


Abstract
Frequently, the complete functional units of biological molecules are assemblies of protein and nucleic acid chains. Stunning examples are the complex structures of ribosomes. Here, we present TopMatch-web, a computational tool for the study of the three-dimensional structure, function and evolution of such molecules. The unique feature of TopMatch is its ability to match the protein as well as nucleic acid chains of complete molecular assemblies simultaneously. The resulting structural alignments are visualized instantly using the high-performance molecular viewer NGL. We use the mitochondrial ribosomes of human and yeast as an example to demonstrate the capabilities of TopMatch-web. The service responds immediately, enabling the interactive study of many pairwise alignments of large molecular assemblies in a single session. TopMatch-web is freely accessible at https://topmatch.services.came.sbg.ac.at.
INTRODUCTION
In the last decades structural biology has revealed the detailed 3D structures of tens of thousands of molecules (1). Recent advances in X-ray crystallography, NMR spectroscopy and electron microscopy enabled the elucidation of huge molecular assemblies. Understanding the molecular basis of biological processes inevitably involves the investigation and comparison of these structures. In particular, the detection of similarities among macromolecules, their parts, and their assemblies provides vital information on their formation, function and evolution.
Prime examples of stunning molecular complexes are the many recently solved ribosomes from diverse organisms and organelles (2–6). These impressive molecular assemblies generally consist of up to one hundred protein chains and several large and small RNA chains. The study of the intricate relationships regarding structure, function and evolution of such molecules requires appropriate structure matching tools.
The development of such tools for molecular assemblies faces a few technical challenges that need to be handled appropriately. The largest molecular complexes are encoded in the mmCIF format so that a program that handles such structures necessarily has to be able to read the corresponding file formats efficiently. Molecular complexes generally consist of polymers of various types, primarily protein and nucleic acid chains. Therefore, structure matching programs should be able to compute alignments of pairs of protein and pairs of nucleic-acid chains. And, most importantly, such a program should be able to compute a complete pairwise match of whole complexes using protein and nucleic acid chains simultaneously.
In the past, there have been only a handful of reports addressing such issues (7–11). In what follows we describe the most recent version of the TopMatch program (7), which handles these requirements efficiently. To achieve these goals we implemented a parser to read and process mmCIF files and a generator to build molecular assemblies from the respective operation expressions. We rebuilt the TopMatch source code to enable the simultaneous processing of protein and nucleic acid chains and we added capabilities for running parallel tasks and threads on multiple processor cores. We endowed TopMatch with an interface to NGL (12), a molecular viewer enabling the rapid visualization of very large molecular assemblies, like complete ribosomes, and their structural similarities in 3D. To express and rank the extent of similarity among structures we use a small number of simple and intuitive quantities, and various interactive features facilitate the investigation and interpretation of structure aligments and superpositions.
Our goal here is to enable the reader to use the TopMatch service immediately and our hope is that the examples presented show that intricate relationships among extremely complex structures like ribosomes can be handled rather easily using appropriate tools. Input and output are described on a simple but sufficient level to run and understand the examples.
STRUCTURE MATCHING BY EXAMPLE
In what follows, we compare two structures of ribosomes found in eukaryotic mitochondria (3): the mitochondrial ribosome from yeast, as resolved by electron microscopy (EM) to a resolution of 3.25 Å (PDB code: 5mrc (5)), and class 1 human mitochondrial ribosome, as resolved by EM to a resolution of 3.5 Å (PDB code: 3j9m (4)). Both structures present spectacular macromolecular complexes assembled from around 80 components each, forming a hetero-78-mer in the case of yeast (75 proteins + 21S rRNA + 15S rRNA + tRNA) and a hetero-85-mer (81 proteins + 16S rRNA + 12S rRNA + tRNAs) in the case of the human mitoribosome. The structural features shared by these assemblies are as interesting as are the differences between them and, clearly, a thorough investigation of them requires comparative analyses on several levels of structural complexity.
Individual protein chains
A strategy commonly employed in the search for structural correlations is the pairwise comparison of individual protein chains. Following this approach would entail the execution of structure comparisons with all pairs of protein chains present in the two ribosomes to reveal the mutual similarities among them. However, the possibility to specify and use complete macromolecular assemblies offers a quick and more convenient way to search for inter-chain correlations in multichain complexes. To illustrate this point, we compare yeast mitoribosomal protein chain mL43 with the complete human mitoribosome. The resulting matches are summarized in Table 1 and depicted in Figure 1. As may be expected, the human orthologue of yeast mL43 (3j9m, chain b) is found as the most similar match. Although the extent of sequence similarity of 23% is low, the thioredoxin-like folds of the mL43 proteins superimpose well, with the exception of some variations in the extended termini (Figure 1, top left). Perhaps more surprising, TopMatch-web reports two additional matches with high similarity to mL43, one located in the small subunit (mS25; Figure 1, bottom right) and the other located in the large subunit (mL53; Figure 1, top right). Since the respective sequence identities of 18% (mS25) and 15% (mL53) are low, these structural relations are not so easily detected by sequence alignment methods. Moreover, when these relations are displayed using the complete assemblies, then the relative spatial location and orientation of these chains is easily recognized (Figure 1, center).
Table 1.
Structure matches of yeast mitoribosomal protein chain mL43 (PDB code: 5mrc, chain 4) to protein chains from the human mitoribosome (PDB code: 3j9m, assembly 1). The table lists the top five results obtained by a structure comparison with TopMatch-web. Superpositions corresponding to the top three matches are shown in Figure 1.
| LEN | QC | TC | SCORE | RMS | SI | Protein chain |
|---|---|---|---|---|---|---|
| (%) | (%) | (Å) | (%) | |||
| 119 | 86.2 | 0.8 | 94.2 | 1.65 | 22.7 | mL43 (3j9m,b) |
| 82 | 59.4 | 0.5 | 68.5 | 1.38 | 18.3 | mS25 (3j9m,AT) |
| 73 | 52.9 | 0.5 | 52.1 | 2.16 | 15.1 | mL53 (3j9m,k) |
| 38 | 27.5 | 0.2 | 26.7 | 2.43 | 7.9 | mL39 (3j9m,7) |
| 39 | 28.3 | 0.3 | 25.8 | 2.59 | 5.1 | uS2m (3j9m,AB) |
LEN, alignment length (=number of equivalent residue pairs); QC, query cover; TC, target cover; SCORE, measure of structural similarity; RMS, root mean square error of superposition; SI, sequence similarity calculated from the structure-based sequence alignment; Protein chain, human protein corresponding to matched region. For nomenclature, see Input section. Calculation of QC, TC and SCORE is explained in the Output section.
Figure 1.
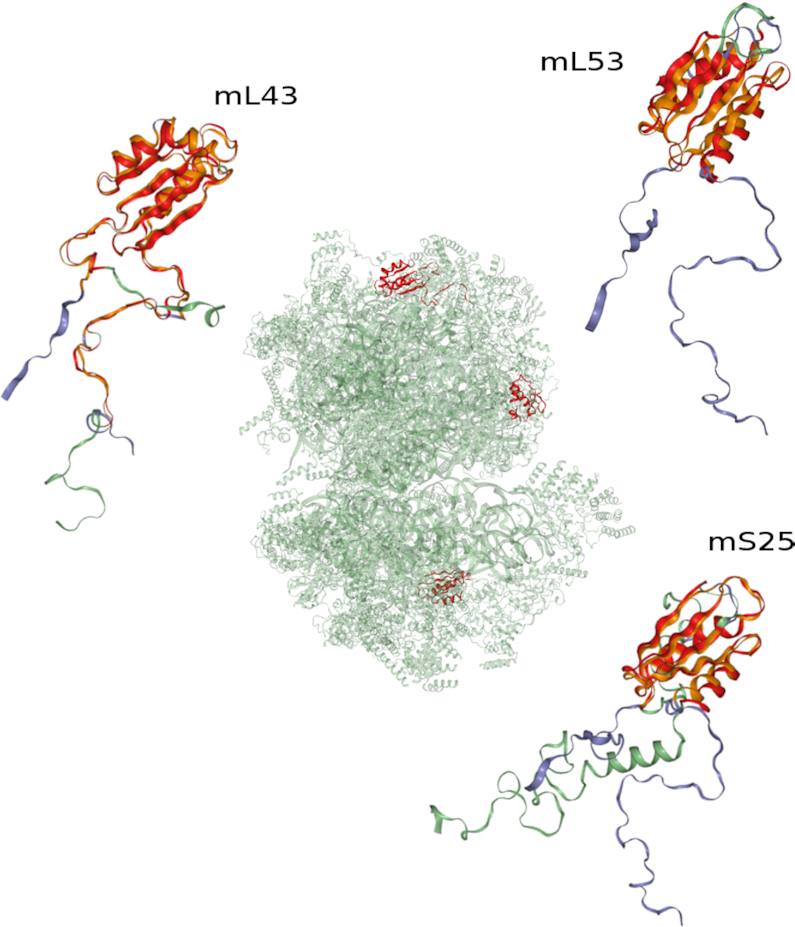
Structure matches of protein chains in eukaryotic mitochondrial ribosomes. The human mitoribosome (green; PDB code: 3j9m, assembly 1) is searched for structure similarities with protein chain mL43 from yeast (blue; PDB code: 5mrc, chain 4). Three superpositions corresponding to the top three structure alignments are shown. Top left: yeast mL43 and human mL43; bottom right: yeast mL43 and human mS25; top right: yeast mL43 and human mL53. Structurally equivalent parts are colored orange (yeast) and red (human). The locations of the matched human protein chains in the mitoribosome are shown on the central mitoribosome complex. For the quantifications of the depicted structure matches, see Table 1.
rRNA
While the ribosomal proteins are numerous and diverse (3), the few RNA chains play an important role in defining the structural core of ribosomes. Using TopMatch finding structural matches among RNA chains is as easy as it is for protein chains. Figure 2 shows the small subunit rRNAs from yeast (∼1500 nucleotides) and human (∼900 nucleotides) mitoribosomes, with 614 pairs of structurally equivalent nucleotides aligned to an RMS of 2.15 Å. In the regions of structural equivalence the sequence similarity is 45% (pairs of identical nucleotides). Both the obtained alignment and the superposition highlight the known rRNA expansion segments in yeast and the known rRNA reduction in human, described as unique structural characteristics of fungal and mammalian mitoribosomes (3).
Figure 2.
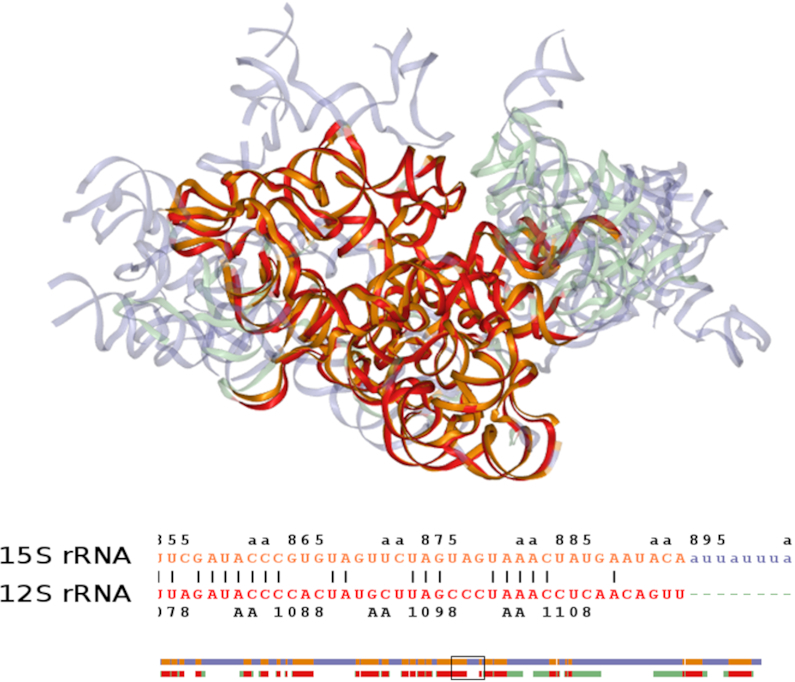
Structure alignment of rRNA from eukaryotic mitochondrial ribosomes. Top: superposition of small-subunit 15S rRNA from yeast (blue; PDB code: 5mrc, chain aa) and human 12S rRNA (green; PDB code: 3j9m, chain AA) according to the top structure alignment obtained from TopMatch-web. Matching parts are colored orange (yeast) and red (human). Bottom: corresponding structure-based sequence alignment (clipped; the complete alignment covers >600 nucleotides and is schematically drawn below, with the clipped part indicated by a rectangle).
Complete ribosomes
Insights into the total extent of structure similarity between two ribosomes can be obtained when all protein and nucleic acid chains are taken into account simultaneously. Figure 3 shows the result of the structure comparison of complete yeast and human mitoribosomes as calculated by TopMatch-web. Protein and RNA chains are simultaneously aligned and superimposed. The matching part consists of 5932 structurally equivalent pairs of amino acids/nucleotides, aligned to an RMS of 3.02 Å. The matching parts cover about one third of the yeast structure (∼18 000 resolved residues) and ∼40% of the human structure (∼15 500 resolved residues), respectively.
Figure 3.

Structure comparison of complete mitochondrial ribosomes. Yeast and human mitoribosome are superimposed according to the simultaneous alignment of all ribosomal protein and RNA chains present in these complexes. Left: yeast mitoribosome (blue; PDB code: 5mrc; hetero-78-mer; matching parts in orange). Right: human mitoribosome (green; PDB code: 3j9m; hetero-85-mer: matching parts in red). Center: superimposed structures, with non-matching parts removed.
The more complex and the larger the structures, the more challenging is their visualization and the distinction of similar and non-similar parts. To ease the exploration of matches of such complexity, we interfaced TopMatch with an interactive molecule viewer (NGL) capable of rapid rendering of even the largest complexes currently known (12). Navigation to specific regions of the compared structures is supported by a direct mapping from the residues shown in the structure-based sequence alignment to the 3D view. In addition, the possibility to highlight contiguous blocks of aligned residues and to control the transparency of unmatched regions facilitate the investigation of alignments (Figure 3).
WEB SERVER USAGE
Input
TopMatch-web requires the atomic coordinates of the structures to be compared. Users can supply coordinates by uploading files in mmCIF format (1,13). However, since all structures in the Protein Data Bank (PDB) are accessible through the server by their PDB codes, it is generally not necessary to upload any files.
The use of the mmCIF format is necessary to handle large molecular complexes. All structures deposited in PDB are available as mmCIF files (1). Approximately 1100 entries are exclusively stored as mmCIF files since they cannot be represented by legacy PDB format. Among these are the largest structures known, including ribosomes and capsids of viruses. In addition there are ∼10 000 biological assemblies that cannot be represented using PDB file format.
All calculations are carried out with the Cα atoms of the protein and the P atoms of the nucleic acid backbones. Hence, TopMatch also works on low resolution structures as long as backbone traces specified by Cα or P atoms are available.
TopMatch-web accepts both author-assigned and PDB-assigned chain identifiers (13), where a comma is used to denote author-assigned chains (e.g. 5mrc,aa) and an underscore character to denote PDB-assigned chains (e.g. 5mrc_WB). Biological assemblies are specified by an @ sign with the respective assembly identifiers appended (e.g. 3j9m@1). A straight PDB code, like 5mrc, specifies the complete asymmetric unit. The TopMatch-web help page provides a detailed account of the rules used to specify chains, models, and molecular assemblies.
Output
Besides the graphical view of the superimposed input structures, the web service provides a table describing the individual structural alignments obtained by a handful of numbers that are either easy to understand or are standard parameters in structural research (see legend to Table 1). The one parameter that needs explanation is the similarity score S (the SCORE of Table 1). This similarity is defined as
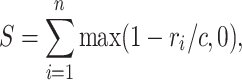 |
where i runs over all pairs of Cα or P atoms that are defined as equivalent by the alignment obtained, n is the number of such pairs, ri is the spatial distance between these two atoms after optimal superposition of the complete molecules, and c = 7 Å is a constant. The definition of S takes into account the extent of similarity (i.e. the number of aligned pairs n), and the average spatial deviation of these pairs (implicitly expressed by the magnitude of the root mean square error RMS of Table 1). For atoms at the same location in space (after optimal superposition of the whole molecules) ri = 0 and the similarity reaches the maximum 1. On the other extreme the similarity is zero when ri ≥ c. Hence the maximum similarity of an alignment of length n is n and the smallest possible similarity is zero. Moreover, the larger the spatial deviations (i.e. the larger the individual ri) the smaller the similarity. The similarity score S is used to rank alignments (e.g. Table 1). Finally, the parameters QC and TC express the percentage of the lengths (i.e. the number of residues) of the query QL and target TL, respectively, covered by the alignment: QC = 100 × LEN/QL and TC = 100 × LEN/TL.
TopMatch-web visualizes the superimposed input structures in 3D using the interactive high-performance molecule viewer NGL (12). By default, the alignment shown corresponds to the alignment of highest similarity S. Clicking on a line in the table provides a quick way to switch from one alignment to another. The matching parts of the molecules displayed are highlighted in red and orange (see Figures 1–3 for examples). Modes of representation and other options are available from a drop down menu on the web page for exploring these parts in more detail.
The 3D view of the molecules is complemented by the corresponding structure-based sequence alignment, which can be displayed on a schematic or detailed level and is linked to the 3D view to facilitate navigation to particular parts of the structures. Alignments and coordinates of superimposed structures can be downloaded in various formats as described on the TopMatch-web help page.
The comparison of the two complete ribosomes, 5mrc@1 and 3j9m@1, takes in the order of five seconds when executed on a typical workstation with forty processor cores. This includes the time required to read and parse the respective mmCIF files. The execution time on the web service is similar but there is some delay due to the transfer of data from the server to the client and the time required to initialize the visualization on the client site.
CONCLUSION
With TopMatch-web it is straightforward to study relationships among the structures of proteins, nucleic acids, and their multi-chain assemblies of even large size and complexity. Hence, this service endows a wide audience with the tools to view, investigate, and grasp the intricate relationships among the biological macromolecules whose structures have been solved to atomic resolution including the most complex structures currently known. TopMatch-web is free and open to all interested users.
DATA AVAILABILITY
We encourage the reader to actively follow the presented examples using these links corresponding to the example figures: Figure 1, Figure 2, Figure 3.
ACKNOWLEDGEMENTS
M.W. designed and implemented the TopMatch web service and selected the mitoribosome examples, M.J.S. developed the TopMatch structure comparison algorithm and implemented the TopMatch engine, M.W. and M.J.S. wrote the paper.
Contributor Information
Markus Wiederstein, Paris-Lodron-University of Salzburg, Department of Biosciences, Hellbrunner Str. 34, 5020 Salzburg, Austria.
Manfred J Sippl, Paris-Lodron-University of Salzburg, Department of Biosciences, Hellbrunner Str. 34, 5020 Salzburg, Austria.
FUNDING
NIH [U01 CA198942 to PI: Peter Rose, in part]. Funding for open access charge: courtesy of AG Peter Lackner, University of Salzburg.
Conflict of interest statement. None declared.
REFERENCES
- 1. Burley S.K., Berman H.M., Bhikadiya C., Bi C., Chen L., Costanzo L.D., Christie C., Duarte J.M., Dutta S., Feng Z. et al.. Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019; 47:D520–D528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Steitz T.A., Moore P.B.. Perspectives on the ribosome. Phil. Trans. R. Soc. B. 2017; 372:20160537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bieri P., Greber B.J., Ban N.. High-resolution structures of mitochondrial ribosomes and their functional implications. Curr. Opin. Struct. Biol. 2018; 49:44–53. [DOI] [PubMed] [Google Scholar]
- 4. Amunts A., Brown A., Toots J., Scheres S.H.W., Ramakrishnan V.. The structure of the human mitochondrial ribosome. Science. 2015; 348:95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Desai N., Brown A., Amunts A., Ramakrishnan V.. The structure of the yeast mitochondrial ribosome. Science. 2017; 355:528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matzov D., Aibara S., Basu A., Zimmerman E., Bashan A., Yap M.-N.F., Amunts A., Yonath A.E.. The cryo-EM structure of hibernating 100S ribosome dimer from pathogenic Staphylococcus aureus. Nat. Commun. 2017; 8:723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sippl M.J., Wiederstein M.. Detection of spatial correlations in protein structures and molecular complexes. Structure. 2012; 20:718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Madej T., Lanczycki C.J., Zhang D., Thiessen P.A., Geer R.C., Marchler-Bauer A., Bryant S.H.. MMDB and VAST+: tracking structural similarities between macromolecular complexes. Nucleic Acids Res. 2014; 42:D297–D303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wiederstein M., Gruber M., Frank K., Melo F., Sippl M.J.. Structure-based characterization of multiprotein complexes. Structure. 2014; 22:1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suzuki H., Kawabata T., Nakamura H.. Omokage search: shape similarity search service for biomolecular structures in both the PDB and EMDB. Bioinformatics. 2016; 32:619–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Minami S., Sawada K., Ota M., Chikenji G.. MICAN-SQ: a sequential protein structure alignment program that is applicable to monomers and all types of oligomers. Bioinformatics. 2018; 34:3324–3331. [DOI] [PubMed] [Google Scholar]
- 12. Rose A.S., Bradley A.R., Valasatava Y., Duarte J.M., Prlić A., Rose P.W.. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics. 2018; 34:3755–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Westbrook J.D., Bourne P.E.. STAR/mmCIF: an ontology for macromolecular structure. Bioinformatics. 2000; 16:159–168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
We encourage the reader to actively follow the presented examples using these links corresponding to the example figures: Figure 1, Figure 2, Figure 3.