

Abstract
Mercury (Hg) is a natural and widespread trace metal, but is considered a priority pollutant, particularly its organic form methylmercury (MMHg), because of human's exposure to MMHg through fish consumption. Pioneering studies showed the methylation of divalent Hg (HgII) to MMHg to occur under oxygen‐limited conditions and to depend on the activity of anaerobic microorganisms. Recent studies identified the hgcAB gene cluster in microorganisms with the capacity to methylate HgII and unveiled a much wider range of species and environmental conditions producing MMHg than previously expected. Here, we review the recent knowledge and approaches used to understand HgII‐methylation, microbial biodiversity and activity involved in these processes, and we highlight the current limits for predicting MMHg concentrations in the environment. The available data unveil the fact that HgII methylation is a bio‐physico‐chemical conundrum in which the efficiency of biological HgII methylation appears to depend chiefly on HgII and nutrients availability, the abundance of electron acceptors such as sulfate or iron, the abundance and composition of organic matter as well as the activity and structure of the microbial community. An increased knowledge of the relationship between microbial community composition, physico‐chemical conditions, MMHg production, and demethylation is necessary to predict variability in MMHg concentrations across environments.
The mercury problem
Mercury (Hg) is a natural and ubiquitous trace metal in the environment that might damage the central nervous system and causes tremors, distorted speech, kidney effects, respiratory failure, dizziness, blurred vision, hallucinations, and even death in severely exposed people (Clarkson and Magos 2006). This pollutant is naturally emitted during episodic events such as volcanic eruptions or ubiquitous weathering of Hg‐containing rocks in the Earth's crust and geothermal activity. Among anthropogenic Hg sources, artisanal and small‐scale gold mining, coal combustion, production of nonferrous metals, cement production, and disposal of wastes containing Hg are of special concern (UNEP 2013). Hg is unique among transition metals due to its high volatility as gaseous elemental Hg (Hg0), with a residence time in the atmosphere of about 6–12 months, allowing for long‐range transport of Hg. Although both Hg0 and inorganic divalent Hg (HgII) are released from many sources through a variety of natural and anthropogenic processes, the reported rise in Hg levels in the biosphere, and in terrestrial and marine systems is a consequence of anthropogenic emissions (Amos et al. 2013; Lamborg et al. 2014; Kocman et al. 2017). In contrast to Hg0 and HgII, direct anthropogenic sources of organic Hg, mono‐methylmercury (MMHg, i.e., CH3Hg+) or dymethylmercury (DMHg, i.e., [CH3]2Hg) are scarce.
The chemical behaviors of the different chemical forms of Hg (i.e., Hg0, HgII, CH3HgI and (CH3)2Hg) play critical roles in the biogeochemical cycling of Hg. Hg0 allows for long‐range transport (Jackson 1997; Pirrone et al. 2009), HgII is the dominant reservoir for Hg in soils and aquatic systems (Fleck et al. 2015; Eklöf et al. 2018), and MMHg is bioconcentrated and biomagnified in aquatic food webs, reaching up to 80–100% of the total‐Hg (THg) measured in fish muscle (Bloom 1992; Mason et al. 2012; Bravo et al. 2014). As a consequence, MMHg exposure through fish consumption is of special concern for human health. A recent study performed in 175 countries, showed that 38% of studied populations (mainly insular and developing nations) were exposed to doses of MMHg above governmental thresholds (Lavoie et al. 2018). Indeed, concentration of Hg in fish is known to repeatedly overpass environmental quality guidelines even in absence of local sources (Depew et al. 2013; Åkerblom et al. 2014; Eagles‐Smith et al. 2016).
Comprehensive evaluations of the chemical and physical processes that govern Hg distribution and fate among the major environmental compartments can be found in the literature (Chételat et al. 2015; Sundseth et al. 2015; Kim et al. 2016; Bjørklund et al. 2017; Dranguet et al. 2017; Paranjape and Hall 2017; Klapstein and Driscoll 2018). Briefly, in the water column HgII can (1) be reduced to Hg0 and reemitted back to the atmosphere, (2) methylated to the organic form MMHg, or (3) bind to organic matter (OM) as well as inorganic particles and directly deposit to bottom sediments. MMHg formed in aquatic ecosystems can also deposit to sediments, be methylated and/or form DMHg. Part of DMHg might be re‐emitted to the atmosphere or again degraded to MMHg, which can also be biotically (Barkay et al. 2003) or abiotically demethylated (Fernández‐Gómez et al. 2013). Although some of the MMHg found in aquatic systems might come from the degradation of DMHg to MMHg, i.e., in oceans (Mason et al. 2012), several studies concluded that most of the MMHg measured in ecosystems was formed in situ or in the surrounding catchment (e.g., soils, wetlands, etc.) and subsequently transported into rivers, lakes (Louis et al. 1996; Eklöf et al. 2012; Bravo et al. 2017), and oceans (Schartup et al. 2015). Abiotic methylation of HgII is possible if suitable methyl donors are present (Celo et al. 2006; Munson et al. 2018). Nevertheless, recent studies have shown that biological HgII methylation is in most environments performed by a variety of microorganisms, carrying the hgcA and hgcB gene cluster (Gilmour et al. 2013, 2018; Parks et al. 2013; Yu et al. 2018). An increasing number of recent studies detailed below have intended to evaluate the biotic HgII methylation by studying the biodiversity and activity of hgcAB+ microorganisms (Gionfriddo et al. 2016; Bravo et al. 2018b,a; Bowman et al. 2019; Jones et al. 2019; Villar et al. 2019). Nevertheless, it is established that net MMHg production also depends on other concomitant processes, including (1) the composition and activity of the whole microbial community that in turn modulate the activity of hgcAB+ microorganisms (Bravo et al. 2018a), (2) physico‐chemistry that controls HgII bioavailability (Schaefer and Morel 2009; Jonsson et al. 2012; Chiasson‐Gould et al. 2014), and uptake in microorganisms (Schaefer et al. 2011), and (3) biotic and abiotic MMHg demethylation (Du et al. 2019).
Recent reviews critically summarized HgII uptake and MMHg efflux in methylating anaerobes, methods and equations to analyze Hg methylation rates (Regnell and Watras 2019) as well as chemotrophic and biotic Hg methylation and demethylation processes by anaerobes and phototrophs (Grégoire and Poulain 2018; Du et al. 2019). In this review, we aim to summarize the main findings on mechanisms responsible for the formation of MMHg, focusing in detail on the new knowledge recently gained on microorganisms involved in MMHg formation. We also aim to highlight pitfalls and limitations impeding the progress in the current understanding, and we propose a road map to overcome these limitations. Indeed, to improve our ability to predict MMHg generation in the environment, a current research priority is to better understand the distribution of methylating populations in the context of the physico‐chemical constraints known to affect MMHg production.
Methylmercury formation is widespread in the environment
Recent advances in the methodology to (1) determine in situ HgII methylation (Jonsson et al. 2014) and (2) identify the organisms involved in this process (Parks et al. 2013; Christensen et al. 2016) have revealed that MMHg can be formed in a wider range of environments than previously identified. Indeed, 30 yr ago, first studies showed that HgII methylation in aquatic systems occurred mainly in sediments and under anaerobic conditions (Compeau and Bartha 1984; Korthals and Winfrey 1987; Pak and Bartha 1998). In general, sediments and sinking particles are a complex matrix of solid phases including clays, quartz, metal oxides (FeOOH, MnO2, AlO3) carbonates, sulfides and a number of other minerals and OM. They provide various microenvironments and habitats to organism populations notably bacteria, archaea, algae, diverse invertebrates, and so forth. To date, biological HgII‐methylation is known to be mediated by species carrying the hgcAB gene cluster (Parks et al. 2013). Because all the identified microorganisms with HgII‐methylating capacity were anaerobes, it was assumed for a long time that MMHg formation was occurring in strictly anoxic environments, i.e., sediments. Nonetheless, several studies revealed that HgII‐methylation can occur in oxygen deficient zones of water column (Eckley et al. 2005; Malcolm et al. 2010), sediments (Drott et al. 2008; Hines et al. 2012; Bouchet et al. 2013; Jonsson et al. 2014; Bravo et al. 2015; Liem‐Nguyen et al. 2016) flooded soils, e.g., wetlands (Louis et al. 1996; Tjerngren et al. 2012; Windham‐Myers et al. 2014) and ponds (Lehnherr et al. 2012; MacMillan et al. 2015; Herrero Ortega et al. 2018). In recent years, HgII‐methylation processes were in addition observed in microenvironments such as periphyton, growing on macrophytes (Cleckner et al. 1999; Mauro et al. 2002; Guimarães et al. 2006; Achá et al. 2011; Hamelin et al. 2011; Bouchet et al. 2018) and settling particles of oxic water columns, including pelagic ocean waters (Monperrus et al. 2007; Cossa et al. 2009; Sunderland et al. 2009;Lehnherr et al. 2011 ; Gascón Díez et al. 2016). Initially, MMHg formation in oxic waters was considered negligible due to high redox and low concentrations of bacteria and nutrients, but studies demonstrated that about 20–40% of the MMHg measured below the surface mixed layer originates from the surface and enters deeper ocean waters (Blum et al. 2013). Similarly, a query of more than 3500 publicly available microbial metagenomes performed by Podar et al. (2015) unveiled the presence of hgcAB‐like genes in sediments and in previously unsuspected environments, including invertebrate digestive tracts, thawing permafrost soils, coastal “dead zones,” soils and extreme environments. Moreover, a recent study assessing 243 metagenomes from the Tara Oceans expedition reported high abundances of hgcAB genes in 77 samples across all oceans (Villar et al. 2019). The progress in genetics (Gilmour et al. 2013; Parks et al. 2013; Podar et al. 2015; Bravo et al. 2018b,a; Liu et al. 2018b; Jones et al. 2019) combined with recent advances in the use of stable isotopes to determine HgII methylation rate constants in sediments (Monperrus et al. 2007; Jonsson et al. 2012; Bravo et al. 2014, 2015), lakes (Eckley and Hintelmann 2006), water columns and oceans (Munson et al. 2018) as well as in sinking particles of marine and lake waters (Lehnherr et al. 2011; Gascón Díez et al. 2016) have demonstrated that the potential for MMHg formation in the environment is widespread across ecosystems.
Toward a better understanding of microbial methylmercury formation
The discovery of hgcAB
Fifty years ago, a decade after the first observation of the Minamata disease in Japan, pioneering research pointed to surface sediments, and bacteria activity as responsible of HgII‐methylation (Jensen and Jernelöv 1969). One of the first studies targeting MMHg and bacteria, evaluated the HgII‐methylation capacity of Pseudomonas fluorescens, Mycobaeterium phlei, Escherichia coli, Aerobacter aerogenes, and Bacillus megaterium over a 7‐day period in pure cultures (Vonk and Sijpesteijn 1973). In the presence of sublethal amounts of HgCl2, tested bacteria produced 49 to 169 ng L−1 d−1 of MMHg in aerobic conditions (Vonk and Sijpesteijn 1973). Another decade later, sulfate‐reducing bacteria (SRB) were eventually identified as major HgII‐methylator in saltmarsh through inhibition of their activity with sodium molybdate and isolation of Desulfovibrio desulfuricans from sediments (Compeau and Bartha 1985).
The relationship between bacterial sulfate reduction and HgII‐methylation was studied adding HgII to anoxic sediment slurries or lake water overlying intact sediment cores collected in Quabbin Reservoir, MA (Gilmour et al. 1992). Comparable profiles of sulfate reduction and HgII‐methylation in sediment cores were reported, further suggesting that HgII‐methylation was linked to this specific bacterial metabolism. Almost 20 yr ago, a correlation between the HgII‐methylation and sulfate reduction rates in sediment of a saltmarsh was also shown (King et al. 2000, 2001). Sulfate reduction was then accepted as the main metabolic pathway related to HgII‐methylation. In 2006, two studies revealed the role of iron‐reducing bacteria (FeRB) on HgII‐methylation in ferruginous conditions (Fleming et al. 2006; Kerin et al. 2006). In 2010, Hamelin et al. further identified methanogens as important HgII‐methylators in lake periphyton. Two laboratory studies later confirmed the efficiency of methanogens in converting HgII to MMHg (Yu et al. 2013; Gilmour et al. 2018). By culturing and isolating HgII‐methylating strains or by using inhibitors of known HgII‐methylators such as molybdate for sulfate‐reduction and BESA for methanogenesis, HgII‐methylation has mainly been then attributed to the action of SRB (Devereux et al. 1996; Pak and Bartha 1998; Hylander 2000; King et al. 2001; Achá et al. 2011, 2012; Yu et al. 2012; Bravo et al. 2016), and in some cases to FeRB (Fleming et al. 2006; Bravo et al. 2015, 2018b) as well as methanogens (Hamelin et al. 2011; Bravo et al. 2018a).
A recent breakthrough in the understanding of the biological HgII‐methylation pathway was the identification of a two‐gene cluster, hgcAB ‐involved in C1 metabolism and the acetyl‐CoA pathway (Qian et al. 2016) required for HgII‐methylation (Parks et al. 2013). The gene hgcA encodes a corrinoid protein that is essential for the biosynthesis of the folate branch of acetyl‐CoA pathway, whereas the gene hgcB encodes a ferredoxin‐like protein thought to be an electron donor to hgcA (Parks et al. 2013). Both provide methyl groups required for HgII methylation, although it is not clear whether MMHg production is a controlled or an accidental metabolic process (Qian et al. 2016). However, deletion of either gene eliminated HgII methylation in Desulfovibrio desulfuricans ND132 (Parks et al. 2013). By directly measuring HgII methylation in several bacterial and archaeal strains encoding hgcAB, Gilmour et al. (2013) confirmed that HgII‐methylation capability could be predicted by the presence of hgcAB in the genome. For the first time, Gilmour et al. (2013) demonstrated HgII‐methylation capability in previously completely unsuspected species including syntrophic, acetogenic, and fermentative Firmicutes.
In recent years, the biodiversity of hgcAB microorganisms was actively studied in contrasting environments. First biodiversity studies using this newly identified gene cluster were based on classical polymerase chain reaction (PCR) amplification with one pair of primers targeting hgcA developed based on the couple of available sequenced genomes of methylating strains at that time (Table 1), followed by cloning and sequencing (Bae et al. 2014; Liu et al. 2014; Schaefer et al. 2014; Smith et al. 2015). In soils of the Florida Everglades, the sequences identified were distributed in diverse phyla, including Deltaproteobacteria, Chloroflexi, Firmicutes, and Methanomicrobia; however, hgcA clone libraries from all sites were dominated by sequences clustering within the order Syntrophobacterales (Bae et al. 2014) (Table 2). By comparing the taxonomically identified hgcA sequences with the activity of SRB (mRNA of dsrB gene), Bae et al. concluded that Syntrophobacterales largely dominated the HgII methylating microbial community of the Florida Everglades (Bae et al. 2014). In the Three Gorges Reservoir in China, PCR amplification and sequencing of hgcA gene resulted in the identification of δ‐Proteobacteria, methanogens and a Clostridia group as putative HgII methylators in this ecosystem. Authors reported in addition a positive correlation between the abundance of hgcA and dsrB genes and MMHg concentrations, suggesting SRB as the main group responsible for HgII methylation in those systems (Luo et al. 2016). In Wanshan Hg mining area of China, the taxonomically annotated sequences were related to δ‐Proteobacteria, Firmicutes, Chloroflexi, and Euryarchaeota (Liu et al. 2014). In temperate and tropical wetland soils, hgcA gene sequences were attributed to δ‐Proteobacteria, Chloroflexi, and Methanomicrobia (Schaefer et al. 2014). In nine rice paddy soils sampled in three mining areas in China, hgcA+ microbes were dominated by Proteobacteria or Euryarcheaota in six and three sites, respectively. Only nine of the 190 operational taxonomic unit (OTUs) found in these rice paddy soils were common to all sites (Liu et al. 2018a).
Table 1.
Published studies on hgcA diversity and quantification in environmental samples. Sequences and position of primers used, amplicon sizes, targets, and used methods are given.
| References | Primer sequences 5′‐ > 3′ | Target | Amplicon size (bp) | Methods |
|---|---|---|---|---|
| Bae et al. 2014 | hgcA_268F GGNRTYAAY RTNTGGTGYGC | hgcA‐hgcB | 888 to 945 | PCR and cloning |
| hgcB_1198R CADGCNCCRCAYTCVATRCA | ||||
| Bowman et al. 2019 | NA | Metagenomics | ||
| Bravo et al. 2018a | As Schaefer et al. 2014 | PCR and high throughput sequencing | ||
| Bravo et al. 2018b | As Schaefer et al. 2014 | PCR and high throughput sequencing | ||
| Bravo et al. 2016 | hgcA_262F GGNRTYAAYRTNTGGTGYGC | hgcA | 650 | qPCR |
| hgcA_912R GGTGTAGGGGGTGCAGCCSGTRWARKT | ||||
| Christensen et al. 2016, 2018 | ORNL‐HgcAB‐uni‐268F AAYGTCTGGTGYGCNGCVGG | hgcA‐hgcB | 818 to 1020 | PCR |
| ORNL‐HgcAB‐uni‐1198R CABGCNCCRCAYTCCATRCA | ||||
| ORNL‐Delta‐HgcA‐181F GCCAACTACAAGMTGASCTWC | hgcA of SRB | 107 | qPCR | |
| ORNL‐Delta‐HgcA‐287R CCSGCNGCRCACCAGACRTT | ||||
| ORNL‐SRB‐firm‐HgcA‐444F TGGDCCGGTDARAGCWAARGATA | hgcA of Firmicutes | 167 | qPCR | |
| ORNL‐SRB‐firm‐HgcA‐610R AAAAGAGHAYBCCAAAAATCA | ||||
| ORNL‐archaea‐HgcA‐184F AAYTAYWCNCTSAGYTTYGAYGC | hgcA of Archae | 125 | qPCR | |
| ORNL‐archaea‐HgcA‐308R TCDGTCCCRAABGTSCCYTT | ||||
| Dranguet et al. 2017 | As Bravo et al. 2016 | qPCR | ||
| Du et al. 2017 | As Schaefer et al. 2014 | qPCR, PCR, and cloning | ||
| Gionfriddo et al. 2016 | NA | Metagenomics | ||
| Lei et al. 2019 | As Christensen et al. 2016 | qPCR | ||
| Liu et al. 2014 | hgcA_626F GGNRTYAAYRTCTGGTGYGC | hgcA | 315 | PCR and cloning, qPCR |
| Liu et al. 2018a | hgcA_941R CGCATYTCCTTYTYBACNCC | |||
|
As Schaefer et al. 2014 hgcA_515F GTGCCAGCMGCCGCGGTAA′ hgcA_806R GGACTACHVGGGTWTCTAAT |
hgcA |
291 |
PCR and cloning qPCR |
|
| Liu et al. 2018b | As Christensen et al. 2016 |
hgcA hgcAB |
qPCR, metagenomics PacBio sequencing |
|
| Ma et al. 2017 | As Bae et al. 2014 | qPCR | ||
| Ndu et al. 2018 | As Christensen et al. 2016 | PCR and cloning, qPCR | ||
| Podar et al. 2015 | NA | hgcAB | Metagenomics | |
| Schaefer et al. 2014 | hgcA_261F CGGCATCAAYGTCTGGTGYGC | |||
| hgcA_912R GGTGTAGGGGGTGCAGCCSGTRWARKT | hgcA | PCR and cloning | ||
| Villar et al. 2019 | NA | Metagenomics | ||
| Vishnivetskaya et al. 2018 | As Christensen et al. 2016 | PCR and cloning, qPCR | ||
| Xu et al. 2019 | As Schaefer et al. 2014 | PCR and high throughput sequencing |
Table 2.
Main characteristics and outcomes of published studies on hgcA biodiversity in environmental samples.
| Method | Studied environment | Number of sequence/reads | Number of OTUs | Dominant group | Dominant Deltaproteobacteria | Reference |
|---|---|---|---|---|---|---|
| PCR and cloning | Florida Everglades | 220 | 168 | Deltaproteobacteria | Syntrophobacterales | Bae et al. 2014 |
| Wetlands | 108 | 40 | Methanomicrobia | Geobacter | Schaefer et al. 2014 | |
| Three gorges reservoir | 151 | 151 | Unidentified | Unidentified | Du et al. 2017 | |
| Rice paddy soils | ∼ 1800 | 190 |
Proteobacteria Euryarchaeota |
Liu et al. 2018a | ||
| Laboratory and sediment slurries | Unidentified | Unidentified | Ndu et al. 2018 | |||
| Rice paddy soils | 5 | ? | Unidentified | Vishnivetskaya et al. 2018 | ||
| Sulfate‐impacted lakes | 300 | 174 |
Geobacteraceae Methanomicrobia Unidentified |
Jones et al. 2019 | ||
| PCR and high throughput sequencing | Boreal lakes | 78,642 | 225 | Deltaproteobacteria | Unidentified | Bravo et al. 2018a |
| Lake Geneva | 741,890 | 356 | Deltaproteobacteria | Unidentified (Geobacter) | Bravo et al. 2018b | |
| Rice paddy soils | Liu et al. 2018a | |||||
| Boreal forest soils | 1,257,577 | 573 | Deltaproteobacteria | Unidentified (Geobacter) | Xu et al. 2019 | |
| Metagenomics | Global | 823,000,000 | Environmental compartment dependent | Podar et al. 2015 | ||
| Antarctic Sea‐ice and brine | Nitrospinae | Gionfriddo et al. 2016 | ||||
| Rice paddy soils | 901,610,484 | Methanoregula spp. | Geobacter | Liu et al. 2018b | ||
| Sulfate‐impacted lakes | 885,923 | 27 | Deltaproteobacteria | Jones et al. 2019 | ||
| Eight sites |
381–102 1 |
Deltaproteobacteria | Christensen et al. 2019 | |||
| Tara gene catalogs oceans | 111,530,851 | 10 | Nitrospinae | Desulfovibrionales | Villar et al. 2019 | |
| Metaproteomics | EFPC and Hinds Creek, TN | 15,270–16,852 | Deltaproteobacteria | Christensen et al. 2019 |
Based on a higher number of sequenced microbial genomes now available, Christensen et al. (2016) developed a broad range hgcAB primer pair that improved the coverage of prior developed primers by 10% (Bae et al. 2014; Schaefer et al. 2014), and several clade‐specific PCR primers to improve amplification of the various members of the HgII‐methylating community (Christensen et al. 2016) (Table 1). In sediments, these clade‐specific primers were useful to detect HgII‐methylating δ‐Proteobacteria and Archaea but failed to detect HgII‐methylating Firmicutes (Christensen et al. 2017).
Recent studies based on hgcAB biodiversity analyzed both hgcA (using one single pair of primers, Table 1) and 16S rRNA genes by high‐throughput illumina sequencing techniques, allowing a deeper sequencing than cloning‐sequencing and hence resulting in a higher number of OTUs. Results evidenced that microbial HgII‐methylating community was composed of members of various clades, including SRB, FeRB, methanogens and syntrophs in temperate and boreal lake sediments (Bravo et al. 2018a) as well as in boreal forest soils (Xu et al. 2019). In boreal lakes, besides the identification of HgII‐methylating methanogens and Geobacteraceae, authors further showed thanks to inhibition of sulfate reduction with molybdate that only 40% of MMHg was dependent on SRB (Bravo et al. 2018a). Another study performed in sediments impacted by a sewage treatment plant showed that HgII methylating Geobacteraceae seemed to have an important role in HgII methylation in sediments showing ferruginous conditions (Bravo et al. 2018b). Importantly, those studies suggested that the differences in the distributions of HgII methylating taxa among the different sites might derive primarily from different species of the same family having different niche requirements (Bravo et al. 2018a). In particular, the high relative abundance of phytoplankton‐derived OM and the presence of specific strains of non‐HgII‐methylating bacteria involved in OM decomposition (e.g., Rhizobiales, Fibrobacterales, Holophalages, etc.) seems to be essential in creating a niche that promotes HgII methylation (Bravo et al. 2018a; Lei et al. 2019). Another study conducted in sulfate‐impacted lakes combined cloning‐sequencing of hgcA with metagenomics targeting hgcA gene and genes involved in other metabolic functions (Jones et al. 2019). This approach yielded after in silico assembly of reads with overlapping sequences in a relatively low number of contigs (27), but revealed a high occurrence of hgcA genes together with genes involved in sulfate‐reduction and fermentation, but also that some abundant hgcA+ microbes were related to uncultivated microbes, such as Aminicenantes, Kiritimatiellaeota, Spirochaetes, as well as completely unidentified microbes. Data showed that potential methylators from uncultivated organisms occurred more abundantly than previously anticipated in these overlooked clades and that they can dominate the methylating community in certain circumstances (Jones et al. 2019).
A recent study conducted in rice paddy soils combining metagenomics illumina sequencing and long‐read PacBio sequencing, which allows overcoming the inherent risk of short‐reads chimeric assembly, revealed the dominance of Geobacter spp. for bacteria and Methanoregula spp. for Archaea (Liu et al. 2018b). These authors hypothesize a syntrophic interaction between both species and in addition reported a significant correlation between Geobacter hgcA+ DNA relative abundance and MMHg concentration in soils (Liu et al. 2018b), supporting an important role of this genus for MMHg production in iron‐rich paddy soils. A recent study in sediments collected in eutrophic lakes showing cyanobacteria blooms in China also reported a correlation between Archae hgcA+ DNA relative abundance and MMHg concentration (Lei et al. 2019). However, several studies that tried to correlate the level of expression of hgcA mRNA with HgII methylation rates (Goñi‐Urriza et al. 2015; Bravo et al. 2016; Christensen et al. 2019) were mostly unsuccessful. For example, in pure cultures of Desulfovibrio dechloroacetivorans BerOc1, the level of expression of hgcA was not correlated with HgII methylation rates (Goñi‐Urriza et al. 2015). Similarity, in sediments collected in a river impacted by effluents from a chlor‐alkali plant, data suggested that physico‐chemistry varied significantly among reservoirs, while functional gene activities, including hgcA, were very similar and did not correlate with MMHg concentrations (Bravo et al. 2016). In contrast, in Hg‐contaminated paddy soils, the hgcAB copy number increased with both increasing THg and MMHg concentrations (Vishnivetskaya et al. 2018).
Despite the differences in primer pairs used in the studies mentioned earlier (Table 1), until now, data globally suggested that in hgcA + δ‐Proteobacteria communities are abundant in surface sediments, but in some sites hgcA + methanogens and other hgcA + uncultivated groups are prevalent (Christensen et al. 2017, 2019; Vishnivetskaya et al. 2018; Bravo et al. 2018a; Jones et al. 2019). Among δ‐Proteobacteria, syntrophs and Geobacter spp. appear more prevalent in hgcA + community than previously expected. Moreover, other groups of hgcA − microbes seem to be of high importance for HgII‐methylating species, certainly by providing some kind of dependence or mutualistic relationship in sediments (Bravo et al. 2018a; Liu et al. 2018b). For example, syntrophs have been shown to modulate HgII methylation of hgcA strains in controlled exposures (Yu et al. 2018). Syntrophy between methanogens or propionate utilizing syntrophs and SRB is hypothesized to enhance methylation in environments devoid of sulfate or where the type and concentration of energy sources are limiting.
Studies listed above focused in freshwaters and therefore the microorganisms processing HgII to MMHg in the ocean are still barely described. Podar et al. (2015) showed that hgcAB appeared to be abundant in marine sediments but they rarely found it in pelagic marine water column, as from 138 metagenome samples analyzed, only seven showed evidence of hgcAB. A recent analysis of 243 seawater metagenome samples from 68 different sites of the Tara Oceans revealed high abundances of hgcAB corresponding to taxonomic relatives of known HgII methylators from Deltaproteobacteria, Firmicutes, and Chloroflexi across all oceans, with the exception of the Arctic that was not studied (Villar et al. 2019). More recently, Bowman et al. (2019), combining PCR amplification and shotgun metagenomics, searched the hgcAB gene cluster in Arctic Ocean seawater without success. Out of all the hgcA‐like genes found in the queries of marine metagenomes, the Nitrospina phylum, a marine nitrite oxidizing bacteria abundant in oxygen‐deficient zones, appeared to be widespread, predominant and likely a key player for MMHg production in the oxic subsurface waters of the global ocean (Villar et al. 2019), including the Arctic (Bowman et al. 2019) as well as Antarctic sea ice–brine–sea water interfaces (Gionfriddo et al. 2016). However, despite metagenomic evidence for the abundance of Nitrospina in the global ocean, the few cultured strains harboring a fused hgcAB‐like gene (Methanococcoides methylutens and Pyrococcus furiosus) were unable to produce MMHg in experimental conditions (Podar et al. 2015; Gilmour et al. 2018). Moreover, there is yet no experimental or observational report on the expression of hgcAB‐like genes in Nitrospina bacteria. As such, an experimental evidence of the HgII methylating capacity in Nitrospina is awaited to confirm their role as important HgII methylators in the global ocean. As MMHg has been detected in the water column of every ocean basin, except for the Indian Ocean (Bowman et al. 2019), it is crucial to unveil the role of the microorganisms involved in both MMHg and degradation in seawaters.
These results and observations reveal the diversity of HgII‐methylating microbial communities’ structure across ecosystems and point for the need of a thorough investigation of their functioning. Notably further work is necessary to better understand the contribution of overlooked microbial groups in HgII methylation, highlighted by the high proportion of unidentified OTUs found in recent studies concerning hgcAB biodiversity (Table 2). However, it would be useful to agree on a standardized protocol to conduct hgcAB biodiversity studies and have an hgcAB open‐access library, as published studies are currently difficult to directly compare due to differences in methods, including primer pairs, alignment algorithms used and depth of sequencing.
HgII methylators are part of a complex microbial community
As described in the previous sections, current knowledge established that MMHg net production was linked to biotic and abiotic variables. Microorganisms behave differently from one system to another due to interactions with the physico‐chemical variables but also with other organisms of their (micro)environment (Andersson et al. 2014; Bravo et al. 2018a). Studies with one strain can only describe the metabolism of this strain in a batch (Andersson et al. 2014), which is useful for a mechanistic understanding of its potential metabolism. However, it cannot be straightforwardly applied for environmental predictions because its metabolism is likely modified by the activity of other microbial groups and the ambient physico‐chemistry. In this sense, one of the most insightful discoveries is the syntrophic HgII methylation recently described in both laboratory (Kerin et al. 2006; Ranchou‐Peyruse et al. 2009) and field studies (Yu et al. 2018). Syntrophy is just a “proof of concept” illustrating the complexity of microbial communities carrying out HgII methylation. It is also important to consider that within a microbial community, besides HgII, some bacteria carrying out the merB gene or other genes yet to be discovered, might demethylate MMHg (Barkay et al. 2003).
Electron donors are also essential for HgII methylation. Different microbial clades are involved in the anaerobic oxidation of OM from complex organic compounds generally that goes through several steps and processes (Gilmour et al. 2013; Bae et al. 2014). For example, an initial hydrolysis of large organic substances is followed by a fermentation of intermediates into smaller organic molecules, such as lactate, propionate, butyrate, acetate, and formate, as well as CO2 and H2. These fermentation products might then be used as electron donors for Geobacterales, Desulfovibrionales, and Syntrophobacterales known to host HgII methylators. HgII methylators thus likely rely on other microorganisms involved in the degradation of large organic compounds. Furthermore, it was also demonstrated that the specific metabolism of one strain may provoke a new metabolism (unknown) in another strain; this is called the Quorum sensing (Lovley and Chapelle 1995). It is therefore very important to better tackle the complexity of microbial communities and describe the compendium of metabolic processes that can affect directly or indirectly HgII methylation.
Physico‐chemistry plays a pivotal role in HgII methylation
Besides the presence and diversity of HgII methylating microbes, HgII methylation depends on the amount of HgII bioavailable for methylation (Schaefer et al. 2011; Jonsson et al. 2014), which is determined by chemical speciation of HgII, solubility of the Hg‐S particles (Hsu‐Kim et al. 2013; Liem‐Nguyen et al. 2017) as well as the availability of electron donors and acceptors for HgII methylating microorganisms (Desrochers et al. 2015). HgII methylation is a bio‐physico‐chemical conundrum because both the amount of HgII available for methylation and the activity of microorganisms involved in the process are determined by multiple physico‐chemical variables such as sulfur (Skyllberg et al. 2003; Drott et al. 2007), iron (Bravo et al. 2015) and OM concentration and speciation (Schartup et al. 2013; Bravo et al. 2017) as well as Eh, pH, nutrient availability, and temperature (Ullrich et al. 2001; Paranjape and Hall 2017) (Fig. 1).
Figure 1.
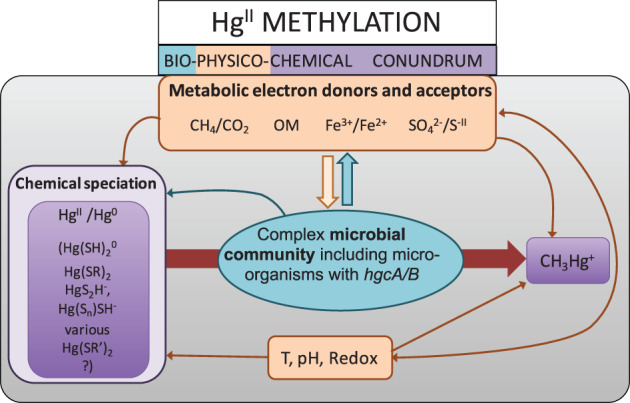
Conceptual summary of the biological and chemical interplays affecting HgII methylation in the environment. Orange boxes and arrows refer to geochemical variables directly affecting microbial activity and Hg speciation. Purple boxes refer to Hg chemical forms. The red arrow indicates the transformation of HgII to CH3Hg+. Blue refers to a compendium of metabolic processes occurring in the environment, among these processes HgII methylation carried out by the hgcAB gene cluster.
Variables affecting HgII chemical speciation
Salinity, sulfur, iron, and OM affect HgII chemical speciation. Sea salt anions may also affect HgII speciation and/or methylation in estuarine and marine environments. In marine waters, HgII forms compounds with chlorine (HgCl3 − and HgCl4 2−) to a greater extent than oxides, that are in turn formed in freshwaters (Mason and Fitzgerald 1993). A lower HgII methylating activity in marine and estuarine sediments than in freshwater sediments has been attributed to the formation of charged chloride but also sulfide complexes, that undergo a slower methylation processes than other HgII forms (Gårdfeldt et al. 2003; Jonsson et al. 2012; Gworek et al. 2016).
In the context of the biological and chemical interplays controlling HgII methylation, sulfur plays a central role by directly affecting HgII speciation and solubility (Jonsson et al. 2012; Graham et al. 2013; Hsu‐Kim et al. 2013; Liem‐Nguyen et al. 2017) and consequently its bioavailability (Schaefer and Morel 2009; Chiasson‐Gould et al. 2014; Schartup et al. 2015; Mazrui et al. 2016). Reactions between HgII and sulfide control the formation of the solid phase metacinnabar, β‐HgS(s) but also aqueous complexes such as Hg(SH)2 0, HgS2H−, and HgS2 2− or polysulfides HgSnSH−(aq) (n = 4–6) (Liem‐Nguyen et al. 2017). Elevated sulfide concentrations, eventually limits HgII bioavailability for methylation (Drott et al. 2007; Hsu‐Kim et al. 2013; Bigham et al. 2017). Conversely low sulfide concentrations might enhance HgII methylation processes (Benoit et al. 1999; Graham et al. 2012). Natural OM often contain thiols, which sulfhydryl group has a high capacity to complex HgII and MMHg (Skyllberg et al. 2006; Skyllberg 2008). Iron plays also a key role on HgII methylation as its reduced form Fe2+ can scavenge sulfide, form stable iron–sulfur compounds (FeS, Fe3S4, or FeS2) and then might let HgII bioavailable for methylation (i.e., ferruginous conditions; Bravo et al. 2015). Each of these HgII complexes, i.e., inorganic sulfides, polysulfides, and OM in aqueous, solid, and adsorbed phases, show different reactivity in the environment. For example, HgII methylation rate constants in estuarine sediments spanned over two orders of magnitude depending on the chemical form: metacinnabar (β—HgS(s)) < cinnabar (α—HgS(s)) < HgII reacted with mackinawite (≡FeS‐HgII) < HgII bonded to natural OM (NOM—HgII) < (Hg(NO3)2(aq)) that is a typical aqueous tracer (Jonsson et al. 2012).
The effect of natural OM on the sediment and pore water‐partitioning coefficient for HgII (Hammerschmidt et al. 2008; Liem‐Nguyen et al. 2016) needs still to be determined. Under low OM and porewater sulfide concentrations, HgII partitioning coefficient becomes a major factor for methylation (Mitchell and Gilmour 2008; Hollweg et al. 2009). Some studies have shown that increases in natural OM concentrations might also raise partitioning coefficients in sediment and pore water and have shown to decrease HgII concentration in the pore water, and thus its bioavailability for uptake by methylating microorganisms (Liem‐Nguyen et al. 2016). In contrast, other studies concluded that OM content did not explain variations in HgII partitioning, most likely only a limited fraction of OM was relevant for Hg complexation or because it is also possible that the composition, rather than amount, of OM controls HgII partitioning (Schartup et al. 2013).
High concentrations of OM decreased HgII bioavailability in laboratory experiments (Chiasson‐Gould et al. 2014) and in marine sediments (Hammerschmidt et al. 2008). Moreover, OM might also be important in determining HgII bioavailability by stabilizing HgS particles at nanoscale that can be methylated by anaerobic bacteria (Graham et al. 2013; Hsu‐Kim et al. 2013). To progress further in the understanding of HgII methylation in future research, it is of the upmost importance to measure and model HgII chemical speciation, which is ultimately determining the HgII availability for methylating bacteria (Fig. 1, violet box).
Variables affecting microbial activity
Sulfur, iron, OM, redox, pH, nutrients, and temperature affect microbial activity. Both sulfur and iron have oxidized forms (SO4 2− and Fe3+, respectively) that can serve as electron acceptor for some HgII methylating bacteria (e.g., SRB and FeRB). Natural OM, besides its strong capacity to bind HgII and influence HgII speciation, controls microbial activity and HgII methylation (Graham et al. 2013; Hsu‐Kim et al. 2013). The molecular composition of OM shows a central role in controlling HgII methylation (Bravo et al. 2017), notably phytoplankton derived OM and fresh humic matter were associated with both high bacterial activity and HgII methylation rates in sediments (Graham et al. 2013; Schartup et al. 2013; Mazrui et al. 2016; Bravo et al. 2017; Christensen et al. 2017; Herrero Ortega et al. 2018). Also, an increase in nutrients, associated to an enhanced algae biomass production, increased MMHg formation in sediments (Bravo et al. 2017; Herrero Ortega et al. 2018). Changes in redox conditions might also affect iron, sulfur and HgII speciation (Liem‐Nguyen et al. 2016), and the activity of the microorganisms (Grégoire and Poulain 2018) and consequently HgII methylation (Fig. 1, orange boxes and arrows). Furthermore, HgII methylation strongly depends on the activity of the whole microbial community (Weber et al. 2006; DeAngelis et al. 2010). Therefore, all the physico‐chemical variables causing increased microbial activity in sediments, such as temperature (Gudasz et al. 2010), might indirectly lead to enhanced HgII methylation (Bravo et al. 2017; Dijkstra et al. 2011).
From all the studies mentioned earlier, we can conclude that both microbial activity and HgII chemical speciation control the formation of MMHg in the environment. Therefore, Hg cycling and in particular, the methylation of HgII is an intricate process regulated by both physico‐chemical and biological constrains that needs holistic approaches to be fully understood.
Knowledge gaps and uncertainties
A high number of unidentified putative HgII methylators
The complexity of microbial communities and its implications for Hg cycling is one of the main current challenges. First biodiversity studies used 16S rRNA gene to investigate the diversity of HgII methylating microbial communities. But biodiversity of this gene cannot provide reliable and robust identification of HgII methylator diversity and abundance, because hgcAB+ strains are too rare (<1%) and not well identified in 16S rRNA databases (<300 species) (Miller and Bassler 2001; Christensen et al. 2019). Recent studies targeting hgcAB are interesting, but these molecular approaches entail several limits. First, as for all microbial biodiversity studies, the DNA extraction protocols need to be well planned to ensure clean subsampling avoiding contamination and may need to be optimized for the efficient recovery of DNA and the elimination of potential inhibitors for PCR and/or sequencing technologies. Indeed, DNA extraction and PCR amplification are known to be prone to artifacts due to any combination of high primer mismatch, low abundance, and/or low DNA extraction efficiency that can significantly affect results (Bravo et al. 2018a; Epp et al. 2019). Although claimed as “universal,” primers inherently show preferences and limitations in covering all species equally among different environmental samples. More specifically, currently available primer pairs for the gene hgcA are predicted to cover 84% (Schaefer et al. 2014) and 94% (Christensen et al. 2016) of the whole biodiversity by PCR‐based approaches. However, within amplified sequences of hgcA a significant proportion cannot be identified above the clade level because of (1) the lack of identified organisms in the databases, and (2) the low conservation of hgcA gene that is not ideal for biodiversity studies. Consequently, a significant proportion of OTUs are attributed to unidentified species. Indeed, identification of hgcA sequences from short reads is made by aligning metagenomics data with a set of known gene sequences isolated from cultivated strains. This approach is prone to inaccuracies, especially if the data are evaluated down to the genus level. For example, in recent studies 62% (Bravo et al. 2018b) and 57% (Bravo et al. 2018a) of the identified OTUs could be taxonomically assigned at the order level. More in detail, different algorithms are available to identify OTUs e.g., simple sequence alignment‐based algorithms (e.g., Basic Local Alignment Search Tool [BLAST]) or profile hidden Markov model (HMM)‐based searches, which are expected to be more sensitive and accurate in identifying homologs. Nonetheless, the HMM model is satisfying for hgcA but not for hgcB that cannot be confidently differentiates from other ferredoxin‐encoding genes due to homology (Christensen et al. 2019). Besides, both primers and algorithms are developed on sequences from cultivated strains and consequently identify those species with a higher efficiency (Podar et al. 2015; Bravo et al. 2018b; Liu et al. 2018b). Moreover, only few hundred sequenced genomes of methylating strains are currently available in databases. Although this number is increasing regularly, databases still do not include reliably microorganisms that cannot be grown in the laboratory. Besides we do not have information on the rates and methylation capabilities of these uncultivated populations and thus we cannot affirm that the genes identified code for MMHg production in these organisms. Further, we cannot evaluate how Hg methylation rates in these organisms compare to other methylators in culture. In sum, available sequences are far from being sufficient to confidently identify all OTUs homologs of hgcA and might not allow identifying unknown microbes. This is a current inherent technical limit that has to be considered when interpreting data.
However, studies conducted on DNA do not reflect the activity of the protein or the enzyme preforming the process, but only the possibility that a strain could sometime methylate HgII. Although the use of RNA should theoretically provide more detailed information on the activity of the organisms involved in HgII, up to now the abundance of hgcAB transcripts could not be correlated to HgII methylation or MMHg concentration (Goñi‐Urriza et al. 2015; Bravo et al. 2016; Vishnivetskaya et al. 2018; Christensen et al. 2019), except in one study (Ledeker and De Long 2013). The precise factors regulating hgcA gene and protein expression are not identified yet but it seems that hgcA gene could be constitutively expressed or regulated by carbon, metabolism, but Hg does not appear to be a key regulator (Goñi‐Urriza et al. 2015; Christensen et al. 2019). Moreover, the correlation between mRNA and protein abundance is not necessarily linear as posttranslational regulations can take place. In addition to the factors regulating hgcA gene expression, it is important to consider that net MMHg production also depends on MMHg demethylation activity. Therefore, the absence of correlation between hgcA gene and MMHg concentration is not surprising and highlights the complexity of predicting MMHg concentration dynamics in the environment.
Currently, the scientific community lacks a straightforward method to directly measure the activity of hgcAB at the protein level. New metaproteomic approaches are currently developed and could in the near future allow a direct analysis of hgcA protein abundance in the environment (Meier et al. 2019), but this approach has been tested only once to study hgcA biodiversity yet (Christensen et al. 2019). Metagenomics, metatranscriptomics, and metaproteomics offer a deeper sequencing and a wealth of information than earlier but they are cost and time‐consuming to conduct analysis of hundreds of samples. This highlights that we still lack of a simple and accurate method to identify HgII‐methylating microbial activity in environmental samples.
Another issue is that there is currently no medium or protocol to reliably culture a whole microbial community including FeRB, SRB, firmicutes, Clostridia, and so forth. Even studies with intact sediments in controlled experimental conditions can only approximate interactions of microbial community in situ and need to be confirmed in the field. For this reason, it is difficult to study and explain interactions of microbial communities through controlled experiments using a single strain. For example, microbial strains showing maximum methylation rates in the laboratory may not be as active in complex consortia under real field conditions. Metagenomics, metatranscriptomics, and metaproteomics could help to progress in the elucidation of the biological metabolism behind HgII methylation in the field. Instead of identifying different taxa, these approaches combine genes coding for all metabolic pathways of the different taxa to describe the potential function of the whole community (metagenomics) or its activity in one compartment (sediment, water, soil, etc.) at the moment of the sampling (metatranscriptomics and metaproteomics). Nonetheless, available metagenomics approaches have seldom identified hgcA sequences (i.e., 63 out of 203 metagenome projects revealed hgcA occurrence; Podar et al. 2015; and 77 out 243, Villar et al. 2019) supporting that new study should be conducted in environments relevant for HgII methylation. Furthermore, metatranscriptomics and metaproteomics are still rarely applied, most likely because they are expensive and time‐consuming. However, although we believe that these approaches are promising and their use will increase in coming years, they will need to be coupled with a detailed analysis of environmental conditions (e.g., physico‐chemistry analysis) to be fully informative.
Are we measuring realistic HgII methylation rates?
Last but not least, besides limitations to directly measure biological process responsible for HgII methylation, there are several pitfalls on the methods used to determine HgII methylation at environmentally relevant HgII concentrations. Several approaches have been used to estimate MMHg formation: using labeled HgII forms with radio‐isotopes (Goñi‐Urriza et al. 2015; Bravo et al. 2016), stable isotopes (Ramlal et al. 1986), by measuring the percentage of total Hg and MMHg (%MMHg) (Hintelmann et al. 2000) or the change in MMHg concentration over time (Drott et al. 2008). The amendment of standards enriched in HgII stable isotope tracers to environmental samples is now widely used in different laboratories because they allow determining HgII methylation rates and MMHg demethylation rates simultaneously. However, it is known that the geochemical form of the HgII isotope used as tracer determines its reactivity (i.e., methylation rate) in the environment. For example, HgII methylation rate constants in estuarine sediments spanned over two orders of magnitude from metacinnabar to a typical aqueous tracer such as Hg(NO3)2(aq) (Jonsson et al. 2012). Therefore, HgII methylation rate constants measured using a highly available tracer might result in an overestimation of the in situ HgII methylation rate, when the tracer is a poor representative of the indigenous HgII chemical forms. The %MMHg and the change of MMHg concentration overtime represent the net MMHg, accounting for MMHg degradation processes and while it might be useful to predict MMHg concentrations in the environment, its use is limited to provide mechanistic understanding of HgII methylation processes.
From this literature review, we observe that after 50 yr of efforts to study HgII methylation through the world, knowledge has greatly increased, but there are still many aspects within the bio‐physico‐chemistry of MMHg formation that need to be unveiled in the natural environment because: (1) there is a lack of techniques and methods for the measurements of the different HgII chemical species available for methylation; (2) the physico‐chemical factors affecting HgII chemistry are still only partly understood; (3) the biological mechanisms involved in the whole bio‐physico‐chemical process of MMHg net production remains to be understood and described more accurately.
How to study methylmercury formation in future research?
Besides current limits inherent to used analysis described earlier, main questions lacking a clear answer concerning the behavior of Hg in environmental systems include: What are all the HgII chemical species available for methylation? How diverse are hgcA + species? How does microbial community consortium impact the amount and speciation of HgII available for methylation? And what is the impact of the hgcA − microbial species on the activity of HgII methylators? Do techniques exist to tackle these questions?
Laboratory studies are essential to identify mechanisms driving HgII methylation but need to include more realistic scenarios and/or be validated under field conditions. Currently, in situ studies to elucidate the biodiversity of HgII methylators and determine the drivers of their activity need to be conducted more widely and in more diverse environments. For in situ studies, HgII speciation and HgII rates need to be determined, and accompanied by the study of the factors controlling the activity of HgII methylating microorganisms. Determination of the methylating activity of natural microbial assemblages in relation to sediment characteristics, specific environmental conditions and the level of Hg contamination needs to be undertaken to validate laboratory‐based measurements and to improve our understanding of Hg cycling in contaminated environments (Fig. 2). In this context, interactions between microbial communities and the physico‐chemistry are key to predict HgII methylation as several studies pointed out that physico‐chemistry rather than the microbial community structure were determining HgII methylation rate constants in lake sediments (Bravo et al. 2016, 2017, 2018a; Liu et al. 2018b). This is likely true but it should not be forgotten that the local physico‐chemistry is also the result of the microbial activity (Bravo et al. 2017, 2018b).
Figure 2.
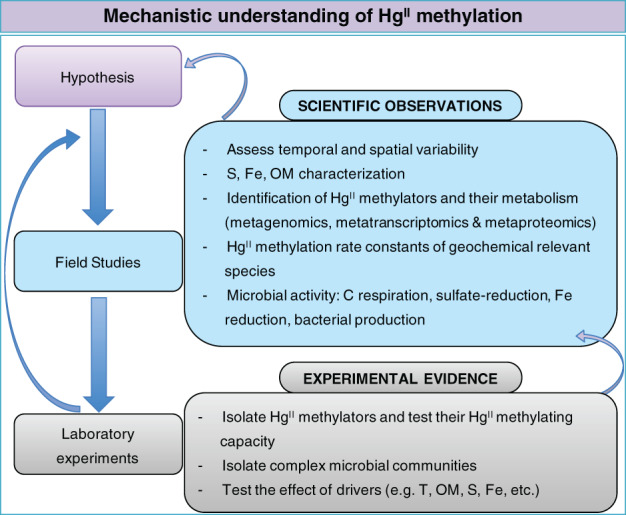
Schematic representation of the proposed conceptual iterative strategy for studying HgII methylation.
We can use existing and future new molecular biology and chemistry techniques to study HgII methylation, but studies have now to put more effort on combining metagenomics, metranscriptomics, and metaproteomics approaches with experiments on bacterial isolates and also complex matrices (i.e., sediments, marine waters, etc.) to explain more in detail environmental mechanisms (Fig. 2). Last, we should keep in mind that MMHg in aquatic systems is the net result of different processes: (1) formation (HgII methylation), (2) degradation (MMHg demethylation), and (3) the inputs and outputs of the system (Fig. 3). Therefore, when studying HgII methylation, we target only a limited part of the whole Hg cycling determining MMHg concentrations in the environment. Future studies need to be invested in studying concomitantly the drivers of MMHg methylation and MMHg demethylation mechanisms in situ as well as the transport of MMHg within the aquatic network, which is indeed highly controlled by hydrological processes (Fig. 3).
Figure 3.
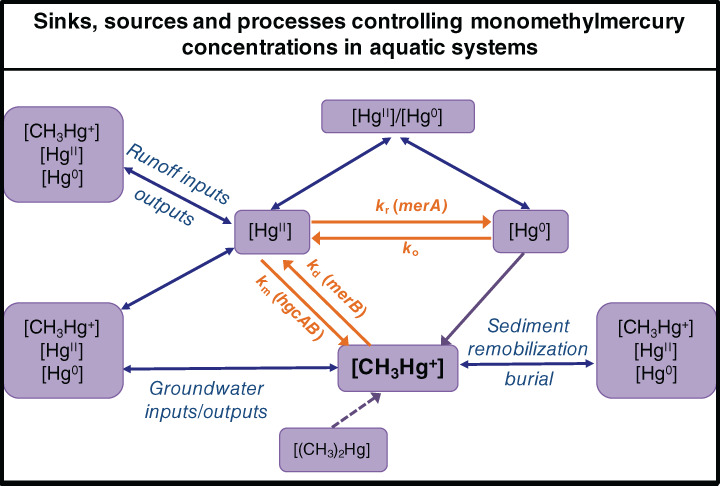
Proposed model to quantify MMHg in freshwater systems. The model considers formation (k m, HgII methylation) and degradation (k d, MMHg demethylation) and the inputs and outputs of MMHg. Processes are represented with orange arrows, known functional genes are shown in brackets. Transport of MMHg to the system or out the system is represented by blue arrows.
Conflict of Interest
None declared.
Acknowledgments
Authors thank Prof. Jeffra Schaeffer (Rutger University, USA) for critical reading of the draft.
AGB acknowledges the European Commission, for the MER‐CURE (Using global marine metagenomics to understand MERcury microbial associated processes: finding a CURE for mercury contaminated environments), an individual Fellowship of the Marie Skłodowska‐Curie Actions.
Author Contribution Statement: The two authors have equally contributed to this work.
Associate editor: Vanessa Hatje
Contributor Information
Andrea G. Bravo, Email: andrea.bravo@icm.csic.es.
Claudia Cosio, Email: claudia.cosio@univ-reims.fr.
References
- Achá, D. , Hintelmann H., and Yee J.. 2011. Importance of sulfate reducing bacteria in mercury methylation and demethylation in periphyton from Bolivian Amazon region. Chemosphere 82: 911–916. doi: 10.1016/j.chemosphere.2010.10.050 [DOI] [PubMed] [Google Scholar]
- Achá, D. , Hintelmann H., and Pabón C. A.. 2012. Sulfate‐reducing bacteria and mercury methylation in the water column of the Lake 658 of the experimental lake area. Geomicrobiol. J. 29: 667–674. doi: 10.1080/01490451.2011.606289 [DOI] [Google Scholar]
- Åkerblom, S. , Bignert A., Meili M., Sonesten L., and Sundbom M.. 2014. Half a century of changing mercury levels in Swedish freshwater fish. Ambio 43: 91–103. doi: 10.1007/s13280-014-0564-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos, H. M. , Jacob D. J., Streets D. G., and Sunderland E. M.. 2013. Legacy impacts of all‐time anthropogenic emissions on the global mercury cycle. Global Biogeochem. Cycles 27: 410–421. doi: 10.1002/gbc.20040 [DOI] [Google Scholar]
- Andersson, M. G. I. , Berga M., Lindström E. S., and Langenheder S.. 2014. The spatial structure of bacterial communities is influenced by historical environmental conditions. Ecology 95: 1134–1140. doi: 10.1890/13-1300.1 [DOI] [PubMed] [Google Scholar]
- Bae, H. S. , Dierberg F. E., and Ogram A.. 2014. Syntrophs dominate sequences associated with the mercury methylation‐related gene hgcA in the water conservation areas of the Florida Everglades. Appl. Environ. Microbiol. 80: 6517–6526. doi: 10.1128/AEM.01666-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkay, T. , Miller S. M., and Summers A. O.. 2003. Bacterial mercury resistance from atoms to ecosystems. FEMS Microbiol. Rev. 27: 355–384. doi: 10.1016/S0168-6445(03)00046-9 [DOI] [PubMed] [Google Scholar]
- Benoit, J. M. , Gilmour C. C., Mason R. P., and Heyes A.. 1999. Sulfide controls on mercury speciation and bioavailability to methylating bacteria in sediment pore waters. Environ. Sci. Technol. 33: 951–957. doi: 10.1021/es9808200 [DOI] [Google Scholar]
- Bigham, G. N. , Murray K. J., Masue‐Slowey Y., and Henry E. A.. 2017. Biogeochemical controls on methylmercury in soils and sediments: Implications for site management. Integr. Environ. Assess. Manag. 13: 249–263. doi: 10.1002/ieam.1822 [DOI] [PubMed] [Google Scholar]
- Bjørklund, G. , Dadar M., Mutter J., and Aaseth J.. 2017. The toxicology of mercury: current research and emerging trends. Environ. Res. 159: 545–554. doi: 10.1016/j.envres.2017.08.051 [DOI] [PubMed] [Google Scholar]
- Bloom, N. S. 1992. On the chemical form of m00ercury in edible fish and marine invertebrate tissue. Can. J. Fish. Aquat. Sci. 49: 1010–1017. doi: 10.1139/f92-113 [DOI] [Google Scholar]
- Bouchet, S. , and others. 2013. MMHg production and export from intertidal sediments to the water column of a tidal lagoon (Arcachon Bay, France). Biogeochemistry 114: 341–358. doi: 10.1007/s10533-012-9815-z [DOI] [Google Scholar]
- Bouchet, S. , and others. 2018. Linking microbial activities and low‐molecular‐weight thiols to Hg methylation in biofilms and periphyton from high‐altitude tropical lakes in the Bolivian Altiplano. Environ. Sci. Technol. 52: 9758–9767. doi: 10.1021/acs.est.8b01885 [DOI] [PubMed] [Google Scholar]
- Bowman, K. L. , Collins R. E., Agather A. M., Lamborg C. H., Hammerschmidt C. R., Kaul D., Dupont C. L., Christensen G. A., and Elias D. A.. 2019. Distribution of mercury‐cycling genes in the Arctic and equatorial Pacific oceans and their relationship to mercury speciation. Limnol. Oceanogr.: 1–11. doi: 10.1002/lno.11310 [DOI] [Google Scholar]
- Bravo, A. G. , Cosio C., Amouroux D., Zopfi J., Chevalley P.‐A., Spangenberg J. E., Ungureanu V.‐G., and Dominik J.. 2014. Extremely elevated methyl mercury levels in water, sediment and organisms in a Romanian reservoir affected by release of mercury from a chlor‐alkali plant. Water Res. 49: 391–405. doi: 10.1016/j.watres.2013.10.024 [DOI] [PubMed] [Google Scholar]
- Bravo, A. G. , Bouchet S., Guédron S., Amouroux D., Dominik J., and Zopfi J.. 2015. High methylmercury production under ferruginous conditions in sediments impacted by sewage treatment plant discharges. Water Res. 80: 245–255. doi: 10.1016/j.watres.2015.04.039 [DOI] [PubMed] [Google Scholar]
- Bravo, A. G. , Loizeau J. L., Dranguet P., Makri S., Björn E., Ungureanu V. G., Slaveykova V. I., and Cosio C.. 2016. Persistent Hg contamination and occurrence of Hg‐methylating transcript (hgcA) downstream of a chlor‐alkali plant in the Olt River (Romania). Environ. Sci. Pollut. Res. 23: 10529–10541. doi: 10.1007/s11356-015-5906-4 [DOI] [PubMed] [Google Scholar]
- Bravo, A. G. , Bouchet S., Tolu J., Björn E., Mateos‐Rivera A., and Bertilsson S.. 2017. Molecular composition of organic matter controls methylmercury formation in boreal lakes. Nat. Commun. 8: 14255. doi: 10.1038/ncomms14255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo, A. G. , and others. 2018a. Methanogens and iron‐reducing bacteria: the overlooked members of mercury‐methylating microbial communities in boreal lakes. Appl. Environ. Microbiol. 84: 0–16. doi: 10.1128/AEM.01774-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo, A. G. , Zopfi J., Buck M., Xu J., Bertilsson S., Schaefer J. K., Poté J., and Cosio C.. 2018b. Geobacteraceae are important members of mercury‐methylating microbial communities of sediments impacted by waste water releases. ISME J. 12: 802–812. doi: 10.1038/s41396-017-0007-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celo, V. , Lean D. R. S., and Scott S. L.. 2006. Abiotic methylation of mercury in the aquatic environment. Sci. Total Environ. 368: 126–137. doi: 10.1016/j.scitotenv.2005.09.043 [DOI] [PubMed] [Google Scholar]
- Chételat, J. , Braune B., Stow J., and Tomlinson S.. 2015. Special issue on mercury in Canada's north: summary and recommendations for future research. Sci. Total Environ. 509–510: 260–262. doi: 10.1016/j.scitotenv.2014.06.063 [DOI] [PubMed] [Google Scholar]
- Chiasson‐Gould, S. A. , Blais J. M., and Poulain A. J.. 2014. Dissolved organic matter kinetically controls mercury bioavailability to bacteria. Environ. Sci. Technol. 48: 3153–3161. doi: 10.1021/es4038484 [DOI] [PubMed] [Google Scholar]
- Christensen, G. A. , and others. 2016. Development and validation of broad‐range qualitative and clade‐specific quantitative molecular probes for assessing mercury methylation in the environment. Appl. Environ. Microbiol. 82: 6068–6078. doi: 10.1128/AEM.01271-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen, G. A. , and others. 2017. Carbon amendments alter microbial community structure and net mercury methylation potential in sediments. Appl. Environ. Microbiol. 84: 1–14. doi: 10.1128/AEM.01049-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen, G. A. , Somenahally A. C., Moberly J. G., Miller C. M., King A. J., Gilmour C. C., Brown S. D., Podar M., Brandt C. C., Brooks S. C., Palumbo A. V., Wall J. D., and Elias D. A., (2018) Carbon amendments alter microbial community structure and net mercury methylation potential in sediments. Applied and Environmental Microbiology 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen, G. A. , and others. 2019. Determining the reliability of measuring mercury cycling gene abundance with correlations with mercury and methylmercury concentrations. Environ. Sci. Technol. 53: 8649–8663. doi: 10.1021/acs.est.8b06389 [DOI] [PubMed] [Google Scholar]
- Clarkson, T. W. , and Magos L.. 2006. The toxicology of mercury and its chemical compounds. Crit. Rev. Toxicol. 36: 609–662. doi: 10.1080/10408440600845619 [DOI] [PubMed] [Google Scholar]
- Cleckner, L. B. , Gilmour C. C., Hurley J. P., and Krabbenhoft D. P.. 1999. Mercury methylation in periphyton of the Florida Everglades. Limnol. Oceanogr. 44: 1815–1825. doi: 10.4319/lo.1999.44.7.1815 [DOI] [Google Scholar]
- Compeau, G. , and Bartha R.. 1984. Methylation and demethylation of mercury under controlled redox, pH and salinity conditions. Appl. Environ. Microbiol. 48: 1203–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compeau, G. C. , and Bartha R.. 1985. Sulfate‐reducing bacteria : principal methylators of mercury in anoxic estuarine sediment. Appl. Environ. Microbiol. 50: 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossa, D. , Averty B., and Pirrone N.. 2009. The origin of methylmercury in open Mediterranean waters. Limnol. Oceanogr. 54: 837–844. doi: 10.4319/lo.2009.54.3.0837 [DOI] [Google Scholar]
- DeAngelis, K. M. , Silver W. L., Thompson A. W., and Firestone M. K.. 2010. Microbial communities acclimate to recurring changes in soil redox potential status. Environ. Microbiol. 12: 3137–3149. doi: 10.1111/j.1462-2920.2010.02286.x [DOI] [PubMed] [Google Scholar]
- Depew, D. , and others 2013. An overview of mercury concentrations in freshwater fish species: a national fish mercury dataset for Canada. Can. J. Fish Aquat. Sci. 451: 436–451. doi: 10.1139/cjfas-2012-033 [DOI] [Google Scholar]
- Desrochers, K. A. N. , Paulson K. M. A., Ptacek C. J., Blowes D. W., and Gould W. D.. 2015. Effect of electron donor to sulfate ratio on mercury methylation in floodplain sediments under saturated flow conditions. Geomicrobiol. J. 32: 924–933. doi: 10.1080/01490451.2015.1035818 [DOI] [Google Scholar]
- Devereux, R. , Winfrey M. R., Winfrey J., and Stahl D. A.. 1996. Depth profile of sulfate‐reducing bacterial ribosomal RNA and mercury methylation in an estuarine sediment. FEMS Microbiol. Ecol. 20: 23–31. doi: 10.1111/j.1574-6941.1996.tb00301.x [DOI] [Google Scholar]
- Dijkstra, P. , Thomas S. C., Heinrich P. L., Koch G. W., Schwartz E., and Hungate B. A.. 2011. Effect of temperature on metabolic activity of intact microbial communities: evidence for altered metabolic pathway activity but not for increased maintenance respiration and reduced carbon use efficiency. Soil Biol. Biochem. 43: 2023–2031. doi: 10.1016/j.soilbio.2011.05.018 [DOI] [Google Scholar]
- Dranguet, P. , Le Faucheur S., and Slaveykova V. I.. 2017. Mercury bioavailability, transformations, and effects on freshwater biofilms. Environ. Toxicol. Chem. 36: 3194–3205. doi: 10.1002/etc.3934 [DOI] [PubMed] [Google Scholar]
- Blum, J. D. , Popp B. N., Drazen J. C., Johnson M. W., and Anela Choy C.. 2013. Methylmercury production below the mixed layer in the North Pacific Ocean. Nat. Geosci. 6: 879–884. doi: 10.1038/ngeo1918 [DOI] [Google Scholar]
- Drott, A. , Lambertsson L., Björn E., and Skyllberg U.. 2007. Importance of dissolved neutral mercury sulfides for methyl mercury production in contaminated sediments. Environ. Sci. Technol. 41: 2270–2276. doi: 10.1021/es061724z [DOI] [PubMed] [Google Scholar]
- Drott, A. , Lambertsson L., Björn E., and Skyllberg U.. 2008. Do potential methylation rates reflect accumulated methyl mercury in contaminated sediments? Environ. Sci. Technol. 42: 153–158. doi: 10.1021/es0715851 [DOI] [PubMed] [Google Scholar]
- Du, H. , Ma M., Sun T., Dai X., Yang C., Luo F., Wang D., and Igarashi Y.. 2017. Mercury‐methylating genes dsrB and hgcA in soils/sediments of the three gorges reservoir. Environ. Sci. Pollut. Res. 24: 5001–5011. doi: 10.1007/s11356-016-8213-9 [DOI] [PubMed] [Google Scholar]
- Du, H. , Ma M., Igarashi Y., and Wang D.. 2019. Biotic and abiotic degradation of methylmercury in aquatic ecosystems: A review. Bull. Environ. Contam. Toxicol. 102: 605–611. doi: 10.1007/s00128-018-2530-2 [DOI] [PubMed] [Google Scholar]
- Eagles‐Smith, C. A. , and others 2016. Spatial and temporal patterns of mercury concentrations in freshwater fish across the Western United States and Canada. Sci. Total Environ. 568: 1171–1184. doi: 10.1016/j.scitotenv.2016.03.229 [DOI] [PubMed] [Google Scholar]
- Eckley, C. S. , and Hintelmann H.. 2006. Determination of mercury methylation potentials in the water column of lakes across Canada. Sci. Total Environ. 368: 111–125. doi: 10.1016/j.scitotenv.2005.09.042 [DOI] [PubMed] [Google Scholar]
- Eckley, C. S. , Watras C. J., Hintelmann H., Morrison K., Kent A. D., and Regnell O.. 2005. Mercury methylation in the hypolimnetic waters of lakes with and without connection to wetlands in northern Wisconsin. Can. J. Fish. Aquat. Sci. 411: 400–411. doi: 10.1139/F04-205 [DOI] [Google Scholar]
- Eklöf, K. , Fölster J., Sonesten L., and Bishop K.. 2012. Spatial and temporal variation of THg concentrations in run‐off water from 19 boreal catchments, 2000‐2010. Environ. Pollut. 164: 102–109. doi: 10.1016/j.envpol.2012.01.024 [DOI] [PubMed] [Google Scholar]
- Eklöf, K. , Bishop K., Bertilsson S., Björn E., Buck M., Skyllberg U., Osman O. A., Kronberg R. M., and Bravo A. G.. 2018. Formation of mercury methylation hotspots as a consequence of forestry operations. Sci. Total Environ. 613–614: 1069–1078. doi: 10.1016/j.scitotenv.2017.09.151 [DOI] [PubMed] [Google Scholar]
- Epp, L. S. , Zimmermann H. H., and Stoof‐Leichsenring K. R.. 2019. Sampling and extraction of ancient DNA from sediments, p. 31–44. In Ancient DNA: Methods and protocols, methods in molecular biology. Springer doi: 10.1007/978-1-4939-9176-1_5 [DOI] [PubMed] [Google Scholar]
- Fernández‐Gómez, C. , Drott A., Björn E., Díez S., Bayona J. M., Tesfalidet S., Lindfors A., and Skyllberg U.. 2013. Towards universal wavelength‐specific photodegradation rate constants for methyl mercury in humic waters, exemplified by a boreal lake‐wetland gradient. Environ. Sci. Technol. 47: 6279–6287. doi: 10.1021/es400373s [DOI] [PubMed] [Google Scholar]
- Fleck, J. A. , and others. 2015. Mercury and methylmercury in aquatic sediment across western North America. Sci. Total Environ. 568: 727–738. doi: 10.1016/j.scitotenv.2016.03.044 [DOI] [PubMed] [Google Scholar]
- Fleming, E. J. , Mack E. E., Green P. G., and Nelson D. C.. 2006. Mercury methylation from unexpected sources: molybdate‐inhibited freshwater sediments and an iron‐reducing bacterium. Appl. Environ. Microbiol. 72: 457–464. doi: 10.1128/AEM.72.1.457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gårdfeldt, K. , and others. 2003. Evasion of mercury from coastal and open waters of the Atlantic Ocean and the Mediterranean Sea. Atmos. Environ. 37: 73–84. doi: 10.1016/S1352-2310(03)00238-3 [DOI] [Google Scholar]
- Gascón Díez, E. , Loizeau J. L., Cosio C., Bouchet S., Adatte T., Amouroux D., and Bravo A. G.. 2016. Role of settling particles on mercury methylation in the oxic water column of freshwater systems. Environ. Sci. Technol. 50: 11672–11679. doi: 10.1021/acs.est.6b03260 [DOI] [PubMed] [Google Scholar]
- Gilmour, C. C. , Henry E. A., and Mitchell R.. 1992. Sulfate stimulation of mercury methylation in freshwater sediments. Environ. Sci. Technol. 26: 2281–2287. doi: 10.1021/es00035a029 [DOI] [Google Scholar]
- Gilmour, C. C. , and others. 2013. Mercury methylation by novel microorganisms from new environments. Environ. Sci. Technol. 47: 11810–11820. doi: 10.1021/es403075t [DOI] [PubMed] [Google Scholar]
- Gilmour, C. C. , Bullock A. L., Mcburney A., and Podar M.. 2018. Robust mercury methylation across diverse methanogenic archaea. mBio 9: 1–13. doi: 10.1128/mBio.02403-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gionfriddo, C. M. , and others. 2016. Microbial mercury methylation in Antarctic Sea ice. Nat. Microbiol. 1: 16127. doi: 10.1038/nmicrobiol.2016.127 [DOI] [PubMed] [Google Scholar]
- Goñi‐Urriza, M. , Corsellis Y., Lanceleur L., Tessier E., Gury J., Monperrus M., and Guyoneaud R.. 2015. Relationships between bacterial energetic metabolism, mercury methylation potential, and hgcA/hgcB gene expression in Desulfovibrio dechloroacetivorans BerOc1. Environ. Sci. Pollut. Res. 22: 13764–13771. doi: 10.1007/s11356-015-4273-5 [DOI] [PubMed] [Google Scholar]
- Graham, A. M. , Aiken G. R., and Gilmour C. C.. 2012. Dissolved organic matter enhances microbial mercury methylation under sulfidic conditions. Environ. Sci. Technol. 46: 2715–2723. doi: 10.1021/es203658f [DOI] [PubMed] [Google Scholar]
- Graham, A. M. , Aiken G. R., and Gilmour C. C.. 2013. Effect of dissolved organic matter source and character on microbial hg methylation in hg‐S‐DOM solutions. Environ. Sci. Technol. 47: 5746–5754. doi: 10.1021/es400414a [DOI] [PubMed] [Google Scholar]
- Grégoire, D. S. , and Poulain A. J.. 2018. Shining light on recent advances in microbial mercury cycling. Facets 3: 858–879. doi: 10.1139/facets-2018-0015 [DOI] [Google Scholar]
- Gudasz, C. , Bastviken D., Steger K., Premke K., Sobek S., and Tranvik L. J.. 2010. Temperature‐controlled organic carbon mineralization in lake sediments. Nature 466: 478–481. doi: 10.1038/nature09383 [DOI] [PubMed] [Google Scholar]
- Guimarães, J. R. D. , Mauro J. B. N., Meili M., Sundbom M., Haglund A. L., Coelho‐Souza S. A., and Hylander L. D.. 2006. Simultaneous radioassays of bacterial production and mercury methylation in the periphyton of a tropical and a temperate wetland. J. Environ. Manage. 81: 95–100. doi: 10.1016/j.jenvman.2005.09.023 [DOI] [PubMed] [Google Scholar]
- Gworek, B. , Bemowska‐Kałabun O., Kijeńska M., and Wrzosek‐Jakubowska J.. 2016. Mercury in marine and oceanic waters—a review. Water Air Soil Pollut. 227: 371. doi: 10.1007/s11270-016-3060-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelin, S. , Amyot M., Barkay T., Wang Y., and Planas D.. 2011. Methanogens: principal methylators of mercury in lake periphyton. Environ. Sci. Technol. 45: 7693–7700. doi: 10.1021/es2010072 [DOI] [PubMed] [Google Scholar]
- Hammerschmidt, C. R. , Fitzgerald W. F., Balcom P. H., and Visscher P. T.. 2008. Organic matter and sulfide inhibit methylmercury production in sediments of New York/New Jersey Harbor. Mar. Chem. 109: 165–182. doi: 10.1016/j.marchem.2008.01.007 [DOI] [Google Scholar]
- Herrero Ortega, S. , and others. 2018. High methylmercury formation in ponds fueled by fresh humic and algal derived organic matter. Limnol. Oceanogr. 63: S44–S53. doi: 10.1002/lno.10722 [DOI] [Google Scholar]
- Hines, M. E. , Poitras E. N., Covelli S., Faganeli J., Emili A., Žižek S., and Horvat M.. 2012. Mercury methylation and demethylation in Hg‐contaminated lagoon sediments (Marano and Grado lagoon, Italy). Estuar. Coast. Shelf Sci. 113: 85–95. doi: 10.1016/j.ecss.2011.12.021 [DOI] [Google Scholar]
- Hintelmann, H. , Keppel‐Jones K., and Evans D.. 2000. Constants of mercury methylation and demethylation rates in sediments and comparison of tracer and ambient mercury availability. Environ. Toxicol. Chem. 19: 2204–2211. doi: 10.1002/etc.5620190909 [DOI] [Google Scholar]
- Hollweg, T. A. , Gilmour C. C., and Mason R. P.. 2009. Methylmercury production in sediments of Chesapeake Bay and the mid‐Atlantic continental margin. Mar. Chem. 114: 86–101. doi: 10.1016/j.marchem.2009.04.004 [DOI] [Google Scholar]
- Hsu‐Kim, H. , Kucharzyk K. H., Zhang T., and Deshusses M. A.. 2013. Mechanisms regulating mercury bioavailability for methylating microorganisms in the aquatic environment: a critical review. Environ. Sci. Technol. 47: 2441–2456. doi: 10.1021/es304370g [DOI] [PubMed] [Google Scholar]
- Hylander, L. 2000. Relationship of mercury with aluminum, iron and manganese oxy‐hydroxides in sediments from the Alto Pantanal, Brazil. Sci. Total Environ. 260: 97–107. doi: 10.1016/S0048-9697(00)00544-1 [DOI] [PubMed] [Google Scholar]
- Jackson, T. a. 1997. Long‐range atmospheric transport of mercury to ecosystems, and the importance of anthropogenic emissions‐a critical review and evaluation of the published evidence. Environ. Rev. 5: 207. doi: 10.1139/er-5-3-4-207 [DOI] [Google Scholar]
- Jensen, S. , and Jernelöv A.. 1969. Biological methylation of mercury in aquatic organisms. Nature 223: 753–754. doi: 10.1038/223753a0 [DOI] [PubMed] [Google Scholar]
- Jones, D. S. , Walker G. M., Johnson N. W., Mitchell C. P. J., Coleman Wasik J. K., and Bailey J. V.. 2019. Molecular evidence for novel mercury methylating microorganisms in sulfate‐impacted lakes. ISME J. 13: 1659–1675. doi: 10.1038/s41396-019-0376-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson, S. , Skyllberg U., Nilsson M. B., Westlund P., Shchukarev A., Lundberg E., and Björn E.. 2012. Mercury methylation rates for geochemically relevant HgII species in sediments. Environ. Sci. Technol. 46: 11653–11659. doi: 10.1021/es3015327 [DOI] [PubMed] [Google Scholar]
- Jonsson, S. , Skyllberg U., Nilsson M. B., Lundberg E., Andersson A., and Bjo E.. 2014. Differentiated availability of geochemical mercury pools controls methylmercury levels in estuarine sediment and biota. Nat. Commun. 5: 4624–4634. doi: 10.1038/ncomms5624 [DOI] [PubMed] [Google Scholar]
- Kerin, E. J. , Gilmour C. C., Roden E., Suzuki M. T., Coates J. D., and Mason R. P.. 2006. Mercury methylation by dissimilatory Iron‐reducing bacteria. Appl. Environ. Microbiol. 72: 7919–7921. doi: 10.1128/AEM.01602-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K.‐H. , Kabir E., and Ara Jahan S.. 2016. A review on the distribution of Hg in the environment and its human health impacts. J. Hazard. Mater. 310: 278–279. doi: 10.1016/j.jhazmat.2016.02.043 [DOI] [PubMed] [Google Scholar]
- King, J. K. , Kostka J. E., Frischer M. E., and Saunders F. M.. 2000. Sulfate‐reducing bacteria methylate mercury at variable rates in pure culture and in marine sediments. Appl. Environ. Microbiol. 66: 2430–2437. doi: 10.1128/AEM.66.6.2430-2437.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, J. K. , Kostka J. E., Frischer M. E., Saunders F. M., and Jahnke R. A.. 2001. A quantitative relationship that demonstrates mercury methylation rates in marine sediments are based on the community composition and activity of sulfate‐reducing bacteria. Environ. Sci. Technol. 35: 2491–2496. doi: 10.1021/es001813q [DOI] [PubMed] [Google Scholar]
- Klapstein, S. J. , and Driscoll N. J. O.. 2018. Methylmercury biogeochemistry in freshwater ecosystems: a review focusing on DOM and photodemethylation. Bull. Environ. Contam. Toxicol. 100: 14–25. doi: 10.1007/s00128-017-2236-x [DOI] [PubMed] [Google Scholar]
- Kocman, D. , Wilson S. J., Amos H. M., Telmer K. H., Steenhuisen F., Sunderland E. M., Mason R. P., Outridge P., and Horvat M.. 2017. Toward an assessment of the global inventory of present‐day mercury releases to freshwater environments. Int. J. Environ. Res. Public Health 14: 138. doi: 10.3390/ijerph14020138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korthals, E. T. , and Winfrey M. R.. 1987. Seasonal and spatial variations in mercury methylation and demethylation in an oligotrophic lake. Appl. Environ. Microbiol. 53: 2397–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamborg, C. H. , and others. 2014. A global ocean inventory of anthropogenic mercury based on water column measurements. Nature 512: 65–68. doi: 10.1038/nature13563 [DOI] [PubMed] [Google Scholar]
- Lavoie, R. A. , Bouffard A., Maranger R., and Amyot M.. 2018. Mercury transport and human exposure from global marine fisheries. Sci. Rep. 8: 1–9. doi: 10.1038/s41598-018-24938-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeker, B. M. , and De Long S. K.. 2013. The effect of multiple primer‐template mismatches on quantitative PCR accuracy and development of a multi‐primer set assay for accurate quantification of pcrA gene sequence variants. J. Microbiol. Methods 94: 224–231. doi: 10.1016/j.mimet.2013.06.013 [DOI] [PubMed] [Google Scholar]
- Lehnherr, I. , Louis V. L. S., Hintelmann H., and Kirk J. L.. 2011. Methylation of inorganic mercury in polar marine waters. Nat. Geosci. 4: 298–302. doi: 10.1038/ngeo1134 [DOI] [Google Scholar]
- Lehnherr, I. , Louis V. L. S., and Kirk J. L.. 2012. Methylmercury cycling in high arctic wetland ponds: controls on sedimentary production. Environ. Sci. Technol. 46: 10523–10531. doi: 10.1021/es300577e [DOI] [PubMed] [Google Scholar]
- Lei, P. , Nunes L. M., Liu Y. R., Zhong H., and Pan K.. 2019. Mechanisms of algal biomass input enhanced microbial Hg methylation in lake sediments. Environ. Int. 126: 279–288. doi: 10.1016/j.envint.2019.02.043 [DOI] [PubMed] [Google Scholar]
- Liem‐Nguyen, V. , Jonsson S., Skyllberg U., Nilsson M. B., Andersson A., Lundberg E., and Björn E.. 2016. Effects of nutrient loading and mercury chemical speciation on the formation and degradation of methylmercury in estuarine sediment. Environ. Sci. Technol. 50: 6983–6990. doi: 10.1021/acs.est.6b01567 [DOI] [PubMed] [Google Scholar]
- Liem‐Nguyen, V. , Skyllberg U., and Björn E.. 2017. Thermodynamic modeling of the solubility and chemical cpeciation of mercury and methylmercury driven by organic thiols and micromolar sulfide concentrations in boreal wetland soils. Environ. Sci. Technol. 51: 3678–3686. doi: 10.1021/acs.est.6b04622 [DOI] [PubMed] [Google Scholar]
- Liu, Y.‐R. , Yu R.‐Q., Zheng Y.‐M., and He J.‐Z.. 2014. Analysis of the microbial community structure by monitoring an Hg methylation gene (hgcA) in paddy soils along an Hg gradient. Appl. Environ. Microbiol. 80: 2874–2879. doi: 10.1128/AEM.04225-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Ma A., Zhuang G., and Zhuang X.. 2018a. Diversity of microbial communities potentially involved in mercury methylation in rice paddies surrounding typical mercury mining areas in China. Microbiology 7: e00577. doi: 10.1002/mbo3.577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y.‐R. , Johs A., Bi L., Lu X., Hu H.‐W., Sun D., He J.‐Z., and Gu B.. 2018b. Unraveling microbial communities associated with methylmercury production in Paddy soils. Environ. Sci. Technol. 52: 13110–13118. doi: 10.1021/acs.est.8b03052 [DOI] [PubMed] [Google Scholar]
- Louis, V. L. S. T. , Rudd J. W. M., Kelly C. a., Beaty K. G., Flett R. J., and Roulet N. T.. 1996. Production and loss of methylmercury and loss of total mercury from boreal forest catchments containing different types of wetlands. Environ. Sci. Technol. 30: 2719–2729. doi: 10.1021/es950856h [DOI] [Google Scholar]
- Lovley, D. R. , and Chapelle F. H.. 1995. Deep subsurface microbial processes. Rev. Geophys. 33: 365–381. doi: 10.1029/95rg01305 [DOI] [Google Scholar]
- Luo, F. , Yang C., Sun T., Igarashi Y., Dai X., Wang D., Ma M., and Du H.. 2016. Mercury‐methylating genes dsrB and hgcA in soils/sediments of the three gorges reservoir. Environ. Sci. Pollut. Res. 24: 5001–5011. doi: 10.1007/s11356-016-8213-9 [DOI] [PubMed] [Google Scholar]
- Ma, M. , Du H., Wang D., Kang S., and Sun T.. 2017. Biotically mediated mercury methylation in the soils and sediments of Nam Co Lake, Tibetan plateau. Environ. Pollut. 227: 243–251. doi: 10.1016/j.envpol.2017.04.037 [DOI] [PubMed] [Google Scholar]
- MacMillan, G. A. , Girard C., Chételat J., Laurion I., and Amyot M.. 2015. High methylmercury in arctic and subarctic ponds is related to nutrient levels in the warming eastern Canadian arctic. Environ. Sci. Technol. 49: 7743–7753. doi: 10.1021/acs.est.5b00763 [DOI] [PubMed] [Google Scholar]
- Malcolm, E. G. , Schaefer J. K., Ekstrom E. B., Tuit C. B., Jayakumar A., Park H., Ward B. B., and Morel F. M. M.. 2010. Mercury methylation in oxygen deficient zones of the oceans: no evidence for the predominance of anaerobes. Mar. Chem. 122: 11–19. doi: 10.1016/j.marchem.2010.08.004 [DOI] [Google Scholar]
- Mason, R. P. , and Fitzgerald W. F.. 1993. The distribution and biogeochemical cycling of mercury in the equatorial Pacific Ocean. Deep. Res. Part I. 40: 1897–1924. doi: 10.1016/0967-0637(93)90037-4 [DOI] [Google Scholar]
- Mason, R. P. , Choi A. L., Fitzgerald W. F., Hammerschmidt C. R., Lamborg C. H., Soerensen A. L., and Sunderland E. M.. 2012. Mercury biogeochemical cycling in the ocean and policy implications. Environ. Res. 119: 101–117. doi: 10.1016/j.envres.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro, J. , Guimares J., Hintelmann H., Watras C., Haack E., and Coelho‐Souza S.. 2002. Mercury methylation in macrophytes, periphyton, and water ‐ comparative studies with stable and radio‐mercury additions. Anal. Bioanal. Chem. 374: 983–989. doi: 10.1007/s00216-002-1534-1 [DOI] [PubMed] [Google Scholar]
- Mazrui, N. M. , Jonsson S., Thota S., Zhao J., and Mason R. P.. 2016. Enhanced availability of mercury bound to dissolved organic matter for methylation in marine sediments. Geochim. Cosmochim. Acta 194: 153–162. doi: 10.1016/j.gca.2016.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier, D. V. , and others. 2019. Microbial metal‐sulfide oxidation in inactive hydrothermal vent chimneys suggested by metagenomic and metaproteomic analyses. Environ. Microbiol. 21: 682–701. doi: 10.1111/1462-2920.14514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, M. B. , and Bassler B. L.. 2001. Quorum sensing in bacteria. Annu. Rev. Microbiol. 55: 165–199. doi: 10.1146/annurev.micro.55.1.165 [DOI] [PubMed] [Google Scholar]
- Mitchell, C. P. J. , and Gilmour C. C.. 2008. Methylmercury production in a Chesapeake Bay salt marsh. J. Geophys. Res. Biogeo. 113: G00C04. doi: 10.1029/2008jg000765 [DOI] [Google Scholar]
- Monperrus, M. , Tessier E., Amouroux D., and Leynaert A.. 2007. Mercury methylation, demethylation and reduction rates in coastal and marine surface waters of the Mediterranean Sea. 107: 49–63. doi: 10.1016/j.marchem.2007.01.018 [DOI] [Google Scholar]
- Munson, K. M. , Lamborg C. H., Boiteau R. M., and Saito M. A.. 2018. Dynamic mercury methylation and demethylation in oligotrophic marine water. Biogeosciences 15: 6451–6460. doi: 10.5194/bg-15-6451-2018 [DOI] [Google Scholar]
- Ndu, U. , Christensen G. A., Rivera N. A., Gionfriddo C. M., Deshusses M. A., Elias D. A., and Hsu‐Kim H.. 2018. Quantification of mercury bioavailability for methylation using diffusive gradient in thin‐film samplers. Environ. Sci. Technol. 52: 8521–8529. doi: 10.1021/acs.est.8b00647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak, K. , and Bartha R.. 1998. Mercury methylation and demethylation in anoxic lake sediments and by strictly anaerobic bacteria. Appl. Environ. Microbiol. 64: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paranjape, A. R. , and Hall B. D.. 2017. Recent advances in the study of mercury methylation in aquatic systems, p. 85–119. In Recent advances in the study of mercury methylation in aquatic systems. doi: 10.1139/facets-2016-0027 [DOI] [Google Scholar]
- Parks, J. M. , and others 2013. The genetic basis for bacterial mercury methylation. Science 339: 1332–1335. doi: 10.1126/science.1230667 [DOI] [PubMed] [Google Scholar]
- Pirrone, N. , and others 2009. Global mercury emissions to the atmosphere from natural and anthropogenic sources In Mercury fate and transport in the global atmosphere: Emissions, measurements and models. Springer doi: 10.1007/s11259-009-9255-y [DOI] [Google Scholar]
- Podar, M. , and others. 2015. Global prevalence and distribution of genes and microorganisms involved in mercury methylation. Sci. Adv. 1: e1500675–e1500675. doi: 10.1126/sciadv.1500675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian, C. , Johs A., Chen H., Mann B. F., Lu X., Abraham P. E., Hettich R. L., and Gu B.. 2016. Global proteome response to deletion of genes related to mercury methylation and dissimilatory metal reduction reveals changes in respiratory metabolism in Geobacter sulfurreducens PCA. J. Proteome Res. 15: 3540–3549. doi: 10.1021/acs.jproteome.6b00263 [DOI] [PubMed] [Google Scholar]
- Ramlal, P. S. , Rudd J. W. M., and Hecky R. E.. 1986. Methods for measuring specific rates of mercury methylation and degradation and their use in determining factors controlling net rates of mercury methylation. Appl. Environ. Microbiol. 51: 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranchou‐Peyruse, M. , Monperrus M., Bridou R., Duran R., Amouroux D., Salvado J. C., and Guyoneaud R.. 2009. Overview of mercury methylation capacities among anaerobic bacteria including representatives of the sulphate‐reducers: implications for environmental studies. Geomicrobiol. J. 26: 1–8. doi: 10.1080/01490450802599227 [DOI] [Google Scholar]
- Regnell, O. , and Watras C. J.. 2019. Microbial mercury methylation in aquatic environments: A critical review of published field and laboratory studies. Environ. Sci. Technol. 53: 4–19. doi: 10.1021/acs.est.8b02709 [DOI] [PubMed] [Google Scholar]
- Schaefer, J. K. , and Morel F. M. M.. 2009. High methylation rates of mercury bound to cysteine by Geobacter sulfurreducens. Nat. Geosci. 2: 123–126. doi: 10.1038/ngeo412 [DOI] [Google Scholar]
- Schaefer, J. K. , Rocks S. S., Zheng W., Liang L., Gu B., and Morel F. M. M.. 2011. Active transport, substrate specificity, and methylation of Hg(II) in anaerobic bacteria. Proc. Natl. Acad. Sci. U. S. A. 108: 8714–8719. doi: 10.1073/pnas.1105781108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, J. K. , Kronberg R.‐M., Morel F. M. M., and Skyllberg U.. 2014. Detection of a key Hg methylation gene, hgcA, in wetland soils. Environ. Microbiol. Rep. 6: 441–447. doi: 10.1111/1758-2229.12136 [DOI] [PubMed] [Google Scholar]
- Schartup, A. T. , Mason R. P., Balcom P. H., Hollweg T. A., and Chen C. Y.. 2013. Methylmercury production in estuarine eediments: role of organic matter. Environ. Sci. Technol. 47: 695–700. doi: 10.1021/es302566w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schartup, A. T. , Ndu U., Balcom P. H., Mason R. P., and Sunderland E. M.. 2015. Contrasting effects of marine and terrestrially derived dissolved organic matter on mercury speciation and bioavailability in seawater. Environ. Sci. Technol. 49: 5965–5972. doi: 10.1021/es506274x [DOI] [PubMed] [Google Scholar]
- Skyllberg, U. 2008. Competition among thiols and inorganic sulfides and polysulfides for Hg and MeHg in wetland soils and sediments under suboxic conditions: illumination of controversies and implications for MeHg net production. J. Geophys. Res. 113: 1–14. doi: 10.1029/2008JG000745 [DOI] [Google Scholar]
- Skyllberg, U. , Qian J., Frech W., Xia K., and Bleam W. F.. 2003. Distribution of mercury, methyl mercury and organic sulphur species in soil, soil solution and stream of a boreal forest catchment. Biogeochemistry 64: 53–76. doi: 10.1023/A:1024904502633 [DOI] [Google Scholar]
- Skyllberg, U. , Bloom P. R., Qian J., Lin C.‐M., and Bleam W. F.. 2006. Complexation of mercury(II) in soil organic matter: EXAFS evidence for linear two‐coordination with reduced sulfur groups. Environ. Sci. Technol. 40: 4174–4180. doi: 10.1021/es0600577 [DOI] [PubMed] [Google Scholar]
- Smith, S. D. , and others. 2015. Site‐directed mutagenesis of hgcA and hgcB reveals amino acid residues important for mercury methylation. Appl. Environ. Microbiol. 81: 3205–3217. doi: 10.1128/AEM.00217-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunderland, E. M. , Krabbenhoft D. P., Moreau J. W., Strode S. A., and Landing W. M.. 2009. Mercury sources, distribution, and bioavailability in the North Pacific Ocean: insights from data and models. Global Biogeochem. Cycles 23. doi: 10.1029/2008GB003425 [DOI] [Google Scholar]
- Sundseth, K. , Pacyna J., Banel A., Pacyna E., and Rautio A.. 2015. Climate change impacts on environmental and human exposure to mercury in the Arctic. Int. J. Environ. Res. Public Health 12: 3579–3599. doi: 10.3390/ijerph120403579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjerngren, I. , Meili M., Björn E., and Skyllberg U.. 2012. Eight boreal wetlands as sources and sinks for methyl mercury in relation to soil acidity, C/N ratio, and small‐scale flooding. Environ. Sci. Technol. 46: 8052–8060. doi: 10.1021/es300845x [DOI] [PubMed] [Google Scholar]
- Ullrich, S. M. , Tanton T. W., Abdrashitova S. A., and Svetlana A.. 2001. Mercury in the aquatic environment: a review of factors affecting methylation. Crit. Rev. Environ. Sci. Technol. 31: 241–293. doi: 10.1080/20016491089226 [DOI] [Google Scholar]
- UNEP . 2013. Global mercury assessment 2013: Sources, emissions, releases, and environmental transport. UNEP 42. doi:DTI/1636/GE
- Villar, E. , Cabrol L., and Heimbürger‐Boavida L.‐E.. 2019. Widespread microbial mercury methylation genes in the global ocean. bioRxiv. doi: 10.1101/648329 [DOI] [PubMed] [Google Scholar]
- Vishnivetskaya, T. A. , and others. 2018. Microbial community structure with trends in methylation gene diversity and abundance in mercury‐contaminated rice paddy soils in Guizhou, China. Environ. Sci. Process. Impacts 20: 673–685. doi: 10.1039/C7EM00558J [DOI] [PubMed] [Google Scholar]
- Vonk, J. W. , and Sijpesteijn A. K.. 1973. Studies on the methylation of mercuric chloride by pure cultures of bacteria and fungi In Studies on the methylation of mercuric chloride by pure cultures of bacteria and fungi. Antonie Van Leeuwenhoek. doi: 10.1007/BF02578894 [DOI] [PubMed] [Google Scholar]
- Weber, K. A. , Achenbach L. A., and Coates J. D.. 2006. Microorganisms pumping iron: Anaerobic microbial iron oxidation and reduction. Nat. Rev. Microbiol. 4: 752–764. doi: 10.1038/nrmicro1490 [DOI] [PubMed] [Google Scholar]
- Windham‐Myers, L. , and others. 2014. Mercury cycling in agricultural and managed wetlands: A synthesis of methylmercury production, hydrologic export, and bioaccumulation from an integrated field study. Sci. Total Environ. 484: 221–231. doi: 10.1016/j.scitotenv.2014.01.033 [DOI] [PubMed] [Google Scholar]
- Xu, J. , and others. 2019. Mercury methylating microbial communities of boreal forest soils. Sci. Rep. 9: 518. doi: 10.1038/s41598-018-37383-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, R. Q. , Flanders J. R., MacK E. E., Turner R., Mirza M. B., and Barkay T.. 2012. Contribution of coexisting sulfate and iron reducing bacteria to methylmercury production in freshwater river sediments. Environ. Sci. Technol. 46: 2684–2691. doi: 10.1021/es2033718 [DOI] [PubMed] [Google Scholar]
- Yu, R.‐Q. , Reinfelder J. R., Hines M. E., and Barkay T.. 2013. Mercury methylation by the methanogen Methanospirillum hungatei . Appl. Environ. Microbiol. 79: 6325–6330. doi: 10.1128/AEM.01556-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, R. Q. , Reinfelder J. R., Hines M. E., and Barkay T.. 2018. Syntrophic pathways for microbial mercury methylation. ISME J. 12: 1826–1835. doi: 10.1038/s41396-018-0106-0 [DOI] [PMC free article] [PubMed] [Google Scholar]