


Abstract
Gene set enrichment analysis has become one of the most frequently used applications in molecular biology research. Originally developed for gene sets, the same statistical principles are now available for all omics types. In 2016, we published the miRNA enrichment analysis and annotation tool (miEAA) for human precursor and mature miRNAs. Here, we present miEAA 2.0, supporting miRNA input from ten frequently investigated organisms. To facilitate inclusion of miEAA in workflow systems, we implemented an Application Programming Interface (API). Users can perform miRNA set enrichment analysis using either the web-interface, a dedicated Python package, or custom remote clients. Moreover, the number of category sets was raised by an order of magnitude. We implemented novel categories like annotation confidence level or localisation in biological compartments. In combination with the miRBase miRNA-version and miRNA-to-precursor converters, miEAA supports research settings where older releases of miRBase are in use. The web server also offers novel comprehensive visualizations such as heatmaps and running sum curves with background distributions. We demonstrate the new features with case studies for human kidney cancer, a biomarker study on Parkinson’s disease from the PPMI cohort, and a mouse model for breast cancer. The tool is freely accessible at: https://www.ccb.uni-saarland.de/mieaa2.
INTRODUCTION
Transcriptomics designates an indispensable set of techniques to study gene expression, often in a genome-wide manner, as the backbone of modern molecular biology and clinical research. The innumerable amount of classical bulk-sequencing datasets is further augmented by the recent advancements in high-resolution single-cell approaches. Since gene expression is constituted by many biological factors, experimental focus has been enlarged to include the regulatory non-coding transcriptome (ncRNAs), i.e. to RNA classes that regulate messenger RNAs (mRNAs) either directly or indirectly. Among these, microRNAs (miRNAs) are small non-coding RNAs, typically 18-25 nucleotides in length, loaded into proteins of the AGO-family to build RNA-induced silencing complexes (RISC) (1). Gene regulation through the RISC complex is facilitated by one or two mature (−5p; −3p) miRNA arms, arising from one or several transcribed precursors (2). Besides other modes of action, activated complexes target preferentially 3′-untranslated regions of mRNAs to induce either catalytic cleavage or translation repression. Hence, profiling miRNA expression contributes to the understanding of gene regulation and potentially portrays cellular states. To date, numerous studies highlight their informative role in disease detection, sub-type classification, or progression, such as for cancer (3), neurodegenerative (4), or metabolic disorders (5) with a variety of bio-specimens (6).
Considering that several thousands of miRNAs have already been discovered, many novel miRNA candidates have been additionally proposed (7), while the total number of human miRNAs is estimated to be 2300 (8). Finding differences in expression for miRNAs is similar to mRNAs and therefore non-trivial. Differential gene expression studies often lead to dozens, hundreds, or even thousands of de-regulated genes. Thus, large scale studies often make use of the functionality of gene set enrichment analysis (GSEA) (9). GSEA can further reduce large amounts of information towards a significant set of molecular functions, biological properties, or pathways of genes. In principle, a user inputs either a set or ordered list of genes and the tool runs the required statistical algorithms and provides background datasets to compare against.
Similar functionality was also implemented for other omics types, including proteomics, metagenomics or epigenomics. An in-depth review of gene set analysis methods for data other than mRNAs demonstrates the increasing interest and demand of the community in respective tools (10). We previously developed a statistical approach tailored for both miRNA precursor and mature miRNA input, the miRNA enrichment analysis and annotation tool (miEAA) (11). Here, we present an update of this tool that includes more categories, supports nine additional species, has new statistical functionality and offers a standardised Application Programming Interface (API) to facilitate the inclusion in modern data analysis workflows (12).
Given the growing interest in miRNAs, other tools with similar functionality to miEAA exist. The pioneering tool providing functionality for miRNA enrichment was TAM (13), which covers in it’s latest version 2.0 (14) as many as 1238 human miRNA categories obtained from manual literature review of ∼9000 scientific manuscripts, along with new query and visualization features. In addition to the over- and under-representation analysis, users can compare the correlation of two miRNA lists under different disease conditions. Another important tool with similar functionality is miSEA (microRNA Set Enrichment Analysis) (15). It facilitates the selection of a large set of microRNA categories, including family classification, disease association, and genome coordinate. Furthermore, custom miRNA sets can be defined by the user. All kinds of enrichment tools rely on high quality sets of miRNA categories that were either obtained by curation of scientific literature or collected from specific databases. For instance, curated miRNA annotations can be obtained from miRBase (16) or miRCarta (17), miRNA–target interactions from miRTarBase (18), miRNA–pathway associations from miRPathDB (19), tissue-specific miRNAs from the human TissueAtlas (20), or miRNA-disease associations from HMDD (21) or MNDR (22), many of which were updated in the last two years. Further specialized annotations like miRNA and transcription factor interactions from TransmiR (23), miRNA sub-cellular localisations collected in RNALocate (24), or extra-cellular circulating miRNAs contained in miRandola (25) provide target categories for comprehensive enrichment analysis.
MATERIALS AND METHODS
In miEAA 2.0, we provide support for ten species whereas the first release of miEAA only supported Homo sapiens, 31 new category sets, and updates to our pre-existing datasets. To unify data preprocessing, we implemented an automated pipeline using Snakemake (26), Python 3.6, and the pandas (27) Python package facilitating data collection and filtering steps. For each species and their corresponding data sources our pipeline performs the same basic process, consisting of downloading the datasets, cleaning and updating the miRNA and precursor identifiers, transforming the results into a Gene Matrix Transposed (GMT) file, and creating background reference sets. Files were copied to the web server without further modification.
Data collection
Novel datasets were obtained to build our enrichment categories, consisting of Gene Ontology (28), miRTarBase 8.0 (18), KEGG (29), miRandola 2017 (25), miRPathDB 2.0 (19), TissueAtlas (20), MNDR v2.0 (22), NPInter 4.0 (30), RNALocate v2.0 (24), SM2miR (31), TAM 2.0 (14) and TransmiR v2.0 (23). Further annotations for cell-type and tissue specific expression of miRNAs and precursors were derived from three dedicated atlas publications (32,33) (10.1101/430561). Other pre-existing datasets have been updated, including HMDD v3.0 (21) and miRBase v22.1 (16). We retained the rest of our pre-existing datasets, namely miRWalk2.0 (34), published age and gender dependent miRNAs and distribution of miRNAs in immune cells (11). Most of the datasets contain miRNAs or precursors for H. sapiens. When available, we also utilise the data to derive categories representing the non-human organisms. Raw datasets were obtained either through a direct download or via an API. In particular, the QuickGO and KEGG datasets are compiled by querying corresponding REST APIs.
Category data preprocessing
First, data from QuickGO was mapped back to miRBase using RNAcentral (35). NCBI Gene was used in conjunction with miRTarBase to produce the indirect annotations. With the aid of the miRBaseConverter R package (36), miRNA and precursor names were translated to the latest version of miRBase. For KEGG Pathways and GO Annotations (direct and indirect through target genes from miRTarBase) we only keep miRNAs for which functional MTI support is available. In the MNDR diseases category set, we exclude HMDD data as it is precursor based, and MNDR is for mature miRNAs. To determine tissue-specific expression we computed the tissue specificity index (20) and applied a threshold filtering at 0.75.
Web server, statistics, and API implementation
The miEAA web server was built using a dockerized Django Web Framework v2.1, which exposes a web-API using the Django REST framework. The celery software was used as the job scheduler. Frontend libraries comprise Highcharts, dataTables, jquery, and Bootstrap. P-value correction methods were implemented using the R stats package. As gene set enrichment analysis (GSEA) implementation we provide an un-weighted variant of the algorithm. This implies the amount by which the running sum is changed in each step is constant, corresponding to a Kolmogorow–Smirnow test. This approach enables to compute the exact P-value without requiring permutations of either the case / control labels, or the miRNA lists (37). As an exception, the static GSEA running sum plots are computed by randomly permuting the test set 100 times and traversing the running sum for each random permutation. If the absolute maximal deviation from zero is positive, miEAA assumes an enrichment on top of the ordered list and results are shown in red colour to denote an enrichment. If the absolute maximal deviation from zero is negative, miEAA assumes an enrichment at the end of the ordered list and results are displayed in green color to denote an inverse enrichment, i.e. a depletion. Alongside our new API we provide a lightweight Python package, as well as a command line interface (CLI) tool, supporting Python 3.5 or higher. These are made freely available through the Python Package Index (pip) and through the ccb-sb conda channel. The already existing miRNA to precursor and miRBase converters were upgraded to miRBase v22.1. The former offers new output modes to simplify the review of ambiguous conversion results and proper down-stream usage.
Case studies
Raw and reads per million miRNA mapping (rpmmm) normalized miRBase v21 precursor counts and metadata of kidney renal clear cell carcinoma case and control samples were obtained from The Cancer Genome Atlas (TCGA). Since multiple sequencing results might be associated with the same sample ID in TCGA, we kept only one result file for each sample by preferring files from H over R over T analytes and selecting the aliquot with the highest plate number and / or lexicographical sorting order. Subsequently, miRNAs with fewer than 5 raw reads in less than 50% of either case or control samples were discarded from the analysis. All remaining miRNA counts were log2-scaled. Effect size was calculated using the implementation of Cohen’s d from the R package effsize. Lists of precursor names, either selected by statistical significance or ordered by effect size, were converted from miRBase v21 to v22.1 using the online miRBase converter feature of miEAA. The list of all precursors from miRBase v21, converted to v22.1, were used as a reference set. The configured parameters included default precursor category sets without the PubMed ID and TransMiR Tissues sets, BH-FDR adjustment to a significance level of 0.05 with independently adjusted P-values per category set, and a minimum of 2 required hits per sub-category.
For the second case study, raw Agilent microarray data and sample metadata was downloaded from NCBI’s GEO using accession ID GSE117000. Array parsing and probe signal processing was performed identically to the description in the first publication of miEAA (11). Subsequently, all counts were quantile-normalized and log2-transformed. All further down-stream analyses were performed analogous to the first case-study described above.
To provide a non-cancer case study we evaluated the performance of miEAA on a high-resolution dataset of small non-coding RNAs in whole blood (38). This dataset is freely available from the Parkinson's Progression Markers Initiative (PPMI) data portal. In summary, for 1600 individuals up to five blood samples from a time frame of over three years were acquired and sequenced for sncRNAs. We quantified all human miRBase v22 precursors from the 4340 sequencing samples. Raw counts were normalized to reads per million (rpm) and precursors were filtered analogously to the criteria defined for the TCGA case study. Next, we compared the miRNA precursor profiles of 2337 Parkinson’s samples to 1538 age-matched controls. For this case study we also mapped back the precursors to miRBase v21 to perform a detailed comparison of enrichment results to TAM 2.0.
RESULTS
Overview on miEAA 2.0
In the following, changes and novelties introduced by the second major release of miEAA are described. Since all annotations of miRNAs to categories and databases are with respect to the miRNA reference database, miRBase, we converted the datasets to match its latest public version 22.1. This also affects the miRBase-version and miRNA-to-precursor converters, the former of which was designed to be fully backwards compatible. Moreover, both ORA and GSEA algorithms accept lists of either precursors or miRNAs, from H. sapiens, Mus musculus, Rattus norvegicus, Arabidopsis thaliana, Bos taurus, Caenorhabditis elegans, Drosophila melanogaster, Danio rerio, Gallus gallus and Sus scrofa. In total, 134 525 categories from 16 published databases/resources are available to test against. A detailed breakdown of the counts by source and organism, on database and category set level, are available from Supplementary Table S1 and S2, respectively. For the precursor annotations, we curated family assignments, re-computed genomic clusters of miRNA genes, updated the chromosomal locations for human, and added all similar categories for other species. We also updated the category set representing PubMed IDs of manuscripts that contributed miRNA entries to miRBase. This feature has both, a biological and technical aspect. From the technical view, miRNAs could have been reported by the original paper due to experimental bias. In case a new input query is enriched for respective miRNAs it could be due to the same kind of bias. From a biological perspective, a study might have found miRNAs in the context of a disease. If such a manuscript is identified in a similar context in miEAA, additional evidence for the validity can be inferred. All species except A. thaliana are annotated with a new category listing high confidence precursors according to miRBase criteria. For human data, we transferred the disease annotations from HMDD to the new major release v3. We added associations from MNDR to allow disease comparisons against HMDD, and incorporated functional RNA interactions from NPInter. Lastly, novel categories such as the cellular localisation of miRNAs and regulatory interactions between miRNAs and transcription factors were incorporated from RNALocate and TransmiR, respectively. For the mature miRNAs, comparable changes apply as for the precursors in the cases of miRBase, MNDR, NPInter, and RNALocate-derived category sets. The gap between annotations of miRNA properties and their function is filled by categories on target genes taken from miRTarBase. Moreover, known miRNA to drug assocations are provided from SM2miR. To facilitate target-based enrichment of molecular pathways or biological function, we computed enrichments on target genes of miRNAs using Gene Ontology and KEGG. As an alternative for end-users, pre-computed significant enrichments of miRNAs associated with pathways provided by miRPathDB were made available for analysis. As the data from miRPathDB already involves a statistical pre-filtering, we implemented a new list of expert categories to highlight the underlying differences. Manually curated classifications from miRandola about known circular or extracellular miRNAs are also integrated. Finally, new annotations for cell-type and tissue-specific precursors and miRNAs have been integrated. Supposedly, the substantially enlarged number of categories might increase the average runtime of our algorithms, especially for the computationally intensive GSEA. Therefore, we profiled and improved our GeneTrail-based implementation to be three times faster, on average (39).
We raised the available number of statistical parameter settings as well. First, users can request unadjusted or adjusted P-values using six published techniques to account for multiple hypothesis testing on the same dataset. In addition to the classical Bonferroni and Benjamini–Hochberg False discovery rate (BH-FDR) procedures, the adjustments proposed by Benjamini-Yekuteli, Hochberg, Holm and Hommel can be selected. Moreover, the default behavior of miEAA to correct P-values database / category set-wise was extended by a P-value pooling approach. In summary, the well-established alternatives for P-value correction can support highly customized research setups where alternate levels of stringency are required (40).
We also evaluated new visualization features for the output of enrichment analyses to provide a simple overview and to improve comprehension. As a result, we made existing graphs interactive and implemented enrichment graphs with simulated background distributions for GSEA as well as automatic word cloud and heatmap plots for all enrichment algorithms. Word clouds display the names of obtained categories while scaling the size of the terms relatively to the number of hits that occurred (on a linear or logarithmic scale) and allow one to qualitatively compare the categories. On top of that, category to miRNA heatmaps depict log-transformed P-values for the hits obtained. This feature permits to compare the similarity of enriched / depleted categories with respect to associated miRNAs or precursors in a simple fashion. The workflow of miEAA and example visualizations are displayed in Figure 1. Finally, we enhanced the general accessibility of miEAA through the implementation of a public API and a Python package, for which more details are provided below.
Figure 1.

miEAA workflow and exemplary results. (A) Each miRNA/precursor enrichment analysis consists of at most five steps. First, users should select whether they want to perform enrichment on precursors or miRNAs. Second, the enrichment algorithm, i.e. either ORA or GSEA must be selected. Next, the desired test set can be defined either through a textbox or a file upload. The fourth step only appears for ORAs where custom background reference sets can be inserted or uploaded. This is optional since miEAA provides pre-computed reference sets for all categories. Lastly, the set of categories and databases as well as statistical parameters should be selected. (B) Typical result view for an ORA. Users can sort, select, filter, and export the obtained enrichment results interactively. Moreover, several visualizations of the results are provided for each run, such as the precursor/miRNA to category heatmap and the category word cloud.
Case study 1: Human kidney renal clear cell carcinoma
As the first case-study of miEAA 2.0, we acquired 591 human miRNA-seq samples from the kidney renal clear cell carcinoma (KIRC) project of TCGA, which can be divided into 520 Primary tumor (PT) and 71 Solid tissue normal (STN) samples. Sample information can be found in Supplementary Table S3. Of the 1881 precursors from miRBase v21, 321 are consistently detected in at least 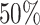 of the samples for each biogroup. Among these, 282 were differentially expressed between PT and STN according to the FDR-adjusted wilcoxon test P-values (P < 0.01). Over-representation analysis of the precursors resulted in 541 significantly enriched and seven significantly depleted (FDR-adjusted; P < 0.05) categories. As shown in Figure 2A, a subset of precursors is ubiquitously present in significant categories, while others seem to be more specific. The top 10 categories sorted by increasing P-value are associated with cancer, including renal cell carcinoma. Also, the observed over expected ratio (123/48.6) indicates a strong enrichment (P = 2.80 × 10−38) of the de-regulated precursors with kidney and other types of cancer. A miRNA set enrichment analysis, using the list of detected precursors and sorted by effect size, revealed 253 enriched and 40 depleted categories. Here, the miRNA gene cluster 147, 189, 704 : 147, 284, 728 on the X chromosome is the most depleted category (P = 8.64 × 10−10), an observation that is in line with the depletion of precursor family hsa-mir-506. Interestingly, the list of highly enriched terms contains many transcription factors, the top 5 being HEY1, WDR5, ELF1, BRD4 and FLI1.
of the samples for each biogroup. Among these, 282 were differentially expressed between PT and STN according to the FDR-adjusted wilcoxon test P-values (P < 0.01). Over-representation analysis of the precursors resulted in 541 significantly enriched and seven significantly depleted (FDR-adjusted; P < 0.05) categories. As shown in Figure 2A, a subset of precursors is ubiquitously present in significant categories, while others seem to be more specific. The top 10 categories sorted by increasing P-value are associated with cancer, including renal cell carcinoma. Also, the observed over expected ratio (123/48.6) indicates a strong enrichment (P = 2.80 × 10−38) of the de-regulated precursors with kidney and other types of cancer. A miRNA set enrichment analysis, using the list of detected precursors and sorted by effect size, revealed 253 enriched and 40 depleted categories. Here, the miRNA gene cluster 147, 189, 704 : 147, 284, 728 on the X chromosome is the most depleted category (P = 8.64 × 10−10), an observation that is in line with the depletion of precursor family hsa-mir-506. Interestingly, the list of highly enriched terms contains many transcription factors, the top 5 being HEY1, WDR5, ELF1, BRD4 and FLI1.
Figure 2.

Web server visualisation of case study results. (A) Category (x-axis) to precursor (y-axis) heatmap with −log10-scaled enrichment P-values for the first case study. (B) GSEA plot with simulated background distributions (green to orange lines) and actual depletion for breast cancer (dark blue line) observed during evaluation of the second case study.
Case study 2: mouse model for breast cancer progression
To showcase the novel support for model organisms in miEAA, we selected a dataset from GEO where circulating miRNAs from a breast-cancer mouse model were measured with microarrays (41). The dataset comprises 36 samples from mutation-carrier (NeuT+) and age-matched wildtype (NeuT–) mice that were collected at the premalignant, preinvasive and invasive stages of the disease. In this particular study, agilent microarrays probed with miRNAs from miRBase v19 were used on mice’s plasma extracted RNA samples. Sample information can be found in Supplementary Table S4. Following a detection threshold procedure similar to our first case study, 212 miRNAs remained for differential expression analysis. Of these, mmu-miR-6243 had to be discarded as a result of mapping the identifiers from miRBase v19 to v22.1, which we performed with the miEAA miRBase version converter. Subsequently, we applied GSEA on the list of miRNAs sorted by decreasing effect size between the premalignant and the invasive stage, for NeuT+ and NeuT- samples separately. Strikingly, the former run returned 311 significant categories, while the latter returned none. Overall, many more categories seemed to be depleted (N = 301) than enriched (N = 9), suggesting a wide-spread up-regulation of molecular pathways as miRNAs get down-regulated in NeuT+. For example, we found Macrophage differentiation (P = 2.54 × 10−5), Vasculature development (P = 1.60 × 10−4), and VEGF signaling pathway (P = 0.0016) to be depleted, which might be a signal for the increased tumor burden of NeuT+ mice at the invasive breast cancer stage. Moreover, we evaluated GSEAs for the comparison of NeuT+ and NeuT- at all three stages. While the first two setups returned a rather unspecific set of categories with all P-values located close to the significance boundary, the last comparison yielded many interesting results. First, observations were in line with the group-wise comparison along the age dimension, because all categories are depleted, i.e. no enrichments at the top of the sorted list. Further, the results show that several dozen conserved miRNAs (P = 4.53 × 10−5) are down-regulated in the NeuT+ model at the invasive stage. More significant categories we found such as exosome (P = 2.31 × 10−5) and circulating (P = 0.0086) miRNAs, breast cancer (P = 0.0094, Figure 2(b)), microRNAs in cancer (P = 0.028), and PI3K-Akt signaling pathway (P = 0.028) can be associated with the research setup of this exemplifying study.
Case study 3: Parkinson’s Biomarkers from PPMI and comparison to TAM 2.0
At last we aimed to test a non-cancer disease (Parkinson’s), to present a direct comparison between TAM 2 and miEAA 2.0. We compared the raw P-values of the tools to exclude an influence of the size of available categories. A direct comparison highlighted 72 hits by both tools (additional 70 reported only by TAM and 144 only by miEAA). Very similar but not exactly matching category names (e.g. Alzheimer’s versus Alzheimers or Carcinoma, Lung, Non-Small-Cell versus Carcinoma, Lung. Non-Small-Cell) had to be matched manually. After matching those, several ambiguously defined categories remained, e.g. Human Immunodeficiency Virus Infection in miEAA and Acquired Immunodeficiency Syndrome in TAM and that had to be mapped. As a result, the overlap increased to 94 hits. Asking whether the overlap between the output of the two tools is larger for the categories with higher significance than expected, we performed a DynaVenn analysis of the result sets ordered by increasing P-value (42). Selecting the 32 most significant miEAA sets and the 30 most significant TAM sets we observed an overlap of 23 categories (P = 10−8), indeed suggesting better comparable results for the most significant categories. Also, when comparing the miRNA hits for the obtained categories we observed very similar results. Alzheimer’s Disease was covered by 10 miRNAs in miEAA and nine in TAM with P-values of 3.31 × 10−4 and 2.19 × 10−3, respectively. We also observed the function category of TAM to be advantageous in this case, revealing direct hits such as Aging, which remained partially hidden in miEAA. On the other hand, miEAA seems to have slight advantages in the disease-associated categories, reporting 176 entries compared to 106 in TAM. This extended list contains among others Parkinson’s Disease which was covered by three miRNAs in TAM and missing the alpha level while being covered by six miRNAs in miEAA and thus being significant (P = 0.019). The full list of results obtained from both tools in direct comparison is shown in Supplementary Table S5. Besides the case study benchmark, we performed a detailed feature comparison with respect to 22 criteria between our tool and TAM that is shown in Supplementary Table S6.
New data export and browsable API
All data, results, and interactive plots shown on the web server are exportable to common data formats. To support the trend towards the development of reproducible and automated data analysis pipelines (12), miEAA hosts a public, browsable API offering the same functionality as the web site, allowing one to access the miRNA converters and statistical algorithms remotely. This functionality is further augmented by a full-featured Python package with API library code and a command-line interface (CLI). For example, a regular workflow as performed on the website can be accomplished with three sequential calls to the web API or one call to the CLI. We provide code examples in the common data science programming languages Python and R to demonstrate this use-case. We also implemented the interface to solve two recurring problems in biological data analysis. First, reproducibility of statistical experiments can be improved, because usage of the versioned API in the context of a workflow manager such as Snakemake (26) or Nextflow fosters self-documenting research setups (43). Second, oftentimes the analysis of miRNA high-throughput data involves the comparison of multiple biogroups, timepoints or other annotation variables. By using our API and the package, multiple runs of miEAA can be performed at ease while minimising the time spent for set up and results aggregation.
DISCUSSION
Statistical tools for biological enrichment analysis are a key to understanding data from high-throughput omics assays. However, the performance primarily depends on the quality of the underlying annotations and the statistical soundness. We show that new developments in the miRNA research field yielded an unprecedented set of biological categories, covering most aspects of miRNA properties and function, with cross-species analysis becoming increasingly important. On the other side, as with every statistical framework applied on biological data, assumptions are not always met and findings should be assessed critically in the light of further validation experiments. The novel release of miEAA attempts to cover these aspects by enhancing the set of available categories both quantitatively and qualitatively as well as through offering more (stringent) approaches for P-value correction. Also, a major limitation of some datasets concerns the availability of mature miRNA identifiers, as only precursor names were available for some of the sources. However, especially in the context of diseases, mature miRNA resolution is preferable to match the biological selectivity for one major miRNA arm being expressed. Datasets incorporated in miEAA were compiled either automatically or manually. The competitor tool TAM uses a fewer number of high-quality annotations. In particular, an advantage of TAM arises from the manual curation of datasets (14). The case study on Parkinson’s disease highlighted the results of miEAA 2.0 and TAM 2.0 to be similar whereas individual advantages in usability, functionality, or scope in the one or the other tool remain.
We have demonstrated the capability of miEAA to yield novel biological results in cancer research. For the kidney renal clear cell carcinoma case study, we found a depletion of the mir-506 precursor family, which has been observed before in other types of cancers (44,45). Many interactions to transcription factors were also found for the up-regulated miRNAs, suggesting an increased regulatory burden due to the exceeding transcriptional up-regulation observed in cancer. For example, HEY1, which is a transcriptional repressor has been characterised to be up-regulated in renal cell carcinomas (46). For the mouse breast cancer progression study, we illustrated the backwards compatibility of miEAA with respect to miRBase. The overall observed depletion of pathway regulating miRNAs in mice agrees with our first case study. Moreover, the significant categories like vasculature development that are associated with morphogenesis, resemble an increased tumor burden of NeuT+ mice, which was previously confirmed with a large human RNA-seq dataset on breast cancer (47). In both case studies, we observed many associations with other types of cancers or diseases. While this may speak for a molecular and biological similarity, a certain publication bias, e.g. for cancer, is a confounding factor that skews the statistics (14).
Establishing a standardized nomenclature is an on-going challenge in miRNA research. Results of the implemented manual converters are more accurate as compared to automated mappings since the naming schemes changed along the different releases. miEAA supports an exact mapping of old (e.g. miR*) to new nomenclature which would be ambiguous using automatic conversion (e.g. hsa-miR-499a-3p could be converted to hsa-miR-499a-3p or hsa-miR-499b-3p). Similar ambiguity issues would arise by performing a case insensitive miRNA to precursor mapping (‘miR’ to ‘mir’), in case multiple precursors with the same miRNA exist (for example hsa-let-7a-5p is annotated in three precursors). Finally, we sought to improve accessibility of miEAA and developed a web-API in combination with a Python package. These features enhance its usability in other applications for miRNA research, for example to annotate functional sub-graphs in regulatory network analysis (48). In conclusion, miEAA 2.0 is a flexible, comprehensive, and highly accessible tool for high-throughput miRNA annotation and enrichment analysis.
DATA AVAILABILITY
miEAA 2.0 is freely available at https://www.ccb.uni-saarland.de/mieaa2. No login is required. Example code for API-usage and the pre-compiled Python package are freely available from https://github.com/Xethic/miEAA-API.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the authors of the utilized GEO dataset for providing their microarray samples to the general public. The results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. We would like to express gratitude towards all specimen donors and research groups involved in the sample acquisition.
Contributor Information
Fabian Kern, Chair for Clinical Bioinformatics, Saarland University, 66123 Saarbrücken, Germany.
Tobias Fehlmann, Chair for Clinical Bioinformatics, Saarland University, 66123 Saarbrücken, Germany.
Jeffrey Solomon, Chair for Clinical Bioinformatics, Saarland University, 66123 Saarbrücken, Germany.
Louisa Schwed, Chair for Clinical Bioinformatics, Saarland University, 66123 Saarbrücken, Germany.
Nadja Grammes, Chair for Clinical Bioinformatics, Saarland University, 66123 Saarbrücken, Germany.
Christina Backes, Chair for Clinical Bioinformatics, Saarland University, 66123 Saarbrücken, Germany.
Kendall Van Keuren-Jensen, Neurogenomics Division, Translational Genomics Research Institute, Phoenix, AZ 85004, USA.
David Wesley Craig, Institute of Translational Genomics, University of Southern California, Los Angeles, CA 90033, USA.
Eckart Meese, Department of Human Genetics, Saarland University, 66421 Homburg, Germany.
Andreas Keller, Chair for Clinical Bioinformatics, Saarland University, 66123 Saarbrücken, Germany; School of Medicine Office, Stanford University, Stanford, CA 94305, USA; Department of Neurology and Neurological Sciences, Stanford University, Stanford, CA 94304, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Michael J. Fox foundation [14446]. Internal funds of Saarland University. Funding for open access charge: Michael J. Fox Foundation for Parkinson's Research.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bartel D.P. Metazoan MicroRNAs. Cell. 2018; 173:20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kern F., Backes C., Hirsch P., Fehlmann T., Hart M., Meese E., Keller A.. What’s the target: understanding two decades of in silico microRNA-target prediction. Brief. Bioinform. 2019; doi:10.1093/bib/bbz111. [DOI] [PubMed] [Google Scholar]
- 3. Cantini L., Bertoli G., Cava C., Dubois T., Zinovyev A., Caselle M., Castiglioni I., Barillot E., Martignetti L.. Identification of microRNA clusters cooperatively acting on epithelial to mesenchymal transition in triple negative breast cancer. Nucleic Acids Res. 2019; 47:2205–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ludwig N., Fehlmann T., Kern F., Gogol M., Maetzler W., Deutscher S., Gurlit S., Schulte C., von Thaler A.K., Deuschle C. et al.. Machine learning to detect Alzheimer’s disease from circulating Non-coding RNAs. Genomics Proteomics Bioinformatics. 2019; 17:430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thomou T., Mori M.A., Dreyfuss J.M., Konishi M., Sakaguchi M., Wolfrum C., Rao T.N., Winnay J.N., Garcia-Martin R., Grinspoon S.K. et al.. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature. 2017; 542:450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Backes C., Meese E., Keller A.. Specific miRNA disease biomarkers in blood, serum and Plasma: Challenges and prospects. Mol. Diagn. Ther. 2016; 20:509–518. [DOI] [PubMed] [Google Scholar]
- 7. Fehlmann T., Backes C., Alles J., Fischer U., Hart M., Kern F., Langseth H., Rounge T., Umu S.U., Kahraman M. et al.. A high-resolution map of the human small non-coding transcriptome. Bioinformatics. 2018; 34:1621–1628. [DOI] [PubMed] [Google Scholar]
- 8. Alles J., Fehlmann T., Fischer U., Backes C., Galata V., Minet M., Hart M., Abu-Halima M., Grässer F.A., Lenhof H.P. et al.. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019; 47:3353–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al.. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mora A. Gene set analysis methods for the functional interpretation of non-mRNA data—genomic range and ncRNA data. Brief. Bioinform. 2019; doi:10.1093/bib/bbz090. [DOI] [PubMed] [Google Scholar]
- 11. Backes C., Khaleeq Q.T., Meese E., Keller A.. MiEAA: MicroRNA enrichment analysis and annotation. Nucleic Acids Res. 2016; 44:W110–W116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perkel J.M. Workflow systems turn raw data into scientific knowledge. Nature. 2019; 573:149–150. [DOI] [PubMed] [Google Scholar]
- 13. Lu M., Shi B., Wang J., Cao Q., Cui Q.. TAM: A method for enrichment and depletion analysis of a microRNA category in a list of microRNAs. BMC Bioinformatics. 2010; 11:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li J., Han X., Wan Y., Zhang S., Zhao Y., Fan R., Cui Q., Zhou Y.. TAM 2.0: Tool for MicroRNA set analysis. Nucleic Acids Res. 2018; 46:W180–W185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Çorapçıoğlu M., Oğul H.. miSEA: microRNA set enrichment analysis. Biosystems. 2015; 134:37–42. [DOI] [PubMed] [Google Scholar]
- 16. Kozomara A., Birgaoanu M., Griffiths-Jones S.. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2018; 47:D155–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Backes C., Fehlmann T., Kern F., Kehl T., Lenhof H.P., Meese E., Keller A.. MiRCarta: A central repository for collecting miRNA candidates. Nucleic Acids Res. 2018; 46:D160–D167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang H.Y., Lin Y.C.D., Li J., Huang K.Y., Shrestha S., Hong H.C., Tang Y., Chen Y.G., Jin C.N., Yu Y. et al.. miRTarBase 2020: updates to the experimentally validated microRNA–target interaction database. Nucleic Acids Res. 2019; 48:D148–D154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kehl T., Kern F., Backes C., Fehlmann T., Stöckel D., Meese E., Lenhof H.P., Keller A.. miRPathDB 2.0: a novel release of the miRNA Pathway Dictionary Database. Nucleic Acids Res. 2020; 48:D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ludwig N., Leidinger P., Becker K., Backes C., Fehlmann T., Pallasch C., Rheinheimer S., Meder B., Stähler C., Meese E. et al.. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016; 44:3865–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang Z., Shi J., Gao Y., Cui C., Zhang S., Li J., Zhou Y., Cui Q.. HMDD v3.0: a database for experimentally supported human microRNA–disease associations. Nucleic Acids Res. 2018; 47:D1013–D1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cui T., Zhang L., Huang Y., Yi Y., Tan P., Zhao Y., Hu Y., Xu L., Li E., Wang D.. MNDR v2.0: an updated resource of ncRNA–disease associations in mammals. Nucleic Acids Res. 2017; 46:D371–D374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tong Z., Cui Q., Wang J., Zhou Y.. TransmiR v2.0: an updated transcription factor-microRNA regulation database. Nucleic Acids Res. 2018; 47:D253–D258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang T., Tan P., Wang L., Jin N., Li Y., Zhang L., Yang H., Hu Z., Zhang L., Hu C. et al.. RNALocate: a resource for RNA subcellular localizations. Nucleic Acids Res. 2016; 45:D135–D138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Russo F., Di Bella S., Vannini F., Berti G., Scoyni F., Cook H.V., Santos A., Nigita G., Bonnici V., Laganà A. et al.. miRandola 2017: a curated knowledge base of non-invasive biomarkers. Nucleic Acids Res. 2017; 46:D354–D359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Köster J., Rahmann S.. Snakemake-a scalable bioinformatics workflow engine. Bioinformatics. 2012; 28:2520–2522. [DOI] [PubMed] [Google Scholar]
- 27. Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J. et al.. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods. 2020; 17:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. The Gene Ontology Consortium The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2018; 47:D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanehisa M., Sato Y., Furumichi M., Morishima K., Tanabe M.. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2018; 47:D590–D595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Teng X., Chen X., Xue H., Tang Y., Zhang P., Kang Q., Hao Y., Chen R., Zhao Y., He S.. NPInter v4.0: an integrated database of ncRNA interactions. Nucleic Acids Res. 2019; 48:D160–D165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu X., Wang S., Meng F., Wang J., Zhang Y., Dai E., Yu X., Li X., Jiang W.. SM2miR: a database of the experimentally validated small molecules’ effects on microRNA expression. Bioinformatics. 2012; 29:409–411. [DOI] [PubMed] [Google Scholar]
- 32. de Rie D., Abugessaisa I., Alam T., Arner E., Arner P., Ashoor H., Åström G., Babina M., Bertin N., Burroughs A.M. et al.. An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat. Biotechnol. 2017; 35:872–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Minami K., Uehara T., Morikawa Y., Omura K., Kanki M., Horinouchi A., Ono A., Yamada H., Ohno Y., Urushidani T.. miRNA expression atlas in male rat. Scientific Data. 2014; 1:140005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dweep H., Gretz N.. MiRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods. 2015; 12:697. [DOI] [PubMed] [Google Scholar]
- 35. The RNAcentral Consortium RNAcentral: a hub of information for non-coding RNA sequences. Nucleic Acids Res. 2018; 47:D221–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu T., Su N., Liu L., Zhang J., Wang H., Zhang W., Gui J., Yu K., Li J., Le T.D.. miRBaseConverter: an R/Bioconductor package for converting and retrieving miRNA name, accession, sequence and family information in different versions of miRBase. BMC Bioinformatics. 2018; 19:514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Keller A., Backes C., Lenhof H.P.. Computation of significance scores of unweighted gene set enrichment analyses. BMC Bioinformatics. 2007; 8:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Marek K., Chowdhury S., Siderowf A., Lasch S., Coffey C.S., Caspell-Garcia C., Simuni T., Jennings D., Tanner C.M., Trojanowski J.Q. et al.. The Parkinson’s progression markers initiative (PPMI)– establishing a PD biomarker cohort. Ann. Clin. Transl. Neur. 2018; 5:1460–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stöckel D., Kehl T., Trampert P., Schneider L., Backes C., Ludwig N., Gerasch A., Kaufmann M., Gessler M., Graf N. et al.. Multi-omics enrichment analysis using the GeneTrail2 web service. Bioinformatics. 2016; 32:1502–1508. [DOI] [PubMed] [Google Scholar]
- 40. Korthauer K., Kimes P.K., Duvallet C., Reyes A., Subramanian A., Teng M., Shukla C., Alm E.J., Hicks S.C.. A practical guide to methods controlling false discoveries in computational biology. Genome. Biol. 2019; 20:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chiodoni C., Cancila V., Renzi T.A., Perrone M., Tomirotti A.M., Sangaletti S., Botti L., Dugo M., Milani M., Bongiovanni L. et al.. Transcriptional profiles and stromal changes reveal bone marrow adaptation to early breast cancer in association with deregulated circulating microRNAs. Cancer Res. 2020; 80:484–498. [DOI] [PubMed] [Google Scholar]
- 42. Amand J., Fehlmann T., Backes C., Keller A.. DynaVenn: web-based computation of the most significant overlap between ordered sets. BMC Bioinformatics. 2019; 20:743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. DI Tommaso P., Chatzou M., Floden E.W., Barja P.P., Palumbo E., Notredame C.. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017; 35:316–319. [DOI] [PubMed] [Google Scholar]
- 44. Li J., Wu H., Li W., Yin L., Guo S., Xu X., Ouyang Y., Zhao Z., Liu S., Tian Y. et al.. Downregulated miR-506 expression facilitates pancreatic cancer progression and chemoresistance via SPHK1/Akt/NF-κB signaling. Oncogene. 2016; 35:5501–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang L., Zhou H., Wei G.. miR-506 regulates cell proliferation and apoptosis by affecting RhoA/ROCK signaling pathway in hepatocellular carcinoma cells. Int. J. Clin. Exp. Pathol. 2019; 12:1163–1173. [PMC free article] [PubMed] [Google Scholar]
- 46. Karim S., Al-Maghrabi J.A., Farsi H.M.A., Al-Sayyad A.J., Schulten H.J., Buhmeida A., Mirza Z., Al-boogmi A.A., Ashgan F.T., Shabaad M.M. et al.. Cyclin D1 as a therapeutic target of renal cell carcinoma- a combined transcriptomics, tissue microarray and molecular docking study from the Kingdom of Saudi Arabia. BMC Cancer. 2016; 16:741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tapia-Carrillo D., Tovar H., Velazquez-Caldelas T.E., Hernandez-Lemus E.. Master regulators of signaling Pathways: An application to the analysis of gene regulation in breast cancer. Front. Genet. 2019; 10:1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fan Y., Siklenka K., Arora S.K., Ribeiro P., Kimmins S., Xia J.. miRNet - dissecting miRNA-target interactions and functional associations through network-based visual analysis. Nucleic Acids Res. 2016; 44:W135–W141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
miEAA 2.0 is freely available at https://www.ccb.uni-saarland.de/mieaa2. No login is required. Example code for API-usage and the pre-compiled Python package are freely available from https://github.com/Xethic/miEAA-API.