


Abstract
Overlapping genes are commonplace in viruses and play an important role in their function and evolution. For these genes, molecular coevolution may be seen as a mechanism to decrease the evolutionary constraints of amino acid positions in the overlapping regions and to tolerate or compensate unfavorable mutations. Tracing these mutational sites, could help to gain insight on the direct or indirect effect of the mutations in the corresponding overlapping proteins. In the past, coevolution analysis has been used to identify residue pairs and coevolutionary signatures within or between proteins that served as markers of physical interactions and/or functional relationships. Coevolution in OVerlapped sequences by Tree analysis (COVTree) is a web server providing the online analysis of coevolving amino-acid pairs in overlapping genes, where residues might be located inside or outside the overlapping region. COVTree is designed to handle protein families with various characteristics, among which those that typically display a small number of highly conserved sequences. It is based on BIS2, a fast version of the coevolution analysis tool Blocks in Sequences (BIS). COVTree provides a rich and interactive graphical interface to ease biological interpretation of the results and it is openly accessible at http://www.lcqb.upmc.fr/COVTree/.
INTRODUCTION
Overlapping genes represent a fascinating evolutionary puzzle, since they encode two or more functionally unrelated proteins from the same DNA or RNA sequence. An immediate consequence of this overlapping structure are the specific constraints to the random mutations of the corresponding proteins, since a single nucleotide substitution may affect the product of both genes simultaneously. In this context, molecular coevolution may be seen as a mechanism to tolerate or compensate unfavorable mutations, decreasing the evolutionary constraints in the overlapping region. Multiple studies of coevolving positions in viral sequences have been essential to understand functionally significant residues (1,2), to predict protein-protein interaction networks (3), to unravel novel mechanisms of viral fusion (4) and to identify drug resistance mutations (5–10) among others.
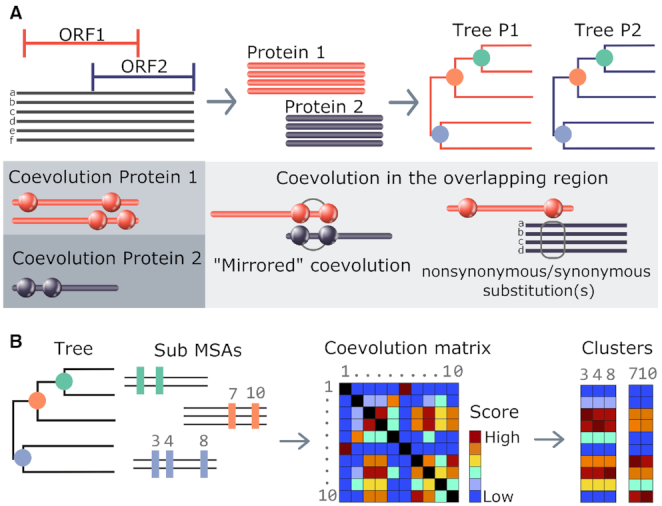
The genomes of most viral species have overlapping genes (11). Their open reading frames (ORF) can belong to the same strand or be located in the complementary one, be completely or partially overlapped, be in phase or shifted. Sequence analysis in overlapping ORFs represents a challenge due to nucleotide sequence changes that may simultaneously affect both overlapped proteins (Figure 1), and up to now, no web server has been dedicated to the coevolution analysis of overlapping proteins. Coevolution in OVerlapped sequences by Tree analysis (COVTree) is a web server that facilitates the analysis of coevolution and mutational impact in overlapped ORFs. To do this, it combines information at the protein and nucleotide levels. To illustrate COVTree importance, we apply it to two overlapping Hepatitis B Virus proteins highlighting novel information.
Figure 1.

Complexity of coevolution patterns in an overlapping region. A mutation of the DNA/RNA sequence might imply two changes at the amino acid level of the two corresponding overlapping proteins (P1 and P2): (A) P1 and P2 lie on the same strand; (B) P1 and P2 lie on opposite strands and a frameshift is present; (C) P1 and P2 lie on opposite strands and are in phase. (D) Relative positioning and mutations of coevolving positions in the overlapped region of two proteins P1 and P2. A cluster of four coevolving positions in P1s alignment shows two sequences maintaining the wild-type residues (red circles) and three displaying mutations on all positions (yellow circles). A mutation in P1 may be coupled by synonymous substitutions in P2 (position 1 in P1); the same non-synonymous substitution (position 2), two non-synonymous substitutions in adjacent positions (positions 3); a variety of non-synonymous substitutions (position 4). Clusters of coevolving positions may contain positions outside the overlapping region (see P2). (E) Clusters of coevolving residues in P1 and P2 over a overlapping region. Note that some of the positions do not overlap.
COVTree PIPELINE
COVTree takes as input a multiple sequence alignment (MSA) of DNA or RNA sequences, a reference sequence in the alignment and the starting and ending positions of two protein coding genes within the alignment (Figure 2A). Their protein genes may overlap in various manners considering the extent of the overlap (partial or complete), the direction of transcription and the ORFs phase (Supplementary Figure S1). Two alignments associated with amino-acid translations of the two protein genes are then generated taking into account the coordinates given by the user. Two associated phylogenetic trees for Protein 1 and Protein 2 are also constructed using FastTree (12) and are used by COVTree to analyze the two protein sequence alignments. COVTree outputs the results of BIS2TreeAnalyzer (10,13) coevolution analysis (Figure 2B) for the two proteins, and an analysis of those coevolving positions in the overlapping region of a protein that affect the other. Through an interactive graphical interface, COVTree highlights amino-acids and nucleotides affected by the overlapping (Figure 1). The details of each step in the COVTree pipeline are given below.
Figure 2.
COVTree workflow. (A) BIS2TreeAnalyzer coevolution analysis: BIS2 is reiterated on all subsets of amino acid sequences corresponding to subtrees of at least 20 sequences of the initial phylogenetic tree. Coevolution matrices, one for each subtree analysis, are produced and clustered. A ‘merged’ set of clusters of coevolving positions is given as output. (B) The input of COVTree is a nucleotide alignment covering two ORFs of interest. Sequences are translated to generate the two protein MSAs. Each protein alignment is used to build a phylogenetic tree. The protein alignment and the phylogenetic tree are analyzed with BIS2TreeAnalyzer (A). COVTree output includes the coevolution analysis of the two proteins, as well as the effect of mutations in one protein over the other, for coevolving positions lying in the overlapping region. Both proteins may show coevolution in ‘mirrored’ positions (‘mirrored’ coevolution; see the ‘COVTree output’ section for a definition) or coevolution in a protein may be accompanied by synonymous/non-synonymous mutations in the other. To distinguish between these two situations, nucleotide information is provided.
From a nucleotide sequence alignment to two amino-acid sequence alignments
To minimize errors in the translation of the nucleotide alignment, possibly caused by out-of-frame indels, COVTree translates each nucleotide sequence using the three possible reading frames of the selected strand.
The translated sequences are compared with the protein reference sequence, and only the sequence which is the most similar to the reference one is kept in the protein MSA.
Clusters of coevolving positions
COVTree uses the coevolution analysis method BIS2TreeAnalyzer (10) to predict clusters of coevolved positions (Figure 2B) in a protein sequence alignment. BIS2TreeAnalyzer is an algorithmic strategy designed to apply BIS (13), a combinatorial coevolution analysis method successfully used in the past on small sets of conserved sequences, to large sets of evolutionary related sequences. BIS2TreeAnalyzer is based on a fast version of BIS, called BIS2 (3,14). In short, BIS2TreeAnalyzer applies BIS2 iteratively to the sub-alignments associated to the subtrees (of at least 20 sequences) of the phylogenetic tree associated to the original sequence alignment. To discover patterns of coevolving positions, that is combinations of specific amino acids observed to occur together in these positions, BIS2TreeAnalyzer computes a coevolution score between all pairs of positions and constructs a coevolution score matrix. Then, the matrix is clusterized using CLAG (15) to identify clusters of positions displaying the same coevolution scores with all other positions in the alignment. A cluster of coevolving positions comprises at least two amino acid patterns. A P-value, previously defined in (10), based on a binomial test and its Bonferroni correction (16), is computed for each pattern (see ‘Help’ page). Clusters with at least one pattern with significant P-value (<0.005) are informed. Typically, in a cluster, one pattern corresponds to wild-type amino acids and the other(s) to mutations. BIS2TreeAnalyzer is available at www.lcqb.upmc.fr/BIS2TreeAnalyzer/. See (10) for its description, validation and comparison with other coevolution analysis methods.
On the size of sub-alignments
BIS2 algorithm is iteratively applied to sub-alignments associated to subtrees of as few as 20 sequences. In this respect, note that BIS2 was originally designed to identify coevolution in small sets of conserved sequences. In (13), the method was applied to several protein families including the AATPase families, recording a high accuracy achieved in eight alignments having <20 sequences. In a study on the genome of the Hepatitis C Virus, it could highlight, in a restricted number of sequences (<50 for each genotype considered), coevolving residues playing a crucial role in conformational changes of the glycoprotein E2 (4). In a study on the Hepatitis B Virus, it found biologically relevant clusters of coevolving positions, responsible for drug resistance, in subtrees of 20 sequences (10).
Identification of coevolving positions in the overlapping region
Based on the clusters of coevolving amino-acid positions in the two protein alignments, COVTree crosses their information to evaluate coevolution signals in the overlapping region of the proteins. The complexity of the problem is illustrated in Figure 1D and E, where the combinatorics of the mutations within the two alignments highlights intricate patterns of coevolution that can emerge. To analyze the effects of mutations in the coevolving position of Protein 1 (Protein 2) over coevolving positions in Protein 2 (Protein 1), COVTree translates the amino-acid sequence alignments into nucleotide sequence alignments and displays the mutated codon in Protein 1 (Protein 2) together with its immediate nucleotidic environment, showing the mutational effect over one or two amino-acid positions in Protein 2 (Protein 1) (Figure 1A–C). The amino-acids coordinates in Protein 2 (Protein 1) are provided.
COVTree INPUT
COVTree input consists of a codon-based MSA of nucleotide sequences in FASTA format. The alignment can be copy/pasted or uploaded as a file. A client-side validation is performed to check the MSA format. If validation fails, the alignment is highlighted in red and a user-friendly message warns about the reason and the location where the format fails. Otherwise, the alignment is shown in green and additional parts of the submission page become available after pressing the ‘Load MSA’ button. A reference sequence has to be set by typing a sequence identifier in the input box, which autocompletes based on the sequence identifiers of the uploaded MSA. For each protein, the user should indicate the coding strand of the protein (direct or complementary) and the ORF coordinates (start and end positions in the reference sequence). The inclusion of the stop codon in the sequence is optional. The selections can be checked by visualizing the corresponding translation of the reference sequence. Also, sequence lengths and the extent of the overlapping region are graphically displayed. Optionally, users can provide an email address to receive notifications about the status of the job and an address to download the results.
COVTree can analyze only one protein or two non-overlapping proteins by accordingly setting the strand, the start and end positions of the proteins. In the first case, the three parameter values should be the same for the two proteins and, in the second case, the end and start positions of the two proteins should not define an overlapping interval. In both cases, the result page ‘Overlap’ should be ignored.
Guidelines on input sequences
We highly recommend taking the amino acid level into account during the alignment of protein-encoding DNA/RNA. This will ensure nucleotides to maintain the correct reading frame for each sequence in the alignment. There are several tools available to generate a codon-based alignment as web servers, such as RevTrans (17), PAL2NAL (18), TranslatorX (19) and software suites as MACSE (20,21).
COVTree OUTPUT
COVTree output is organized in three dedicated pages, two for the proteins (tag buttons ‘Protein 1’ and ‘Protein 2’) and one for the overlapping region (‘Overlap’). A ‘Download results’ tag button is also present.
The two pages dedicated to the proteins display an interactive table reporting the BIS2TreeAnalyzer coevolution analysis (Figure 3A). Each row of the table corresponds to a cluster of coevolving positions and each column represents a property of a cluster, namely: the cluster identifier, the name of the subtree of sequences where the cluster is found, the list of coevolving positions in the cluster, the number of sequences displaying the same coevolution pattern (two or more numbers are given, whose sum corresponds to the total number of sequences labeling the subtree), the amino acid patterns found in the sequences and the P-value corresponding to each pattern. Clusters (rows) can be filtered and sorted by any of their properties (columns) without page refreshes. Also, it is possible to sort multiple properties simultaneously by holding down the Shift key and clicking a second, third or fourth column header. Clusters can be filtered out by upper bounds on the P-value; for instance, if the upper bound is a P-value < 0.05, the table will show only those clusters with at least one amino acid pattern matching the criteria. When a cluster is selected by clicking its row, the subtree and sub-alignments where the cluster was found are shown in different panels. In the subtree panel (Figure 3C), the FastTree scale bar for branch lengths is shown at the top of the tree and the exact distance is shown in a pop-up window when the mouse hovers over a branch. The sub-alignment panel (Figure 3B) shows a sequence logo representation, indicating the conservation of amino-acids in the protein sequence and the alignment of the protein sequences colored by physico-chemical properties. One or several positions in the sequence alignment can be selected, by clicking on the position number(s), to facilitate the reading of the amino acids distribution in the positions. At last, a plot describes, for each alignment position, how many times the position is found in distinguished BIS2TreeAnalyzer clusters. By passing the mouse over a bar plot, the names of all clusters containing the position is given. An interval in the plot can be zoomed by selecting it with the mouse.
Figure 3.

COVTree interactive page presenting protein coevolution analysis. (A) Table of clusters of coevolving positions, where each row describes a BIS2TreeAnalyzer cluster (see text). By selecting a cluster (green row: cluster 6 associated to subtree 30), the sub-alignment of sequences where the cluster is detected and its subtree are shown on a dedicated sequence alignment panel (B) and on a dedicated subtree panel (C). Colors in the sequence logo representation correspond to physico-chemical amino acid properties. The user can select one or more specific positions in the alignment (see position 322 highlighted in red). The names of the sequences are reported on the left of the alignment. Their positioning in the tree (C) can be highlighted (see sequences containing a proline in position 322 in red). Panels B and C show, by default, the results corresponding to the first cluster of the table. (D) Zoom of the histogram of all coevolving positions in the protein. As illustrated for position 322, the identifiers of the clusters containing the position are highlighted in red when the position is selected.
The third page corresponds to the description of coevolution signals found in the overlapping region. It provides two tables, one for each protein, complementing the information already present in the protein’s pages and the visualization of the coevolved positions in Protein 1 and Protein 2 mapped on the reference nucleotide sequence. The first table corresponds to Protein 1, where each row describes a coevolving position p of Protein 1 belonging to the overlapping region. Columns indicate the cluster identifier, the subtree identifier, the coevolving position p, the number of sequences in which coevolution was detected, the amino acid found in p, the nucleotide change(s) responsible for the mutations at the coevolved site p, the amino acids on the overlapping positions in Protein 2 and the names of these positions. The ‘Nucleotides’ column reports the nucleotide change(s) responsible for the mutations at the coevolved sites; the central codon corresponds to the amino acid of Protein 1, whereas the fragment encodes the overlapping residues for Protein 2. The nucleotide sequence allows distinguishing between non-synonymous (amino-acid-altering) and synonymous (silent) substitutions. In particular, it allows to read off synonymous substitutions in one gene causing amino acid changes in the other one. Also, by selecting a particular row of the table, the subtree in which the cluster containing p was detected is shown in the ‘Subtree’ panel. A cross comparison between coevolving positions reported in the two tables allows the user to identify whether coevolution signals in a protein ‘mirror’ those in the other protein (Figure 2A). This is the case when the positions in Protein 2 (Protein 1) affected by mutations in Protein 1 (Protein 2) are found as coevolving positions in the table of Protein 2 (Protein 1) for the same set of sequences. An interactive map of all coevolved positions in Protein 1 and Protein 2 on the reference nucleotide sequence allows the user to access, at once, all coevolving positions in Protein 1 and Protein 2, at the nucleotide and amino acid levels, as well as the clusters identifying each position.
The user can reason on ‘overlapping coevolving positions’, that is positions in Protein 1 and Protein 2 sharing at least one nucleotide at the DNA/RNA level. These positions can be easily identified in the two tables and in the map. Furthermore, (s)he might want to explore whether pairs of overlapping coevolving positions form a ‘mirrored coevolution’, that is whether there exists a set of nucleotide sequences 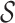 identifying a pair of coevolving positions p1, p2 in Protein 1, and a pair of coevolving positions
identifying a pair of coevolving positions p1, p2 in Protein 1, and a pair of coevolving positions 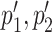 in Protein 2 where
in Protein 2 where 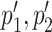 overlap p1, p2, respectively. An interactive table allows the user to select the pairs of subtrees supporting coevolution of p1, p2 and
overlap p1, p2, respectively. An interactive table allows the user to select the pairs of subtrees supporting coevolution of p1, p2 and 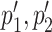 , to quickly test whether they comprise the same sequences at the nucleotide level and to learn how many overlapping coevolving positions they share. The two corresponding sub-alignments will help the user to verify that the patterns of amino acids in p1, p2 and in
, to quickly test whether they comprise the same sequences at the nucleotide level and to learn how many overlapping coevolving positions they share. The two corresponding sub-alignments will help the user to verify that the patterns of amino acids in p1, p2 and in 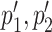 correspond to the same sequences.
correspond to the same sequences.
The ‘Download results’ tag button allows for the direct download of a tar folder collecting the coevolution analyses of the two proteins. For each protein, COVTree provides the FASTA files with the nucleotide and amino-acid alignments, the COVTree table with all statistically significant clusters of coevolving positions involving or not the overlapping region, the sub-alignments corresponding to each cluster (in FASTA format) and their corresponding subtrees (in Newick tree format). Note that a CSV file is downloadable for each table displayed in the COVTree interface. Data will be removed from the server storage space one month after the end of the job.
A NOTEWORTHY EXAMPLE OF ‘MIRRORED’ COEVOLUTION: COVTREE ANALYSIS OF THE HBV POLYMERASE AND SURFACE PROTEINS
Hepatitis B Virus (HBV) infection is a major cause of acute and chronic hepatitis B. HBV presents a complex genome organization (Figure 4A), none of the four genes is free of overlapping regions and the region encoding the virus envelope protein (surface antigen or HBsAg) is completely embedded in the gene coding the viral polymerase Pol (22). In particular, the S domain of the HBsAg contains the major B-cell epitope, known as ‘a’ determinant. Mutations in and around the ‘a’ determinant may result in (i) escape of vaccine-induced immunity, (ii) escaping anti-HBV immunoglobulin therapy and (iii) cause diagnostic problems due to false-negative results in serological tests (23). On the other hand, Pol is the target of antiviral therapy with nucleos(t)ide analogs. However, treatment may fail due to the emergence of drug-resistant mutants. Primary drug resistance mutations refer to amino acid change(s) that result in reduced susceptibility to an antiviral agent, whereas the compensatory mutations in Pol restore replication defects associated with primary drug resistance. In (10), known primary and secondary mutations in Pol have been demonstrated to coevolve together and have been related to a number of other Pol mutations. The overlapped region of S and Pol is of particular interest because a single substitution in the nucleotide sequence may simultaneously affect the structure and function of the surface antigen and the polymerase.
Figure 4.

COVTree analysis of HBV proteins Pol and S. (A) Structure of the HBV genome. (B) Schema of the mutations of coevolving positions in Pol and S. A mirrored coevolution is identified for Pol positions 552 (rt204), 567 (rt219) and S positions 369 (s195), 384 (s210). The number of sequences supporting the amino acid patterns are indicated (see C). (C) COVTree tables for the analysis of residue 552 in Protein 1. This position appears in a cluster with positions 528 (rt180), 552 (rt204), 567 (rt 219) and 596 (rt248) (see B, top), and its mutation influence coevolving positions 369 and 370 in S (center). Vice versa, position 369 in S influences positions 551 and 552 (bottom). (D) Visual representation of coevolved positions in Pol and S mapped on the reference nucleotide sequence. Positions occurring in larger number of clusters are colored with darker tones: red for Pol, blue for S. A white background is used for positions that do not coevolve.
COVTree allows to perform coevolution analysis of the proteins S and Pol separately (Figure 4B and C; Supplementary Table S1):
a cluster of four positions in the RT domain of Pol was detected. The mutation detected at position rt204 corresponds to a well known primary drug resistance mutation, whereas mutations at rt180 and rt219 are known to compensate the effect of primary mutations;
the three positions s100, s195 and s210 form a cluster of coevolving positions in S;
and to go further with the analysis of mutations within the overlapping region (Figure 4C and D; Supplementary Figures S2–6). COVTree allows to verify that:
the set of sequences where coevolution, in (i) and (ii) above, is found for Pol and S is the same;
nucleotide mutations in positions s195 and s210 directly interfere with positions rt204 and rt219 by inducing amino acid mutations.
From (a) and (b), one concludes that the overlapping regions of S and Pol in the HBV genome presents mirrored coevolution, described by COVTree with specific patterns of amino acids, the number of sequences showing the mutational patterns and the P-values of the clusters. Drug resistance rtM204V in Pol corresponds to sI195M in S and affects its antigenicity (24) (see Figure 4C and D). Also, for rtL180M, a known drug resistance compensatory mutation, COVTree results at nucleotide level highlight a mutation in Pol which produces a synonymous substitution in S (Supplementary Figure S2). Mutation rtS219A is also a known drug resistance compensatory mutation in Pol, which produces a non-synonymous substitution in S (Supplementary Figure S3).
DISCUSSION
The large range of overlap frequencies across single- and double-stranded RNA and DNA genomes suggests that gene overlapping is a common evolutionary trait that provides flexible genome structures in all virus families (11). Hence, it is expected that coevolution of amino acid positions in viral sequences should be often used as a mechanism to avoid the constraints imposed by the overlapping structure. Then, COVTree becomes a particularly important analysis tool to unravel these constraints and the molecular coevolution signals that are exploited by the virus and that define its complex mutational landscape. COVTree is the first interactive web server providing the end-users with a highly intuitive graphic view of coevolved positions in pairs of protein sequences and in their overlap. It offers an interactive platform to easily trace, on relevant phylogenetic trees and alignments, the mutations that lead to coevolution signals and it allows for an in-depth analysis of coevolution in the overlapping region of protein pairs with a zoom into the nucleotide sequence. The interplay between information at nucleotide and protein level is made explicit by a precise mapping provided to the user together with statistical values supporting the signals. The server has no restrictions on protein length and on the maximum number of sequences in the alignment. COVTree functionality is unique in guiding the user toward the understanding of constrained evolution of overlapped genes.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Diego Vadell for his helpful advice on various technical issues.
Contributor Information
Elin Teppa, Sorbonne Université, UPMC Univ Paris 06, CNRS, IBPS, UMR 7238, Laboratoire de Biologie Computationnelle et Quantitative (LCQB), 75005 Paris, France.
Diego J Zea, Sorbonne Université, UPMC Univ Paris 06, CNRS, IBPS, UMR 7238, Laboratoire de Biologie Computationnelle et Quantitative (LCQB), 75005 Paris, France.
Francesco Oteri, Sorbonne Université, UPMC Univ Paris 06, CNRS, IBPS, UMR 7238, Laboratoire de Biologie Computationnelle et Quantitative (LCQB), 75005 Paris, France.
Alessandra Carbone, Sorbonne Université, UPMC Univ Paris 06, CNRS, IBPS, UMR 7238, Laboratoire de Biologie Computationnelle et Quantitative (LCQB), 75005 Paris, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
French National Agency for Research against HIV and Hepatitis (ANRS) [CSS4 ECTZ25224 2017-19]; French Government, “Programme d'Investissement d'Avenir" (LABEX CALSIMLAB) [ANR-11-LABX-0037-01 and ANR-11-IDEX-0004-02]. Funding for open access charge: Agence Nationale de la Recherche (ANR ChromaLight Grant)
Conflict of interest statement. None declared.
REFERENCES
- 1. Le L., Leluk J.. Study on phylogenetic relationships, variability, and correlated mutations in M2 proteins of influenza virus A. PLoS One. 2011; 6:e22970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jain J., Mathur K., Shrinet J., Bhatnagar R.K., Sunil S.. Analysis of coevolution in nonstructural proteins of chikungunya virus. Virol. J. 2016; 13:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Champeimont R., Laine E., Hu S.-W., Penin F., Carbone A.. Coevolution analysis of hepatitis C virus genome to identify the structural and functional dependency network of viral proteins. Sci. Rep. 2016; 6:26401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Douam F., Fusil F., Enguehard M., Dib L., Nadalin F., Schwaller L., Hrebikova G., Mancip J., Mailly L., Montserret R. et al.. A protein coevolution method uncovers critical features of the hepatitis C virus fusion mechanism. PLoS Pathog. 2018; 14:e1006908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Handel A., Regoes R.R., Antia R.. The role of compensatory mutations in the emergence of drug resistance. PLoS Comput. Biol. 2006; 2:e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rhee S.-Y., Liu T.F., Holmes S.P., Shafer R.W.. HIV-1 subtype B protease and reverse transcriptase amino acid covariation. PLoS Comput. Biol. 2007; 3:e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bloom J.D., Gong L.I., Baltimore D.. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science. 2010; 328:1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. González-Ortega E., Ballana E., Badia R., Clotet B., Esté J.A.. Compensatory mutations rescue the virus replicative capacity of VIRIP-Resistant HIV-1. Antivir. Res. 2011; 92:479–483. [DOI] [PubMed] [Google Scholar]
- 9. Tanaka M.M., Valckenborgh F.. Escaping an evolutionary lobster trap: drug resistance and compensatory mutation in a fluctuating environment. Evolution. 2011; 65:1376–1387. [DOI] [PubMed] [Google Scholar]
- 10. Teppa E., Nadalin F., Combet C., Zea D., David L., Carbone A.. Coevolution analysis of amino-acids reveals diversified drug resistance solutions in viral sequences: a case study of hepatitis B virus. Virus Evol. 2020; 6:veaa006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schlub T.E., Holmes E.C.. Properties and abundance of overlapping genes in viruses. Virus Evol. 2020; 6:veaa009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Price M.N., Dehal P.S., Arkin A.P.. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One. 2010; 5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dib L., Carbone A.. Protein fragments: functional and structural roles of their coevolution networks. PloS One. 2012b; 7:e48124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oteri F., Nadalin F., Champeimont R., Carbone A.. BIS2Analyzer: a server for co-evolution analysis of conserved protein families. Nucleic Acids Res. 2017; 45:W307–W114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dib L., Carbone A.. CLAG: an unsupervised non hierarchical clustering algorithm handling biological data. BMC Bioinformatics. 2012a; 13:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bonferroni C. Teoria statistica delle classi E calcolo delle Probabilitá. Pubblicazioni Del R Istituto Superiore Di Scienze Economiche E Commerciali Di Firenze. 1936; 8:3–62. [Google Scholar]
- 17. Wernersson R., Pedersen A.G.. RevTrans: multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003; 31:3537–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suyama M., Torrents D., Bork P.. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006; 34:W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abascal F., Zardoya R., Telford M.J.. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010; 38:W7–W13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ranwez V., Harispe S., Delsuc F., Douzery E.J.P.. MACSE: multiple alignment of coding sequences accounting for frameshifts and stop codons. PLoS One. 2011; 6:e22594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Löytynoja A. Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 2014; 1079:155–170. [DOI] [PubMed] [Google Scholar]
- 22. Mizokami M., Orito E., Ohba K., Ikeo K., Lau J.Y., Gojobori T.. Constrained evolution with respect to gene overlap of hepatitis B virus. J. Mol. Evol. 1997; 44:S83–S90. [DOI] [PubMed] [Google Scholar]
- 23. Lazarevic I. Clinical implications of hepatitis B virus mutations: recent advances. World J. Gastroenterol. 2014; 20:7653–7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lacombe K., Boyd A., Lavocat F., Pichoud C., Gozlan J., Miailhes P., Lascoux-Combe C., Vernet G., Girard P.M., Zoulim F.. High incidence of treatment‐induced and vaccine‐escape hepatitis B virus mutants among human immunodeficiency virus/hepatitis B–infected patients. Hepatology. 2013; 58:912–922. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.