphyromonas gingivalis and its enzymatic machinery for peptide citrullination may contribute to the generation of autoimmune anti‐citrullinated peptide antibodies. Taken together, comorbidities including periodontitis and/or dysbiotic microbiomes can have a major impact across different medical conditions.
3. PERIODONTITIS IS A COMPLEX CHRONIC INFLAMMATORY DISEASE
Periodontitis is considered a chronic inflammatory disease, and can be defined as a multicausal, complex, chronic inflammatory disorder.17, 63, 64, 65 Periodontitis is a chronic inflammatory disease exhibiting immune dysregulation (aberrant immune function) at its base, involving multiple causal components, which interplay simultaneously (as outlined above for other chronic inflammatory diseases).11 In Figure 1 we have provided a schematic drawing illustrating that the blueprint of the host response could determine immune fitness, which can either have normal tolerance and homeostasis with the dental biofilm, or an aberrant host response leading to an imbalance with the dental biofilm resulting in inflammation‐driven destruction of periodontal tissues, ie, periodontitis. In 2003, experimental work in rabbits overexpressing 15‐lipoxygenase types I and II demonstrated that inflammation is the driving force behind neutrophil‐mediated tissue degradation and alveolar bone loss in P. gingivalis‐induced periodontitis.66 Controlling excess inflammation with lipoxins showed dramatically reduced to no periodontal bone loss despite the ligature and P. gingivalis challenge to the periodontal tissues and bone.
Figure 1.
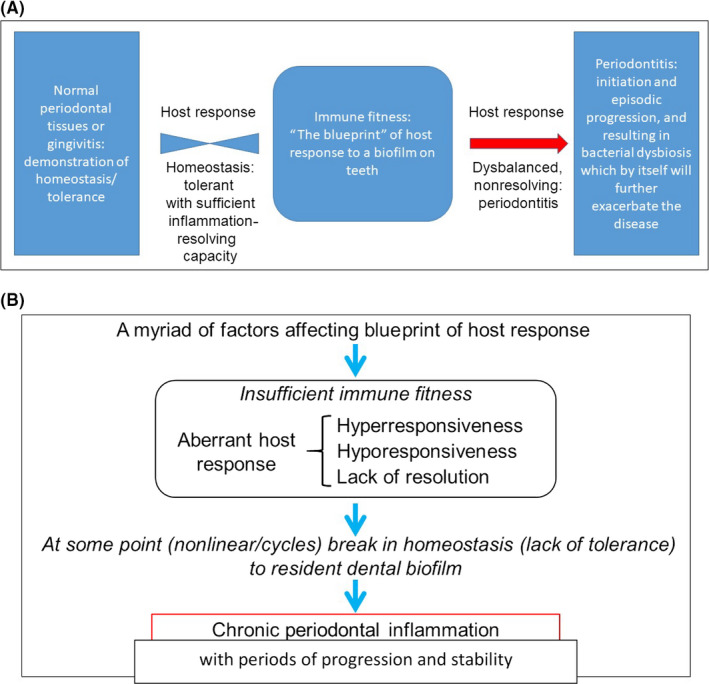
Panel (A) shows how immune fitness of the host determines the host response to the dental biofilm, which can either be symbiosis and homeostasis, or an aberrant host response leading to an imbalance resulting in inflammation‐driven destruction of periodontal tissues, ie, periodontitis. Panel (B) summarizes the complexity of periodontitis
In analogy to other chronic inflammatory diseases, we would expect that periodontitis behaves in a nonlinear fashion.17 Although we have limited evidence for periodontitis, we propose that the causes and effects may be disproportional to each other and that the disease progression rate fluctuates, or rather, can move from a homeostatic state or resolving state to an active state and back.67, 68, 69 The nonlinearity in periodontitis is seen in daily practice, where the disease often reveals its heterogeneity in the clinical course between any 2 patients. In periodontitis, it has long been established that the disease can occur in bursts of activity followed by periods of quiescence or stability,70, 71, 72 although the bouts of disease progression may occur in micromillimeters, which are not easily measured clinically. Also, the periodontitis disease state and disturbance of the homeostasis may change throughout life, where aging, epigenetic modifications, and comorbidities alter immune function.73, 74, 75, 76 These factors are further outlined below.
Interestingly, we understand now that the aberrant host response leading to periodontal inflammation can induce a microbial dysbiosis in the submarginal and subgingival regions. The normal submarginal and subgingival biofilm is diverse and may contain over 1,000 different species. With modern DNA techniques, the resident microbiome has been studied and in health an abundance of gram‐positive species can be found. Also, in health, traces of the well‐known periodontal pathogens are present.77, 78 In this way, they are termed symbionts, natural members of the submarginal and subgingival tooth biofilm. Normally their expansion is outcompeted by species that are not dependent on proteinaceous and blood metabolite materials such as hemin (an iron source), which are growth requirements for anaerobic gram‐negative species. However, when the host responds to the normal resident biofilm as it accumulates, then this in turn triggers inflammation, altering the microbial ecology and dysbiosis. The pathologic and dysfunctional immune response can actually induce an “ecological catastrophe”, a self‐feeding cycle of escalating dysbiosis.65, 79, 80, 81, 82 Specifically, the inflammatory reactions result in increased gingival crevicular fluid containing collagen breakdown products and the full range of host immune factors, including immunoglobulins, complement, serum proteins, cytokines, and chemokines, as well as increased amounts of immune cell remnants (after apoptosis of, for example, polymorphonuclear leukocytes), cells from desquamated pocket epithelium and, importantly, abundant collagen peptides from the degradation of gingival collagen by matrix metalloproteineases. Also, because of increased capillary permeability, serum exudates are found in inflamed pockets providing essential nutrients. Moreover, in inflammatory conditions, an anoxic environment develops, an additional factor driving proliferation of anaerobic bacteria.77, 78 Seminal experiments in rabbits have proven the above reasoning; experimentally induced periodontitis was associated with increased amounts of gram‐negative, anaerobic, and proteolytic bacteria, while treatment of the periodontal inflammatory lesions with the potent proresolving mediator resolvin E1 resulted in healing of the periodontal lesions and complete regression of these bacteria.79 In similar experimental settings reported by Lee et al in 2016,83 when analyzing the microbiome of periodontitis lesions after treatment with proresolving mediators it was shown that resolution of inflammation reversed the periodontal dysbiosis. Thus the altered ecological conditions transform some symbionts to pathobionts, ie, commensals that under conditions of disrupted homeostasis have the potential to cause disease.65, 78, 79 The outgrowth of the pathobionts (the most well‐known example is P. gingivalis) can further induce and worsen the host inflammatory responses.77, 78 Indeed, a transcriptomic analysis of subgingival microbiomes in periodontitis showed elevated expression of genes for proteolytic enzymes and for iron acquisition, as well as for lipopolysaccharide synthesis, highly suggestive that in periodontal inflammation many asaccharolytic, anaerobic, and gram‐negative bacteria exploit the ecological changes for their nutritional and expansive needs.78, 84 The bacterial biomass of human periodontitis‐associated biofilms increases with increasing periodontal inflammation. Thus the selective outgrowth of these inflammatory pathobionts can perpetuate periodontal inflammation resulting in a vicious cycle for disease progression, where the dysbiosis and inflammation reinforce each other.63, 64, 65, 77, 78, 79, 83, 85, 86, 87, 88, 89 In Figure 2, we have schematically illustrated the vicious cycle in periodontitis.
Figure 2.
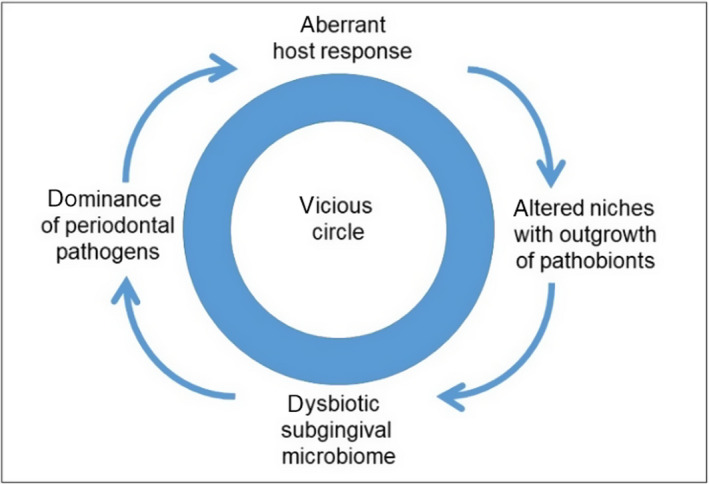
The vicious cycle of the “ecological catastrophe” driven by an aberrant host response in periodontitis
The paradigm, as outlined above, is also current for inflammatory bowel diseases, where dysregulated immune responses can induce intestinal dysbiosis that perpetuates the inflammation and disease, with reduction in microbial diversity and transformation of symbionts into pathobionts.90, 91, 92 The dysbiosis in the gut is associated with changes in gut permeability (“leaky gut”), leading to invasion of bacteria into the mucosal lining. We speculate that a similar phenomenon occurs in the gingiva; bacterial invasion and persistence have been described as critical events in the pathogenesis of periodontitis (“leaky gums”).93, 94, 95 In particular, the pathobionts P. gingivalis and Aggregatibacter actinomycetemcomitans have been shown to possess these characteristics.
4. THE ABERRANT HOST RESPONSE IN PERIODONTITIS
The literature in the periodontal field indicates that the aberrant immune response is in fact a hyperactive immune response; this is the central component of the pathobiology in periodontitis.63, 64, 65, 78, 85, 86, 87, 88, 89, 96, 97, 98, 99, 100 Thus the excessive inflammatory reactions lead to the dysbiotic changes discussed above, concomitant with periodontal tissue and alveolar bone breakdown. Polymorphonuclear neutrophils are the most likely cells to contribute substantially to destruction of periodontal tissues, and their hyper‐functionality has been found in periodontitis, in particular in aggressive periodontitis.64, 85, 86, 87, 88, 89, 101, 102, 103 Neutrophils release elevated levels of tissue‐destructive enzymes and substances such as reactive oxygen species, lysozyme, collagenases, and elastase. In addition, they produce proinflammatory cytokines and chemokines that add to the continued chronicity of the inflammation; these immune mediators will leak into the circulation and add to the systemic proinflammatory actions of periodontitis.100 For example, it was shown in ex vivo experiments that plasma samples from periodontitis patients were effective in enhancing superoxide production by neutrophils from healthy donors104; in this way a typical feedback loop or vicious cycle is present, where hyperactive neutrophils are also primed in an endocrine manner. Hyperactive neutrophils can also contribute to bone destruction through the stimulation of osteoclastic activity, in particular when they are in close proximity to the alveolar bone; most likely, neutrophils participate in the chemotactic recruitment of Th17 cells, which mediate and direct the activity of osteoclasts.105
Periodontitis patients showing regular relapses and nonresponsiveness to therapy are often characterized as having refractory periodontitis. Their neutrophils have been shown to have increased potential to produce oxygen radicals. In addition, for these refractory patients, increased phagocytosis by neutrophils was observed. This might be associated with higher intrinsic intracellular activity of the nicotinamide adenine dinucleotide phosphate oxidase system.98 A genetic polymorphism was identified in the nicotinamide adenine dinucleotide phosphate oxidase gene106; however, it is not known whether this genetic mutation is coupled to the increased production of the enzyme.
Interestingly, in periodontitis, increased influx of B‐cells and plasma cells is observed.107, 108 The plasma cell‐dominated lesions, which presumably produce antibodies, can be seen as a way for the immune system to attempt to neutralize the pathobionts that have proliferated in the dental biofilm. However, we can also hypothesize that the polyclonal B‐cell activation in the periodontal lesions represents a frustrated cellular immune system that has not found an effective way to reduce B cell/plasma cell activity and therefore shows overreactive antibody production.
Notably, various ex vivo whole blood cell culture systems have been employed to investigate immune reactivity to lipopolysaccharide preparations in periodontitis.109, 110 Most likely the cells responsive to stimuli in these systems represent the cells of the monocyte/macrophage lineage, which are also antigen‐presenting cells.111, 112 Collectively, these studies have shown higher reactivity in whole blood cell cultures from patients with aggressive or chronic periodontitis, as evidenced by elevated levels of key cytokines produced in the cultures. This was also suggested when employing isolated monocytes from peripheral blood samples.113 In some studies, monocytic hyperresponsiveness was partly attenuated after periodontal therapy,110, 112 while others did not observe the reduction.114 The various studies cited here confirm possible intrinsic aberrant reactivity with the ultimate result that heightened inflammatory reactions feed the proteolytic species in the dental biofilm.
Although the majority of the literature indicates that neutrophils are hyperreactive in periodontitis, in particular in severe, early onset forms, at the same time investigators have observed reduced neutrophil functions.100 In particular, reduced chemotaxis and Ca2+ uptake seems to be a comorbidity associated with primed neutrophils.96, 100, 101, 102 Further, aberrations in phagocytosis, respiratory burst, and intracellular killing have been reported.115 Rare mutations leading to rare forms of periodontitis (Papillon‐Lefèvre syndrome, leukocyte adhesion defect‐syndrome, and other congenital diseases with rampant periodontitis) can be regarded as forms of periodontal inflammation where specific neutrophil functions are altered or absent.100 Another aspect of the immune system that can be less functional in periodontitis is the decreased production of immunoglobulins.101, 116 This is especially seen in smokers, further explaining how smoking reduces immune fitness.
How hypoactivity of parts of the immune system, such as leukocyte adhesion deficiency, actually results in excessive inflammation with concomitant conversion of a normal microbiome to a dysbiotic microbiota, is becoming clearer. Moustopoulous and co‐workers have demonstrated that an aberrant host response is overcompensated by other parts of the immune system.117 For example, where neutrophils in the leukocyte adhesion deficiency syndrome are unable to exit the circulation to phagocytize bacteria, the system is compensated by Th17 cells and interleukin‐17 production.
5. EVIDENCE FOR FAILURE OF RESOLUTION PATHWAYS
Inflammatory responses are protective biological processes designed to eliminate the harmful stimuli and promote return of the affected tissue to its preinflammatory state and function (homeostasis). There are a series of anti‐inflammatory mediators, including tissue inhibitors of metalloproteinases, which are important tissue‐destroying enzymes released by macrophages during the inflammatory response. There is also an inhibitor set of mediators that results in active resolution of inflammation.118 In the past, it was believed that inflammatory resolution was a passive event that resulted from the dilution of chemokine gradients over time, thus reducing the chemotaxis of leukocytes to the site of injury, and production of anti‐inflammatory mediators. However, evidence from studies performed over the last few decades has shown that it is a carefully orchestrated active process.119 The identification of specific proresolving pathways and lipid mediators introduced a paradigm shift and opened a new window to understanding the resolution of inflammation.119 It is now widely appreciated that resolution of inflammation represents a sequence of overlapping events during which proinflammatory mediators induce the generation of specialized proresolving mediators that stimulate their receptor targets.26 The specialized proresolving mediators are lipoxins, resolvins, protectins, and maresins, which originate from the enzymatic conversion of omega‐3 polyunsaturated fatty acids in the human diet. In other words, the signals that regulate the resolution of acute inflammatory responses are tightly interrelated with the mediators which initiate these responses. Thus the peak of an acute inflammatory response is considered the beginning of resolution.120
Resolution of inflammation is a well‐orchestrated active process mediated by a variety of specialized proresolving mediators. Resolvin E1 is biosynthesized from eicosapentaenoic acid and selectively interacts with specific receptors to inhibit further leukocyte infiltration and cytokine/chemokine generation, to induce the apoptosis of neutrophils and their removal by macrophages to restore tissue homeostasis. In periodontitis, as well as in other inflammatory conditions, inflammation fails to resolve and results in chronic pathology. Recent studies have detected lower levels of specialized proresolving mediators in the stimulated whole blood of localized aggressive periodontitis patients suggesting a defect, which might contribute to the failure of resolution.121 A growing body of in vitro and in vivo evidence points to the effects of resolvin E1 and other specialized proresolving mediators on different cell types in regulating the resolution of periodontal inflammation. To date, no study has addressed the issue of the tissue expression levels of specialized proresolving mediators in gingival biopsies retrieved from chronic gingivitis patients, as opposed to periodontitis patients.119 With this important piece of information lacking, it is as yet unknown whether a robust engagement of the resolution pathways can efficiently prevent the conversion of gingivitis to periodontitis.
To compensate for rapid metabolism or dilution of proresolving lipid mediators, specialized and stable proresolving mediator analogs have been constructed as mimetics of endogenous resolution, offering new therapeutic approaches. In addition, incorporation of specialized proresolving mediators in microparticles and the employment of nano‐proresolving medicines have provided a new possibility for local delivery at the site of inflammation.122 Although promising results have been obtained in animal studies, the efficacy of this experimental data has yet to be established in human clinical trials. Human studies involving omega‐3 fatty acid supplementation as a source of polyunsaturated fatty acid and low‐dose aspirin as adjunct to periodontal treatment provide promising results and indicate a synergistic effect of these agents in periodontal treatment.123 Up to now there have been no long‐term randomized clinical trials investigating the clinical benefits of omega‐3 fatty acids vs other broadly used pharmacological agents, such as antibiotics, as an adjunct to periodontal treatment in humans. Furthermore, large scale in vivo and ex vivo studies evaluating the effects of treatment with specialized proresolving mediators in individuals with periodontitis may shed more light on the complex molecular mechanisms involved in the resolution of periodontal inflammation. Further studies are warranted to elucidate if resolvin E1, alone or in combination with other specialized proresolving mediators, is effective for elimination of periodontal inflammation and regeneration of lost tissues in humans, as was seen in various animal models.79, 83
6. WHICH FACTORS CO‐CONTRIBUTE TO DYSREGULATION OF IMMUNE FITNESS IN PERIODONTITIS?
As explained above for other chronic inflammatory diseases, multiple factors can contribute to the blueprint of immune fitness at a certain time/moment, and the dysregulation of immune fitness in periodontitis occurs by the total of all risk factors and their interplay. The onset of periodontal inflammation is triggered by the microbial communities in the dental biofilm; without microorganisms there is little or no gingival inflammation. Above, we have already concluded that the inflammatory reactions in the periodontal tissues can derail from normal and tolerant to destructive. At that point, the change in ecosystem allows the outgrowth of pathobionts, which subsequently, in a vicious cycle, can worsen the inflammatory reactions and allow the disease to become chronic.
We group the causal risk factors for periodontitis into the following main clusters: (a) the subgingival bacterial biofilm on both the tooth root surface and on the epithelial lining; (b) genetic risk factors and epigenetic modifications; (c) lifestyle‐related risk factors; (d) systemic diseases; and (e) miscellaneous factors. The 5 main causal clusters for periodontitis can be brought together into a pie chart model (Figure 3) (based on17 and modified from124, 125, 126).
Figure 3.
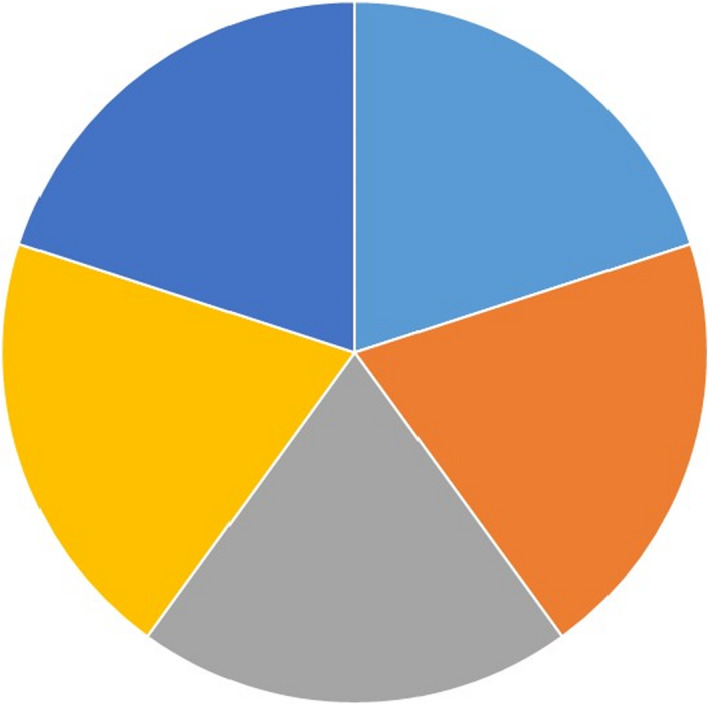
A generic multi‐causality model for periodontitis, where 5 clusters of causal (risk) factors are playing a role simultaneously, (epi)genetic factors (light blue), lifestyle factors (orange), comorbidities (systemic diseases) (gray), microbial communities, ie, dental biofilms (yellow) and other factors (tooth and dention related and stochasticity) (dark blue). Notably, for each individual periodontitis patient, the relative contribution of the 5 clusters of causal factors varies and needs to be estimated, and as such, theoretically, for each patient, a unique pie chart can be created. Adapted from17 based on124, 125, 126
In Figure 3 we present a generic multi‐causality model for periodontitis, where each of the 5 causal components have an equal contribution. However, we should emphasize, that for each individual periodontitis case, the relative contribution (ie, size of piece of pie) of each of the 5 clusters of causal factors varies and needs to be estimated based on all available clinical data. At this point clinical judgement is required. Nevertheless, in general, older patients with periodontitis are considered to have a major contribution from expansion of pathobionts, possibly induced by increasing inflammation with age, unfavorable lifestyle factors, and often concomitant with another chronic inflammatory disease. On the other hand, periodontitis in younger patients, for example, in those suffering from early onset periodontitis, can be linked to a greater extent to genetic factors.17
Below we summarize knowledge on the genetic risk factors for periodontitis. Risk factors from the clusters “lifestyle” (such as smoking, stress, and diet), “systemic diseases” (such as diabetes, rheumatoid arthritis, and other autoimmune diseases), and “miscellaneous” (such as dentition and tooth‐related factors, but also stochastic factors) are not reviewed here, but have been reviewed elsewhere.127 At the onset, we want to stress that the identification of the myriad of risk factors playing a role in the whole pathobiology of periodontitis is hampered by the fact that it is so difficult to identify and study one factor among the others simultaneously playing a role. Each of the factors individually, including the genetic factors discussed below, is not a “black and white” determinant; we stress again that the mono‐causality concept is obsolete and that the disease is complex and nonlinear.
The genetic blueprint of an individual is very dependent on the type and function of genomic variants in multiple genetic loci: (a) multiple genes play a role (polygenic) (gene‐gene interactions); (b) not all disease cases have the same genetic risk variants; and (c) multiple genetic variations are present at the same time. All these genetic variations potentially modify the host response, which is also not at a constant level. We discussed earlier that the host response is trying to maintain homeostasis with its environment, and can cycle between remission or relapse states where the genetic variations may cause modifications in the immune system, while immune fitness is also affected by lifestyle factors and certain systemic diseases. Furthermore, epigenetic changes of DNA and mutations during a lifetime may further modify an individual's susceptibility to periodontitis.
On the basis of epidemiological studies in twins and in family studies with a high rate of early onset periodontitis, it can be concluded that in younger patients the genetic contribution may be as large as 50% to the total sum of causal factors, while in older patients the genetic contribution to the total of all causes is at most 25%.17, 128, 129, 130, 131 Similar to other complex chronic diseases, it is important to realize that a multitude of genetic variations (probably >100) are involved and therefore the disease is called polygenic.132 Currently, for periodontitis, the genetic factors associated with or actually contributing to the pathogenesis have only been identified to a limited extent and are often poorly validated. Research into specific genetic polymorphisms is hampered by the fact that genetic variants, which are associated with periodontitis in, for example, Caucasians, may not be associated with disease in other ethnic populations such as Asians, Brazilians, and Africans.133 Although a considerable overlap of genetic variants (often single nucleotide polymorphisms) between the different races and ethnicities is expected,134 it is important to realize that there might also be population‐specific risk gene variants.132
The risk genes or genomic noncoding regions (loci) for periodontitis, with the variants or single nucleotide polymorphisms within them, have been identified either by the candidate gene approach or by genome‐wide association studies.18 Many candidate gene studies have been performed in periodontitis yielding varied and often contradictory results. Because of the absence of sufficient statistical power, and also because of the absence of correction for multiple testing, false positive results are a common problem.17, 18, 132, 133 Recently, we have seen the sharing of data and patient samples between research groups to further explore and to discover new candidate genes, or to extend the results of genome‐wide associated studies.135, 136 Other issues also exist. For example, earlier studies often included just 1 or a few candidate single nucleotide polymorphisms within the genetic locus of interest, but today, confirming whether a given single nucleotide polymorphism has an association with periodontitis requires exploring the complete genetic locus including upstream and downstream regulatory regions and/or neighboring loci; and requires a robust mechanistic explanation on the functional consequences of a given genetic variant. Furthermore, the phenotype classification of periodontitis and control subjects has not been consistent across the various studies. Also, many studies have not taken into account lifestyle factors that could play a role in the periodontitis phenotype, or the presence of comorbidities.
Currently, variants in at least 65 genes have been suggested as being associated with periodontitis. However, the number of genetic variants proposed to be associated with periodontitis is very dependent on the applied criteria in the original discovery studies and in systematic reviews. For systematic reviews, questions such as what is considered a sufficient number of cases and controls that were included in the original studies, what were the phenotypic definitions, and whether the investigators have also looked into replication/validation of single or pilot studies, are important. Results of genome‐wide association studies are often more reliable as they include 100s or up to 1000s of periodontitis patients, apply strict significance levels to avoid false positive results because of multiple testing, and mostly contain a validation or replication cohort.18, 133, 135, 137, 138, 139 The genes having variant alleles (minor alleles) or haplotypes associated with both aggressive and chronic periodontitis, or only with aggressive periodontitis, or only with chronic periodontitis, or just with unspecified periodontitis, are summarized in various current reviews.3, 17, 129, 140, 141
Interestingly, reports have found pleiotropy between periodontitis and cardiovascular diseases142; the same genetic variants have been observed as being associated with both cardiovascular diseases and periodontitis.139, 142, 143, 144, 145 This is an intriguing finding because a common genetic background for coronary artery disease and periodontitis could be interpreted as similarly aberrant host responses during inflammatory processes, irrespective of where they take place. Notably, it is now well accepted that atherosclerotic plaques can be regarded as inflammatory lesions, and that atherosclerotic cardiovascular disease can be regarded as an inflammatory disease.146, 147, 148, 149, 150 Similar host immune reactions and pathobiology responses could be hypothesized to be directed to the bacteria and bacterial antigens that are transmigrated from the periodontium, intestines, and other mucosal surfaces into macrophages/foam cells residing in atherosclerotic plaques.151
One of the first and best replicated genetic loci associated with coronary artery disease is the CDKN2B‐AS1 locus.17, 139, 142, 143 The CDKN2B‐AS1 locus is a regulatory region and does not contain a protein‐encoding gene. It is a long noncoding antisense RNA also known as ANRIL. Importantly, it appears to be from a highly pleiotropic genetic region (chromosome 9, p21.3), as it is also associated with type 2 diabetes, ischemic stroke, and Alzheimer's disease. Since 2009, it has been reported that certain genetic variations in CDKN2B‐AS1 are also consistently associated with periodontitis.17, 142, 143, 145, 152 Its function and role has recently been further investigated and found to be related to regulation of gene expression.153 Interestingly, a pilot study identified that one of the genetic variants in the CDKN2B‐AS1 locus is associated with the extent of elevated levels of C‐reactive protein in periodontitis, but details of the possible pathway have not yet been established.154
Further, a conserved noncoding element within CAMTA1 upstream of VAMP3, first identified as a genetic susceptibility locus for coronary artery disease, was also found to be associated with periodontitis.144, 155 Interestingly, previously, a genome‐wide association study suggested that the VAMP3 locus was associated with a higher probability of subgingival overgrowth of periodontal pathogens.156 Another coronary artery disease risk locus PLG was also found to be associated with periodontitis. There is now evidence for PLG as a shared genetic risk factor of coronary artery disease and periodontitis.144 Plasminogen is converted into plasmin, which can dissolve the fibrinogen fibers that entangle the blood cells in a blood clot, in a process called fibrinolysis. The plasminogen‐plasmin axis has an important function in tissue degradation and control of the blood coagulation system. Interestingly, bacteria (including P. gingivalis) can convert plasminogen to plasmin, and this complex is highly proteolytic and can possibly inactivate plasmin inhibitors, causing uncontrolled plasmin activity. Yet another shared risk locus for coronary artery diseases and periodontitis has been identified. Based on a genome‐wide association meta‐analysis of individuals of north European ancestry, it is a haplotype block at the VAMP8 locus.143
Despite the strong significance of the shared genetic variants in the VAMP3 and VAMP8 loci, it is not clear whether they are the causative variants and what the functional consequences are. The VAMP3 and VAMP8 variants encode for vesicle‐associated membrane proteins and are simultaneously expressed in various cell types, including mast cells, adipocytes, and in the secretory granules of platelets. These vesicle‐associated membrane proteins may play a role in membrane trafficking and the release of inflammatory mediators from platelets during coagulation, pathogen recognition, aggregation, and wound healing.143 Adipocytes may also be part of biological pathways that connect glucose and fatty acid metabolism steps with immune responses via the vesicle‐associated membrane proteins.143, 144, 155
Collectively, these shared genetic factors suggest mechanistic links or immunologic commonalities between coronary artery disease, periodontitis, diabetes, metabolic syndrome, obesity, and inflammation. The impairment of the regulatory pathways by genetic factors may be a common pathogenic denominator of at least coronary artery disease and periodontitis.142, 143, 153, 155 We hypothesize that aberrant inflammatory reactivity, determined in part by genetic variants in the loci CDKN2B‐AS1 (ANRIL), PLG, CAMTA1/VAMP3, and VAMP8, could explain in part the epidemiological link between periodontitis and cardiovascular diseases.157, 158, 159, 160, 161 Thus the shared genes suggest that periodontitis per se is not actually causally related to atherosclerosis, but rather both conditions are sequelae of similar (the same?) aberrant inflammatory pathways, which contribute to the pathogenesis of both periodontitis and cardiovascular disease.
One other important aspect needs to be noted. The human genome, and genetic and epigenetic variants or mutations, can also contribute to microbial colonization and infection patterns (the gene‐environmental axis).162 From several genetically modified mouse models, we have observed that when apparent key genes are silenced or knocked out, a dysfunctional immune system is present, and with the ensuing inflammation and periodontal tissue destruction, dental biofilm can proliferate and increase markedly in biomass.163 It is important to note that the mouse gut microbiome can be influenced by genetic modifications.164 Spor and colleagues concluded that the overall architecture of host genetics impacts the diversity of the microbiome of the gut.165 The concept of infectogenomics in the periodontal field was introduced in 2009 and several papers have found genetic variants to be associated with the level of some bacterial species.156, 166, 167, 168, 169 The latest review on this topic lists all gene‐bacterial interactions extensively; however, the results need to interpreted with care, as already explained above, because false positive associations can be present as a result of a lack of statistical power.167 Future studies should show statistical adjustment, to minimize for the false positives that are common in genetic as well as microbiome studies.
Finally, an additional important feature playing a role in modifying the genetic blueprint of host responses to the dental biofilm is epigenetic modification of coding and noncoding DNA.74, 170, 171, 172, 173, 174 This emerging field will likely yield new information in relation to the susceptibility to periodontitis and subsequent persisting inflammatory reactions in periodontitis.171 Several studies suggest that smoking, inflammatory processes in the periodontal tissues, and the microbiome compositions adjacent to the sulcular/pocket epithelial cells, may induce aberrant epigenetic modifications of genomic DNA with functional consequences.170, 171, 175, 176 For example, based on gingival biopsies from chronic periodontitis patients, hyper‐methylation and hypo‐methylation of the promoter regions of the genes encoding tumor necrosis factor‐alpha and interferon‐gamma, respectively, were inversely associated with their expression.177, 178 Generally, hyper‐methylation in gene‐promoter regions seems to correlate with gene silencing, while hypo‐methylation is associated with increased gene expression.173 Similarly, in a pilot study in 15 patients with aggressive periodontitis and 10 controls, the methylation patterns for 22 inflammatory candidate genes in gingival biopsies were quantified,179 and reduced methylation was found for the genes CCL25 and IL17C in periodontitis compared with control gingival biopsies. This hypo‐methylation pattern was suggested as being associated with an increase of the expression of these genes having a pro‐inflammatory effect. In earlier studies, Barros and Offenbacher observed that the periodontal tissues are epigenetically modified in particular at the biofilm‐gingiva interface around the teeth.171 The authors suggest that epigenetics may be partly responsible for the cycling of chronic periodontal lesions from hyper‐inflammation to hypo‐inflammation and resolution, and alterations in the epigenome can reveal the molecular basis for certain risk exposures.171 Interestingly, variance or major differences in subgingival microbiome from one individual to the next may result in differential epigenetic changes on various genes in the exposed tissues.171 The clinically important suggestion is that epigenetic modification of periodontal tissues may explain why periodontal disease preferentially persists at one specific periodontal site relative to another and why these local tissues will not fully repair or regenerate, thus persisting as a long‐term clinical management problem.171 The interesting clinical insight from these studies is that, potentially, excisional periodontal surgery can eliminate the epigenetically modified tissues yielding “normal” periodontal tissues after healing, presumably able to maintain homeostasis with the dental biofilm.
Rather than studying the local epigenome, Shaddox et al174 studied peripheral white blood cells for epigenetic signatures in children and adolescents of African‐American ethnicity. They found that there were significant differences in the DNA methylation status in the localized aggressive periodontitis patients compared with healthy controls; the authors reported differences for both hyper‐ and hypo‐methylation patterns in genes that are part of the toll‐like receptor signaling pathways.174 These patterns correlated with lipopolysaccharide‐stimulated inflammatory cytokines, suggesting the functionality of the methylation patterns and possibly explaining systemic susceptibility for periodontitis174 and the previously observed hyperresponsive phenotype in localized aggressive periodontitis.109
7. CONCLUSIONS
Periodontitis is a complex chronic inflammatory disease with nonlinear progression that is caused by various factors each playing a role simultaneously and interacting with each other. The various factors determine the immune fitness of a subject. The host exists in a symbiotic relationship with the oral microbiome to maintain homeostasis. Loss of homeostasis results from loss of the host balance and an aberrant host response. This aberrant host response can manifest as a hyper‐ or hyporesponsiveness and/or lack of sufficient resolution of inflammatory reactions. The consequent chronic inflammation elicits changes in the ecology of the subgingival environment providing favorable conditions for the overgrowth of pathobionts that further propagate periodontal inflammation. The factors that determine immune fitness include: (a) genetic factors and epigenetic factors; (b) lifestyle factors; (c) comorbidities; (d) local or dental factors and factors that act randomly; and (e) pathobionts in a dysbiotic subgingival biofilm. Variants in at least 65 genes to date have been suggested as being associated with periodontitis based on genome‐wide association studies and candidate gene case control studies. Interestingly, reports have found pleiotropy between periodontitis and cardiovascular diseases. To date, 4 genetic loci are shared between coronary artery disease and periodontitis. The shared genes suggest that periodontitis is not causally related to atherosclerotic diseases, but rather both conditions are sequelae of similar (the same?) aberrant inflammatory pathways. In addition to variations in genomic sequences, epigenetic modifications of DNA can affect the genetic blueprint of the host responses. Further studies are required to verify and expand our knowledge base before final cause and effect conclusions can include specific genetic markers.
Loos BG, Van Dyke TE. The role of inflammation and genetics in periodontal disease. Periodontol 2000. 2020;83:26–39. 10.1111/prd.12297
REFERENCES
- 1. Armitage G. Development of a classification system for periodontal diseases and conditions. Ann Periodontol. 1999;4:1‐6. [DOI] [PubMed] [Google Scholar]
- 2. Delatola C, Loos BG, Levin E, Laine ML. At least three phenotypes exist among periodontitis patients. J Clin Periodontol. 2017;44:1068‐1076. [DOI] [PubMed] [Google Scholar]
- 3. Fine DH, Patil AG, Loos BG. Classification and diagnosis of aggressive periodontitis. J Clin Periodontol. 2018;45(Suppl 20):S95‐S111. [DOI] [PubMed] [Google Scholar]
- 4. Morelli T, Moss KL, Beck J, et al. Derivation and validation of the periodontal and tooth profile classification system for patient stratification. J Periodontol. 2017;88:153‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Papapanou PN, Sanz M, Buduneli N, et al. Periodontitis: consensus report of workgroup 2 of the 2017 World Workshop on the classification of periodontal and peri‐implant diseases and conditions. J Clin Periodontol. 2018;45(Suppl 20):S162‐S170. [DOI] [PubMed] [Google Scholar]
- 6. Tonetti MS, Greenwell H, Kornman KS. Staging and grading of periodontitis: Framework and proposal of a new classification and case definition. J Clin Periodontol. 2018;45(Suppl 20):S149‐S161. [DOI] [PubMed] [Google Scholar]
- 7. Caton JG, Armitage G, Berglundh T. A new classification scheme for periodontal and peri‐implant diseases and conditions ‐ Introduction and key changes from the 1999 classification. J Periodontol. 2018;89:S1‐S8. [DOI] [PubMed] [Google Scholar]
- 8. Eke PI, Thornton‐Evans GO, Wei L, Borgnakke WS, Dye BA, Genco RJ. Periodontitis in US adults. National Health and Nutrition Examination Survey 2009‐2014. J Am Dent Assoc. 2018;149:576‐588 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kassebaum NJ, Bernabe E, Dahiya M, Bhandari B, Murray CJ, Marcenes W. Global burden of severe periodontitis in 1990‐2010: a systematic review and meta‐regression. J Dent Res. 2014;93:1045‐1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Norderyd O, Koch G, Papias A, et al. Oral health of individuals aged 3‐80 years in Jonkoping, Sweden during 40 years (1973‐2013). II. Review of clinical and radiographic findings. Swed Dent J. 2015;39:69‐86. [PubMed] [Google Scholar]
- 11. Te Velde AA, Bezema T, van Kampen AH, et al. Embracing complexity beyond systems medicine: a new approach to chronic immune disorders. Front Immunol. 2016;7:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Andreassen OA, Desikan RS, Wang Y, et al. Abundant genetic overlap between blood lipids and immune‐mediated diseases indicates shared molecular genetic mechanisms. PLoS ONE. 2015;10:e0123057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ellinghaus D, Jostins L, Spain SL, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease‐specific patterns at shared loci. Nat Genet. 2016;48:510‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gustafsson M, Nestor CE, Zhang H, et al. Modules, networks and systems medicine for understanding disease and aiding diagnosis. Genome Med. 2014;6:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu JX, Thomas CE, Brunak S. Network biology concepts in complex disease comorbidities. Nat Rev Genet. 2016;17:615‐629. [DOI] [PubMed] [Google Scholar]
- 16. Sivakumaran S, Agakov F, Theodoratou E, et al. Abundant pleiotropy in human complex diseases and traits. Am J Hum Genet. 2011;89:607‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Loos BG, Papantonopoulos G, Jepsen S, Laine ML. What is the contribution of genetics to periodontal risk? Dent Clin North Am. 2015;59:761‐780. [DOI] [PubMed] [Google Scholar]
- 18. Vaithilingam RD, Safii SH, Baharuddin NA, et al. Moving into a new era of periodontal genetic studies: relevance of large case‐control samples using severe phenotypes for genome‐wide association studies. J Periodontal Res. 2014;49:683‐695. [DOI] [PubMed] [Google Scholar]
- 19. Riccio P, Rossano R. Nutrition facts in multiple sclerosis. ASN Neuro. 2015;7 1-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trefois C, Antony PM, Goncalves J, Skupin A, Balling R. Critical transitions in chronic disease: transferring concepts from ecology to systems medicine. Curr Opin Biotechnol. 2015;34:48‐55. [DOI] [PubMed] [Google Scholar]
- 21. Tanaka K, Hamada K, Nakayama T, et al. Risk for cardiovascular disease in Japanese patients with rheumatoid arthritis: a large‐scale epidemiological study using a healthcare database. Springerplus. 2016;5:1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuman M. The chronic diseases require nonlinear mathematical description. Adv Complement ALT Med. 2018;1:1‐4. ACAM.000514 [Google Scholar]
- 23. Philippe P, Mansi O. Nonlinearity in the epidemiology of complex health and disease processes. Theor Med Bioeth. 1998;19:591‐607. [DOI] [PubMed] [Google Scholar]
- 24. Trachana K, Bargaje R, Glusman G, Price ND, Huang S, Hood LE. Taking systems medicine to heart. Circ Res. 2018;122:1276‐1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ajeganova S, Huizinga T. Sustained remission in rheumatoid arthritis: latest evidence and clinical considerations. Ther Adv Musculoskelet Dis. 2017;9:249‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro‐resolving superfamily of mediators. J Clin Invest. 2018;128:2657‐2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune‐mediated diseases. Nat Rev Genet. 2013;14:661‐673. [DOI] [PubMed] [Google Scholar]
- 28. Klein K, Gay S. Epigenetics in rheumatoid arthritis. Curr Opin Rheumatol. 2015;27:76‐82. [DOI] [PubMed] [Google Scholar]
- 29. Raghuraman S, Donkin I, Versteyhe S, Barres R, Simar D. The emerging role of epigenetics in inflammation and immunometabolism. Trends Endocrinol Metab. 2016;27:782‐795. [DOI] [PubMed] [Google Scholar]
- 30. Sorrentino R. Genetics of autoimmunity: an update. Immunol Lett. 2014;158:116‐119. [DOI] [PubMed] [Google Scholar]
- 31. Pan W, Zhu S, Yuan M, et al. MicroRNA‐21 and microRNA‐148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol. 2010;184:6773‐6781. [DOI] [PubMed] [Google Scholar]
- 32. Loddo I, Romano C. Inflammatory bowel disease: genetics, epigenetics, and pathogenesis. Front Immunol. 2015;6:551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. O'Connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295‐312. [DOI] [PubMed] [Google Scholar]
- 34. Langlais D, Fodil N, Gros P. Genetics of infectious and inflammatory diseases: overlapping discoveries from association and exome‐sequencing studies. Annu Rev Immunol. 2017;35:1‐30. [DOI] [PubMed] [Google Scholar]
- 35. Ai TL, Solomon BD, Hsieh CS. T‐cell selection and intestinal homeostasis. Immunol Rev. 2014;259:60‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arpaia N, Campbell C, Fan X, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T‐cell generation. Nature. 2013;504:451‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Campbell C, Dikiy S, Bhattarai SK, et al. Extrathymically generated regulatory t cells establish a niche for intestinal border‐dwelling bacteria and affect physiologic metabolite balance. Immunity. 2018;48:1245‐1257 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gefen T, Geva‐Zatorsky N. What came first: the microbiota or the Tr(egg) cells? Immunity. 2018;48:1072‐1074. [DOI] [PubMed] [Google Scholar]
- 39. Ivanov II, Honda K. Intestinal commensal microbes as immune modulators. Cell Host Microbe. 2012;12:496‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short‐chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Alvarez C, Rojas C, Rojas L, Cafferata EA, Monasterio G, Vernal R. Regulatory T lymphocytes in periodontitis: a translational view. Mediators Inflamm. 2018;2018:7806912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ananthakrishnan AN. Environmental risk factors for inflammatory bowel diseases: a review. Dig Dis Sci. 2015;60:290‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang K, Yang SM, Kim SH, Han KH, Park SJ, Shin JI. Smoking and rheumatoid arthritis. Int J Mol Sci. 2014;15:22279‐22295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Neuman MG, Nanau RM. Inflammatory bowel disease: role of diet, microbiota, life style. Transl Res. 2012;160:29‐44. [DOI] [PubMed] [Google Scholar]
- 45. Parkes GC, Whelan K, Lindsay JO. Smoking in inflammatory bowel disease: impact on disease course and insights into the aetiology of its effect. J Crohns Colitis. 2014;8:717‐725. [DOI] [PubMed] [Google Scholar]
- 46. Rom O, Avezov K, Aizenbud D, Reznick AZ. Cigarette smoking and inflammation revisited. Respir Physiol Neurobiol. 2013;187:5‐10. [DOI] [PubMed] [Google Scholar]
- 47. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191‐1197. [DOI] [PubMed] [Google Scholar]
- 48. Spite M, Claria J, Serhan CN. Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab. 2014;19:21‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. West CE, Renz H, Jenmalm MC, et al. The gut microbiota and inflammatory noncommunicable diseases: associations and potentials for gut microbiota therapies. J Allergy Clin Immunol. 2015;135:3‐13; quiz 14. [DOI] [PubMed] [Google Scholar]
- 50. Claesson MJ, Jeffery IB, Conde S, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488:178‐184. [DOI] [PubMed] [Google Scholar]
- 51. Cohen S, Janicki‐Deverts D, Doyle WJ, et al. Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc Natl Acad Sci USA. 2012;109:5995‐5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kiecolt‐Glaser JK, Derry HM, Fagundes CP. Inflammation: depression fans the flames and feasts on the heat. Am J Psychiatry. 2015;172:1075‐1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Morgan N, Irwin MR, Chung M, Wang C. The effects of mind‐body therapies on the immune system: meta‐analysis. PLoS ONE. 2014;9:e100903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Segerstrom SC, Miller GE. Psychological stress and the human immune system: a meta‐analytic study of 30 years of inquiry. Psychol Bull. 2004;130:601‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gibney SM, Drexhage HA. Evidence for a dysregulated immune system in the etiology of psychiatric disorders. J Neuroimmune Pharmacol. 2013;8:900‐920. [DOI] [PubMed] [Google Scholar]
- 56. Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16:22‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Triposkiadis F, Giamouzis G, Parissis J, et al. Reframing the association and significance of co‐morbidities in heart failure. Eur J Heart Fail. 2016;18:744‐758. [DOI] [PubMed] [Google Scholar]
- 58. Oliveira Mde F, Rocha Bde O, Duarte GV. Psoriasis: classical and emerging comorbidities. An Bras Dermatol. 2015;90:9‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Avina‐Zubieta JA, Thomas J, Sadatsafavi M, Lehman AJ, Lacaille D. Risk of incident cardiovascular events in patients with rheumatoid arthritis: a meta‐analysis of observational studies. Ann Rheum Dis. 2012;71:1524‐1529. [DOI] [PubMed] [Google Scholar]
- 60. Jagpal A, Navarro‐Millan I. Cardiovascular co‐morbidity in patients with rheumatoid arthritis: a narrative review of risk factors, cardiovascular risk assessment and treatment. BMC Rheumatol. 2018;2:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Innala L, Moller B, Ljung L, et al. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: a five year prospective study. Arthritis Res Ther. 2011;13:R131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nikitakis NG, Papaioannou W, Sakkas LI, Kousvelari E. The autoimmunity‐oral microbiome connection. Oral Dis. 2017;23:828‐839. [DOI] [PubMed] [Google Scholar]
- 63. Bartold P, Van Dyke T. Periodontitis: a host‐mediated disruption of microbial homeostasis. Unlearning learned concepts. Periodontol 2000. 2013;62:203‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bartold PM, Van Dyke TE. Host modulation: controlling the inflammation to control the infection. Periodontol 2000. 2017;75:317‐329. [DOI] [PubMed] [Google Scholar]
- 65. Hajishengallis G. Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol. 2014;35:3‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Serhan CN, Jain A, Marleau S, et al. Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15‐lipoxygenase and endogenous anti‐inflammatory lipid mediators. J Immunol. 2003;171:6856‐6865. [DOI] [PubMed] [Google Scholar]
- 67. Papantonopoulos G, Takahashi K, Bountis T, Loos BG. Using cellular automata experiments to model periodontitis: A first theoretical step towards understanding the nonlinear dynamics of periodontitis. Int J Bifurcation Chaos. 2013;23(3):1350056. 1‐17. [Google Scholar]
- 68. Papantonopoulos G, Takahashi K, Bountis T, Loos BG. Aggressive periodontitis defined by recursive partitioning analysis of immunologic factors. J Periodontol. 2013;84:974‐984. [DOI] [PubMed] [Google Scholar]
- 69. Papantonopoulos G, Takahashi K, Bountis T, Loos BG. Mathematical modeling suggests that periodontitis behaves as a non‐linear chaotic dynamical process. J Periodontol. 2013;84:e29‐e39. [DOI] [PubMed] [Google Scholar]
- 70. Goodson JM, Haffajee AD, Socransky SS. The relationship between attachment level loss and alveolar bone loss. J Clin Periodontol. 1984;11:348‐359. [DOI] [PubMed] [Google Scholar]
- 71. Graves DT, Oates T, Garlet GP. Review of osteoimmunology and the host response in endodontic and periodontal lesions. J Oral Microbiol. 2011;3 5304-5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jeffcoat MK, Reddy MS. Progression of probing attachment loss in adult periodontitis. J Periodontol. 1991;62:185‐189. [DOI] [PubMed] [Google Scholar]
- 73. Ebersole JL, Graves CL, Gonzalez OA, et al. Aging, inflammation, immunity and periodontal disease. Periodontol 2000. 2016;72:54‐75. [DOI] [PubMed] [Google Scholar]
- 74. Larsson L, Castilho RM, Giannobile WV. Epigenetics and its role in periodontal diseases: a state‐of‐the‐art review. J Periodontol. 2015;86:556‐568. [DOI] [PubMed] [Google Scholar]
- 75. Levine RS. Obesity, diabetes and periodontitis‐a triangular relationship? Br Dent J. 2013;215:35‐39. [DOI] [PubMed] [Google Scholar]
- 76. Sonnenschein SK, Meyle J. Local inflammatory reactions in patients with diabetes and periodontitis. Periodontol 2000. 2015;69:221‐254. [DOI] [PubMed] [Google Scholar]
- 77. Cugini C, Klepac‐Ceraj V, Rackaityte E, Riggs JE, Davey ME. Porphyromonas gingivalis: keeping the pathos out of the biont. J Oral Microbiol. 2013;5 1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hajishengallis G. The inflammophilic character of the periodontitis‐associated microbiota. Mol Oral Microbiol. 2014;29:248‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hasturk H, Kantarci A, Goguet‐Surmenian E, et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo . J Immunol. 2007;179:7021‐7029. [DOI] [PubMed] [Google Scholar]
- 80. Marsh PD. Microbial ecology of dental plaque and its significance in health and disease. Adv Dent Res. 1994;8:263‐271. [DOI] [PubMed] [Google Scholar]
- 81. Marsh PD. Are dental diseases examples of ecological catastrophes? Microbiology. 2003;149:279‐294. [DOI] [PubMed] [Google Scholar]
- 82. Marsh PD, Zaura E. Dental biofilm: ecological interactions in health and disease. J Clin Periodontol. 2017;44(Suppl 18):S12‐S22. [DOI] [PubMed] [Google Scholar]
- 83. Lee CT, Teles R, Kantarci A, et al. Resolvin E1 reverses experimental periodontitis and dysbiosis. J Immunol. 2016;197:2796‐2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Duran‐Pinedo AE, Chen T, Teles R, et al. Community‐wide transcriptome of the oral microbiome in subjects with and without periodontitis. ISME J. 2014;8:1659‐1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Van Dyke TE. Cellular and molecular susceptibility determinants for periodontitis. Periodontol 2000. 2007;45:10‐13. [DOI] [PubMed] [Google Scholar]
- 86. Van Dyke TE. Control of inflammation and periodontitis. Periodontol 2000. 2007;45:158‐166. [DOI] [PubMed] [Google Scholar]
- 87. Van Dyke TE. Inflammation and periodontal diseases: a reappraisal. J Periodontol. 2008;79:1501‐1502. [DOI] [PubMed] [Google Scholar]
- 88. Van Dyke TE. The management of inflammation in periodontal disease. J Periodontol. 2008;79:1601‐1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Van Dyke TE, Kornman KS. Inflammation and factors that may regulate inflammatory response. J Periodontol. 2008;79:1503‐1507. [DOI] [PubMed] [Google Scholar]
- 90. Chow J, Mazmanian SK. A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe. 2010;7:265‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pigneur B, Sokol H. Fecal microbiota transplantation in inflammatory bowel disease: the quest for the holy grail. Mucosal Immunol. 2016;9:1360‐1365. [DOI] [PubMed] [Google Scholar]
- 93. Baek KJ, Ji S, Kim YC, Choi Y. Association of the invasion ability of Porphyromonas gingivalis with the severity of periodontitis. Virulence. 2015;6:274‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ji S, Choi YS, Choi Y. Bacterial invasion and persistence: critical events in the pathogenesis of periodontitis? J Periodontal Res. 2015;50:570‐585. [DOI] [PubMed] [Google Scholar]
- 95. Van der Velden U. What exactly distinguishes aggressive from chronic periodontitis: is it mainly a difference in the degree of bacterial invasiveness? Periodontol 2000. 2017;75:24‐44. [DOI] [PubMed] [Google Scholar]
- 96. Agarwal S, Huang JP, Piesco NP, Suzuki JB, Riccelli AE, Johns LP. Altered neutrophil function in localized juvenile periodontitis: intrinsic or induced? J Periodontol. 1996;67(Suppl 3S):337‐344. [DOI] [PubMed] [Google Scholar]
- 97. Figueredo CM, Gustafsson A. Increased amounts of laminin in GCF from untreated patients with periodontitis. J Clin Periodontol. 2000;27:313‐318. [DOI] [PubMed] [Google Scholar]
- 98. Johnstone AM, Koh A, Goldberg MB, Glogauer M. A hyperactive neutrophil phenotype in patients with refractory periodontitis. J Periodontol. 2007;78:1788‐1794. [DOI] [PubMed] [Google Scholar]
- 99. Matthews JB, Wright HJ, Roberts A, Cooper PR, Chapple IL. Hyperactivity and reactivity of peripheral blood neutrophils in chronic periodontitis. Clin Exp Immunol. 2007;147:255‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Nicu EA, Loos BG. Polymorphonuclear neutrophils in periodontitis and their possible modulation as a therapeutic approach. Periodontol 2000. 2016;71:140‐163. [DOI] [PubMed] [Google Scholar]
- 101. Johannsen A, Susin C, Gustafsson A. Smoking and inflammation: evidence for a synergistic role in chronic disease. Periodontology. 2000;2014(64):111‐126. [DOI] [PubMed] [Google Scholar]
- 102. Kantarci A, Oyaizu K, Van Dyke TE. Neutrophil‐mediated tissue injury in periodontal disease pathogenesis: findings from localized aggressive periodontitis. J Periodontol. 2003;74:66‐75. [DOI] [PubMed] [Google Scholar]
- 103. Ryder MI. Comparison of neutrophil functions in aggressive and chronic periodontitis. Periodontol 2000. 2010;53:124‐137. [DOI] [PubMed] [Google Scholar]
- 104. Dias IH, Matthews JB, Chapple IL, Wright HJ, Dunston CR, Griffiths HR. Activation of the neutrophil respiratory burst by plasma from periodontitis patients is mediated by pro‐inflammatory cytokines. J Clin Periodontol. 2011;38:1‐7. [DOI] [PubMed] [Google Scholar]
- 105. Pelletier M, Maggi L, Micheletti A, et al. Evidence for a cross‐talk between human neutrophils and Th17 cells. Blood. 2010;115:335‐343. [DOI] [PubMed] [Google Scholar]
- 106. Nibali L, Parkar M, Brett P, Knight J, Tonetti MS, Griffiths GS. NADPH oxidase (CYBA) and FcgammaR polymorphisms as risk factors for aggressive periodontitis: a case‐control association study. J Clin Periodontol. 2006;33:529‐539. [DOI] [PubMed] [Google Scholar]
- 107. Berglundh T, Donati M, Zitzmann N. B cells in periodontitis: friends or enemies? Periodontol 2000. 2007;45:51–66. [DOI] [PubMed] [Google Scholar]
- 108. Fujihashi K, Kono Y, Beagley KW, Yamamoto M, McGhee JR, Mestecky J, Kiyono H. Cytokines and periodontal disease: immunopathological role of interleukins for B cell responses in chronic inflamed gingival tissues. J Periodontol. 1993;64(5 Suppl):400–406. [PubMed] [Google Scholar]
- 109. Shaddox L, Wiedey J, Bimstein E, et al. Hyper‐responsive phenotype in localized aggressive periodontitis. J Dent Res. 2010;89:143‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Shaddox LM, Goncalves PF, Vovk A, et al. LPS‐induced inflammatory response after therapy of aggressive periodontitis. J Dent Res. 2013;92:702‐708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Fokkema SJ, Loos BG, Slegte C, van der Velden U. A type 2 response in lipopolysaccharide (LPS)‐stimulated whole blood cell cultures from periodontitis patients. Clin Exp Immunol. 2002;127:374‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Fokkema SJ, Loos BG, van der Velden U. Monocyte‐derived RANTES is intrinsically elevated in periodontal disease while MCP‐1 levels are related to inflammation and are inversely correlated with IL‐12 levels. Clin Exp Immunol. 2003;131:477‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bajestan MN, Radvar M, Afshari JT, Naseh MR, Arab HR. Interleukin‐6 production by cultured peripheral blood monocytes before and after stimulation by E. coli lipopolysaccharide in Iranian patients with aggressive periodontitis. Med Sci Monit. 2006;12:CR393‐CR396. [PubMed] [Google Scholar]
- 114. Radvar M, Tavakkol‐Afshari J, Bajestan MN, Naseh MR, Arab HR. The effect of periodontal treatment on IL‐6 production of peripheral blood monocytes in aggressive periodontitis and chronic periodontitis patients. Iran J Immunol. 2008;5:100‐106. [DOI] [PubMed] [Google Scholar]
- 115. Tapashetti RP, Sharma S, Patil SR, Guvva S. Potential effect of neutrophil functional disorders on pathogenesis of aggressive periodontitis. J Contemp Dent Pract. 2013;14:387‐393. [DOI] [PubMed] [Google Scholar]
- 116. Loos BG, Roos MT, Schellekens PT, van der Velden U, Miedema F. Lymphocyte numbers and function in relation to periodontitis and smoking. J Periodontol. 2004;75:557‐564. [DOI] [PubMed] [Google Scholar]
- 117. Moutsopoulos NM, Konkel J, Sarmadi M, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL‐17‐driven inflammatory bone loss. Sci Transl Med. 2014;6:229ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti‐inflammatory and pro‐resolution lipid mediators. Nat Rev Immunol. 2008;8:349‐361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Balta MG, Loos BG, Nicu EA. Emerging concepts in the resolution of periodontal inflammation: a role for Resolvin E1. Front Immunol. 2017;8:1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Recchiuti A, Serhan CN. Pro‐resolving lipid mediators (SPMs) and their actions in regulating mirna in novel resolution circuits in inflammation. Front Immunol. 2012;3:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Fredman G, Oh SF, Ayilavarapu S, Hasturk H, Serhan CN, Van Dyke TE. Impaired phagocytosis in localized aggressive periodontitis: rescue by Resolvin E1. PLoS ONE. 2011;6:e24422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Van Dyke TE, Hasturk H, Kantarci A, et al. Proresolving nanomedicines activate bone regeneration in periodontitis. J Dent Res. 2015;94:148‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. El‐Sharkawy H, Aboelsaad N, Eliwa M, et al. Adjunctive treatment of chronic periodontitis with daily dietary supplementation with omega‐3 Fatty acids and low‐dose aspirin. J Periodontol. 2010;81:1635‐1643. [DOI] [PubMed] [Google Scholar]
- 124. Heaton B, Dietrich T. Causal theory and the etiology of periodontal diseases. Periodontol. 2000;2012(58):26‐36. [DOI] [PubMed] [Google Scholar]
- 125. Lopez R, Hujoel P, Belibasakis GN. On putative periodontal pathogens: an epidemiological perspective. Virulence. 2015;6:249‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Rothman K, Greenland S. Causation and causal inference in epidemiology. Am J Public Health. 2005;95(Suppl. 1):S144‐S150. [DOI] [PubMed] [Google Scholar]
- 127. Reynolds MA. Modifiable risk factors in periodontitis: at the intersection of aging and disease. Periodontol 2000. 2014;64:7‐19. [DOI] [PubMed] [Google Scholar]
- 128. Laine ML, Jepsen S, Loos BG. Progress in the identification of genetic factors in periodontitis. Curr Oral Health Rep. 2014;1:272‐278. [Google Scholar]
- 129. Laine ML, Crielaard W, Loos BG. Genetic susceptibility to periodontitis. Periodontology. 2000;2012(58):37‐68. [DOI] [PubMed] [Google Scholar]
- 130. Michalowicz BS, Diehl SR, Gunsolley JC, et al. Evidence of a substantial genetic basis for risk of adult periodontitis. J Periodontol. 2000;71:1699‐1707. [DOI] [PubMed] [Google Scholar]
- 131. Teumer A, Holtfreter B, Volker U, et al. Genome‐wide association study of chronic periodontitis in a general German population. J Clin Periodontol. 2013;40:977‐985. [DOI] [PubMed] [Google Scholar]
- 132. Schäfer AS, Van der Velden U, Laine ML, Loos BG. Genetic susceptibility to periodontal disease: new insights and challenges In: Lindhe J, Lang NP, eds. Clinical Periodontology and Implant Dentistry. Chichester, UK: Wiley Blackwell; 2015:290‐310. [Google Scholar]
- 133. Schäfer A, Jepsen S, Loos BG. Periodontal genetics: a decade of genetic association studies mandates better study designs. J Clin Periodontol. 2011;38:103‐107. [DOI] [PubMed] [Google Scholar]
- 134. Goncalves PF, Harris TH, Elmariah T, Aukhil I, Wallace MR, Shaddox LM. Genetic polymorphisms and periodontal disease in populations of African descent: a review. J Periodontal Res. 2018;53:164‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Munz M, Richter GM, Loos BG et al. Meta‐analysis of genome wide association studies. Eur J Hum Genet. 2019;27:102-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Offenbacher S, Divaris K, Barros SP, et al. Genome‐wide association study of biologically informed periodontal complex traits offers novel insights into the genetic basis of periodontal disease. Hum Mol Genet. 2016;25:2113‐2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Divaris K, Monda KL, North KE, et al. Exploring the genetic basis of chronic periodontitis: a genome‐wide association study. Hum Mol Genet. 2013;22:2312‐2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Munz M, Willenborg C, Richter GM, et al. A genome‐wide association study identifies nucleotide variants at SIGLEC5 and DEFA1A3 as risk loci for periodontitis. Hum Mol Genet. 2017;26:2577‐2588. [DOI] [PubMed] [Google Scholar]
- 139. Schaefer AS, Richter GM, Dommisch H, et al. CDKN2BAS is associated with periodontitis in different European populations and is activated by bacterial infection. J Med Genet. 2011;48:38‐47. [DOI] [PubMed] [Google Scholar]
- 140. Nibali L, Di Iorio A, Tu YK, Vieira AR. Host genetics role in the pathogenesis of periodontal disease and caries. J Clin Periodontol. 2017;44(Suppl 18):S52‐S78. [DOI] [PubMed] [Google Scholar]
- 141. Vieira AR, Albandar JM. Role of genetic factors in the pathogenesis of aggressive periodontitis. Periodontol 2000. 2014;65:92‐106. [DOI] [PubMed] [Google Scholar]
- 142. Aarabi G, Zeller T, Seedorf H, et al. Genetic susceptibility contributing to periodontal and cardiovascular disease. J Dent Res. 2017;96:610‐617. [DOI] [PubMed] [Google Scholar]
- 143. Munz M, Richter GM, Loos BG, et al. Genome‐wide association meta‐analysis of coronary artery disease and periodontitis reveals a novel shared risk locus. Sci Rep. 2018;8:13678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Schaefer AS, Bochenek G, Jochens A, et al. Genetic evidence for plasminogen as a shared genetic risk factor of coronary artery disease and periodontitis. Circ Cardiovasc Genet. 2015;8:159‐167. [DOI] [PubMed] [Google Scholar]
- 145. Schaefer AS, Richter GM, Groessner‐Schreiber B, et al. Identification of a shared genetic susceptibility locus for coronary heart disease and periodontitis. PLoS Genet. 2009;5:e1000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Beck JD, Eke P, Heiss G, et al. Periodontal disease and coronary heart disease: a reappraisal of the exposure. Circulation. 2005;112:19‐24. [DOI] [PubMed] [Google Scholar]
- 147. Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol. 2009;27:165‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685‐1695. [DOI] [PubMed] [Google Scholar]
- 149. Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868‐874. [DOI] [PubMed] [Google Scholar]
- 150. Roivainen M, Viik‐Kajander M, Palosuo T, et al. Infections, inflammation, and the risk of coronary heart disease. Circulation. 2000;101:252‐257. [DOI] [PubMed] [Google Scholar]
- 151. Dave S, Van Dyke T. The link between periodontal disease and cardiovascular disease is probably inflammation. Oral Dis. 2008;14:95‐101. [DOI] [PubMed] [Google Scholar]
- 152. Ernst FD, Uhr K, Teumer A, et al. Replication of the association of chromosomal region 9p21.3 with generalized aggressive periodontitis (gAgP) using an independent case‐control cohort. BMC Med Genet. 2010;11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Hubberten M, Bochenek G, Chen H, et al. Linear isoforms of the long noncoding RNA CDKN2B‐AS1 regulate the c‐myc‐enhancer binding factor RBMS1. Eur J Hum Genet. 2019;27:80‐89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Teeuw WJ, Laine ML, Bizzarro S, Loos BG. A lead ANRIL polymorphism is associated with elevated CRP levels in periodontitis: a pilot case‐control study. PLoS ONE. 2015;10:e0137335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Bochenek G, Hasler R, El Mokhtari NE, et al. The large non‐coding RNA ANRIL, which is associated with atherosclerosis, periodontitis and several forms of cancer, regulates ADIPOR1, VAMP3 and C11ORF10. Hum Mol Genet. 2013;22:4516‐4527. [DOI] [PubMed] [Google Scholar]
- 156. Divaris K, Monda KL, North KE, et al. Genome‐wide association study of periodontal pathogen colonization. J Dent Res. 2012;91:21S‐28S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Dietrich T, Webb I, Stenhouse L, et al. Evidence summary: the relationship between oral and cardiovascular disease. Br Dent J. 2017;222:381‐385. [DOI] [PubMed] [Google Scholar]
- 158. Friedewald VE, Kornman KS, Beck JD, et al. The American Journal of Cardiology and Journal of Periodontology Editors’ Consensus: periodontitis and atherosclerotic cardiovascular disease. Am J Cardiol. 2009;104:59‐68. [DOI] [PubMed] [Google Scholar]
- 159. Kholy KE, Genco RJ, Van Dyke TE. Oral infections and cardiovascular disease. Trends Endocrinol Metab. 2015;26:315‐321. [DOI] [PubMed] [Google Scholar]
- 160. Lockhart PB, Bolger AF, Papapanou PN, et al. Periodontal disease and atherosclerotic vascular disease: does the evidence support an independent association?: a scientific statement from the American Heart Association. Circulation. 2012;125:2520‐2544. [DOI] [PubMed] [Google Scholar]
- 161. Schenkein HA, Loos BG. Inflammatory mechanisms linking periodontal diseases to cardiovascular diseases. J Clin Periodontol. 2013;40(Suppl 14):S51‐S69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Barros SP, Williams R, Offenbacher S, Morelli T. Gingival crevicular fluid as a source of biomarkers for periodontitis. Periodontol 2000. 2016;70:53‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. de Vries TJ, Andreotta S, Loos BG, Nicu EA. Genes critical for developing periodontitis: lessons from mouse models. Front Immunol. 2017;8:1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Larsson E, Tremaroli V, Lee YS, et al. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut. 2012;61:1124‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol. 2011;9:279‐290. [DOI] [PubMed] [Google Scholar]
- 166. Cavalla F, Biguetti CC, Melchiades JL, et al. Genetic association with subgingival bacterial colonization in chronic periodontitis. Genes. 2018;9:E271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Kaur G, Grover V, Bhaskar N, Kaur RK, Jain A. Periodontal infectogenomics. Inflamm Regen. 2018;38:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Nibali L, Di Iorio A, Onabolu O, Lin GH. Periodontal infectogenomics: systematic review of associations between host genetic variants and subgingival microbial detection. J Clin Periodontol. 2016;43:889‐900. [DOI] [PubMed] [Google Scholar]
- 169. Nibali L, Donos N, Henderson B. Periodontal infectogenomics. J Med Microbiol. 2009;58:1269‐1274. [DOI] [PubMed] [Google Scholar]
- 170. Barros SP, Hefni E, Nepomuceno R, Offenbacher S, North K. Targeting epigenetic mechanisms in periodontal diseases. Periodontol 2000. 2018;78:174‐184. [DOI] [PubMed] [Google Scholar]
- 171. Barros SP, Offenbacher S. Modifiable risk factors in periodontal disease: epigenetic regulation of gene expression in the inflammatory response. Periodontol 2000. 2014;64:95‐110. [DOI] [PubMed] [Google Scholar]
- 172. Larsson L. Current concepts of epigenetics and its role in periodontitis. Curr Oral Health Rep. 2017;4:286‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Lindroth A, Park Y. Epigenetic biomarkers: a step forward for understanding periodontitis. J Periodontal Implant Sci. 2013;43:111‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Shaddox LM, Mullersman AF, Huang H, Wallet SM, Langaee T, Aukhil I. Epigenetic regulation of inflammation in localized aggressive periodontitis. Clin Epigenetics. 2017;9:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Cho YD, Kim PJ, Kim HG, et al. Transcriptomics and methylomics in chronic periodontitis with tobacco use: a pilot study. Clin Epigenetics. 2017;9:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Diomede F, Thangavelu SR, Merciaro I, et al. Porphyromonas gingivalis lipopolysaccharide stimulation in human periodontal ligament stem cells: role of epigenetic modifications to the inflammation. Eur J Histochem. 2017;61:2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Zhang S, Barros SP, Moretti AJ, et al. Epigenetic regulation of TNFA expression in periodontal disease. J Periodontol. 2013;84:1606‐1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Zhang S, Crivello A, Offenbacher S, Moretti A, Paquette DW, Barros SP. Interferon‐gamma promoter hypomethylation and increased expression in chronic periodontitis. J Clin Periodontol. 2010;37:953‐961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Schulz S, Immel UD, Just L, Schaller HG, Glaser C, Reichert S. Epigenetic characteristics in inflammatory candidate genes in aggressive periodontitis. Hum Immunol. 2016;77:71‐75. [DOI] [PubMed] [Google Scholar]