


Abstract
Microbial association networks are frequently used for understanding and comparing community dynamics from microbiome datasets. Inferring microbial correlations for such networks and obtaining meaningful biological insights, however, requires a lengthy data management workflow, choice of appropriate methods, statistical computations, followed by a different pipeline for suitably visualizing, reporting and comparing the associations. The complexity is further increased with the added dimension of multi-group ‘meta-data’ and ‘inter-omic’ functional profiles that are often associated with microbiome studies. This not only necessitates the need for categorical networks, but also integrated and bi-partite networks. Multiple options of network inference algorithms further add to the efforts required for performing correlation-based microbiome interaction studies. We present MetagenoNets, a web-based application, which accepts multi-environment microbial abundance as well as functional profiles, intelligently segregates ‘continuous and categorical’ meta-data and allows inference as well as visualization of categorical, integrated (inter-omic) and bi-partite networks. Modular structure of MetagenoNets ensures logical flow of analysis (inference, integration, exploration and comparison) in an intuitive and interactive personalized dashboard driven framework. Dynamic choice of filtration, normalization, data transformation and correlation algorithms ensures, that end-users get a one-stop solution for microbial network analysis. MetagenoNets is freely available at https://web.rniapps.net/metagenonets.
INTRODUCTION
Microbial ecosystem is inherently complex owing to the plurality of the microbes residing under the inter-play of various confounding factors or environmental conditions (1). Metagenomics, the study of genomic material acquired from environmental samples, which targets microbial DNA to decipher taxonomic and functional attributes of collected samples, has obtained a significant boost with the advent of next- generation sequencing technologies (2). Obtaining structural or compositional insights into various microbial assemblages has always remained one of the primary objectives of most metagenomic studies (2). However, another question that interests the microbiome researchers pertains to microbial community dynamics, i.e. how various microbes correlate or associate with each other in a metagenomic environment under study (3,4). Even though multiple strategies are available for mining microbe-microbe associations, e.g. evidence based relationship mining and function driven associations (4–6), a commonly used microbial interaction mining approach aims at probing correlations between the occurrence (abundance) profile of microbes detected in an environment (6). Such networks are therefore also termed as co-occurrence networks and are frequently employed in metagenomic research studies (7). However. given the complexity of microbial ecosystems and technical aspects associated with network/graph theory approaches, researchers often face multiple challenges in performing a meaningful network analysis (8). These challenges may be classified into three groups:
Lengthy workflow for microbial network analysis
A typical workflow for network inference generally involves (a) abundance data filtration to remove spurious or irrelevant features (4,9), (b) choosing from multiple data normalization and transformation strategies to account for inter-sample biases, confounding factors, compositionality etc. (10,11), (c) choosing among multitude of correlation inference methods to derive network files (correlation matrix, adjacency matrix, edge-lists etc.) (4), (d) employing graph theory algorithms to compute network characteristics (like global network properties, local centrality measures etc.) using the said network files (12,13) and (e) use a visualization tool to view the the networks (14).
Meta-data introduces additional complexity
Availability of comprehensive meta-data associated with metagenomic studies puts an additional layer of complexity to the problem of inferring and probing microbial association networks (15). For a given environment, there can be multiple levels of meta-data groups or categories (like geography as an environment can have countries as groups). This gives rise to a need for individually processing networks for each of such groups. In addition, quite often continuous meta-data (like BMI, age) are also collected, and researchers are therefore interested in probing correlations of microbial abundances with such continuous data points (or covariates) as well (15,16).
Inter-omic data further increases complexity
It is not uncommon for metagenomic studies to have one or more ‘associated’ inter-omic abundance profile (16,17). For example, a shotgun metagenomics study can not only provide the researchers with microbial abundance profile, but also the abundances of various functional units (like enzymes, GO, COG, genes etc.). Related inter-omic studies on same set of samples (like transcriptome profile) can also become a closely associated inter-omic data. The inferred functions for 16S studies are another example of inter-omic profile associated with microbial abundance datasets. Availability of such secondary datasets often lead to the requirement of mining correlation of microbes with such inter-omic units (like functions, genes etc.). The outcomes of such correlations are often visualized in the form of ‘inter-omic integrated networks’ and 'bi-partite networks’ (17,18). The process of achieving the same for each meta-data category (and corresponding group) is therefore expected to be complex and tedious.
For example, for a population of samples collected from various body sites of healthy individuals as well as those affected by a disorder, following questions pertaining to a typical microbial community dynamics study may stand relevant:
What are the microbial co-occurrence patterns specific to healthy and affected individuals?
What are the association patterns specific to individual body sites in healthy as well as affected individuals? How do these networks compare in terms of the interactions and various network properties?
Is there any correlation between the occurrence of certain microbes with age or BMI or weight or any other 'continuous' trait of the individuals in all/any category of network?
Given a metabolic profile of the given samples, is there any association between the occurrence of a microbe or a group of microbes with the profile of a metabolite or a group of metabolites of interest?
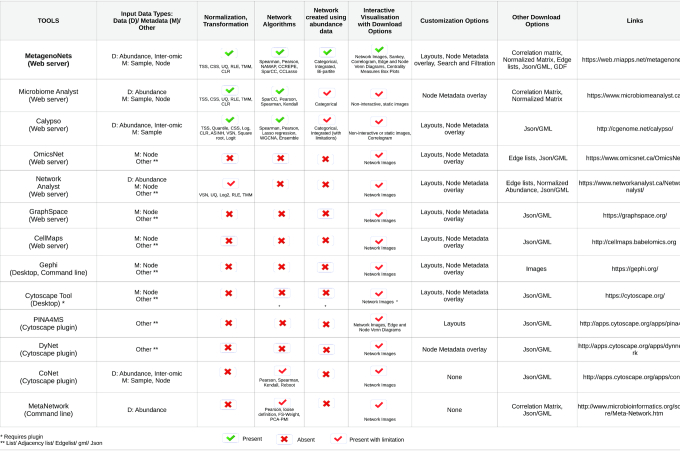
Many of these questions enthuse researchers and finding answers to them requires concerted efforts. In the current state of the art, a typical study on microbial correlation networks requires dependency on stand-alone generic softwares, plugins, locally installed programs as well as knowledge of advance programming (19–21). Limited number of available web-applications are either too specialized for other research areas or offer minimal functionalities (22–24). Furthermore, currently there is a lack of webservers which allow inter-omic correlation network analysis and meaningful visualization to address such questions. Table 1 provides a comprehensive comparison of the scope and features of various tools in the network biology space (including those specifically used for microbiome research) in the current state of the art.
Table 1.
Comparison of the scope and key features of various tools in the network biology space (including those specifically used for microbiome research) in the current state of the art. Links to access the tools have been provided in the last column of the table
|
We present MetagenoNets, a web-based modular framework, developed with the aim of easing the process of inferring and analyzing correlation driven microbial association networks. Following features of MetagenoNets are expected to be of significant value addition in the space of microbiome network analysis:
Accepts small to large microbial feature tables (occurrence or abundance profile) along with multi-level meta-data. Provision for secondary feature tables (like functional profile) allows deeper insights for an integrated analysis.
Offers frequently used data normalization strategies and transformation methods.
Provision for feature reduction through prevalence and occurrence-based filters.
Availability of correlation driven network inference methods frequently used by researchers.
Intelligent categorization of meta-data into categorical and continuous data types.
Provision for categorical, integrated and bi-partite network generation and visualization.
Interactive visualizations for all networks, network properties and correlation scores.
Compositional comparison of categorical networks through interactive Venn diagrams.
Registration independent personalized dashboard system for privacy, traceability, collaboration.
DATA FORMAT, INPUT PARAMETERS AND METHODS
Abundance data
MetagenoNets accepts two types of abundance datasets: (i) primary input data; (ii) secondary input data. Primary input data is essentially a (tab or comma) delimited multi-variate abundance table representing abundances of various operational taxonomic units (OTUs) obtained either from shotgun metagenomic studies or from various de-novo or reference based taxonomic classifiers which are used in 16S studies. BIOM files may also be submitted. It is mandatory to provide a primary input dataset to MetagenoNets. Secondary input data, also a multi-variate abundance table (or BIOM file), is an optional input data type. This table may contain inter-omic features (like pathway abundances, metabolite abundances etc.) for the samples provided in the primary input data.
Meta-data
MetagenoNets accepts two types of metadata, (i) primary metadata, (ii) node metadata. Primary metadata is a (tab or comma) delimited file containing multiple columns of sample information. Each column of metadata file, representing various classes of environments (e.g. disease state, geography etc.), contains the names of various sub-classes or the quantum of co-variate (e.g. BMI or age) corresponding to all samples in the population. Node metadata, which is optional, contains meta-information pertaining to the features present in the primary input data (e.g. phylum affiliation of different microbial genera). This metadata information is employed to dynamically customize the colors of the nodes of the network(s). In case of rich BIOM file types, metadata is automatically extracted from the uploaded input BIOM file.
Filtration parameters
MetagenoNets provides the end-users with provisions for filtering sparse features through prevalence and occurrence-based filtration criteria. While prevalence refers to the minimum abundance at which a given feature must be present in a sample, occurrence refers to the minimum number of samples in which the given feature must prevail at the prevalence threshold (23). These parameters can be adjusted both before and after development of personal dashboard (i.e. the analysis workspace, as described later).
Normalization and transformation methods
Most of the popular data normalization and transformation methods used for microbiome datasets are offered in MetagenoNets (Supplementary Table S1). While ‘Total Sum Scaling’ (TSS), ‘Cumulative Sum Scaling’ (CSS) and Quartile normalization constitute the set of normalization techniques, ‘Relative Log Expression’ (RLE, as implemented in edgeR package), ‘Trimmed Mean of M values’ (TMM) and ‘Centered-Log Ratio’ (CLR) are the popular transformation methods available for the end-users (23,25). The choice of changing the normalization or transformation method is available dynamically, allowing the flexibility of testing various strategies in a single workspace or personal dashboard (without the need for re-uploading the data). It is pertinent to note that these methods are applied only on the primary input data.
Correlation inference algorithms
Previously, researchers were reliant on classical correlation metrics like Spearman or Pearson correlation coefficient for assessing relevant associations between the microbes/OTUs. With the awareness about compositional nature of microbiome datasets, algorithms like CCREPE/ReBoot (26), SPARCC (27,28), CCLasso (29) and NAMAP (30) are not only preferred but are rather recommended for a meaningful analysis. Each of these computationally and statistically intensive algorithms have their own challenges pertaining to installation, data preparation and workflow (23). Apart from classical correlation metrics, MetagenoNets is equipped with the above-mentioned algorithms and also includes other popularly used bootstrapped variants of classical Spearman/Pearson correlation-based methods (4). Dynamic choice of statistical significance (P-value, q-value), iteration threshold and other algorithm tuning parameters is offered to the end-users.
UPLOAD PROCESS AND TASK MANAGEMENT SYSTEM
MetagenoNets follows a sequential widget-based task submission workflow. Step 1 pertains to upload of all types of data available with the end-users. Given the registration-free framework, it is mandatory to provide an easy to identify ‘job label’ in this step. This job label is mapped to the unique and personalized (eight characters) dashboard identifier specific to the task initiated by the user. The same can be accessed through the job history page of MetagenoNets. A live status terminal is provided to keep track of any errors in the submission. Step 2 provides a summary of the data statistics as uploaded by the user. It is pertinent to note that MetagenoNets automatically infers the categorical and continuous metadata types in the supplied meta-data file. The statistics summary provides quantitative information pertaining to all classes of input as well as metadata. Step 3 allows the end-users to provide initial choice of data filtration, normalization/transformation parameters that may be applied on the primary input data (i.e. the feature or OTU abundance profile) for developing the personal dashboard. Step 4 provides a global summary of all the inputs/choices and seeks approval from the end-users for development of personal dashboard (i.e. analysis workspace). Once the approval is granted, dashboard is instantly developed, and the user is given the option to access the same. Job history page is simultaneously updated with the job label and corresponding dashboard identifier for accessing the workspace later. Job search widget provided in the job history section also allows the user to access any dashboard using the unique identifier shared by a remote collaborator.
PERSONAL DASHBOARD AND RESULTS
Modules of MetagenoNets
The four modules in MetagenoNets, each designated for a specific set of analyses and visualizations, are provided in the framework of a personal workspace, called dashboard. These modules include: (i) categorical networks, (ii) integrated networks, (iii) Venn diagrams and (iv) properties.
Categorical networks module
This module allows inference and visualization of various category- specific networks, wherein categories are derived automatically from the categorical metadata classes detected by MetagenoNets. For example, for the class pertaining to ‘State of health’, the categories of ‘Disordered’ and ‘Healthy’ may be present. Networks can not only be dynamically inferred using various algorithms (and associated parameters), but can also be visualized using interactive and customizable network diagrams and correlograms (heatmap of correlation matrix) for each category of interest. The options to incorporate various centrality measures and node metadata affiliation in the visualizations further add to the overall utility of this module (Figure 1A). Results of this module can be downloaded in the form of high-resolution images as well as re-usable textual data (edge-lists, correlation matrices, jsons etc.).
Figure 1.

A summary of various visualizations generated by different modules of MetagenoNets. (A) Categorical networks and corresponding correlograms for each group of metadata class (i.e. disease condition). Node are colored according to their phylum affiliation and sized according to their degree. (B) Integrated bi-partite networks and sankey plots, probing correlations between microbial occurrence and abundance of branched chain amino acid (BCAA), lipopolysaccharide biosynthesis (LPS) and methyerythritol phosphate pathway-1 function. (C) Node composition and edge composition Venn diagrams all the groups of networks in the meta-data class of disease condition. (D) Network centrality measures for each group and their comparisons using the grouped box plot. Degree centrality and clustering coefficient have been compared in both the groups (categories) of networks.
Integrated networks module
This module allows inference and visualization of integrated and bi-partite networks. Integrated networks represent correlations between mixed feature types like taxa (or microbes), functions (e.g. metabolites) and continuous meta-data classes (e.g. age), such that intra-feature-type as well as inter-feature-type associations are allowed. Bi-partite networks on the other hand allow only inter- feature-type correlation mining. MetagenoNets allows end-users to select the functional (or secondary features) as well as continuous meta-data classes using a search enabled widget, to probe the possibility of correlation(s) of the searched features with the primary feature set (taxa). Apart from inheriting all functionalities of Categorical networks module, this module of MetagenoNets allows visualization of bi-partite networks through intuitive Sankey diagrams (31), wherein taxa are aligned along the left axis while the co-variates and functions are aligned along the right axis (Figure 1B).
Venn diagram module
This module aims at probing compositional comparisons between related networks of a given class of meta-data. The compositional comparison refers to identification of the sets of identical or exclusive nodes as well as edges between related categories of networks. This is achieved by offering automated generation of node composition Venn diagrams and edge composition Venn diagrams (32). Users can select any meta-data class of interest and probe intersecting or exclusive nodes and edges across all categories of networks in that class (Figure 1C). The dynamic choice of classes, network algorithms and other parameters is offered in this module as well.
Properties module
Exploring and comparing centrality measures associated with various networks is a rational and frequently followed approach in network biology. Properties module allows exploration of popular centrality measures, namely, degree, clustering coefficient, closeness, betweenness, eccentricity and coreness (33). Two types of results are provided by this module: (i) a tabulated listing of the centrality measures for all nodes of a selected network, with the functionality of searching, sorting, filtering and exporting the results; and (ii) a trend-line embedded grouped box plot view for global comparison of a selected centrality measure across all categories of networks in a chosen class of meta-data (Figure 1D). Like all other modules, the dynamic choice of classes, network algorithms and related parameters is available in this module as well.
A troubleshooting module for formatting data according to the requirements of MetagenoNets has also been provided. Apart from cleaning the data for the presence of special characters, NA (or missing) values, this module allows extraction of abundance data and metadata from various types of BIOM files.
DEMOS AND CASE STUDY
Four ready to execute demos have been hosted on MetagenoNets server at https://web.rniapps.net/metagenonets/demos.php. One of the demos corresponding to the dashboard ID: 1a52c9c2 pertain to the use case aimed at studying microbial (and inter-omic) association patterns in Inflammatory Bowel Disease (IBD) which includes Crohn's disease (CD) and Ulcerative colitis (UC). Taxonomic and functional profiles corresponding to the metagenomic study (downloadable from MetagenoNets demo page) were obtained from the Inflammatory Bowel Disease Multi’omics Database pertaining to HMP2 (https://ibdmdb.org/) (34,35).
We considered the zeroth day sample corresponding to all the subjects in the study (76 IBD: 48 CD, 28 UC; 24 non-IBD). Spearman's correlation coefficient, as employed previously by the authors of the study, was used for association mining (34,35). Categorical networks pertaining to IBD samples indicated an apparent increase in the network density in comparison to the non-IBD category. Node meta-data mapping with respect to phylum affiliation of the microbes indicated an enrichment of high degree nodes belonging to phylum Bacteroidetes and decrease in those belonging to phyla Firmicutes and Actinobacteria in IBD samples (Figure 1A). It has previously been reported that members belonging to phyla Firmicutes and Actinobacteria produce beneficial metabolites like SCFAs that help in maintaining colon health and the integrity of gut lumen (36). Similarly, higher abundance of phylum Bacteroidetes has been reported to be associated with various metabolic disorders (36). Additionally, we used the search widget of Integrated networks module to select a deleterious pathway reported to be enriched in IBD category of samples (lipopolysaccharide biosynthesis pathway or LPS) and a beneficial pathway enriched in non-IBD category (Branched chain amino acid biosynthesis pathway or BCAA), to infer important microbes associated with these functions and how such associations vary across different categories of networks (Figure 1B). It may be noted that while LPS is a known hallmark of low-grade systemic inflammation, BCAAs are known to promote gut health as reported in the previous studies (37). Bi-partite networks and Sankey plots, as generated through integrated networks module, indicated that while there was no significant correlation between any microbe and LPS in non-IBD samples, Escherichia coli (a Gram-negative bacterium) was found to be positively correlated with LPS in IBD category. Additionally, while BCAA biosynthesis was seen to be positively correlated to most of the commensal and beneficial bacterial strains in non-IBD samples, such correlations were observed to disappear and shift to pathogenic strains in IBD category. A positive correlation between methylerythritol phosphate (MEP) and Alistipes putredinis in non-IBD, as reported by the authors as an over-transcribed function of A. putredinis (36,37), was also observed. Correlograms generated for both categorical and integrated networks indicated the strength of the above discussed correlations (Figure 1B).
A comparison of node and edge compositions of each category of network, using the Venn diagrams module, indicated that most of the nodes were common between IBD and non-IBD networks, while the Venn diagram for edges indicated the presence of disease-condition specific exclusive edges. This affirms that although the set of nodes between the IBD and non-IBD specific categorical networks was almost similar, their mutual associations underwent a significant change or dysbiosis according to the disease condition (Figure 1C). In addition, the average clustering coefficient across IBD and non-IBD categories of networks, as obtained through the properties module, also indicated the emergence of closely clustered communities in IBD as compared to non-IBD. The major players in each category of network were also identified from the properties table, wherein Alistipes shahii was observed to have highest degree and closeness in IBD network while, Dorea formicigenerans showed highest degree and closeness in non-IBD network (Figure 1D).
IMPLEMENTATION
MetagenoNets can be freely accessed by the researchers at https://web.rniapps.net/metagenonets. Its back-end is primarily based on Python and C++. Data visualizations are based on Cytoscape.js (38), jVenn (32), D3.js (39) and in-house customizations of the same for better user experience. Server connections are established using PHP and the front-end design is based on HTML, CSS and Javascript. The platform has been tested with Mozilla Firefox, Chrome, Opera and Safari. Supplementary Table S1 provides description and references to the publications/ source codes of various algorithms employed in MetagenoNets.
FUTURE DIRECTIONS
Use of statistical tests (like Kruskal-Wallis and Wilcoxon tests) and marker feature detection algorithms (like classification and regression based methods) for feature reduction is a common strategy adopted by microbiome researchers (23). Such feature reduction strategies can particularly be helpful in generating integrated networks using statistically relevant primary and secondary features. Currently, MetagenoNets only employs the standard occurrence and prevalence-based feature filtration approach and relies on availability of relevant primary and secondary input datasets. Future version of MetagenoNets is planned to include the utility of aforementioned feature reduction strategies. In addition, given the popular use of ordination-based dimensionality reduction (on primary feature set) coupled with co-inertia analysis and procrustes analysis for inter-omic correlation inferences, future version of MetagenoNets will implement these methods as well (17). Many features of MetagenoNets are a result of the continuous feedback and requests by the existing userbase, and we expect this tool to continue evolving beyond planned development strategies as well.
DISCUSSION
Network analysis is commonly used in microbiome research. However, researchers need to follow a lengthy workflow to perform even a simple correlation analysis and visualization. Need for inter-omic association mining using the secondary datasets or co-variates in the meta-data, further complicates the process of inferring the correlations and generating meaningful visualizations. We have developed MetagenoNets, a web-based application, to reduce the time and effort needed to conduct such analyses. The inclusion of multiple algorithms and data management methods in this tool enables the researchers to explore and employ appropriate strategy suitable to the nature of their data (and associated meta-data). In addition, the provision for multiple interactive visualization techniques and real time choice of algorithms in the framework of a modular workspace, ensures that the end-users can approach the problem of microbial correlation analysis in a logical progression. Although inter-omic correlation analysis is rather a much needed approach for microbial network analyses, it has rarely been represented in this field. MetagenoNets has made initial attempts in automating the inference and suitable visualization of integrated and bi-partite networks. Future versions of MetagenoNets will focus on expanding the scope of inter-omic correlation mining, apart from expanding the general scope of microbial association mining using microbiome datasets.
DATA AVAILABILITY
MetagenoNets application is freely available at http://web.rniapps.net/metagenonets.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank the Bio-Sciences R&D Division team and collaborators for testing MetagenoNets and their constructive feedback from time to time.
Contributor Information
Sunil Nagpal, Bio-Sciences R&D Division, TCS Research, Pune, Maharashtra 411013, India.
Rashmi Singh, Bio-Sciences R&D Division, TCS Research, Pune, Maharashtra 411013, India.
Deepak Yadav, Bio-Sciences R&D Division, TCS Research, Pune, Maharashtra 411013, India.
Sharmila S Mande, Bio-Sciences R&D Division, TCS Research, Pune, Maharashtra 411013, India.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
TCS Research Ltd (Tata Consultancy Services, Pune, India). Funding for open access charge: TCS Research Ltd (Tata Consultancy Services, Pune, India).
Conflict of interest statement. Authors are salaried employees of TCS Research Ltd (Tata Consultancy Services, Pune, India). Tata Consultancy Services promotes fundamental and applied research in BioSciences research division of TCS Research. This tool is an outcome of one of such research efforts.
REFERENCES
- 1. Koskella B., Hall L.J., Metcalf C.J.E.. The microbiome beyond the horizon of ecological and evolutionary theory. Nat. Ecol. Evol. 2017; 1:1606–1615. [DOI] [PubMed] [Google Scholar]
- 2. Mande S.S., Mohammed M.H., Ghosh T.S.. Classification of metagenomic sequences: methods and challenges. Brief. Bioinform. 2012; 13:669–681. [DOI] [PubMed] [Google Scholar]
- 1. HilleRisLambers J., Adler P.B., Harpole W.S., Levine J.M., Mayfield M.M.. Rethinking community assembly through the lens of coexistence theory. Annu. Rev. Ecol. Evol. Syst. 2012; 43:227–248. [Google Scholar]
- 4. Faust K., Raes J.. Microbial interactions: from networks to models. Nat. Rev. Microbiol. 2012; 10:538–550. [DOI] [PubMed] [Google Scholar]
- 5. Srivastava D., Baksi K.D., Kuntal B.K., Mande S.S.. “EviMass”: a literature evidence-based miner for human microbial associations. Front. Genet. 2019; 10:849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tandon D., Haque M.M., Mande S.S.. Inferring intra-community microbial interaction patterns from metagenomic datasets using associative rule mining techniques. PLoS One. 2016; 11:e0154493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Faust K., Sathirapongsasuti J.F., Izard J., Segata N., Gevers D., Raes J., Huttenhower C.. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012; 8:e1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Röttjers L., Faust K.. From hairballs to hypotheses–biological insights from microbial networks. FEMS Microbiol. Rev. 2018; 42:761–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raes J., Foerstner K.U., Bork P.. Get the most out of your metagenome: computational analysis of environmental sequence data. Curr. Opin. Microbiol. 2007; 10:490–498. [DOI] [PubMed] [Google Scholar]
- 10. Gloor G.B., Macklaim J.M., Pawlowsky-Glahn V., Egozcue J.J.. Microbiome datasets are compositional: and THIS IS NOT OPTIOnal. Front. Microbiol. 2017; 8:2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li H. Microbiome, metagenomics, and high-dimensional compositional data analysis. Annu. Rev. Stat. Appl. 2015; 2:73–94. [Google Scholar]
- 12. Layeghifard M., Hwang D.M., Guttman D.S.. Disentangling interactions in the microbiome: a network perspective. Trends Microbiol. 2017; 25:217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Layeghifard M., Hwang D.M., Guttman D.S.. Beiko RG, Hsiao W, Parkinson J. Constructing and analyzing microbiome networks in R. Microbiome Analysis: Methods and Protocols. 2018; NY: Springer; 243–266.Methods in Molecular Biology. [DOI] [PubMed] [Google Scholar]
- 14. Kuntal B.K., Mande S.S.. Visual exploration of microbiome data. J. Biosci. 2019; 44:119. [PubMed] [Google Scholar]
- 15. Duvallet C., Gibbons S.M., Gurry T., Irizarry R.A., Alm E.J.. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat. Commun. 2017; 8:1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang D., Armour C.R., Hu C., Mei M., Tian C., Sharpton T.J., Jiang Y.. Microbiome multi-omics network analysis: statistical considerations, limitations, and opportunities. Front. Genet. 2019; 10:995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chong J., Xia J.. Computational approaches for integrative analysis of the metabolome and microbiome. Metabolites. 2017; 7:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McHardy I.H., Goudarzi M., Tong M., Ruegger P.M., Schwager E., Weger J.R., Graeber T.G., Sonnenburg J.L., Horvath S., Huttenhower C. et al.. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome. 2013; 1:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Faust K., Raes J.. CoNet app: inference of biological association networks using Cytoscape [version 2; peer review: 2 approved]. F1000Res. 2016; 5:1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brohée S., Faust K., Lima-Mendez G., Vanderstocken G., van Helden J.. Network analysis tools: from biological networks to clusters and pathways. Nat. Protoc. 2008; 3:1616–1629. [DOI] [PubMed] [Google Scholar]
- 21. Callahan B.J., Sankaran K., Fukuyama J.A., McMurdie P.J., Holmes S.P.. Bioconductor workflow for microbiome data analysis: from raw reads to community analyses [version 2; peer review: 3 approved]. F1000Res. 2016; 5:1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xia J., Benner M.J., Hancock R.E.W.. NetworkAnalyst - integrative approaches for protein–protein interaction network analysis and visual exploration. Nucleic Acids Res. 2014; 42:W167–W174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dhariwal A., Chong J., Habib S., King I.L., Agellon L.B., Xia J.. MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017; 45:W180–W188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zakrzewski M., Proietti C., Ellis J.J., Hasan S., Brion M.-J., Berger B., Krause L.. Calypso: a user-friendly web-server for mining and visualizing microbiome–environment interactions. Bioinformatics. 2017; 33:782–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weiss S., Xu Z.Z., Peddada S., Amir A., Bittinger K., Gonzalez A., Lozupone C., Zaneveld J.R., Vázquez-Baeza Y., Birmingham A. et al.. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017; 5:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Faust K., Sathirapongsasuti J.F., Izard J., Segata N., Gevers D., Raes J., Huttenhower C.. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012; 8:e1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Friedman J., Alm E.J.. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012; 8:e1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Watts S.C., Ritchie S.C., Inouye M., Holt K.E.. FastSpar: rapid and scalable correlation estimation for compositional data. Bioinformatics. 2019; 35:1064–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fang H., Huang C., Zhao H., Deng M.. CCLasso: correlation inference for compositional data through Lasso. Bioinformatics. 2015; 31:3172–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yadav D., Ghosh T.S., Mande S.S.. Global investigation of composition and interaction networks in gut microbiomes of individuals belonging to diverse geographies and age-groups. Gut Pathogens. 2016; 8:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Riehmann P., Hanfler M., Froehlich B.. Interactive Sankey diagrams. IEEE Symposium on Information Visualization, 2005. 2005; INFOVIS 2005; 233–240. [Google Scholar]
- 32. Bardou P., Mariette J., Escudié F., Djemiel C., Klopp C.. jvenn: an interactive Venn diagram viewer. BMC Bioinformatics. 2014; 15:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pavlopoulos G.A., Secrier M., Moschopoulos C.N., Soldatos T.G., Kossida S., Aerts J., Schneider R., Bagos P.G.. Using graph theory to analyze biological networks. BioData Mining. 2011; 4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Das P., Babaei P., Nielsen J.. Metagenomic analysis of microbe-mediated vitamin metabolism in the human gut microbiome. BMC Genomics. 2019; 20:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schirmer M., Franzosa E.A., Lloyd-Price J., McIver L.J., Schwager R., Poon T.W., Ananthakrishnan A.N., Andrews E., Barron G., Lake K. et al.. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat Microbiol. 2018; 3:337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Qin P., Zou Y., Dai Y., Luo G., Zhang X., Xiao L.. Characterization a novel butyric acid-producing bacterium collinsellaaerofaciens Subsp. Shenzhenensis Subsp. Nov. Microorganisms. 2019; 7:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nie C., He T., Zhang W., Zhang G., Ma X.. Branched Chain Amino Acids: Beyond nutrition metabolism. Int. J. Mol. Sci. 2018; 19:954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Franz M., Lopes C.T., Huck G., Dong Y., Sumer O., Bader G.D.. Cytoscape.js: a graph theory library for visualisation and analysis. Bioinformatics. 2016; 32:309–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bostock M., Ogievetsky V., Heer J.. D3 data-driven documents. IEEE Trans. Vis. Comput. Graph. 2011; 11:2301–2309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MetagenoNets application is freely available at http://web.rniapps.net/metagenonets.