


Abstract
Single-cell omics enables researchers to dissect biological systems at a resolution that was unthinkable just 10 years ago. However, this analytical revolution also triggered new demands in ‘big data’ management, forcing researchers to stay up to speed with increasingly complex analytical processes and rapidly evolving methods. To render these processes and approaches more accessible, we developed the web-based, collaborative portal ASAP (Automated Single-cell Analysis Portal). Our primary goal is thereby to democratize single-cell omics data analyses (scRNA-seq and more recently scATAC-seq). By taking advantage of a Docker system to enhance reproducibility, and novel bioinformatics approaches that were recently developed for improving scalability, ASAP meets challenging requirements set by recent cell atlasing efforts such as the Human (HCA) and Fly (FCA) Cell Atlas Projects. Specifically, ASAP can now handle datasets containing millions of cells, integrating intuitive tools that allow researchers to collaborate on the same project synchronously. ASAP tools are versioned, and researchers can create unique access IDs for storing complete analyses that can be reproduced or completed by others. Finally, ASAP does not require any installation and provides a full and modular single-cell RNA-seq analysis pipeline. ASAP is freely available at https://asap.epfl.ch.
INTRODUCTION
Single-cell omics is a recent field that started to bloom in 2013–15 with the advent of commercially available single-cell RNA-seq (scRNA-seq) protocols (1,2). At its origin, platforms could process hundreds of cells at a time, whose corresponding transcriptomes could still be handled by traditional bulk RNA-seq bioinformatics tools. In a very short amount of time, however, sample sizes exploded (3). This is exemplified by recent efforts aiming to create ‘cell atlases’ for entire tissues (4,5) or organisms (6–9) at resolutions and scales that are more difficult to handle computationally (>100k or even millions of cells and thus transcriptomes). As the field evolves, so are the underlying analytical approaches and tools, making it increasingly more difficult to see ‘the forest through the methodological trees’ and to select the proper analysis pipeline (10,11). The latter is also in part dictated by the size of the focal dataset, with powerful tools now emerging that aim to handle single-cell omics datasets in a scalable manner (12). However, these tools have yet to take firm root in the field, especially with researchers who have so far been accustomed to working with smaller-sized datasets (∼1–10k cells). The urgency for these tools to become widely implemented is illustrated by recent cell atlasing projects, which clearly demonstrate the need for both scalable computing power and analytical approaches (10). Finally, novel single-cell omics approaches such as single-cell ATAC-seq (scATAC-seq) are rapidly emerging, posing additional problems in terms of data management and integration (13).
To reduce the complexity of single-cell omics analyses, we developed ASAP (14), enabling standardized analyses that can be run in minutes by any user without requiring significant computing power. The entire, canonical scRNA-seq pipeline is available in ASAP, and can be summarized into 8 consecutive steps: (i) filtering low quality cells and lowly expressed genes, (ii) normalization across cells, (iii) scaling and covariate removal (such as read depth, mitochondrial content, etc.), (iv) computing highly variable genes of interest, (v) performing dimension reduction using PCA followed by high dimensional methods such as t-SNE or UMAP, (vi) clustering of cells to identify subpopulations, (vii) differential expression analysis to identify marker genes of identified subpopulations and (viii) functional enrichment of these marker genes into pathways or cell types. Of course, all these steps are parametrizable, and we acknowledge that it may be difficult to find one fixed pipeline that will fit all dataset types (10). A trained bioinformatician tends to therefore tune the parameters to the dataset of interest. Given this, ASAP allows users to choose from a panel of tools, thereby providing guiding tutorials to help researchers with their selection of the correct tools or parametrization for their datasets.
Here, we report a major ASAP upgrade, with several substantial improvements such as a completely remodeled user interface, a fully Dockerized (15,16) system, and the internal implementation of the .loom file format. We chose this format since it substantially increased the scalability of the tools used to perform out-of-RAM computations, allowing the analysis of high-dimensional datasets of virtually any size. This new format also enhances the communication between existing portals such as SCope (17), the Human Cell Atlas (7), and many others that are adopting the same file format for storing complete analyses into one single file.
RESULTS AND METHODS
Implementation overview
ASAP is now using the Docker technology (15,16) to make the whole platform modular and versioned (Figure 1). Docker containers separate the main website (the Ruby-on-rails web server code) from the running jobs (R, Python and Java bioinformatics tools), enabling jobs to run on a different machine than the main server hosting the web application. Moreover, this architecture allows the asap_run container (hosting the versioned bioinformatics tools) to be dispatched to many external machines for enhanced computing power, and maybe in the future, to the cloud.
Figure 1.

ASAP architecture. The ASAP application is a docker-compose-based Ruby-on-Rails application. ASAP implements web-sockets (using Redis and Cable containers) for an interactive display of results at the client end. Analyses launched by users are submitted to a scheduler that will run third party software (Python, R, Java) in versioned docker containers ASAP_run:vM, enabling scalability and reproducibility of the platform. The scheduler also ensures that the number of cores that are used on the machine and the level of RAM used on the machine are not exceeding hardware capacities. The ASAP_core database stores users, projects and job stats (for benchmarking the tools) and is thus not versioned. A versioned ASAP_data_vN (currently v5) database stores external public data on genes, gene sets and future ontologies. Results of analyses are written on a fast-access disk (NVME) shared by the Ruby-on-Rails and the ASAP_run:vM docker containers. Projects that are not accessed for a long period are automatically saved on an object storage system (through a CRON job) for saving space on the fast access NVME disk.
Since the single-cell community is very active, and new methods appear or are upgraded almost on a monthly basis, this architecture allows an easier versioning of the portal with each asap_run container encapsulating its own tool versions. This will enhance reproducibility and retro-compatibility with previous studies. The Dockerized architecture also keeps all tool versions fixed for a given global version of ASAP, thus all listed tools are embedded at a fixed version and correspond to a single versioned Docker.
.loom files
.loom files represent a standardized file format for storing/handling single-cell datasets. It was proposed and developed by the Linnarsson Lab (http://loompy.org/). .loom files are HDF5 (Hierarchical Data Format) files following certain constraints in terms of group/dataset names and types. They allow for very efficient computation and access to row/columns of datasets, thus greatly enhancing the scalability of computational methods. The matrices can be chunked, which allows out-of-RAM computation by processing the data ‘chunk by chunk’. The new version of ASAP now internally handles .loom files for every project. When a user submits a dataset (plain text, archive, 10×, etc.), it is automatically transformed into a .loom file during the parsing step. This step also computes basic statistics (number of detected genes, ratio mitochondrial content, depth, etc.) that are immediately added to the parsed .loom file and available downstream, such as for example when coloring plots during visualization.
Web application
The ASAP web application is developed with Ruby-on-Rails (RoR). The backend is implemented as a PostgreSQL relational database. The frontend uses different JavaScript libraries and is set to enable front-end scalability with big datasets. Specifically, (i) scattergl plots from plotly.js (18) to render dimension reduction plots scalable; (ii) pako-inflate.js (https://github.com/nodeca/pako) to compress big integer arrays between the client and the server and (iii) an adapted version of JQuery (https://jquery.com/) file input for scalable file uploads. Other important javascript libraries that are used include Cytoscape.js (19,20) to generate a graphical display of the analysis pipeline composition or of Jquery autocomplete for gene selection in the visualization tool.
As mentioned previously (see Figure 1), the ASAP web application runs in a Docker container called asap_web. Together with other containers for the (i) websockets (Cable, Redis containers), (ii) PostgreSQL server and (iii) Puma web server, they are embedded in a docker-compose that guarantees independence with respect to the hosting system and that could facilitate further migration / deployment of the system.
Reproducibility
The ASAP server incorporates a versioning system that ensures full reproducibility of the analyses that are carried out on the web application. This release handles new projects and retro-compatibility of old projects starting from version 4 (v4). When starting a project, users have the option to use the stable version of their choice (i.e. v4 or v5 at the moment).
Version stability is enabled by two key components of the system: (i) all external (project-independent) data are stored in a versioned PostgreSQL relational database asap_data_vN; and (ii) all scripts and executables are installed with the necessary dependencies in an r-base docker container asap_run:vM that is available on Dockerhub (https://hub.docker.com/r/fabdavid/asap_run/tags).
Note that for a given global ASAP version, versions of the docker container and of the relational database M and N can be different, since the database or the docker container are not necessarily updated each time.
For every run, we also provide the user with the exact list of commands that was used to produce the output (using the Docker module). Therefore, all steps are completely reproducible, and a default pipeline can be readily implemented using Docker and the scripts generated by ASAP. A global script is also dynamically generated for each project, so users can reproduce their complete analysis locally on their machine/server. The script loads the right version of the docker container and of the relational database and runs the whole pipeline, as designed by the users.
Execution of analyses
On the ASAP server, the different analysis scripts and executables are run within the asap_run_vN docker containers by a scheduler that evaluates if the system can accept a new analysis at a given time. The scheduler assesses the status of the system (checking the load on the machine and the number of free CPUs). For each analysis, the amount of RAM required, and the execution time are monitored and stored; this information is then available to the users through the interface.
Operations requiring a minimal amount of resources, such as unarchiving projects, are directly launched (without waiting) on a queue through DelayedJob, a RoR module that allows to run a piece of code asynchronously.
Referencing and searching ASAP projects
Identifiers from GEO (21) or ArrayExpress (22) can be associated manually to an ASAP project. If users publish the results of an ASAP analysis, they can also provide the PubMed ID of the article (at the same time as setting the project as ‘public’). If a project is loaded from the Human Cell Atlas (HCA) Data Coordination Platform (DCP), then GEO and ArrayExpress identifiers are automatically associated to the ASAP project. From these identifiers (assigned manually or automatically), information from GEO, ArrayExpress and BioProject (23) (mainly literature references, description and identifiers) is automatically extracted and associated to the ASAP project.
In addition, an instance of SunSpot/SolR runs on the RoR application and provides an efficient search engine to retrieve lists of projects that are associated with any GEO, ArrayExpress or BioProject project, based on identifiers or free-text descriptions.
Input
ASAP can handle read/UMI count matrices in several formats: (i) plain-text files (compressed or not), (ii) archives of text files (compressed or not), (iii) .loom files or (iv) .h5 files produced by the 10× CellRanger pipeline (https://github.com/10XGenomics/cellranger). When the data finishes uploading on the server, ASAP starts to parse the file and shows a snapshot (preview) of the dataset (10 first rows, 10 first columns) as well as cell/gene names (Figure 2). This allows the user to change some parsing options, such as the separator or the column id containing the gene names, without having to re-upload the dataset.
Figure 2.

Dataset preview after its upload in ASAP. After uploading a file (of any type), ASAP shows a preview of the main count matrix (10 first rows/columns), as well as genes and cell names. It also shows an icon with the type of file that is recognized automatically. Therefore, the user has the possibility to change the parsing options if needed (delimiter, header, …). In this page, the user can name the project, choose an organism from the ∼500 organisms available from Ensembl, and choose the version to run on (here, v4 is the latest stable version (default) and v5 is still in beta).
Users can also choose to create a new project from data hosted by the HCA DCP. This feature uses an API provided by the HCA (Matrix Service API) to query the available datasets. The user can choose specific datasets for import into ASAP, and the HCA API will automatically generate a .loom file containing all selected cells (Figure 3). Finally, a new project is created on ASAP with the imported .loom file, with which the user can start analysis and visualization.
Figure 3.

Dataset download from the Human Cell Atlas Matrix Service API. Users can query the Matrix Service API of the Human Cell Atlas (HCA) from the ASAP ‘New Project’ page. They will see a list of projects from which the Matrix Service can generate count matrices in the form of .loom files (.fastq and other raw sequencing files are automatically filtered out). The user can then choose a project and the HCA API will automatically send a .loom file to ASAP. The latter file will be parsed automatically, thus creating a ‘ready to analyze’ project in ASAP. Importantly, all metadata sent from the HCA are automatically imported along with the .loom file, and will be readily available in ASAP (such as sequencing platform, tissue of origin, etc.).
Internally, all inputs are transformed into .loom files as a common format for all steps. Of course, the users can download the .loom files for their projects and also load them into R (using loomR, https://github.com/mojaveazure/loomR) or Python (using loompy, http://loompy.org/). Of note, since .loom files are essentially normed HDF5 files, they can potentially be loaded with any other programming language as well.
Ensembl and gene set database
In its last version (v5), ASAP incorporates information from the Ensembl (24) ‘vertebrates’ database v54 to v99 and from Ensembl ‘genomes’ v5 to v46. The ASAP_data_v5 database contains 16 734 890 genes with unique Ensembl identifiers for 551 different species. During file parsing, all genes are mapped to the database version chosen by the user (v4 or v5), with the latest stable one always being pre-selected. This mapping is not necessary for most of the steps included in ASAP, but can provide additional information in the result tables, or when hovering on the dynamic plots. It is mostly needed during the last step of the analysis (cell type annotation/functional enrichment), when ASAP needs to relate differentially expressed genes (or marker genes) to gene sets such as GO (25), KEGG (26), Drugbank (27) or cell type annotation databases (28–30).
Available tools, bioinformatics scripts and executables
Since the initial implementation of ASAP, several tools were added, and obsolete tools were removed for this major upgrade. Currently, ASAP hosts tools in Python, R, and Java.
The parsing and filtering steps are performed in Java, which we found to be both much faster than R or Python as well as scalable to any dataset (implemented to take advantage of the .loom format and the chunking of the count matrices). In addition, for the Cell Filtering step, we implemented dynamic plots for selecting the best thresholds according to major QC metrics: number of detected genes, number of UMIs/reads, ratio of reads mapping to protein-coding genes, ratio of mitochondrial reads, and ratio of ribosomal reads (Figure 4). The user can see the plots, select the best thresholds for each of them, and visualize the resulting number of filtered cells interactively, prior to validation, which will produce a novel .loom file filtered according to the different thresholding parameters.
Figure 4.

Interactive cell filtering step enables users to set various thresholds for QC. The cell filtering step features interactive plots for filtering out outlier cells that do not pass certain quality controls (QC). In all panels, a point is a cell. Of note, when a threshold is selected in one of the five panels, all other panels are automatically refreshed so the user can see the retained cells (green) and the ones that were filtered out (grey). A recap of the final number of selected vs. filtered out genes is available in the top bar. (A) Number of UMI/read counts per cell (sorted in descending order). This plot is similar to the plot generated by CellRanger in the 10x pipeline. Users can select a minimum number of UMI/reads per cell. (B) Number of UMIs/Read counts vs number of detected genes. (C). Ratio of reads that maps to mitochondrial genes (vs all mapped reads). This feature uses the Ensembl database to know on which chromosome the genes are mapping, so only genes that are mapped to our Ensembl database are considered. (D, E) Similar to C. but using the biotype of the genes from Ensembl to know if the reads map to a protein-coding gene (D), or to a ribosomal gene (E).
The highly variable gene calculation is using tools from three packages: M3Drop (31), Seurat (32) and Scanpy (12). Of note, only the one from Scanpy is scalable to >100k cells. Also, for these methods, the user is able to see the resulting curve and highlight genes of interest by hovering on the cell (Figure 5). In the subsequent visualization step, PCA (Incremental PCA) is implemented in Python and is parallelized and scalable. The UMAP (33) and t-SNE (34) methods from Seurat are implemented as well and are scalable when run on the results of the PCA. A parallelized version of t-SNE was also added from the Scanpy package in ASAP v5.
Figure 5.

Calculation and interactive visualization of Highly Variable Genes and M3Drop. Different methods in ASAP are available to select highly variable genes. All methods produce an interactive plot where the user can hover the cells to see their characteristics (rectangle box tooltip). Here, we see the output of two methods. On the left panel, highly variable genes are calculated from Seurat (v2) using the Brennecke et al. method (50). On the right panel is the output of the M3Drop method, more specifically the Depth-Adjusted Negative Binomial (DANB) model, which is tailored for datasets quantified using unique molecular identifiers (UMIs).
Many clustering methods are implemented, mostly in R (Seurat, SC3 (35), k-means) and should be run on the results of the PCA for scalability purposes. Similarly, many differential expression methods were implemented in R (Seurat, limma (36), DESeq2 (37)) or re-implemented by us in Java for the purpose of scalability (Wilcoxon-ASAP). Only Seurat and our homemade Wilcoxon methods are scalable.
Finally, we have also developed in Java the functional enrichment step using a simple Fisher's Exact Test, thereby considering the correct background for not inflating the resulting P-values. This method is scalable as well to any dataset.
Outputs
For most steps, the main output is a newly annotated .loom file. For example, when generating a dimension reduction output, the initial .loom file is modified with an additional column attribute containing the ‘cells vs. components’ result matrix. In addition, the user can visualize this data directly in the browser as an interactive plot. Internally, the server will extract the column attributes from the .loom file and generate a JSON file that will be sent to the client and that can be visualized using plotly.js scattergl. The WebGL version was chosen because it allows the plotting of millions of cells in a timely manner.
Different steps have different outputs. For some steps, such as detecting highly variable genes, the output is a filtered .loom file and a dynamic plot showing the interpolation that was produced during the calculation. Other steps such as the differential expression or the functional enrichment steps produce sorted tables of statistically significant genes/gene sets. These tables have dynamic links to external databases such as GO (25) or Ensembl (24).
The main visualization step is the dimension reduction (using PCA, t-SNE (34) or UMAP (33)). This step allows the user to visualize the dataset in 2D or 3D. The 2D view can be tuned in different ways. First, the user can color the cells according to external metadata (such as sex, library type, depth, batch etc.), clustering results, or gene expression (Figure 6). The plot is also dynamic, so the user can select cells of interest to create new metadata on which additional operations can be performed, such as a novel differential expression calculation. Finally, the user can also annotate the clusters according to marker genes (with a cell type for example), either from this view or directly from the ‘Marker Gene’ view in the differential expression step.
Figure 6.

UMAP visualization of an HCA public project involving 780k cells with coloring options. After dimension reduction, the user can see 2D and 3D plots of the dataset. Here, we show an ASAP project that was created using the HCA Matrix service feature (see Figure 3) involving ∼780k cells from human bone marrow + cord blood. The pipeline was run until the UMAP step which is what is visualized in the top and bottom panels. In each panel, on the right, we opened the ‘Controls’ view which allows the user to change the appearance of the plot (size of the points, colors, etc..) and to manage any clustering results (and eventually annotate clusters). (A) Here, we show coloring by the number of detected genes. This shows a region which seems to have much more detected/expressed genes which can be a biological result or may represent doublets. (B) Here, the cells are colored using an annotation that was imported from the HCA: ‘derived_organ_parts_ontology’. We can clearly see the coloring of the two organ parts that compose the dataset: bone marrow and cord blood, which highlights a need for better integration of both datasets. One way would be to use Seurat or MNN methods to remove the batch effect between the two organ parts, this is currently in development (see Discussion).
Estimation of time and RAM for each tool
With this new version, we developed a novel tool to predict the computing time and maximum RAM that will be required by a job, before running it. To achieve this, we store certain characteristics of jobs that were run by users in a separate versioned database. These include the size of the dataset (number of cells/rows) that was used as input and the time/RAM that was required by the job, providing the run was successful. Currently, the prediction is only made based on the size of the dataset, but in the future, we may consider adding method parameters as well (in case they have a strong effect on the overall computing time and RAM usage predictions). We use two simple linear models that are trained on these datasets for every tool that is present in ASAP: (i) time ∼ nbcells * nbgenes and (ii) ram ∼ nbcells * nbgenes. These models are recomputed daily using a CRON task and are stored as .Rdata files for fast prediction in the UI.
Project sharing
A key feature of our upgraded ASAP web application is the interactivity and collaboration possibilities. To implement this, we established a project sharing system, allowing concurrent access to the same project. Users can share their projects with other ASAP users (or send an email to a novel user who will need to register) to allow accessing the same project simultaneously. We set up right permissions, so that the owner of a project can control his/her projects in terms of visibility, modification, and further sharing. There is also the possibility to render a project public, or to clone a project. Public projects are associated with a unique ASAP-ID that can be listed in a publication and that can be used for enhanced reproducibility in published papers. Symmetrically, the PMID of the published work can be entered in the details of the ASAP project and the reference will then be displayed on the project page.
Once a project is open, any change in the status of analyses is transmitted to the user through Websockets (ActiveCable in Ruby-on-Rails). This feature enables interactive, collaborative projects, since any modification to a project by any of the sharing users is indicated to the others in real time.
DISCUSSION AND FUTURE IMPLEMENTATIONS
Single-cell omics technologies are increasingly applied in both biological as well as clinical research to identify new cell types and to uncover cellular dynamics during development or disease (e.g. tumor heterogeneity). Conventional pipelines tend to require hours/days of work by a trained bioinformatician to deliver meaningful results. ASAP’s main goal is to aid with the interpretation of these data since the whole pipeline can be run in minutes, providing on-the-go visualization, identification of new cell or disease populations by clustering, differential expression analysis and enrichment. With ASAP, we strive to build a centralized platform to store single-cell projects and their complete analyses in a shareable and reproducible fashion. The interface of ASAP is designed to be user-friendly and provides versatility with a library of state-of-the-art tools that are documented. Tutorials thereby guide the user through the different steps of the analysis. Users can easily upload their dataset and readily start working with it through interactive plots and output tables without previous analysis experience.
Given the desire of the research community at large to render single-cell analyses more accessible, several other interactive visualization tools or platforms have been developed in parallel (38). Building on a recent pre-print overviewing these tools (38), we compared the new version of ASAP (2020) presented in this manuscript to the original one (ASAP 2017) (14), and to the other state-of-the art tools that are currently available (see Table 1). Here, we mostly focused on tools with a web interface, thus disregarding (i) software such as the BioTuring Single-Cell Browser (Bbrowser) or the Loupe cell browser, and (ii) packages such as Seurat (32) or scanpy (12). Other portals, such as the Single-Cell Expression Atlas (39) are only meant to visualize public datasets, and thus are not designed for user-specific datasets. Conversely, we can also mention two Galaxy servers that simplify the processing pipeline but do not support an interactive visualization of the results: (a) a common server providing a single-cell analysis workflow (https://singlecell.usegalaxy.eu/), (b) and another one specifically designed for the analysis of data from the Human Cell Atlas initiative (https://humancellatlas.usegalaxy.eu/). The latter is connected to the HCA Matrix service to import datasets, and relies on the UCSC Cell Browser (Table 1) or the Single-cell Expression Atlas (39) for visualization.
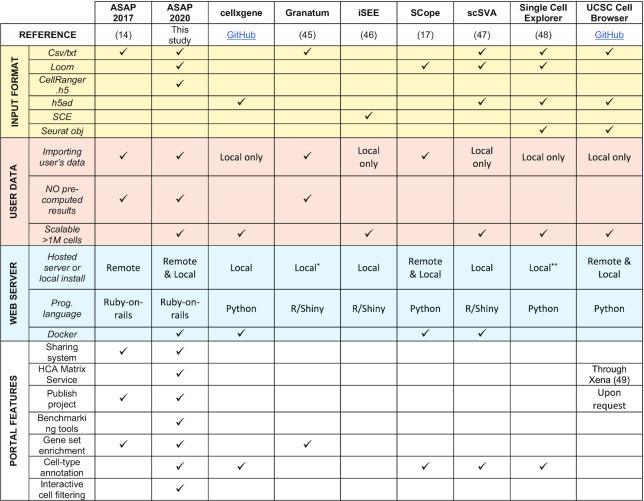
Table 1.
Overview of state-of-the-art web portals supporting single-cell RNA-seq data analysis and interactive visualization. Two versions of ASAP were compared to state-of-the-art tools. Docker indicates whether a docker image with the tool is provided by the developers. HCA: Human Cell Atlas. Benchmarking tools: The ability to monitor all the tools on the platform for computing time and/or RAM usage. Cell-type annotation: The ability to interactively annotate clusters/cell types
|
*A remote website was also available but seemed to mostly serve as an example, since no job queuing system was implemented, the website became inaccessible every time a step was launched
**SingleCellExplorer ‘Click here to Launch’ remote server was not functioning at the time of this paper
As we can see in Table 1, ASAP is amongst the first portals that are directly linked to the Human Cell Atlas (7). In addition, most portals are in essence visualization tools that require external pipelines to analyze a dataset, which can then be visualized in the respective portal (see ‘NO precomputed results’ in Table 1). Few portals therefore support a complete end-to-end analysis of the data within a web user interface, and the ones that do tend to require a local or cloud installation. In contrast, and as indicated in (38): ‘ASAP is a comprehensive hosting platform and as such it does not require a local or cloud installation’. Consequently, and contrary to most available portals, ASAP users can perform all the desired analyses directly within the portal, and do not have to consider installation prerequisites. Moreover, given ASAP’s multi-user functionalities, users can share their analysis projects with others in an interactive and modular fashion, which is currently unique to ASAP (see Table 1, ‘Sharing system’).
We are also currently working with the Fly Cell Atlas (FCA) consortium (https://flycellatlas.org/) to generate a central repository for atlas-like initiatives. In particular, we are collaborating with the Scope (17) portal to develop new methods for crowd annotation of clusters into cell types. Indeed, we believe that the next important demand in the single-cell field will be the ability to implement accurate cell annotations (40,41). Currently, this is still a great, outstanding challenge that requires hours of manual annotation and literature review. To address this, we plan to use the available user base of ASAP and SCope to create a crowd-based annotation of cells though an individual curation and voting system, thereby reinforcing correct cluster annotations. This will lead to the creation of a public database that will record cell identity features (such as marker genes) from personal projects as well as from those hosted by atlas-like initiatives (such as the HCA or the FCA). Thereafter, we plan to use this database for the interactive and automated annotation of cells.
Finally, we would like to point out that scATAC-seq datasets from 10x (CellRanger output) can in principle also be loaded into ASAP. For now, they can only be processed with the same scRNA-seq pipeline, i.e. no specific methods have so far been added such as cisTopic (42) or other motif enrichment analysis tools. However, the user can still perform UMAP/t-SNE and/or clustering, which can already be insightful. This shows the modular capacity of ASAP, which potentially offers a platform that will be able to include a more specific scATAC-seq data analysis workflow in the future.
We also plan to add an integration feature with the goal of integrating datasets and of correcting for batch effects. We are aware of existing techniques that support such integration, such as MNN (43) or Seurat (32,44), and are benchmarking them on high-dimensional datasets to select the most relevant method.
DATA AVAILABILITY
ASAP is freely available at https://asap.epfl.ch. It is an open source software whose source code is deposited in two GitHub repositories: (i) the R/Python/Java scripts are deposited in https://github.com/DeplanckeLab/ASAP and are available as a ready-to-use Docker container at https://hub.docker.com/r/fabdavid/asap_run/tags and (ii) the server code is available at https://github.com/fabdavid/asap2_web.
ACKNOWLEDGEMENTS
We would like to thank the ASAP user community for providing great feedback and interesting discussions on how to best evolve ASAP. We also thank Peter L. Hliva for technical help in the implementation of certain visualizations and Alex R. Lederer who helped us reviewed this manuscript.
Contributor Information
Fabrice P A David, Institute of Bioengineering, School of Life Sciences, École Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland; Swiss Institute of Bioinformatics, CH-1015 Lausanne, Switzerland; BioInformatics Competence Center, École Polytechnique Fédérale de Lausanne (EPFL), Switzerland.
Maria Litovchenko, Institute of Bioengineering, School of Life Sciences, École Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland; Swiss Institute of Bioinformatics, CH-1015 Lausanne, Switzerland.
Bart Deplancke, Institute of Bioengineering, School of Life Sciences, École Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland; Swiss Institute of Bioinformatics, CH-1015 Lausanne, Switzerland.
Vincent Gardeux, Institute of Bioengineering, School of Life Sciences, École Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland; Swiss Institute of Bioinformatics, CH-1015 Lausanne, Switzerland.
FUNDING
Chan Zuckerberg Initiative (CZI) grant for collaborative computational tools [2018-182612 (5022)]; Precision Health & related Technologies grant [PHRT-502]; Swiss National Science Foundation (SNSF) project grant [310030_182655]; institutional support by the EPFL (Open Science Fund). Funding for open access charge: Open Science fund from the EPFL. The open access publication charge for this paper has been waived by Oxford University Press – NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wang Y., Navin N.E.. Advances and applications of single-cell sequencing technologies. Mol. Cell. 2015; 58:598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hu Y., An Q., Sheu K., Trejo B., Fan S., Guo Y.. Single cell multi-omics technology: methodology and application. Front. Cell Dev. Biol. 2018; 6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Svensson V., Vento-Tormo R., Teichmann S.A.. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc. 2018; 13:599–604. [DOI] [PubMed] [Google Scholar]
- 4. Hung R.J., Hu Y., Kirchner R., Liu Y., Xu C., Comjean A., Tattikota S.G., Li F., Song W., Ho Sui S. et al.. A cell atlas of the adult Drosophila midgut. PNAS. 2020; 117:1514–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aizarani N., Saviano A., Sagar Mailly, L., Durand S., Herman J.S., Pessaux P., Baumert T.F., Grun D.. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature. 2019; 572:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cao J., Packer J.S., Ramani V., Cusanovich D.A., Huynh C., Daza R., Qiu X., Lee C., Furlan S.N., Steemers F.J. et al.. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science (New York, N.Y.). 2017; 357:661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Regev A., Teichmann S.A., Lander E.S., Amit I., Benoist C., Birney E., Bodenmiller B., Campbell P., Carninci P., Clatworthy M. et al.. The human cell atlas. eLife. 2017; 6:doi:10.7554/eLife.27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Han X., Wang R., Zhou Y., Fei L., Sun H., Lai S., Saadatpour A., Zhou Z., Chen H., Ye F. et al.. Mapping the mouse cell atlas by microwell-seq. Cell. 2018; 172:1091–1107. [DOI] [PubMed] [Google Scholar]
- 9. Schaum N., Karkanias J., Neff N.F., May A.P, Quake S.R., Wyss-Coray T., Darmanis S., Batson J., Botvinnik O., Chen M.B. et al.. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018; 562:367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Luecken M.D., Theis F.J.. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 2019; 15:e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rostom R., Svensson V., Teichmann S.A., Kar G.. Computational approaches for interpreting scRNA-seq data. FEBS Lett. 2017; 591:2213–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wolf F.A., Angerer P., Theis F.J.. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018; 19:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cusanovich D.A., Hill A.J., Aghamirzaie D., Daza R.M., Pliner H.A., Berletch J.B., Filippova G.N., Huang X., Christiansen L., DeWitt W.S. et al.. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell. 2018; 174:1309–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gardeux V., David F.P.A., Shajkofci A., Schwalie P.C., Deplancke B.. ASAP: a web-based platform for the analysis and interactive visualization of single-cell RNA-seq data. Bioinformatics. 2017; 33:3123–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boettiger C. An introduction to Docker for reproducible research. SIGOPS Oper. Syst. Rev. 2015; 49:71–79. [Google Scholar]
- 16. Merkel D. Docker: lightweight Linux containers for consistent development and deployment. Linux J. 2014; 2014:Article 2. [Google Scholar]
- 17. Davie K., Janssens J., Koldere D., De Waegeneer M., Pech U., Kreft L., Aibar S., Makhzami S., Christiaens V., Bravo Gonzalez-Blas C. et al.. A single-cell transcriptome atlas of the aging drosophila brain. Cell. 2018; 174:982–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sievert C., Parmer C., Hocking T., Chamberlain S., Ram K., Corvellec M., Despouy P.. plotly: Create interactive web graphics via ‘plotly. js’. R package version. 2017; 4:110. [Google Scholar]
- 19. Ono K., Demchak B., Ideker T.. Cytoscape tools for the web age: D3.js and Cytoscape.js exporters [version 2; peer review: 2 approved]. F1000Research. 2014; 3:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lopes C.T., Franz M., Kazi F., Donaldson S.L., Morris Q., Bader G.D.. Cytoscape Web: an interactive web-based network browser. Bioinformatics. 2010; 26:2347–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barrett T., Troup D.B., Wilhite S.E., Ledoux P., Rudnev D., Evangelista C., Kim I.F., Soboleva A., Tomashevsky M., Marshall K.A. et al.. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009; 37:D885–D890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brazma A., Parkinson H., Sarkans U., Shojatalab M., Vilo J., Abeygunawardena N., Holloway E., Kapushesky M., Kemmeren P., Lara G.G. et al.. ArrayExpress–a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 2003; 31:68–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barrett T., Clark K., Gevorgyan R., Gorelenkov V., Gribov E., Karsch-Mizrachi I., Kimelman M., Pruitt K.D., Resenchuk S., Tatusova T. et al.. BioProject and BioSample databases at NCBI: facilitating capture and organization of metadata. Nucleic Acids Res. 2012; 40:D57–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hubbard T., Barker D., Birney E., Cameron G., Chen Y., Clark L., Cox T., Cuff J., Curwen V., Down T. et al.. The ensembl genome database project. Nucleic Acids Res. 2002; 30:38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. et al.. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000; 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kanehisa M., Goto S.. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wishart D.S., Knox C., Guo A.C., Cheng D., Shrivastava S., Tzur D., Gautam B., Hassanali M.. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008; 36:D901–D906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Franzen O., Gan L.M., Bjorkegren J.L.M.. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database. 2019; 2019:doi:10.1093/database/baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X., Lan Y., Xu J., Quan F., Zhao E., Deng C., Luo T., Xu L., Liao G., Yan M. et al.. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019; 47:D721–D728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Diehl A.D., Meehan T.F., Bradford Y.M., Brush M.H., Dahdul W.M., Dougall D.S., He Y., Osumi-Sutherland D., Ruttenberg A., Sarntivijai S. et al.. The cell ontology 2016: enhanced content, modularization, and ontology interoperability. J. Biomed. Semantics. 2016; 7:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Andrews T.S., Hemberg M.. M3Drop: dropout-based feature selection for scRNASeq. Bioinformatics. 2019; 35:2865–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A.. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015; 33:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McInnes L., Healy J., Saul N., Großberger L.. Umap: uniform manifold approximation and projection. J. Open Source Softw. 2018; 3:861. [Google Scholar]
- 34. Maaten L.v.d, Hinton G.. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008; 9:2579–2605. [Google Scholar]
- 35. Kiselev V.Y., Kirschner K., Schaub M.T., Andrews T., Yiu A., Chandra T., Natarajan K.N., Reik W., Barahona M., Green A.R. et al.. SC3: consensus clustering of single-cell RNA-seq data. Nat. Methods. 2017; 14:483–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Law C.W., Chen Y., Shi W., Smyth G.K.. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014; 15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Çakır B., Prete M., Huang N., van Dongen S., Pir P., Kiselev V.Y.. Comparison of visualisation tools for single-cell RNAseq data. 2020; bioRxiv doi:07 February 2020, preprint: not peer reviewed 10.1101/2020.01.24.918342. [DOI] [PMC free article] [PubMed]
- 39. Papatheodorou I., Moreno P., Manning J., Fuentes A.M., George N., Fexova S., Fonseca N.A., Fullgrabe A., Green M., Huang N. et al.. Expression Atlas update: from tissues to single cells. Nucleic Acids Res. 2020; 48:D77–D83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pliner H.A., Shendure J., Trapnell C.. Supervised classification enables rapid annotation of cell atlases. Nat. Methods. 2019; 16:983–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hou R., Denisenko E., Forrest A.R.R.. scMatch: a single-cell gene expression profile annotation tool using reference datasets. Bioinformatics. 2019; 35:4688–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bravo Gonzalez-Blas C., Minnoye L., Papasokrati D., Aibar S., Hulselmans G., Christiaens V., Davie K., Wouters J., Aerts S.. cisTopic: cis-regulatory topic modeling on single-cell ATAC-seq data. Nat. Methods. 2019; 16:397–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Haghverdi L., Lun A.T.L., Morgan M.D., Marioni J.C.. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018; 36:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Butler A., Hoffman P., Smibert P., Papalexi E., Satija R.. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018; 36:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhu X., Wolfgruber T.K., Tasato A., Arisdakessian C., Garmire D.G., Garmire L.X.. Granatum: a graphical single-cell RNA-Seq analysis pipeline for genomics scientists. Genome Med. 2017; 9:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rue-Albrecht K., Marini F., Soneson C., Lun A.T.L.. iSEE: Interactive SummarizedExperiment Explorer [version 1; peer review: 3 approved]. F1000Research. 2018; 7:741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tabaka M., Gould J., Regev A.. scSVA: an interactive tool for big data visualization and exploration in single-cell omics. 2019; bioRxiv doi:06 January 2019, preprint: not peer reviewed 10.1101/512582. [DOI]
- 48. Feng D., Whitehurst C.E., Shan D., Hill J.D., Yue Y.G.. Single Cell Explorer, collaboration-driven tools to leverage large-scale single cell RNA-seq data. BMC Genomics. 2019; 20:676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Goldman M., Craft B., Hastie M., Repečka K., Kamath A., McDade F., Rogers D., Brooks A.N., Zhu J., Haussler D.. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. 2019; bioRxiv doi:05 March 2019, preprint: not peer reviewed 10.1101/326470. [DOI] [PMC free article] [PubMed]
- 50. Brennecke P., Anders S., Kim J.K., Kolodziejczyk A.A., Zhang X., Proserpio V., Baying B., Benes V., Teichmann S.A., Marioni J.C. et al.. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods. 2013; 10:1093–1095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
ASAP is freely available at https://asap.epfl.ch. It is an open source software whose source code is deposited in two GitHub repositories: (i) the R/Python/Java scripts are deposited in https://github.com/DeplanckeLab/ASAP and are available as a ready-to-use Docker container at https://hub.docker.com/r/fabdavid/asap_run/tags and (ii) the server code is available at https://github.com/fabdavid/asap2_web.