


Abstract
To increase the number of value-added chemicals that can be produced by metabolic engineering and synthetic biology, constructing metabolic space with novel reactions/pathways is crucial. However, with the large number of reactions that existed in the metabolic space and complicated metabolisms within hosts, identifying novel pathways linking two molecules or heterologous pathways when engineering a host to produce a target molecule is an arduous task. Hence, we built a user-friendly web server, novoPathFinder, which has several features: (i) enumerate novel pathways between two specified molecules without considering hosts; (ii) construct heterologous pathways with known or putative reactions for producing target molecule within Escherichia coli or yeast without giving precursor; (iii) estimate novel pathways with considering several categories, including enzyme promiscuity, Synthetic Complex Score (SCScore) and LD50 of intermediates, overall stoichiometric conversions, pathway length, theoretical yields and thermodynamic feasibility. According to the results, novoPathFinder is more capable to recover experimentally validated pathways when comparing other rule-based web server tools. Besides, more efficient pathways with novel reactions could also be retrieved for further experimental exploration. novoPathFinder is available at http://design.rxnfinder.org/novopathfinder/.
INTRODUCTION
One of the main focuses in metabolic engineering and synthetic biology is to construct biosynthetic pathways for producing value-added compounds. A large amount of metabolites can be derived from nature, and it is estimated that there are at least 1 060 000 metabolites in all plants, without considering microbes and fungi (1). Although many successful cases of bioproduction of value-added chemicals have been reported in recent decades (2–7), they only represent a small proportion, it is necessary to broaden the range of producible compounds by expanding the metabolic space. To address this point, computational tools could aid in the experimental process at the design stage of engineering cycle (8). The main limitations for expanding metabolic space are underestimation of side enzymatic activities and the incompleteness of reaction databases (9). An enormous amount of enzymes are present in nature; however, only a small proportion of them have been well characterized (10). Some enzymes are highly specific, whereas others are promiscuous. For example, it is estimated that 37% of Escherichia coli K12 enzymes have promiscuous activity when the substrates are similar to their main known substrates in structure (11). The promiscuity of enzyme paves the way toward the rational construction of pathways with putative reactions derived from chemical transformations. To explore enzyme versatility, many high-quality enzyme and biological reaction databases, such as KEGG (12), Rhea (13), RxnFinder (14) and BRENDA (10) have been established to provide extensive information. Based on enzyme and reaction databases, chemical transformations can be extracted that represent the same structural changes at the reaction center when one or more reactions occur. Retrosynthesis algorithms are utilized to enumerate a series of orderly chemical transformations linking target molecules to simpler chemical building blocks (15–17).
The novel pathway construction tools can be classified into two categories according to whether or not they are chassis-related. Tools in the first category mainly focus on the construction of novel pathways between two specified compounds without considering hosts. For instance, based on characteristic RDM patterns in the KEGG database, PathPred, a web-based server, can predict plausible pathways of multistep reactions starting from a query compound (18). BNICE utilized the Enzyme Commission classification to formulate the enzyme reaction rule, which can predict pathways with novel reactions (19). Based on subgraph mining, ReactionMiner, a java-based package, can predict a series of biochemical transformations linking two molecules (20). Similarly, Masaaki developed a recursive supervised approach to link two molecules by using a reaction-filling framework (21). Tools in the second category can retrieve pathways for producing target molecules within a specified host. For example, rePrime&novoStoic is an optimization-based novel-pathway construction framework that integrates existing reactions and chemical transformations (22). By integrating constraint-based reconstruction and analysis (COBRA) (23) with GEM-model (24), GEM-path identified 245 novel pathways for producing 20 large volume compounds when engineering E. coli within four steps (25). RetroPath2.0, an open source workflow, could perform a retrosynthesis search from chassis to target by using chemical transformations and then rank pathways based on enzyme promiscuity (9). All of the aforementioned tools in the two categories can help to predict novel pathways by using chemical transformations. However, many of them do not provide a user-friendly web server, making it infeasible for users fully utilize the results, especially experimental researchers who are not experienced in programming. For example, the results from GEM-path consist of 245 pathways for producing 20 targets (25), and ReactionMiner (20) is a java-based package that can be downloaded from GitHub. Second, chemical transformations utilized in some tools only contain reactant-product pairs. For example, when processing multisubstrate reactions, a practical solution in RetroPath2.0 is to model enzymatic promiscuity for only one substrate at a time (9), and PathPred utilizes main RDM pairs (18). Third, Comprehensive evaluation methods to estimate pathways is also essential due to hundreds of novel pathways derived from chemical transformations. For example, enzyme promiscuity plays an essential role in novel pathway construction, most of the aforementioned tools do not quantify enzyme promiscuity by utilizing annotated sequences of enzymes, except RetroPath2.0 (9). We have summarized these tools in Table 1.
Table 1.
Retrosynthesis tools for the construction of novel pathways
| Web Server | Specified chassis | Specified precursor | OCSa | Theoretical yield | Multiple-steps | EPPSb | Thermodynamic feasibility | |
|---|---|---|---|---|---|---|---|---|
| PathPred (18) | ✓ | – | ✓ | – | – | ✓ | – | – |
| BNICE(ATLAS) (30) | ✓ | – | ✓ | – | – | ✓ | – | ✓ |
| ReactionMiner (20) | – | – | ✓ | – | – | ✓ | – | – |
| Masaaki (21) | – | – | ✓ | – | – | 4(at most) | – | – |
| rePrime&novoStoic (22) | – | ✓ | ✓ | ✓ | ✓ | ✓ | – | ✓ |
| GEM-path (25) | – | ✓ | – | – | ✓ | 4(at most) | – | ✓ |
| RetroPath2.0 (9) | – | ✓ | – | – | – | ✓ | ✓ | – |
| novoPathFinder | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
aOCS: overall stoichiometric conversions.
bEPPS: enzyme promiscuity penalty score.
To solve the issues mentioned above, in this article, we developed a user-friendly web server, novoPathFinder, to predict novel pathways for metabolic engineering. Compared with other novel pathway design tools, the proposed web server has several features: (i) it not only allows for the design of novel pathways for target production from specified precursors but can also identify heterologous novel pathways when engineering E. coli or yeast without providing precursors; (ii) due to the integration of chemical transformations and genome-scale metabolic models, novoPathFinder supports the calculation of overall stoichiometric conversions (26) and growth-coupled theoretical yield in real-time under customized growth conditions of hosts; and (iii) novel pathways predicted by novoPathFinder can be evaluated based on several criteria, including thermodynamic feasibility (27), enzyme promiscuity penalty score (9), Synthetic Complex Score (SCScore) (28) and LD50 (29) of intermediates, pathway length, overall stoichiometric conversions (26) and theoretical yield. According to the results, novoPathFinder is more capable to recover experimentally validated pathways when comparing other rule-based web server tools. Besides, more efficient pathways with novel reactions could also be retrieved for further experimental exploration. Thus, it is a convenient web server tool with full functionality to aid rational prediction of novel pathways.
MATERIALS AND METHODS
Workflow of novoPathFinder
Figure 1 shows the workflow of novoPathFinder. First, by using the reaction rule extraction method mentioned below, the reaction rule repository was constructed on the basis of valid reactions in Rhea. Second, expanded metabolic spaces containing known/putative reactions were built to link nodes in the known compound repository. Next, the retrosynthesis algorithm could be performed in real time to retrieve novel pathways for target molecule production, and the results could be evaluated with multicategories (e.g. thermodynamic feasibility analysis, theoretical yield, enzyme promiscuity and overall stoichiometric conversion), among which theoretical yield and thermodynamic feasibility analysis can be calculated in real time under customized physical conditions. Finally, Django, CSS, JavaScript and HTML were utilized to visualize novel pathways found in this platform, and all data tables utilized in novoPathFinder are stored in the PostgreSQL database.
Figure 1.

Workflow of novoPathFinder.
Data resources
novoPathFinder makes full use of several data resources. (i) Compounds: Due to the reaction-filling framework used in novoPathFinder, metabolites in KEGG COMPOUND (12) and ChEBI (31) are used as valid nodes when constructing expanded reaction spaces. (ii) Reactions: Without considering transport reactions and reactions that involved genetic compounds, there are 20 942 one-way reactions in the Rhea database (release: 105) (13), which served as referenced data for the extraction of chemical transformations. (iii) Enzyme: The relationship between enzyme classification and reaction entries is obtained from the Rhea database, and enzyme sequences are obtained from the reviewed part of UniProt (32). (iv) GEM-model: GEM-models for Escherichia coli K12 MG1655 (ID: iML1515) and Saccharomyces cerevisiae S288c (ID: iMM904) are obtained from BiGG databases (24).
Extraction of reaction rule
The same structure change in the reaction center at a defined bound-distance when one or more reactions occurred can be represented by a reaction rule (Figure 2). During this process, defining the reaction center is essential. In this study, we defined the reaction center as atoms whose circumjacent bounds changed when a reaction occurred. The structure change can be coded by several methods (33,34), and the SMIRKS pattern (https://www.daylight.com/dayhtml/doc/theory) was used in this study. The process of reaction rule abstraction can be divided into two parts: (i) Identification of Atom-Atom Mapping (AAM): AAM can label all atoms in substrates and then track their positions in products (35). Because AAM is an NP-hard problem (36), the more complex the reaction is, the more time it will take to calculate, especially for some reactions that involved cofactors (e.g. NADPH, NADP, ATP). To this point, our practical solution is to manually identify some typical stereotype transformations (Table 2) and remove them from the reactions. For example, as shown in Figure 2, by removing a typical stereotype transformation (n ATP →n ADP + n pi) from a reaction (A + nATP → B + C+ nADP + npi), the reaction can be converted to a simplified reaction (A → B + C) before identifying AAM. The putative reaction (A′ → B′ + C′) predicted by the reaction rule extracted from the simplified reaction will be completed later by adding cofactors. As a result, the final putative reaction is A′ + nATP → B′ + C′+ nADP + npi. (ii): Extraction of the reaction rule: Based on labeled atoms from AAM, we extracted reaction rules at a defined bound distance. Similar to previous research (37–39), to enable sensitivity and specificity, we set the bond distance to 1 (radius = 1), which means that we only consider the structure change at a distance of one bound from the reaction center.
Figure 2.

Generation of putative reaction from known reactions. (A) Workflow of generating putative reaction from a known reaction with typical stereotype transformation. (B) Workflow of generating putative reaction from a known reaction without typical stereotype transformation. (C) Extraction of reaction rules from known reactions.
Table 2.
A list of typical stereotype transformations
| Typical stereotype transformations |
|---|
| S-adenosyl-l-methionine< = > S-adenosyl-l-homocysteine |
| NADPH < = > NADP |
| NADH < = > NAD |
| ATP < = > AMP + ppi |
| ATP < = > ADP + pi |
| FMNH2 < = > FMN |
| FAD < = > FADH2 |
Expand reaction space
A repertory consisting of known metabolites (excluding currency compounds and compounds that involve R and *) from KEGG COMPOUND (12) and ChEBI (31) was first established. By using reaction rules, every compound in the repertory was regarded as a parent node to produce child nodes. One of the main challenges in expanding the reaction space is how to handle the thousands of these child nodes for the next iteration. To address this point, we first calculated the similarity between the substrate and the product from 15,937 RDM patterns (main pair) in the KEGG database. According to the results, we set the similarity threshold value to 0.1875, covering 15 155 main pairs (∼95% of 15 937), which means that if any one of the compounds in a product set (products from a rule-based reaction) is similar to the target with a similarity score <0.1875, the product set will be removed from the child nodes. Second, novoPathFinder utilized a reaction-filling framework to link nodes in the known metabolite repertory through known or putative reactions. For example, to shrink the scale of the child nodes and to construct chassis-related reaction spaces (e.g. E. coli), our practical solution is to reserve the rule-based reactions, of which at most one child node is E. coli nonnative, meaning that any other cosubstrate is derived from E. coli. Here, we use the term of cosubstrate because the pathway search algorithm is retrosynthesis. By using native compounds in yeast, we constructed an expanded reaction space for yeast in the same way. To construct a universal reaction space without considering chassis hosts, we counted the occurrence frequency for every metabolite in the BiGG database, which contains 84 genome-scale metabolic models (24). Next, we regarded metabolites with occurrence frequencies >42 times (84/2) as sink compounds, meaning that these compounds exist in most chassis hosts and could be regarded as cosubstrates just as the native compounds within E. coli or yeast. By employing these strategies, every compound in the metabolite repertory could be linked to its child nodes through rule-based reactions.
Retrosynthesis algorithm
Based on the different reaction spaces established before, a retrosynthesis algorithm is used to construct novel pathways for producing target molecules. Starting from a target molecule, reactions that involve the target molecule as the main product are obtained from the related reaction space. Reactants in the reaction set, excluding currency metabolites, serve as candidates to be randomly selected for the next iteration. One challenge in retrosynthesis algorithms is handling reactions with multiple substrates. To predict novel pathways without considering chassis hosts, an algorithm (40) (Equation 1) is utilized to calculate MCS (Maximum Common Substructure: the largest substructure that appears in both structures) score between every reactant in a single multi-reactant reaction and the target molecule, and then, we choose a reactant with the maximum MCS score to compose the candidates set for the next iteration. However, when considering a chassis host (E. coli or yeast) instead of choosing the reactant with the maximum MCS score, our practical solution is to choose the heterologous metabolite to compose the candidates set for the next iteration. The retrosynthesis algorithm proceeds until the customized iterations are reached, and each iteration stops when a predefined step or the precursor/sink metabolites are reached.
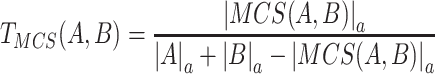 |
(1) |
Pathway evaluation criteria
To evaluate the predicted pathways, novoPathFinder utilizes several methods, including overall stoichiometric conversions, theoretical yield, thermodynamic feasibility analysis, enzyme promiscuity penalty score, synthetic complexity score and LD50 of intermediates. (i) The overall stoichiometric conversion (e.g. aA + cC < = > bB + dD) can abstract the global elemental balance sheet for the chemical changes including metabolites, ions and free energy (26). With predicted pathways, an automatic procedure is utilized in novoPathFinder to calculate the overall stoichiometric conversion. (ii) With the integration of the genome-scale metabolic model, the maximum theoretical yield can be calculated under customized growth conditions by using Equations (2–6), where the meanings of each parameter have been described in our previous studies (41,42). (iii) The thermodynamic feasibility of the reaction direction can be significantly affected by cellular conditions. By using eQuilibrator (27), the standard Gibbs energy for each reaction in predicted pathways can be calculated under customized physical conditions, including pH, ionic strength and temperature. For metabolite concentrations, we utilized 1 mM for all reactants. (iv) For known reactions, a default enzyme promiscuity penalty score of 0 was set in novoPathFinder, while for putative reactions, the enzyme promiscuity penalty score was calculated using an approach from RetroPath2.0 (9) on the basis of the enzyme sequence from UniProt (32). (v) Based on fragment contributions and the complexity penalty, the synthetic complexity score (SCScore) of intermediates in predicted pathways can be calculated by using a method from a previous study (28). The SCScore ranges between 0 and 5, for which lower scores reflect lower complexity of the compound. Besides, the LD50 of intermediates can also be calculated (29). The penalty score of the whole pathway can be calculated using Equation (7).
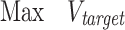 |
(2) |
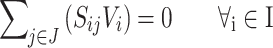 |
(3) |
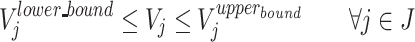 |
(4) |
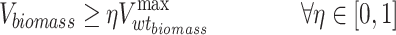 |
(5) |
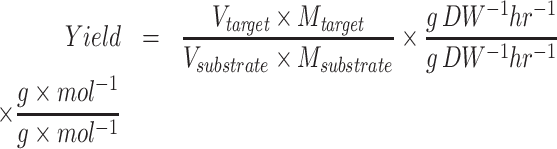 |
(6) |
Sets
I = (i | 1,…, N): set of metabolites.
J = (j | 1,…, M): set of reactions.
Vj: carbon flux of the jth reaction.
η: percentage that the minimum growth rate of the mutant type accounts for the maximum growth rate of the WT type.
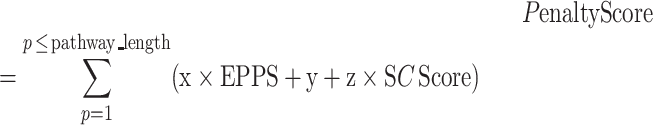 |
(7) |
 |
RESULT AND DISCUSSION
Construction of reaction rule repository and reaction spaces
By utilizing biological reactions from Rhea, we extracted chemical transformations to construct a reaction rule repository. As a result, there were 4996 reaction rules covering 20 942 one-way reactions from Rhea, in which transport reactions and reactions involving genetic or structure-unclear compounds were excluded. Based on reaction rules, we next predicted edges consisting of known or putative reactions to link nodes in the known compound repository, which contains 57 512 unique metabolites from KEGG and ChEBI. Next, we constructed reaction spaces aiming at different categories, including the E. coli-based/yeast-based module and no-hosts. As a result, each reaction space built in this research is >10 times larger than Rhea. More specifically, the reaction space of no-hosts contains 255 817 reactions, and the E. coli-based reaction space contains 255 322 reactions. The yeast-based reaction space contains 234 757 reactions.
Case study
We utilized novoPathFinder to retrieve novel pathways for producing various value-added metabolites by engineering hosts (E. coli K-12 MG 1655 or S. cerevisiae) and no-hosts specified but with a precursor. To demonstrate its efficiency, we summarized the results and then compared them with pathways obtained from the literature or other rule-based web server tools, such as PathPred (18) and ATLAS (30). To retrieve novel pathways by using other web server tools, we utilized their default parameters, except maximum pathway length. As a result, we discovered that novoPathFinder not only recovered the experimentally validated pathways that in many cases other web server tools could not recover but also identified more efficient pathways that contain putative steps.
Production of vanillin
Vanillin, a primary flavoring agent, is widely used in foods, beverages, and pharmaceuticals. Due to the scarcity of vanillin and the expense of its extraction, significant advancements have been made toward elucidating its biosynthetic pathways in the last two decades (43). In novoPathFinder, we first retrieved novel pathways for producing vanillin from tyrosine within five steps without considering hosts. We found that the experimentally validated pathway (five steps) could be recovered (Figure 3A) (44). In addition, novel pathways with fewer steps could also be identified in novoPathFinder (Figure 3B). The first pathway in Figure 3B contains two putative steps (50% of the whole pathway), while the shortest pathways retrieved by ATLAS contains three putative reactions (75% of whole pathway) and a known reaction, and no results could be retrieved by PathPred. Previous studies have claimed that novel pathways with less putative steps are more favored in metabolic engineering. Thus far, for the case of vanillin production, novoPathFinder outperformed ATLAS and PathPred. Second, we retrieved heterologous pathways for the production of vanillin within E. coli and yeast (Figure 3C, D). As a result, heterologous novel pathways with three steps and native precursors in E. coli could be automatically retrieved by using the E. coli-based module in novoPathFinder, and only two steps are needed when engineering yeast according to results from the yeast-based module. Next, the maximum theoretical yield of each pathway within the two hosts was calculated under the hosts’ specified growth conditions. As a result, in the case of E. coli, the heterologous novel pathway (pathway-2 in Figure 3C) achieved the highest theoretical yield (0.130 g/g glucose) comparing with the other two pathways. In the case of yeast, pathway-3 in Figure 3D shows that only two heterologous reactions are needed to produce vanillin; however, pathway-3 achieved less theoretical yield (0.044 g/g glucose) than pathway-2, which yielded 0.055 g/g glucose. Further experimental exploration should be performed to prove the in silico solutions.
Figure 3.

Search results for vanillin production in novoPathFinder. (A) Experimental pathway recovered by novoPathFinder. (B) Pathways for producing vanillin from tyrosine without considering hosts. (C, D): Heterologous pathways and theoretical yield under specified conditions for producing vanillin within E. coli and yeast.
Production of value-added metabolites from Farnesyl-PP
Farnesyl PP is an intermediate in the mevalonate pathway used in many organisms. Due to its economic viability and long-term sustainability, the biosynthesis of value-added metabolites from Farnesyl PP has gained much attention (45). Using Farnesyl PP as a precursor, we retrieved biosynthetic pathways for the production of two value-added metabolites, lycopene and artemisinic acid. As one of the major carotenoids, lycopene has received much attention due to its beneficial biological and pharmaceutical activities (46). Artesinin is highly effective against malaria, but it is in short supply. Artemisinic acid could be regarded as an immediate precursor for effectively producing artemisinin (47).
First, we utilized novoPathFinder to search pathways for lycopene production within four steps without considering hosts. The results are shown in Figure 4, in which the top-ranked (pathway-1) was the experimental pathway (46). We also retrieved pathways using PathPred and ATLAS. PathPred could not recover the experimental pathway, and only one pathway (three putative steps) was obtained within four steps; ATLAS could not identify pathways for lycopene production from Farnesyl PP within four steps. On the other hand, we applied novoPathFinder to search heterologous pathways for lycopene production when engineering E. coli and S. cerevisiae. As a result, in the case of E. coli, the experimental pathway (pathway-1-A in Figure 4), whose theoretical yield is 0.080 g/g glucose, and native precursor (farnesyl PP) could be automatically retrieved. In the yeast-based module, a known two-step heterologous pathway (pathway-1-B in Figure 4) with a theoretical yield of 0.071 g/g glucose and a native precursor (GGDP) was found.
Figure 4.

Three pathways found in novoPathFinder for lycopene production from farnesyl PP. Pathway-1-A: Heterologous pathway retrieved by using E. coli-based module. Pathway-1-B: Heterologous pathway retrieved by using yeast-based module.
Second, we searched novel pathways for producing artemisinic acid from Farnesyl PP in novoPathFinder within five steps without considering hosts. As a result, seven pathways were retrieved, and the top-ranked pathway was the experimentally validated pathway, which contains two known steps (47), in which Farnesyl PP was first converted to amorpha-4,11-diene and then converted to artemisinic acid (Figure 5). We also searched its pathways in PathPred and ATALS by setting the maximum steps to five; however, no pathway was found in either tool. Next, we searched heterologous pathways for artemisinic acid production in novoPathFinder when engineering chassis hosts, E. coli and S. cerevisiae. The precursor (Farnesyl PP) could be automatically found in both hosts, and the two hosts shared the same heterologous pathway (Figure 5). The theoretical yield was 0.091 g/g glucose in E. coli and 0.083 g/g glucose in yeast.
Figure 5.

Search results for artemisinic acid production in novoPathFinder.
Production of cannabidiol
Cannabidiol, an important substance of cannabinoids, has been approved as a prescription drug for a range of diseases (e.g. epilepsy, autoimmune disorders) (48). A recent study constructed a biosynthetic pathway for cannabidiol production (49). Using novoPathFinder, we first searched biosynthetic pathways for cannabidiol production from hexanoyl-CoA without considering hosts within five steps. Ten pathways were found, which contained not only experimentally validated pathways (Figure 6A) but also more efficient pathways with fewer steps (three steps or four steps), as shown in Figure 6B. More specifically, the three-step pathway in Figure 6 shows that 5-penthylresorcinol could be converted to cannabigerol, and then, cannabigerol could be converted to cannabidiol directly, both steps are putative. However, four steps are needed to achieve such conversion in the experimental pathway (Figure 6A). We also searched the heterologous pathways needed for cannabidiol production when engineering chassis hosts using E. coli-based and yeast-based modules in novoPathFinder. The top-ranked pathways in the two hosts and the maximum theoretical yield under specified growth conditions are shown in Figure 6. Next, we searched its pathways in PathPred and ATALS by setting the maximum pathway length to five; however, no pathways were found with either tool.
Figure 6.

Search results for cannabidiol production in novoPathFinder. (A) Experimental pathway recovered by novoPathFinder. (B) Two top-ranked pathways retrieved in novoPathFinder from hexanoyl-CoA. (C) Heterologous pathways retrieved by using E.coli-based and yeast-based modules.
DISCUSSION
A user-friendly web server named novoPathFinder was developed in this research with the following three objectives: (i) to enumerate novel pathways between two specified molecules without considering hosts; (ii) to construct heterologous pathways with known or putative reactions for producing target molecules within E. coli or yeast without giving precursors and (iii) to estimate novel pathways considering several categories, including enzyme promiscuity, SCScore and LD50 of intermediates, overall stoichiometric conversions, pathway length, theoretical yield and thermodynamic feasibility. Instead of extracting reaction rules based on Enzyme Commission nomenclature, a data-driven method based on 20 942 one-way reactions from Rhea was utilized in novoPathFinder to generate reaction rules. As a result, 4996 reaction rules were extracted to link 57 512 metabolites in KEGG and ChEBI. Meanwhile, by using chemical transformations, three expanded reaction spaces targeting different categories were constructed. Each reaction space built in this research is more than 10 times larger than the known reaction repository, Rhea database. Specifically, the no-host reaction space contains 255 817 reactions, the E. coli-based reaction space contains 255 322 reactions, and the yeast-based reaction space contains 234 757 reactions. By using the random-based retrosynthetic algorithm, novoPathFinder could not only recover experimental pathways but also identify more efficient pathways containing novel reactions for target production. By integrating the chassis-based reaction space with GEM-model, heterologous pathways containing novel reactions for target molecule production within E. coli or yeast without a predefined precursor could be identified, and then, growth-coupled theoretical yield could be calculated under customized growth conditions (e.g. main carbon source, oxygen condition and biomass) by using FBA. Every chemical transformation between two molecules in novel pathways is supported by one or more reactions, the corresponding reference reaction in Rhea and the promiscuity penalty score are provided, and the thermodynamic feasibility under customized physiological states (e.g. pH, ionic strength, temperature) can be calculated in real time.
A main piece of evidence demonstrating the efficiency of a novel pathway design tool is whether the tool can recover the experimental pathway. However, because the expanded reaction spaces contain tens or hundreds of thousands of novel reactions derived from chemical transformations, they are much larger than known biological reaction repositories. Thus, it is more difficult to recover the known pathways in comparison with other tools that focus on pathway construction with known reactions. In this research, we provided several example cases to elaborate that novoPathFinder is more capable of recovering experimentally validated pathways than other rule-based web server tools (e.g. ATLAS and PathPred). In addition, more efficient pathways with novel reactions could also be retrieved for further experimental exploration.
novoPathFinder presented in this research is a versatile online tool for metabolic engineering. It not only supports the exploration of novel pathways between two specified compounds without considering hosts but can also construct heterologous novel pathways when producing targets within E. coli or yeast. Moreover, the feasibility of each pathway can be evaluated from multiple aspects, including the penalty score of enzyme promiscuity, growth-coupled theoretical yield and thermodynamic feasibility, among which the latter two can be calculated in real-time under customized physical conditions. However, some limitations should also be considered. Due to the exponential growth of child nodes generated by reaction rules from a parent compound when using the retrosynthetic method, we utilized a reaction-filling framework for the sake of computation time. Taking novel compounds into consideration when constructing biosynthetic pathways would help build much larger reaction spaces to connect more compounds. In addition, the FBA algorithm was utilized in this research to calculate growth-coupled theoretical yield; however, due to the lack of kinetic parameters and regulatory parameters, the result from FBA may be inconsistent with in vivo experiments. The integration of kinetic information with GEM-models would improve this phenomenon. Even with the current limitations, novoPathFinder performs well in recovering experimental pathways and identifying more efficient pathways that should be experimentally explored. novoPathFinder is a convenient web server tool with full functionality that paves the way for more rational design strategies in metabolic engineering.
Contributor Information
Shaozhen Ding, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Shanghai Institutes for Biological Sciences, University of Chinese Academy of Sciences, Shanghai 200031, People's Republic of China.
Yu Tian, School of Biology and Pharmaceutical Engineering, Wuhan Polytechnic University, Wuhan, Hubei 430023, China.
Pengli Cai, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Shanghai Institutes for Biological Sciences, University of Chinese Academy of Sciences, Shanghai 200031, People's Republic of China; Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308, People's Republic of China.
Dachuan Zhang, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Shanghai Institutes for Biological Sciences, University of Chinese Academy of Sciences, Shanghai 200031, People's Republic of China.
Xingxiang Cheng, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Shanghai Institutes for Biological Sciences, University of Chinese Academy of Sciences, Shanghai 200031, People's Republic of China.
Dandan Sun, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Shanghai Institutes for Biological Sciences, University of Chinese Academy of Sciences, Shanghai 200031, People's Republic of China.
Le Yuan, Department of Biology and Biological Engineering, Chalmers University of Technology, Kemivägen 10, SE412 96 Gothenburg, Sweden.
Junni Chen, Wuhan LifeSynther Science and Technology Co. Limited, Wuhan 430070, People's Republic of China.
Weizhong Tu, Wuhan LifeSynther Science and Technology Co. Limited, Wuhan 430070, People's Republic of China.
Dong-Qing Wei, State Key Laboratory of Microbial Metabolism (Shanghai Jiao Tong University), Shanghai 200240, China.
Qian-Nan Hu, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Shanghai Institutes for Biological Sciences, University of Chinese Academy of Sciences, Shanghai 200031, People's Republic of China.
FUNDING
National Key Research and Development Program of China [2019YFA0904300, 2018YFA0900700, 2017YFC1601702]; National Natural Science Foundation of China [31700081, 31570092]; Scientific Research Conditions and Technical Support System Program [ZSYS-016]; CAS STS program [QYZDB-SSW-SMC012]; International Partnership Program of Chinese Academy of Sciences of China [153D31KYSB20170121]; Natural Science Foundation of Tianjin [15JCYBJC54300]. Funding for open access charge: National Key Research and Development Program of China [2019YFA0904300, 2018YFA0900700, 2017YFC1601702].
Conflict of interest statement. None declared
REFERENCES
- 1. Nakamura Y., Afendi F.M., Parvin A.K., Ono N., Tanaka K., Morita A.H., Sato T., Sugiura T., Altaf-Ul-Amin M., Kanaya S.. KNApSAcK metabolite activity database for retrieving the relationships between metabolites and biological activities. Plant Cell Physiol. 2014; 55:e7. [DOI] [PubMed] [Google Scholar]
- 2. Ajikumar P.K., Xiao W.-H., Tyo K.E.J., Stephanopoulos G.. Isoprenoid pathway optimization for taxol precursor overproduction in Escherichia coli. Science. 2010; 330:70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Atsumi S., Hanai T., Liao J.C.. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nat. Lett. 2008; 451:86–90. [DOI] [PubMed] [Google Scholar]
- 4. Hanai T., Atsumi S., Liao J.C.. Engineered synthetic pathway for isopropanol production in Escherichia coli. Appl. Environ. Microbiol. 2007; 73:7814–7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Menzella H.G., Reisinger S.J., Welch M., Kealey J.T., Kennedy J., Reid R., Tran C.Q., Santi D.V.. Redesign, synthesis and functional expression of the 6-deoxyerythronolide B polyketide synthase gene cluster. J. Ind. Microbiol. Biotechnol. 2006; 33:22–28. [DOI] [PubMed] [Google Scholar]
- 6. Steen E.J., Kang Y., Bokinsky G., Hu Z., Schirmer A., McClure A., B S., Cardayre D., Keasling J.D.. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nat. Lett. 2010; 463:559–563. [DOI] [PubMed] [Google Scholar]
- 7. Zhang W., Li Y., Tang1 Y.. Engineered biosynthesis of bacterial aromatic polyketides in Escherichia coli. Proc. Natl Acad. Sci. U.S.A. 2008; 105:20683–20688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carbonell P., Currin A., Jervis A.J., Rattray N.J.W., Swainston N., Yan C., Takano E., Breitling R.. Bioinformatics for the synthetic biology of natural products: integrating across the Design–Build– Test cycle. Nat. Prod. Rep. 2016; 33:925–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Delépinea B., Duigouc T., Carbonelld P., Faulon J.-L.. RetroPath2.0: A retrosynthesis workflow for metabolic engineers. Metab. Eng. 2018; 45:158–170. [DOI] [PubMed] [Google Scholar]
- 10. Jeske L., Placzek S., Schomburg I., Chang A., Schomburg D.. BRENDA in 2019: a European ELIXIR core data resource. Nucleic Acids Res. 2019; 47:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nam H., Lewis N.E., Lerman J.A., Lee D.-H., Chang R.L., Kim D., Palsson B.O.. Network context and selection in the evolution to enzyme specificity. Science. 2012; 337:1101–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K.. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017; 45:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morgat A., Lombardot T., Axelsen K.B., Aimo L., Niknejad A., Hyka-Nouspikel N., Coudert E., Pozzato M., Pagni M., Moretti S. et al.. Updates in Rhea – an expert curated resource of biochemical reactions. Nucleic Acids Res. 2017; 4:D415–D418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu Q.-N., Deng Z., Hu H., Cao D.-S., Liang Y.-Z.. RxnFinder: biochemical reaction search engines using molecular structures, molecular fragments and reaction similarity. Bioinformatics. 2011; 27:2465–2467. [DOI] [PubMed] [Google Scholar]
- 15. Lin G.-M., Warden-Rothman R., Voigt C.A.. Retrosynthetic design of metabolic pathways to chemicals not found in nature. Curr. Opin. Syst. Biol. 2019; 14:82–107. [Google Scholar]
- 16. Hadadi N., Hatzimanikatis V.. Design of computational retrobiosynthesis tools for the design of de novo synthetic pathways. Curr. Opin. Chem. Biol. 2015; 28:99–104. [DOI] [PubMed] [Google Scholar]
- 17. Carbonell P., Planson A.-G.l., Faulon J.-L.. Retrosynthetic design of heterologous pathways. Methods Mol. Biol. 2013; 985:149–173. [DOI] [PubMed] [Google Scholar]
- 18. Moriya Y., Shigemizu D., Hattori M., Kanehisa M.. PathPred: an enzyme-catalyzed metabolic pathway prediction server. Nucleic Acids Res. 2010; 38:138–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hatzimanikatis V., Li C., Ionita J.A., Henry C.S., Broadbelt L.J.. Exploring the diversity of complex metabolic networks. Bioinformatics. 2005; 21:1603–1609. [DOI] [PubMed] [Google Scholar]
- 20. Sankar A., Ranu S., Raman K.. Predicting novel metabolic pathways through subgraph mining. Bioinformatics. 2017; 15:3955–3963. [DOI] [PubMed] [Google Scholar]
- 21. Kotera M., Tabei Y., Yamanishi Y., Goto S.. Metabolome-scale prediction of intermediate compounds in multistep metabolic pathways with a recursive supervised approach. Bioinformatics. 2014; 30:165–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kumar A., Wang L., Ng C.Y., Maranas C.D.. Pathway design using de novo steps through uncharted biochemical spaces. Nat. Commun. 2018; 9:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schellenberger J., Que R., Fleming R.M.T., Thiele I., Orth J.D., Feist A.M., Zielinski D.C., Bordbar A., Lewis N.E., Rahmanian S. et al.. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2.0. Nat. Protoc. 2011; 6:1290–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. King Z.A., Lu J., Dräger A.D., Miller P., Federowicz S., Lerman J.A., Ebrahim A., Palsson B.O., Lewis N.E.. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2015; 44:515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Campodonico M.A., Andrews B.A., Asenjo J.A., Palsson B.O., Feist A.M.. Generation of an atlas for commodity chemical production in Escherichia coli and a novel pathway prediction algorithm, GEM-Path. Metab. Eng. 2014; 25:140–158. [DOI] [PubMed] [Google Scholar]
- 26. Chowdhury A., Maranas C.D.. Designing overall stoichiometric conversions and intervening metabolic reactions. Sci. Rep. 2015; 5:16009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Flamholz A., Noor E., Bar-Even A., Milo R.. eQuilibrator—the biochemical thermodynamics calculator. Nucleic Acids Res. 2011; 40:770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Caille S., Cui S., Faul M.M., Mennen S.M., Tedrow J.S., Walker S.D.. Molecular complexity as a driver for chemical process innovation in the pharmaceutical industry. J. Org. Chem. 2019; 84:4583–4603. [DOI] [PubMed] [Google Scholar]
- 29. Dong J., Wang N.N., Yao Z.J., Zhang L., Cheng Y., Ouyang D., Lu A.P., Cao D.S.. ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J. Cheminformatics. 2018; 10:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hadadi N., Hafner J., Shajkofci A., Zisaki A., Hatzimanikatis V.. ATLAS of biochemistry: a repository of all possible biochemical reactions for synthetic biology and metabolic engineering studies. ACS Syntth. Biol. 2016; 5:1155–1166. [DOI] [PubMed] [Google Scholar]
- 31. Hastings J., Owen G., Dekker A., Steinbeck C.. ChEBI in 2016: improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016; 44:D1214–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Consortium T.U. UniProt: a worldwide hub of protein knowledg. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oh M., Yamada T., Hattori M., Goto S., Kanehisa M.. Systematic analysis of enzyme-catalyzed reaction patterns and prediction of microbial biodegradation pathways. J.Chem.Inf. Model. 2007; 47:1702–1712. [DOI] [PubMed] [Google Scholar]
- 34. Dugundji J., Ugi I.. An algebraic model of constitutional chemistry as a basis for chemical computer programs. Comput. Chem. 1973; 39/1:19–64. [Google Scholar]
- 35. Willighagen E.L., Mayfeld J.W., Alvarsson J., Berg A., Carlsson L., Jeliazkova N., Kuhn S., Pluskal T., Rojas-Chertó M., Spjuth O. et al.. The Chemistry Development Kit (CDK) v2.0: atom typing, depiction, molecular formulas, and substructure searching. J. Cheminformatics. 2017; 9:33–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen W.L., Chen D.Z., Taylor K.T.. Automatic reaction mapping and reaction center detection. WIRES Comput. Sci. 2013; 3:560–593. [Google Scholar]
- 37. Sivakumar T.V., Giri V., Park J.H., Kim T.Y., Bhaduri A.. ReactPRED: a tool to predict and analyze biochemical reactions. Bioinformatics. 2016; 32:3522–3524. [DOI] [PubMed] [Google Scholar]
- 38. MHS S., M P., MP4 W.. Planning chemical syntheses with deep neural networks and symbolic AI. Nature. 2018; 555:604–610. [DOI] [PubMed] [Google Scholar]
- 39. Baylon J.L., Cilfone N.A., Gulcher J.R., Chittenden T.W.. Enhancing retrosynthetic reaction prediction with deep learning using multiscale reaction classification. J. Chem. Inf. Model. 2019; 59:673–688. [DOI] [PubMed] [Google Scholar]
- 40. Yuan L., Tian Y., Ding S., Liu Y., Chen F., Zhang T., Tu W., Chen J., Hu Q.-N.. PrecursorFinder: a customized biosynthetic precursor explorer. Bioinformatics. 2018; 35:1603–1604. [DOI] [PubMed] [Google Scholar]
- 41. Ding S., Cai P., Yuan L., Tian Y., Hu Q.-N.. CF-targeter: a rational biological cell factory targeting platform for biosynthetic target chemicals. ACS Syntth. Biol. 2019; 8:2280–2286. [DOI] [PubMed] [Google Scholar]
- 42. Ding S., Liao X., Tu W., Wu L., Tian Y., Sun Q., Chen J., Hu Q.-N.. EcoSynther: a customized platform to explore the biosynthetic potential in E. coli. ACS Chem. Biol. 2017; 12:2823–2829. [DOI] [PubMed] [Google Scholar]
- 43. Kundu A. Vanillin biosynthetic pathways in plants. Planta. 2017; 245:1068–1079. [DOI] [PubMed] [Google Scholar]
- 44. Ni J., Tao F., Du H., Xu P.. Mimicking a natural pathway for de novo biosynthesis: natural vanillin production from accessible carbon sources. Sci. Rep. 2015; 5:296–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gershenzon J., Dudareva N.. The function of terpene natural products in the natural world. Nat. Chem. Biol. 2007; 3:408–414. [DOI] [PubMed] [Google Scholar]
- 46. Alper H., Jin Y.-S., Moxley J.F., Stephanopoulos G.. Identifying gene targets for the metabolic engineering of lycopene biosynthesis in Escherichia coli. Metab. Eng. 2005; 7:155–164. [DOI] [PubMed] [Google Scholar]
- 47. Ro D.-K., Paradise E.M., Ouellet M., Fisher K.J., Newman K.L., Ndungu J.M., Ho K.A., Eachus R.A., Ham T.S., Kirby J. et al.. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006; 7086:940–943. [DOI] [PubMed] [Google Scholar]
- 48. Pisanti S., Malfitano A.M., Ciaglia E., Lamberti A., Ranieri R., Cuomo G., Abate M., Faggiana G., Proto M.C., Fiore D. et al.. Cannabidiol: State of the art and new challenges for therapeutic applications. Pharmacol. Ther. 2017; 175:133–150. [DOI] [PubMed] [Google Scholar]
- 49. Luo X., Reiter M.A., d’Espaux L., Wong J., Denby C.M., Lechner A., Zhang Y., Grzybowski A.T., Harth S., Lin W. et al.. Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature. 2019; 567:123–126. [DOI] [PubMed] [Google Scholar]