


Abstract
Anti-CRISPR (Acr) proteins encoded by (pro)phages/(pro)viruses have a great potential to enable a more controllable genome editing. However, genome mining new Acr proteins is challenging due to the lack of a conserved functional domain and the low sequence similarity among experimentally characterized Acr proteins. We introduce here AcrFinder, a web server (http://bcb.unl.edu/AcrFinder) that combines three well-accepted ideas used by previous experimental studies to pre-screen genomic data for Acr candidates. These ideas include homology search, guilt-by-association (GBA), and CRISPR-Cas self-targeting spacers. Compared to existing bioinformatics tools, AcrFinder has the following unique functions: (i) it is the first online server specifically mining genomes for Acr-Aca operons; (ii) it provides a most comprehensive Acr and Aca (Acr-associated regulator) database (populated by GBA-based Acr and Aca datasets); (iii) it combines homology-based, GBA-based, and self-targeting approaches in one software package; and (iv) it provides a user-friendly web interface to take both nucleotide and protein sequence files as inputs, and output a result page with graphic representation of the genomic contexts of Acr-Aca operons. The leave-one-out cross-validation on experimentally characterized Acr-Aca operons showed that AcrFinder had a 100% recall. AcrFinder will be a valuable web resource to help experimental microbiologists discover new Anti-CRISPRs.
INTRODUCTION
Acr (anti-CRISPR) proteins were first discovered in 2013 in Pseudomonas phages and prophages (1). Acr encoding genes often form operons with HTH (helix-turn-helix) domain-containing transcription suppressor genes (2,3) that encode Aca (Acr associated) proteins (4,5). These short Acr proteins (<200 aa) are made by phages/viruses and other mobile genetic elements to inhibit the CRISPR-Cas (clustered regularly interspersed short palindromic repeats [CRISPR]—CRISPR-associated genes) systems of their prokaryotic hosts for successful invasion and survival. Therefore, Acrs can turn off the CRISPR-Cas system of their hosts, and thus are ‘naturally occurring off-switch’ of CRISPR–Cas systems. Hence, anti-CRISPRs have great potential to serve as regulators/modulators of CRISPR–Cas genome editing tools for safer and more controllable genome engineering (6–9).
The research of anti-CRISPR is very young and growing at a remarkable rate. Since the first anti-CRISPR paper published in 2013 (1), there have been ∼130 papers published and available in PubMed as of February 2020, but only four were exclusively bioinformatics work (10–13). Clearly, there is a lack of bioinformatics resources for anti-CRISPRs, although most of the 56 experimentally characterized Acr protein families (Supplementary Table S1) had been identified with the help of bioinformatics (7,13,14). At present, the only three peer-reviewed bioinformatics resources (Table 1) for anti-CRISPRs include two online databases anti-CRISPRDB (12) and CRISPRminer (11), as well as a Google Doc for unifying anti-CRISPR nomenclature (https://tinyurl.com/anti-CRISPR) (13). The anti-CRISPRDB collects and presents experimentally characterized Acr proteins and their homologs on the web. The CRISPRminer focuses on CRISPR–Cas systems but also has an anti-CRISPR annotation module, which contains experimentally characterized Acr proteins, their homologs and genomic neighborhoods. In addition, a standalone program called Self-Targeting Spacer Searcher (STSS) was developed to detect if any spacers in a CRISPR array of a genome target a protospacer in the same genome, a phenomenon known as CRISPR spacer self-targeting (15). The self-targeting idea, i.e., bacterial genomes having CRISPR spacers and their targets (i.e., protospacers) in the same genome, has also been applied to searching for new Acrs (16).
Table 1.
Overview of current bioinformatics tools for Acr research
| Name | Resource provided | Features | Input | Output |
|---|---|---|---|---|
| anti-CRISPRDB | Database | Experimentally characterized Acrs and their homologs and BLAST search | NA | NA |
| CRISPRminer | Database | Experimentally characterized Acrs and their homologs and genomic context | NA | NA |
| Acr nomenclature | Google spreadsheets | Experimentally characterized Acrs and Acas nomenclature | NA | NA |
| Self-Targeting Spacer Searcher | Standalone package | Workflow for self-targeting spacer identification | List of genomes | Self-targeting spacers |
| AcrCatalog | Database | Predicted Acrs from decision tree ML classifier + heuristic filtering | NA | NA |
| AcRanker | Web server and standalone package | XGBoost ML classifier using AA biases | Protein sequences | Ranked protein list (no Acr subtype) |
| AcrFinder | Web server and standalone package | Workflow combining Homology + GBA + Self-targeting and user-friendly website | Protein or DNA sequences | Acr-Aca operons (with Acr subtype) |
Although all the above four bioinformatics resources (Table 1) can facilitate the research of anti-CRISPRs, none of them can automatically identify Acr proteins from given genomes. This task is difficult because all the 56 experimentally characterized Acr protein families (https://tinyurl.com/anti-CRISPR) are very divergent in sequence and most do not contain any known Pfam domains (17). We recently performed a large-scale survey of thousands of Acr homologs in 75 000+ bacterial genomes and suggested combining three computational approaches in order to improve the sensitivity of bioinformatics Acr discovery (10). These three approaches include: (i) sequence homology search; (ii) finding the more conserved Acr-associated (Aca) homologs that often sit next to Acr genes first and then searching the Aca gene neighborhood for Acr candidates (guilt-by-association or GBA approach); and (iii) searching for Acr candidates in genomes with self-targeting spacers. Particularly, the GBA and self-target approaches have contributed to the discovery of most of the 56 published Acr proteins.
After November 2019, two preprints became available online in bioRxiv, which reported using machine learning algorithms for bioinformatics discovery of new Acr proteins (18,19) (Table 1). AcRanker (19) (published when AcrFinder was under review) used amino acid compositions of known Acr proteins (positive data) and non-Acr proteins (negative data) to train an XGBoost classifier, which can then be used to predict new Acr proteins given a proteome input. AcrCatalog (18) defined and combined eight sequence features, the most important of which include self-targeting and directon (small protein encoding operons) protein features, and trained a random forest (RF) classifier for new Acr prediction. Millions of proteins from prokaryotic viruses and pro-viruses of the GenBank databases were examined using this RF classifier, and thousands of new Acr families were predicted. These pre-computed Acr families were used to build an online database called AcrCatalog, while the RF classifier itself is unavailable.
Here, as a follow-up to our previous work (10), we developed a new standalone software package (https://github.com/HaidYi/acrfinder) and a web server (http://bcb.unl.edu/AcrFinder), AcrFinder, to allow for automated genome mining for Acr-Aca operons. Compared to AcRanker, the only tool that also provides a standalone package and web server (Table 1), AcrFinder offers new utilities that: (i) allow not only protein but also nucleotide sequence file as input, (ii) identify not only Acrs but also their genomic neighborhood (e.g. the operons that also contain Acas), (iii) provide an Acr and Aca sequence databases, (iv) integrate Acr homology search, GBA and self-targeting in one software, and (v) provide a more user-friendly web interface with much more appealing graphic representation of the genomic context of operons that contain the predicted Acr genes.
DATABASES OF ACR AND ACA
We downloaded the Fasta sequences of all experimentally characterized Acrs from https://tinyurl.com/anti-CRISPR (13) to form the Acr database. As shown in Figure 1 and Supplementary Table S1, among the 56 Acr proteins, 52 are shorter than 200 aa (44 are shorter than 150 aa); 32 have their encoding genes co-localized adjacent to Aca genes and all these Aca genes are shorter than 150 aa. All the 56 Acr genes are located in short-gene operons meaning all genes are on the same strand (i.e. running in the same direction), encode short proteins (<200 aa), and most intergenic distances are <150 bp. Using the Acr database, we have further built an Aca database.
Figure 1.
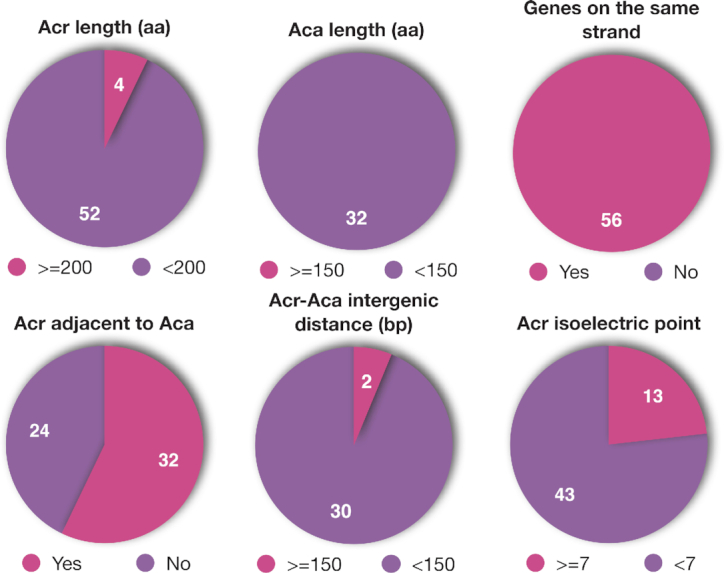
Sequence properties of 56 experimentally characterized Acr proteins and their genomic context. Numbers in the pies are the number of proteins or loci: (1) 52 out of the 56 Acr proteins are shorter than 200 aa; (2) all the 32 Aca proteins are shorter than 150 aa; (3) all the 56 Acr proteins are located in genomic operons with all the genes in the operon running in the same direction (on the same strand); (4) 32 out of the 56 Acr genes have neighboring Aca genes; (5) 30 Acr-Aca operons have all intergenic distances < 150 bp; (6) 43 out of the 56 Acr proteins have isoelectric point < 7. The detailed information can be found in Supplementary Table S1.
We used Acr homologous gene neighborhood (GBA) to identify HTH-domain containing proteins, as described in our recent paper (10). In other words, genes that encode HTH proteins and form short-gene operons with Acr homologs were identified as Acas. Briefly, the 56 Acr protein sequences were used as baits to DIAMOND (20) blast against five different databases: (i) NCBI RefSeq bacterial genomes (21); (ii) NCBI RefSeq archaeal genomes (21); (iii) assembled viral/proviral contigs of the JGI IMG/VR database (22); (iv) assembled human virome contigs of HuVirDB (23); and (v) assembled human gut virome contigs of GVD (24). Acr homologs (E-value < 1e−2 and sequence length < 200 aa) in these databases were located in the genomic contigs and the Acr gene neighborhood was examined. Aca candidates were then identified within the Acr operons (all genes are on the same strand and shorter than 200 aa) meeting the following criteria: (i) length < 200 aa; (ii) contain a Pfam HTH domain (E-value < 1e−2 and HTH coverage > 0.5); (iii) distance between the Aca candidate and the Acr homolog ≤ 3 genes. Aca candidate proteins from the five databases were combined as the Aca-GBA-DB.
The Aca-GBA-DB was further supplemented with 42 published Aca proteins (named Aca-Pub-DB) to form the final Aca database. The 42 published Aca proteins include 29 Acas surrounding the experimentally characterized Acr proteins plus 13 Aca proteins identified in (25). In our evaluation experiments, the Aca database (Aca-GBA-DB + Aca-Pub-DB) was filtered to obtain different smaller sets of Aca proteins corresponding to subsets of the 56 Acrs (see below).
WORKFLOW
Given a new genome in nucleotide Fasta sequences, AcrFinder will call gene prediction programs to generate a protein sequence file and a GFF (general feature format with gene position information in the contigs) file. It also allows users to submit a protein sequence file plus a GFF file as input. As shown in Figure 2, the workflow contains two independent routes. One route is Acr homology search (red arrows in Figure 2), which uses the 56 Acr proteins (http://bcb.unl.edu/AcrFinder/Download/database/Known_AcrDB.faa) as the query and the input protein sequences as the subject for a DIAMOND search. If an Acr homolog is found, the genomic operon that contains the Acr homolog will be extracted, and its subtype will be inferred according to its Acr query in the Acr database.
Figure 2.

AcrFinder workflow. Two major routes are included: (i) Acr homology search; once Acr homolog is found, the gene neighborhood is examined to only keep those that are located in short-gene operons. (ii) Aca GBA route contains three major steps (described in the main text). The resulting Acr-Aca operons are classified into three groups with different confidence levels.
The other route (blue arrows in Figure 2) combines Aca homology search, GBA, and self-targeting approaches, and contains three major steps. In step 1, proteins of the Aca database (http://bcb.unl.edu/AcrFinder/Download/database/AcrFinder_AcaDB.faa) will be DIAMOND blasted against a protein Fasta file that contains proteins encoded by short-gene operons that meet a set of specific criteria (Figure 2). The resulting Aca operons will be further filtered to remove those that have non-Aca proteins containing CDD functional domains (26) (except phage, HTH, and other mobilome domains). The reason is that most known Acr proteins do not have conserved functional domains and tend to be located next to mobile genetic elements (MGEs). In step 2, the filtered Aca operons will be further examined to look for putative MGEs in the neighborhood, which relies on the homology search against the PHASTER (pro)phage database (27) or against the mobilome position-specific scoring matrix models of the CDD database (26).
In step 3, CRISPRCasFinder (28) will be run on the input nucleotide Fasta file to identify high-confidence (level 3 and 4) CRISPR-Cas loci. If no CRISPR-Cas loci are found, AcrFinder will exit and produce no output from this route. Otherwise, the spacers of CRISPR arrays will be searched against the self-genome (with CRISPR-Cas loci masked) using BLASTn for identical self-targeting hits. If there are self-targeted protospacers found, then the Aca operons resulted from step 1 will be examined to see if they are within 5,000bp up- or down-stream of the self-targeted protospacers. If yes, these Aca operons will likely encode Acr proteins, and thus be labeled as ‘high confidence’ Acr-Aca operons, and inferred to target the CRISPR-Cas locus with the self-targeting spacer. If not, they will be labeled as ‘medium confidence’ Acr-Aca operons, and inferred to target the CRISPR-Cas locus with the self-targeting spacer. If there are no self-targeted protospacers found in the genome, then the Aca operons resulted from step 1 will be labeled as ‘low confidence’ Acr-Aca operons. If any of the Acr-Aca operons also contain homologs of known Acr proteins, they will also be indicated in the result.
PERFORMANCE EVALUATION
AcrFinder describes a bioinformatics workflow. The Acr homology search route only looks for homologs of known Acrs. We have evaluated the performance of the Aca GBA route by leave-one-out experiments. Specifically, out of the 56 experimentally characterized Acr proteins, eight (AcrIF4, AcrIF6, AcrIE4-F7, AcrIIA2, AcrIIA3, AcrIIA4, AcrIIA12, AcrIIA21) are from genomes with complete CRISPR-Cas systems and have neighboring Aca proteins (Supplementary Table S1). For each (denoted as A) of these eight Acrs in the Acr database, we created a new Aca database by removing Aca candidates derived from A (see above). AcrFinder was then run with this filtered Aca database on the genome that contains A to see if it can be found. The result (see Table 2 and its expanded version Supplementary Table S2) shows that all the eight Acrs can be identified in their corresponding leave-one-out experiments (i.e. recall = 100%). As expected, the size of the Acr gene neighborhood and the minimum number of prophage hits are two important parameters that affect the recall. When required to have at least one prophage hit within 10 genes up- and down-stream of the Acr gene, seven of the eight tested genes can be found by AcrFinder (recall = 87.5%).
Table 2.
Leave-one-out evaluation of AcrFinder on genomes containing 8 Acr proteins*
| Prophage hits within n genes up- or down-stream | Min. # of prophage hits | Found positives | Total positives | Recall |
|---|---|---|---|---|
| n = 5 | 1 | 5 | 8 | 62.5% |
| 0 | 8 | 8 | 100.0% | |
| n = 10 | 1 | 7 | 8 | 87.5% |
| 0 | 8 | 8 | 100.0% |
* Detailed experiment results can be found in Supplementary Table S2.
In addition to the true positive Acr and its associated operon, AcrFinder also found more Acr-Aca operons in each genome (Supplementary Table S3). Are these all false positives and can we calculate a precision for AcrFinder? To calculate a precision, one has to create a reliable negative Acr dataset so as to clearly define false positives and true negatives. AcRanker built the negative Acr dataset by excluding all proteins in a proteome that share > 40% sequence identity to all known Acr proteins. The negative dataset was only used for training the AcRanker classifier but was not used for calculating a precision (19). The reason is that the 56 experimentally characterized Acr families only represent a very tiny fraction of all the possible Acr families that exist in prokaryotes and their viruses (7), and a genome can encode multiple Acr proteins that share no sequence similarity. Indeed, we previously have found that one bacterial genome can contain multiple Acr homologs present in different operons (10). The recent AcrCatalog paper (18) also made similar observation in viral genomes. Even different experimentally characterized Acrs can be present in one genome. For example, as shown in Supplementary Table S1, Listeria monocytogenes J0161 (GCF_000168635.1) contains two Acr-Aca operons (AcrIIA2-AcrIIA1 and AcrIIA4-AcrIIA1) that are distant from each other in the genome; Moraxella bovoculi 58069 (GCA_000988605.1) has four different Acr genes in one operon (AcrVA1, AcrVA2, AcrIC1 and AcrVA3). Therefore, like AcRanker, we decide not to calculate a precision. However, we provide all the predicted operons ranked in three levels according to whether they have self-targeting spacer targets adjacent to the Acr-Aca operons, or do not have self-targeting spacer in the genome at all. If any of the operon also contains homologs of known Acr proteins, it will also be indicated. We also filter the Acr-Aca operons to make sure none of the Acr candidates in the operons contain conserved CDD domains (Figure 2).
Additionally, to compare with AcRanker (19), we have built a smaller Aca-GBA-DB using only 18 experimentally characterized Acr proteins (AcrIE1 to AcrIE4, AcrIF1 to AcrIF10, and AcrIIA1, AcrIIA2, AcrIIA4, AcrIIA5) as the baits for GBA finding Aca candidates, and a smaller Aca-Pub-DB with Acas adjacent to only these 18 experimentally characterized Acr proteins. We chose these 18 Acr proteins because the AcRanker web server was also trained on these proteins. Therefore, using the same training set we can equitably compare the performance of AcrFinder and AcRanker. We ran AcrFinder and AcRanker on genomes that contain four recently characterized Acrs that also have neighboring Acas, namely AcrIE4-F7, AcrIIA3, AcrIIA12 and AcrIIA21. As AcRanker ranks all the proteins in the input proteomes, the result (Table 3) shows the four Acr proteins were ranked 78th, 10th, 5th and 159th in their corresponding protein lists. Therefore, two of the four tested Acr proteins were ranked in the top 10. Unlike AcRanker, AcrFinder finds short-gene operons that contain at least one Aca homolog in a genome with CRISPR–Cas systems. Table 3 shows that AcrFinder was able to find Acr-Aca operons of two (AcrIE-IF7 and AcrIIA3) of the four tested Acrs. Hence, on the independent dataset, AcRanker and AcrFinder had similar performance. It should be noted that AcrFinder also correctly inferred the Acr subtypes for the tested proteins, while AcRanker only ranked the proteins without subtype inference (Table 1 and Table 3).
Table 3.
Independent evaluation of AcrFinder and AcRanker on genomes containing 4 Acr proteins (not in the training set)
| Acr family | AcrIE4-F7 | AcrIIA3 | AcrIIA12 | AcrIIA21 |
|---|---|---|---|---|
| Acr ID | WP_064584002.1 | WP_014930691.1 | WP_003731276.1 | WP_000384271.1 |
| Neighboring Aca ID | WP_064584003.1 | WP_014930689.1 | WP_003722518.1 | WP_000134666.1 |
| GCF ID | GCF_001654435.1 | GCF_000210795.2 | GCF_009807465.1 | GCF_002197205.1 |
| Proteome size | 6716 | 2822 | 2938 | 2153 |
| CRISPR-Cas subtype(s) | TypeIF | TypeIIA + TypeIB | TypeIB | TypeIIA |
| AcRanker rank | 78th | 10th | 5th | 159th |
| AcrFinder subtype | AcrIE4-IF7 | AcrIIA3 | - | - |
| Total # of AcrFinder predicted loci* | 5 | 10 | 10 | 6 |
* AcrFinder condition: up- or down-stream prophage hits n = 10, Min. # of prophage hits = 1, DIAMOND search mode = –more-sensitive and E-value < 0.01 and query coverage > 0.8.
UTILITIES
AcrFinder is provided as a standalone program and a web server. Genome sequences in fna, gff and faa formats are taken as input. Only one fna file as input is also acceptable; in that case, the gff and faa file will be generated by running Prodigal (29). Genomes of Archaea, Bacteria, and (pro)Viruses are all allowed. (pro)Viruses will not run CRISPRCasFinder (28), MGE search and CDD filtering; Archaea will run CRISPRCasFinder with a special Archaea flag (-ArchaCas). The AcrFinder standalone program (https://github.com/HaidYi/acrfinder) outputs a folder, where two files and three sub-folders are found. The two files contain the homology-based and GBA-based search results. The three sub-folders include: (i) input files; (ii) CRISPRCasFinder result files; (iii) all the intermediate result files.
On the AcrFinder web server, the job submission page has an option to let the users try out the sample data (Figure 3A). A help page is available to provide the detailed instructions on how to use the web server, particularly the interpretation of the data in the result page. A typical bacterial genome submission will finish ∼2 min. A result web link and a job ID are provided while the job is running. The result page has data tables to show the member genes in the identified Acr-Aca operon, as well as the genomic positions, strand, sequence, length, isoelectric point, molecular weight, if adjacent to MGE/prophage, if match with known Acr or Aca proteins, and if adjacent to self-targeting CRISPR spacers. Jbrowse is used to graphically display the gene neighborhood. Figure 3 shows the result page of an example bacterial genome input as a case study.
Figure 3.

AcrFinder web server case study. The URL of this case study is http://bcb.unl.edu/AcrFinder/result.php?jobid=1583809584. In this case study, we submitted the fna, faa, and gff files of the RefSeq bacterial genome assembly (GCF_000210795.2), which is known to encode AcrIIA3. (A) is the job submission page, where users can choose different parameters (default values are shown in the text fields). Clicking on ‘Run An EXAMPLE’ will initiate this case study job, which will take ∼2 minutes to finish. The result page will contain five major sections: (B) is the Guilt-by-Association result in a table, which has 17 columns with a variety of information including the inferred Acr subtype (the screenshot only shows the left nine columns); (C) is the JBrowse view of the GBA loci (genes in the loci are highlighted in yellow background); (D) is the Homology-based Acr search result in a table, which has 12 columns with a variety of information including the best known Acr homolog (the screenshot only shows the left six columns); (E) is the JBrowse view of the homology-based loci (genes in the loci are highlighted in yellow background); (F) is the result of CRISPRCasFinder result in a table (parsed to keep only high confidence CRISPR-Cas loci).
DISCUSSION
Anti-CRISPR (Acr) proteins are now being employed to develop various biotechnological tools with significant applications (8,30–32). Bioinformatics sequence analysis has assisted the discovery of most of the 56 known Acr families, which target 9 of over 30 different CRISPR-Cas subtypes (33). No bioinformatics tools were available for automated genome mining for new Acr proteins until November 2019, when AcRanker became online (19). AcRanker is the only tool currently available to allow online data submission. However, hosted on PythonAnywhere the AcRanker web server has very limited functions. Users can only upload a protein Fasta sequence file and the result is returned as a three-column file (sequence ID, rank, and score). Unfortunately, any Fasta sequences can be ranked even if they are from unrelated sources (e.g. plants or animals or false sequences), and no help/readme is provided to help understand what the score means and what cutoff value should use.
AcrFinder provides a web service that surpasses and differs significantly from AcRanker and AcrCatalog in many ways (Table 1), in particular the tabular and graphic representation of the genomic contexts of Acr-Aca operons with a lot of useful information to users (Figure 3). AcRanker suggested identifying prophages first and then submitting prophage regions for better ranking. AcrCatalog (18) implemented a number of heuristic filters before and after the RF classifier prediction, such as prophage identification, HTH search, and self-targeting spacer search. All these pre- and post-filtering steps require advanced bioinformatics skills but are not provided within AcRanker and AcrCatalog classifiers. They are, however, implemented and fully automated in AcrFinder's GBA route, together with an Acr homology search function. Within AcrFinder's standalone software and web server, various parameters can also be adjusted and explored by users with different levels of computer skills to achieve better and more meaningful Acr predictions. To rank the predicted Acr-Aca operons, in addition to the three levels of confidence, we have also provided two metrics that could be useful to users: (i) Acr homology identity (Acr_Hit|pident column, Figure 3B); (ii) Aca homology identity and E-value (Acr/Aca column, Figure 3B). The first metric is useful when there are homologs of known Acrs in the predicted operons. The second metric applies to all identified operons as they have to encode at least one protein homologous to Aca. Another indirect metric is in the MGE/Prophage column of Figure 3B, which provides the BLAST E-value of any encoded proteins in an operon that are homologous to an MGE or prophage.
AcrFinder also has limitations. First, it relies on Aca references to locate short-gene operons, where new Acr candidates are potentially present. However, as shown in Supplementary Table S1, some of the more recently characterized Acrs do not have Acas in proximity (e.g. AcrVA genes and many AcrIIA genes found in lytic phages or themselves containing an HTH domain). Second, AcrFinder requires that genomes have complete CRISPR-Cas systems to be mined for Acr-Aca operons. However, we and others have recently found that Acr homologs are present in genomes without complete CRISPR-Cas systems (10,34). Therefore, AcrFinder will not be able to find operons without Acas within or Acr-Aca operons from genomes without complete CRISPR-Cas systems. It should be mentioned that these limitations are due to the design of AcrFinder workflow, which intends to reduce false positives. As mentioned above, AcrCatalog implemented a number of filters that include the presence of HTH proteins in the Acr gene neighborhood and the presence of CRISPR-Cas and self-targeting spacers in the target genome, which were shown to be extremely critical to remove false positives (18). Our future improvement of AcrFinder will incorporate novel machine learning or deep learning algorithms in the pipeline that can partially overcome these limitations, as well as develop a quantitative measure (e.g. a score or P-value) to better rank the predicted Acr-Aca operons.
We plan to update AcrFinder at least once a year, to add newly characterized Acr sequences into the Acr database and create the new Aca-GBA-DB and Aca-Pub-DB to form the updated Aca database.
Supplementary Material
ACKNOWLEDGEMENTS
This work was partially completed utilizing the Holland Computing Center of the University of Nebraska, which receives support from the Nebraska Research Initiative.
Contributor Information
Haidong Yi, Department of Computer Science, University of North Carolina at Chapel Hill, NC, USA; College of Computer Science, Nankai University, Tianjin, China.
Le Huang, College of Computer Science, Nankai University, Tianjin, China.
Bowen Yang, Nebraska Food for Health Center, Department of Food Science and Technology, University of Nebraska - Lincoln, Lincoln, NE, USA.
Javi Gomez, Department of Computer Science, Northern Illinois University, DeKalb, IL, USA.
Han Zhang, College of Artificial Intelligence, Nankai University, Tianjin, China.
Yanbin Yin, Nebraska Food for Health Center, Department of Food Science and Technology, University of Nebraska - Lincoln, Lincoln, NE, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation (NSF) CAREER award [DBI-1933521]; United States Department of Agriculture (USDA) award [58–8042-9–089]; start-up grant of UNL [2019-YIN to Y.Y.]; National Natural Science Foundation of China [31728013, 61973174 to H.Z.]. Funding for open access charge: NSF CAREER award [DBI-1933521].
Conflict of interest statement. None declared.
REFERENCES
- 1. Bondy-Denomy J., Pawluk A., Maxwell K.L., Davidson A.R.. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013; 493:429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Birkholz N., Fagerlund R.D., Smith L.M., Jackson S.A., Fineran P.C.. The autoregulator Aca2 mediates anti-CRISPR repression. Nucleic Acids Res. 2019; 47:9658–9665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stanley S.Y., Borges A.L., Chen K.H., Swaney D.L., Krogan N.J., Bondy-Denomy J., Davidson A.R.. Anti-CRISPR-Associated proteins are crucial repressors of Anti-CRISPR transcription. Cell. 2019; 178:1452–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Borges A.L., Davidson A.R., Bondy-Denomy J.. The discovery, mechanisms, and evolutionary impact of Anti-CRISPRs. Ann. Rev. Virol. 2017; 4:37–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bondy-Denomy J. Protein inhibitors of CRISPR-Cas9. ACS Chem. Biol. 2018; 13:417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pawluk A., Amrani N., Zhang Y., Garcia B., Hidalgo-Reyes Y., Lee J., Edraki A., Shah M., Sontheimer E.J., Maxwell K.L. et al.. naturally occurring off-switches for CRISPR-Cas9. Cell. 2016; 167:1829–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pawluk A., Davidson A.R., Maxwell K.L.. Anti-CRISPR: discovery, mechanism and function. Nat. Rev. Microbiol. 2018; 16:12–17. [DOI] [PubMed] [Google Scholar]
- 8. Nakamura M., Srinivasan P., Chavez M., Carter M.A., Dominguez A.A., La Russa M., Lau M.B., Abbott T.R., Xu X., Zhao D. et al.. Anti-CRISPR-mediated control of gene editing and synthetic circuits in eukaryotic cells. Nat. Commun. 2019; 10:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shin J., Jiang F., Liu J.J., Bray N.L., Rauch B.J., Baik S.H., Nogales E., Bondy-Denomy J., Corn J.E., Doudna J.A.. Disabling Cas9 by an anti-CRISPR DNA mimic. Sci. Adv. 2017; 3:e1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yin Y., Yang B., Entwistle S.. Bioinformatics identification of Anti-CRISPR loci by using homology, Guilt-by-Association, and CRISPR Self-Targeting spacer approaches. mSystems. 2019; 4:e00455–e00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang F., Zhao S., Ren C., Zhu Y., Zhou H., Lai Y., Zhou F., Jia Y., Zheng K., Huang Z.. CRISPRminer is a knowledge base for exploring CRISPR-Cas systems in microbe and phage interactions. Commun. Biol. 2018; 1:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dong C., Hao G.F., Hua H.L., Liu S., Labena A.A., Chai G., Huang J., Rao N., Guo F.B.. Anti-CRISPRdb: a comprehensive online resource for anti-CRISPR proteins. Nucleic Acids Res. 2018; 46:D393–D398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bondy-Denomy J., Davidson A.R., Doudna J.A., Fineran P.C., Maxwell K.L., Moineau S., Peng X., Sontheimer E.J., Wiedenheft B.. A unified resource for tracking Anti-CRISPR names. CRISPR J. 2018; 1:304–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stanley S.Y., Maxwell K.L.. Phage-Encoded Anti-CRISPR defenses. Annu. Rev. Genet. 2018; 52:445–464. [DOI] [PubMed] [Google Scholar]
- 15. Watters K.E., Fellmann C., Bai H.B., Ren S.M., Doudna J.A.. Systematic discovery of natural CRISPR-Cas12a inhibitors. Science. 2018; 362:236–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rauch B.J., Silvis M.R., Hultquist J.F., Waters C.S., McGregor M.J., Krogan N.J., Bondy-Denomy J.. Inhibition of CRISPR-Cas9 with bacteriophage proteins. Cell. 2017; 168:150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. El-Gebali S., Mistry J., Bateman A., Eddy S.R., Luciani A., Potter S.C., Qureshi M., Richardson L.J., Salazar G.A., Smart A. et al.. The Pfam protein families database in 2019. Nucleic Acids Res. 2019; 47:D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gussow A.B., Shmakov S.A., Makarova K.S., Wolf Y.I., Bondy-Denomy J., Koonin E.V.. Vast diversity of anti-CRISPR proteins predicted with a machine-learning approach. 2020; bioRxiv doi:24 January 2020, preprint: not peer reviewed 10.1101/2020.01.23.916767. [DOI] [PMC free article] [PubMed]
- 19. Eitzinger S., Asif A., Watters K.E., Iavarone A.T., Knott G.J., Doudna J.A., Afsar Minhas F.U.A.. Machine learning predicts new anti-CRISPR proteins. Nucleic Acids Research. 2020; gkaa219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Buchfink B., Xie C., Huson D.H.. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015; 12:59–60. [DOI] [PubMed] [Google Scholar]
- 21. Tatusova T., DiCuccio M., Badretdin A., Chetvernin V., Nawrocki E.P., Zaslavsky L., Lomsadze A., Pruitt K.D., Borodovsky M., Ostell J.. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016; 44:6614–6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paez-Espino D., Chen I.A., Palaniappan K., Ratner A., Chu K., Szeto E., Pillay M., Huang J., Markowitz V.M., Nielsen T. et al.. IMG/VR: a database of cultured and uncultured DNA Viruses and retroviruses. Nucleic Acids Res. 2017; 45:D457–D465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Soto-Perez P., Bisanz J.E., Berry J.D., Lam K.N., Bondy-Denomy J., Turnbaugh P.J.. CRISPR-cas system of a prevalent human gut bacterium reveals hyper-targeting against phages in a human virome catalog. Cell Host Microbe. 2019; 26:325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gregory A.C., Zablocki O., Howell A., Bolduc B., Sullivan M.B.. The human gut virome database. 2019; bioRxiv doi:02 July 2019, preprint: not peer reviewed 10.1101/655910. [DOI] [PMC free article] [PubMed]
- 25. Marino N.D., Zhang J.Y., Borges A.L., Sousa A.A., Leon L.M., Rauch B.J., Walton R.T., Berry J.D., Joung J.K., Kleinstiver B.P. et al.. Discovery of widespread type I and type V CRISPR-Cas inhibitors. Science. 2018; 362:240–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marchler-Bauer A., Derbyshire M.K., Gonzales N.R., Lu S., Chitsaz F., Geer L.Y., Geer R.C., He J., Gwadz M., Hurwitz D.I. et al.. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015; 43:D222–D226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arndt D., Grant J.R., Marcu A., Sajed T., Pon A., Liang Y., Wishart D.S.. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016; 44:W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Couvin D., Bernheim A., Toffano-Nioche C., Touchon M., Michalik J., Neron B., Rocha E.P.C., Vergnaud G., Gautheret D., Pourcel C.. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018; 46:W246–W251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hyatt D., Chen G.L., Locascio P.F., Land M.L., Larimer F.W., Hauser L.J.. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010; 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bubeck F., Hoffmann M.D., Harteveld Z., Aschenbrenner S., Bietz A., Waldhauer M.C., Borner K., Fakhiri J., Schmelas C., Dietz L. et al.. Engineered anti-CRISPR proteins for optogenetic control of CRISPR-Cas9. Nat. Methods. 2018; 15:924–927. [DOI] [PubMed] [Google Scholar]
- 31. Johnston R.K., Seamon K.J., Saada E.A., Podlevsky J.D., Branda S.S., Timlin J.A., Harper J.C.. Use of anti-CRISPR protein AcrIIA4 as a capture ligand for CRISPR/Cas9 detection. Biosens. Bioelectron. 2019; 141:111361. [DOI] [PubMed] [Google Scholar]
- 32. Hirosawa M., Fujita Y., Saito H.. Cell-Type-Specific CRISPR activation with MicroRNA-Responsive AcrllA4 switch. ACS Synth. Biol. 2019; 8:1575–1582. [DOI] [PubMed] [Google Scholar]
- 33. Makarova K.S., Wolf Y.I., Iranzo J., Shmakov S.A., Alkhnbashi O.S., Brouns S.J.J., Charpentier E., Cheng D., Haft D.H., Horvath P. et al.. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020; 18:67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shehreen S., Chyou T.Y., Fineran P.C., Brown C.M.. Genome-wide correlation analysis suggests different roles of CRISPR-Cas systems in the acquisition of antibiotic resistance genes in diverse species. Philos. Trans. R Soc. Lond. B Biol. Sci. 2019; 374:20180384. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.