


Abstract
A mixed Protein Structure Network (PSN) and Elastic Network Model-Normal Mode Analysis (ENM-NMA)-based strategy (i.e. PSN-ENM) was developed to investigate structural communication in bio-macromolecules. Protein Structure Graphs (PSGs) are computed on a single structure, whereas information on system dynamics is supplied by ENM-NMA. The approach was implemented in a webserver (webPSN), which was significantly updated herein. The webserver now handles both proteins and nucleic acids and relies on an internal upgradable database of network parameters for ions and small molecules in all PDB structures. Apart from the radical restyle of the server and some changes in the calculation setup, other major novelties concern the possibility to: a) compute the differences in nodes, links, and communication pathways between two structures (i.e. network difference) and b) infer links, hubs, communities, and metapaths from consensus networks computed on a number of structures. These new features are useful to identify commonalties and differences between two different functional states of the same system or structural-communication signatures in homologous or analogous systems. The output analysis relies on 3D-representations, interactive tables and graphs, also available for download. Speed and accuracy make this server suitable to comparatively investigate structural communication in large sets of bio-macromolecular systems. URL: http://webpsn.hpc.unimore.it.
INTRODUCTION
The representation of biomolecular structures as networks of interacting amino acids/nucleotides is ever increasingly employed to investigate and elucidate complex phenomena such as protein folding and unfolding, protein stability, the role of structurally and functionally important residues, protein-protein and protein–DNA interactions, as well as intraprotein and interprotein communication and allosterism (1–25). These studies rely on methods that differ in the set of graph construction rules. Methods were first developed on proteins and successively updated to deal with nucleic acids and to include small molecules or ions, when in complex with the considered macromolecular system. In a protein structure graph (PSG), each amino acid residue is represented as a node and these nodes are connected by edges based on the strength of non-covalent interactions between residues. The interaction strength is often based on geometric criteria (e.g. distance cutoff) and can be used as an objective cutoff to build the PSG (3). Edges can be weighted using force field-based interaction energies, leading to the protein energy networks (PENs) (26,27).
Tools useful to simplify the representation and analysis of protein structures by 2D/3D visualization of residue interaction networks (RINs) are available as a downloadable software (http://www.rinalyzer.de (28)) or as a webserver (RING, http://protein.bio.unipd.it/ring/ (29,30)) that allows network visualization directly in the browser or in Cytoscape (31), or as a protein contact atlas (http://www.mrc-lmb.cam.ac.uk/pca/ (32)).
The employment of atomistic Molecular Dynamics (MD) trajectories instead of a single structure provides a dynamic description of the network as links break and form with atomic fluctuations. Dynamic networks can be inferred by employing time averages of the interaction strength cutoff for PSG building, time averages of the interaction energy for edge weighting, frequency cutoffs for link formation and hub definition, and cross-correlation of atomic motions to search for the shortest communication pathways (7,10,13,18,19,21,23–25,33–35). The majority of the resources for network analysis on MD trajectories are software packages such as Wordom (36), PSN-Ensemble (37), the PyMOL plugin xPyder (38), MD-TASK (39), PyInteraph (40) and gRINN (41). The latter two allow for PEN computation. The MDN web portal (42) and the updated version of the NAPS webserver (43) allow network analyses on atomistic MD trajectories provided by the user.
The evidence that functional dynamics of proteins relies on highly cooperative, low frequency, global/essential modes caused the diffusion of methods like the Normal Mode Analysis (NMA) able to infer such collective modes (44). The observed robustness of global modes with respect to details in atomic coordinates or specific interatomic interactions and their insensitivity to the specific energy functions and parameters that define the force field provided support to the development of simplified, i.e. coarse-grained (CG), descriptions of protein structures such as the Elastic Network Models (ENM) (44–46). The latter rely on the fact that the property that apparently dominates the shape of global modes is the network of inter-residue contacts, which is a purely geometric quantity defined by the overall shape or native contact topology of the protein. In recent years, ENM-based NMA (ENM-NMA), contributed significantly to improving our understanding of the collective dynamics of a number of allosteric proteins (44,46).
The graph-based approach proposed by Vishveshwara and coworkers and defined as Protein Structure Network (PSN) analysis (3) is the one that we implemented in the freely available Wordom software (36), which basically computes network features (e.g. nodes, hubs, links, communities etc.) and shortest communication pathways on MD trajectories. In a development of the method intended for high throughput investigations, the PSG is computed on a single high resolution structure rather than an MD trajectory and information on system dynamics (i.e. cross-correlation of atomic motions) is supplied by ENM-NMA (47). The approach, hereafter indicated as PSN-ENM, is powerful and fast but complex in the setup and analysis steps. To foster wide usage, the method was implemented in a webserver (webPSN) that allows the user to easily set the calculation, perform post-processing analyses, and both visualize and download numerical and 3D representations of the output (48). The server relies on Wordom and an auxiliary code (PSNtools). Cross-correlations of atomic motions from ENMs are also employed by the more recent ProSNEx webserver as an option for edge weighting (49).
webPSN was originally designed to compute single-structure PSN analyses (48). Here we propose an important extension of the server features. The server is now able to (a) compute the differences in nodes, links, and communication pathways between two structures (i.e. PSN difference) and (b) infer links, hubs, communities, and metapaths from consensus networks computed on a number of structures (Figure 1). Computation of consensus networks allows inferring common structural communication features in homologous systems sharing the same functionality (21,25) or even only the fold. On the other hand, computation of network differences is particularly useful to identify commonalties and differences in the structural communication of two functionally different states of the same system (21,25,47).
Figure 1.
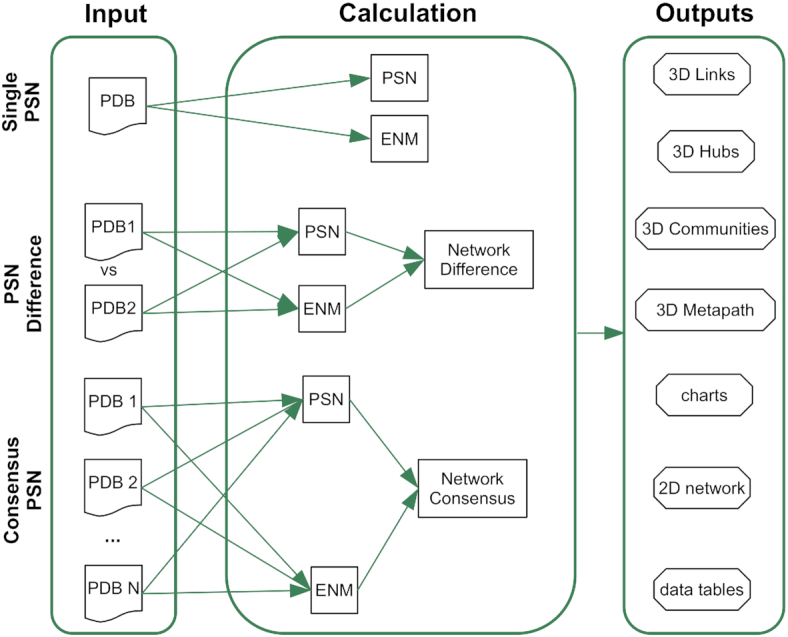
Flowchart of the webPSN processes. A schematic view of webPSN input, calculations, and output is provided here.
All computations can now be performed also on nucleic acids and can handle any ion or small molecule. Here, the improvements and novelties of webPSN along with a couple of example application cases are described. We discuss as well the unique features of webPSN with respect to two more recent webservers, NAPS ((43,50)) and ProSNEx (49), which, similar to webPSN, implement the PSN analysis method (3).
MATERIALS AND METHODS
Workflow of the PSN-ENM approach
The first step in PSN analysis consists in computing the PSG, i.e. an ensemble of nodes and links on a single high-resolution structural model (Figure 1). Building of the PSG is carried out by means of the PSN module implemented in Wordom (36). Details on the methodology have been published elsewhere (36,47,48), and are provided both as Supplementary Methods and web documentation. A number of network features including hubs (nodes connected by at least four links (see Supplementary Methods)) or node communities, i.e. sets of highly interconnected nodes such that nodes belonging to the same community are densely linked to each other and poorly connected to nodes outside the community (see Supplementary Methods), are computed as well.
Building the PSG provides the basis to search for the shortest paths between pairs of nodes, i.e. linked nodes connecting two extremities. The server starts computing the shortest communication pathways between all node pairs selected for calculation on job submission and retains only those pathways, in which at least one central node holds correlated motions with either one of the two path extremities. The server provides a global metapath made up by the most recurrent links in the pool of filtered paths. Metapaths represent a coarse/global picture of the structural communication in the considered system. In the result page, the user can filter those paths that begin and end at given residue pair(s) or that pass through a residue. Such a path filtering provides a novel metapath and is particularly recommended when some information on residues involved in structural communication is available.
The input
Here a brief description of the input in the original version of the server is provided (48). webPSN required a protein structure from the Protein Data Bank (PDB) format as an input. The user could either specify the PDB ID for retrieval from the PDB (http://www.rcsb.org) or upload a structure of interest from the local computer. The server permitted also to upload more than one structure instrumental in the parametrization of not yet parametrized residues. Those residues selected for the computation of PSG and communication pathways were interactively highlighted on the protein structure in the Jmol applet. Depending on the users’ expertise, a basic or an advanced input mode could be chosen. It was possible to execute the whole PSN-ENM procedure, which led to the computation of both PSG and communication pathways, or compute just the PSG. Advanced setting was available as well for computations of PSG, ENM, and shortest pathways.
The output
Here, a brief description of the output in the original version (48) of the server is provided. Results could be both interactively visualized on the web page and downloaded. Web 3D visualizations (i.e. by the Jmol applet) included PSG, hub distribution, metapaths, and the centers of the three most populated path clusters.
An interactive table listed all pathways with a number of associated indices, such as: (a) path length; (b) mean square distance fluctuations between all node pairs in a path (MSDFp), an index of path stiffness (47,48); (c) hub content; (d) fraction of user-defined relevant amino acid nodes; (e) correlation score, i.e. the ratio between number of correlated residues in the path and path length (the latter excludes the two extremities); (f) average similarity score (according to the clustering method) between the path and each path in the cluster; and (g) number of the cluster the path belongs to. The table could be sorted by any of these indices, or filtered according to cutoff values chosen by the user. Any predicted pathway could be visualized on the 3D structure by clicking the path itself in the table.
The output page showed also the distribution of: (a) path length; (b) MSDFp index; (c) hub content; (d) correlation score; (e) node frequency in paths; and (f) frequency of four-amino acid fragments in the predicted paths. A table was shown as well listing the number of nodes, hubs, and links in the PSG or in each node cluster, as well as the number of paths, nodes, and links in those paths.
Downloads were available both for the 3D representations of the output (as pymol and VMD scripts) and the numerical data (as csv and plain text files). The downloadable zip files also contained an output manual explaining the meaning of output files and numerical data.
New features of the webPSN server
The original version of webPSN has been significantly updated. Changes concern the calculation approach/setup, the whole style of the server, the input/output and the downloads.
Changes in the computation approach/setup are summarized here and in Results. Glycines are now included in the calculation. The server relies on an internal database of the normalization factors for the 20 standard amino acids and the 8 standard nucleotides (i.e. dA, dG, dC, dT, A, G, C, and U), as well as >30 000 biologically relevant small molecules and ions from the PDB (see Supplementary Methods). Additionally, the server automatically identifies un-parametrized molecules in the submitted PDB file and calculates their normalization factors transparently (see Supplementary Methods). All residues selected for PSG calculation in the submission stage are automatically used as extremities for path search. Computation of the ENM correlation of atomic motions for path filtering now relies only on one ENM method, i.e. the one that describes the system as Cα-atom coordinates interacting by a Hookean harmonic potential, with the force constant inversely proportional to the distance between the interacting particles (see Supplementary Methods) (45). Other changes in the procedure of PSG and metapath prediction were based on the outcome of benchmarks on five protein systems with known amino acid residues implicated in allosterism. Details are provided in Results.
Major changes in the input/output are the following: (a) the 3D-visualization now relies on the PV viewer (http://dx.doi.org/10.5281/zenodo.20980); (b) modalities to select the residues for computation; (c) the submission stage has been now simplified and fully automated with all the calculation parameters already properly pre-set; (d) PSN analysis can be carried out on a single structure (single PSN), on two structures to infer differences/commonalities between links, hubs, or metapaths (PSN difference), or on a set of homologous/analogous proteins/nucleic acids to infer consensus links, hubs, communities, or metapaths (consensus PSN) (Figure 2); (e) the output network elements, i.e. links, hubs, node communities, and metapaths, are simultaneously visualized on the 3D structure and in a table interacting with the 3D structure (Figure 2); (f) it is possible to interactively filter the pool of paths according to a sub-selection of residues as the extreme or middle position in a pathway, which leads to a novel metapath; and (g) comprehensive tables summarizing the most relevant network parameters for PSG or global and filtered metapaths are displayed as well in the browser.
Figure 2.

Network comparisons by webPSN. Examples of 3D-visualization of consensus PSN and PSN difference are shown here, using arrestins as an example case. This figure also summarizes the different types of analysis output provided by the server. All images here are snapshots of the webserver output pages.
Network comparisons rely on a positional labeling either (a) automatically performed based on established sequence or structure alignment methods or (b) provided by the user. In this context, amino acid residues or nucleotides are labeled according to their position in a multiple-sequence alignment rather then by their sequential numbering. This serves to identify structurally equivalent residues in homologous or analogous systems. Structurally equivalent small molecules or ions must receive the same label by the user, to be properly considered in network comparisons.
The user can provide a list of functionally important residues that are mapped onto the output network. Information on residue conservation from the ConSurf server/database (51) can enter the output analysis if the user uploads the ConSurf output file on job submission (i.e. as ‘External Values’).
Plots have changed as well and include: (a) 2D metapath representation; (b) distribution of the correlation score in the path pool; (c) distribution of the average interaction strength in the path pool; (d) hub distribution (as a percentage) in the path pool; (e) distribution of path length; and (f) distribution of the percentage of correlated residues in the path pool.
Downloadable output files have accordingly changed as well.
RESULTS
We illustrate the results of benchmarks, which led to the update of the PSN-ENM approach, and two example-application cases, which can be also found on the webPSN server.
Benchmarks of the updated PSN-ENM approach
Changes in the PSN-ENM methodology/parameters implemented in the updated version of the webserver relied on benchmarks, which evaluated the ability of the approach to predict amino acid residues likely involved in allosteric communication. A comprehensive database of allosteric proteins and modulators, ASD (52), was used as a source of information from in vitro experiments on allosteric residues for four out of the five considered proteins. Those proteins comprised: (a) the PDZ domain from the synaptic protein PSD-95 (PDZ3) in complex with its ligand peptide (PDB: 1BE9) (53,54); (b) the vitamin D receptor (VDR) bound to an agonist (PDB: 1IE9) (55–57); (c) the human Cystathionine β-Synthase (CBS) bound to Pyridoxal 5-Phosphate (PLP) (PDB: 1M54) (58); (d) the off- and on-states of Bruton's tyrosine kinase (Btk) (PDBs: 3GEN and 3K54, respectively) (59); and (e) dimeric caspase-1 (Csp1) in the on-state when the active site is occupied (PDB: 2HBQ) and in the off-state when the active site is empty (PDB: 1SC1) or when the enzyme is bound to an allosteric ligand at the dimer interface (PDB: 2FQQ) (60) (Figure 3).
Figure 3.
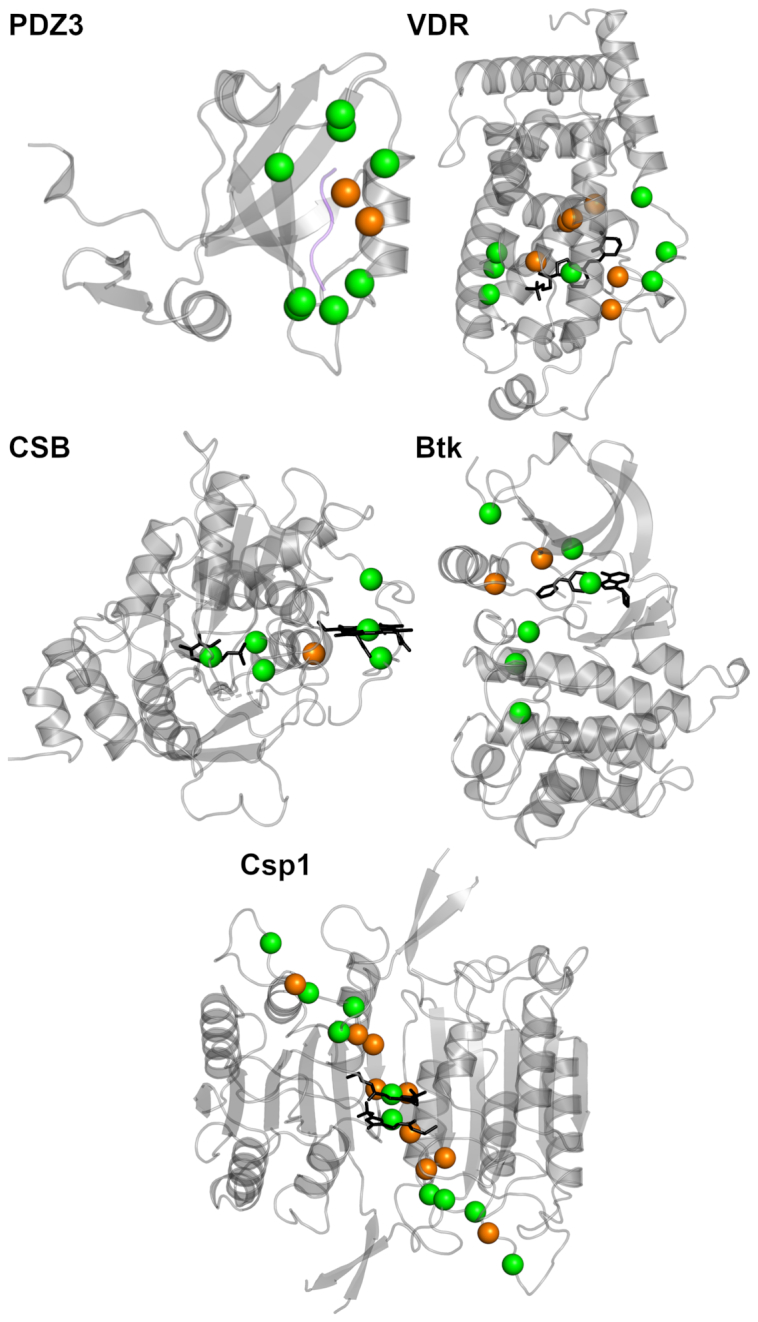
Benchmarks of the PSN-ENM method. The five protein systems used to validate the PSN-EMN method are shown, represented as cartoons. The spheres centred on the Cα-carbon atoms represent the amino acid residues likely implicated in allosteric communication, according to in vitro experiments; they are defined as ‘active residues’ here below. The ‘active residues’ participating as nodes in the metapath are green, whereas those not included in the metapath are orange. In detail, for PDZ3, the active residues are: G322, F325, G329, G330, G335, V362, H372, A376, K380, and V386; sensitivity, specificity, and J-index are, respectively: 0.80, 0.89, and 0.69. For VDR, the active residues are: Y143, Y147, F150, I238, I271, R274, W286, Y295, H397, Y401, V418, F422, and the ligand; sensitivity, specificity, and J-index are, respectively: 0.62, 0.99, and 0.61. For CSB, the active residues are: T257, T260, R266, C52, H65, Heme, and PLP; sensitivity, specificity, and J-index are, respectively: 1, 0.99, and 0.99. For Btk, the active residues are: W395, L460, T474, M449, F540, H519, D579, and the small-molecule inhibitor; sensitivity, specificity, and J-index are, respectively: 1, 0.88, and 0.88. Finally, for Csp1, in its homodimeric state, the active residues in each protomer are: S339, N337, D336, T334, S333, S332, R286, T388, E390, and the allosteric inhibitor; sensitivity, specificity, and J-index are, respectively: 0.5, 0.92, and 0.42.
Validation of the PSN-ENM method relied on comparison of those residues indicated as implicated in allosteric communication by in vitro experiments (listed in the legend of Figure 3) and the nodes making up the metapath. All possible combinations of the following network parameters were probed concerning the different stages of metapath prediction. As for the amino acid residues constituting the extremities of the paths, all amino acids, or a subset of amino acids, or a pair of amino acids were probed. As for the PSG, all resulting clusters were iteratively connected by the link(s) with the highest sub-Icritic interaction strength, i.e. cluster merge; no merging was probed as well. As for the number of ENM eigenvectors employed to compute the correlation of motions, a variable number or all possible eigenvectors were probed. As for the shortest communication pathway-search stage, link weighting by cross-correlation of motions or by interaction strength, or both, or no weighting were probed. As for the filtering stage, different motion correlation cutoffs were probed. Furthermore, different recurrence cutoffs (i.e. minimum % of paths a link must be present in to be part of the resulting metapath) and two different ways to compute recurrence were probed.
The Youden's index (J-index), combining in a single number sensitivity and specificity (61–63), was used to evaluate the predictive ability of the method. The J-index averaged over the indices of the five considered systems (i.e. by automatically selecting the best performer state if more than one state per protein was present) was used to select the computation conditions to implement in the updated version. The average sensitivity, specificity, and J-index for the selected conditions were, respectively, 0.78, 0.93, and 0.72, whereas the three indices per system are listed in the legend of Figure 3. Those results were achieved by using the most distant pairs of allosteric residues for the shortest communication path search. Modifications of the original default setup (48) included: (a) the application of cluster merging by sub-optimal Imin while computing the PSG to compensate, at least in part, for the lack of side chain fluctuations; (b) employment of 10 instead of 50 ENM-eigenvectors as they were sufficient to describe almost the entirety of total variance while accounting for higher correlated motions; (c) increase of the motion correlation coefficient cutoff for path filtering from 0.6 to 0.7 to strengthen the predictive ability; and (d) increase of link recurrence for metapath building from 5% to 10% to strengthen the predictive ability as well.
Example application cases
Here examples of network comparisons (PSN difference and consensus PSN) on a protein system and of single PSN on a protein-nucleic acid complex are reported. Example cases of network comparisons concern arrestins, a small family of proteins involved in regulation of signal transduction (64–66). They quench G protein activation by binding to and sterically hampering G protein coupled receptors (GPCRs). They mediate GPCR internalization and trafficking, and initiate further rounds of signaling by scaffolding other signaling proteins. Arrestin-1, also called visual arrestin, is specialized for opsins, while arrestin-2 and arrestin-3 recognize the other GPCRs. Arrestins share a β-sandwich architecture and an immunoglobulin-like topology made of one N-terminal (comprising β-strands I–X and the connecting loops (67)) and one C-terminal (comprising β-strands XI–XX and the connecting loops (67)) domains (ND and CD, respectively). Each domain is, thus, made of a seven-stranded β-sandwich made of a four-stranded β-sheet packed against a three-stranded β-sheet (Figure 2). The two domains are related by an intramolecular pseudo two-fold rotation axis, which changes with arrestin activation. The side-by-side placement of the domains gives rise to the central crest, composed of the finger loop (V–VI loop), a key receptor-binding element, the middle loop (VIII–IX loop) and the C-loop (XV–XVI loop), both stabilizing elements for inactive basal-state arrestin (66). The interface of the ND and CD is stabilized by hydrophobic interactions and hydrogen bond networks including the well-described polar core, a network of buried charged residues. In addition to the inter-domain rearrangement, the ND and central loops show large structural changes associated with arrestin activation, which is also accompanied by conformational changes in the so called ‘lariat loop’ (XVII–XVIII loop).
In this case example, network features shared in common by the inactive states of arrestin-1 (PDB: 1CF1 (67)), arrestin-2 (PDB: 1G4M (68)), and arrestin-3 (PDB: 3PZD (69)) have been inferred (i.e. consensus PSN), by using a positional labeling derived from sequence alignment (67). In the off-state of arrestins, hubs distribute on the β-strands in the ND and at the interface between the two domains (Figure 2). The inactive state is also characterized by a number of small node communities distributed on the strands of both domains. Inter-domain communities are almost absent, which is in line with the two domains acting as independent dynamic units in the transition from the inactive to the active states. Most recurrent links in the consensus metapath essentially involve the ND and the CD portions proximal to the inter-domain interface. Structural differences related to arrestin activation were inferred by computing the PSN difference between the crystal structures of the inactive (PDB: 1CF1) and active (PDB: 5W0P (70)) states of arrestin-1 (Figure 2). The two states share only five hubs in common (green spheres), indicative of significant structural divergences. While in the inactive state hubs distribute on both domains (orange spheres), in the active state they tend to crowd in the ND (violet spheres), which may be in line with a higher mobility of the latter state. In line with hub distribution, the inactive and active states of the two arrestins exhibit marked differences in their structural communication. Indeed, only three links are shared in common by the metapaths of the two states. Metapath links tend to equi-distribute in both domains. However, considering only links with recurrence ≥20%, the percentages of links in the ND is higher for the active state than the inactive one, likely due to the interactions made by the C-tail and to the integrity of the polar core in the inactive state.
As already stated above, all calculations can now be done also on nucleic acids. Aminoacyl-tRNA synthetases catalyze the formation of an aminoacyl-AMP from an amino acid and ATP, prior to the aminoacyl transfer to tRNA (71). The productive complex between ATP-bound glutamyl-tRNA synthetase (GluRS) and tRNA (PDB: 1N77 (71)) was used as an example case. GluRS is made of five domains: (a) the Rossmann fold (catalytic domain); (b) the connective peptide (or acceptor binding); (c) the stem contact fold; and (d) two anticodon binding (71). Snapshots of 3D representation of the output are shown in Figure 4. The ATP molecule and all residues in the protein and in tRNA were used to build the PSG and to search for the shortest communication pathways (Figure 4A). Those hubs most strongly linked (i.e. holding the highest average interaction strength of their links; colored red) involve almost all tRNA bases (including also the C34, U35, and C36 anticodon triplet and the last two bases, C75 and A76, in the acceptor stem) and the catalytic site of GluRS, including ATP (Figure 4B). The biggest node community (made of 83 nodes, 156 links, and 72 hubs) involves almost the entire tRNA and the catalytic site of GluRS (Figure 4C). The second node community, significantly smaller than the first community (made of 11 nodes, 15 links, and 8 hubs; colored green), concerns the interface between the anticodon loop and GluRS. In line with the shape of the biggest community, the global metapath inferred from all predicted pathways delineates a communication between the anticodon loop and the catalytic site of GluRS via the tRNA structure (Figure 4D). A similar metapath is obtained from the subset of pathways holding the anticodon C34 and ATP, as well as all residues within a sphere of 3-Å radius (from the centroids of the selected residues) as extremities (i.e. following an interactive path filtering) (Figure 4E). Thus, the productive conformation of the ternary complex involving ATP, GluRS and tRNA seems to favor the allosteric communication between the anticodon region of tRNA and the catalytic site of the protein.
Figure 4.

Structural communication in a protein-nucleic acid complex. Snapshots of the 3D output visualization are shown. They concern single PSN calculation on a productive ternary complex between ATP-bound GluRS and tRNA. (A) All residues included in calculation of the PSG and the shortest communication pathways are shown. The image of the GluRS-ATP-tRNA complex is a snapshot of the webserver input page, where the graphical representation (i.e. cartoons for the protein and the nucleic acid and ball-and-sticks for ATP) and coloring are the viewer defaults. (B) Hubs are shown as spheres centered on the Cα-atoms and colored according to the average interaction strength of their links. A color legend is available on the webserver. (C) The nodes and link communities are shown, colored according to their size, with red indicating the biggest community. (D) The global metapath is shown, with color indicating link recurrence. A color legend is available on the webserver. (E) The displayed metapath was inferred from the subset of pathways holding the anticodon C34 and ATP as well as all residues within a sphere of 3-Å radius (from the centroids of the selected residues) as extremities (i.e. following an interactive path filtering). All images in panels B-E are snapshots of the webserver output page, where GluRS is gray, tRNA is white, and ATP is colored by atom type.
DISCUSSION
Biomolecular function depends on structural communication and the entailed dynamics, especially those motions that occur relatively slowly and require long timescale computations (72). However, computational analyses using established techniques with well-chosen approximations have continually proven to be of great value in investigating interesting biophysical and biochemical problems with relatively modest demand in terms of computer power (44,72). In this framework, we coupled ENM-NMA with the graph theory-based PSN analysis in a so-called mixed PSN-ENM approach to incorporate protein dynamics in the structural communication when dealing with single crystallographic structures (47). The approach was implemented in the webPSN server (48).
A number of applications of the PSN analysis highlighted the importance of comparing one or more structure networks to infer the structural effects of missense mutations (13,21,24), identify differences in the structural communication between two different functional states of the same protein (22,25,47), or the structural communication signatures in a number of homologous or analogous proteins (22). The ability to compare two or more networks inferred from PSN-ENM on single structural models is an important novel feature of the webPSN server. The latter has been radically re-styled and the approach made more robust than the previous version. Moreover, the present version is easier to use, since all calculation parameters are now set as optimized defaults. Another relevant novelty of the updated server is its extension to nucleic acids taken alone or in complex with proteins.
Following the publication of the webPSN server (48) other two webservers dealing with single-PSN analysis were published, NAPS (43,50) and ProSNEx (49). A number of edge-weighting options (e.g. by edge strength, or interaction energy, or cross-correlation of atomic motions) are available for input setting at both webservers. Our benchmarks indicated that including edge weighting either by interaction strength, or cross-correlation of atomic motions, or both in pathway search did not improve the predictive ability of the method. Therefore, none of the three options was set as a default. Unique features and added values of webPSN compared to NAPS and ProSNEx include: (a) structure-dependent and user-independent setting of the calculation parameters and approach, whose predictive ability was validated on a number of systems; (b) the possibility to include all possible small molecules and ions in the PSG; (c) a user-independent incorporation of information on system's dynamics into prediction of communication pathways on single structures; (d) the possibility to predict the relevant residues in the structural communication and to identify allosteric sites in an unbiased manner, by automatically computing the shortest communication pathways among all node pairs in the PSG (leading to a global pool of communication pathways), which is a fundamental requirement when the allosteric sites are unknown; (e) the possibility to extract from the global pool a subset of paths that begin and/or end, or pass through user-selected residues (i.e. a path filtering action in the output analysis page), which becomes a relevant option when some information on the allosteric residues is available; (f) computation of difference and consensus networks; (g) extension to nucleic acids of the same computational approach employed for proteins; (h) publication quality graphs; and (i) improved speed.
Speed and accuracy make this server suitable to comparatively infer the fingerprints of structural communication, including allosterism, in large sets of bio-macromolecular systems.
DATA AVAILABILITY
The webPSN server is freely available at http://webpsn.hpc.unimore.it
Supplementary Material
Notes
Present address: Michele Seeber, Department of Physics, Informatics, and Mathematics, University of Modena and Reggio Emilia, Modena 41125, Italy.
Contributor Information
Angelo Felline, Department of Life Sciences, University of Modena and Reggio Emilia, Modena 41125, Italy.
Michele Seeber, Department of Life Sciences, University of Modena and Reggio Emilia, Modena 41125, Italy.
Francesca Fanelli, Department of Life Sciences, University of Modena and Reggio Emilia, Modena 41125, Italy; Center for Neuroscience and Neurotechnology, University of Modena and Reggio Emilia, Modena 41125, Italy.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
MIUR (Ministero dell’Istruzione, dell’Università e della Ricerca Scientifica) Italy [PRIN2017]; University of Modena and Reggio Emilia grants [FAR-2018 to FF.]. Funding for open access charge: MIUR [PRIN2017] or University of Modena and Reggio Emilia [FAR2018].
Conflict of interest statement. None declared.
REFERENCES
- 1. Vendruscolo M., Paci E., Dobson C.M., Karplus M.. Three key residues form a critical contact network in a protein folding transition state. Nature. 2001; 409:641–645. [DOI] [PubMed] [Google Scholar]
- 2. Amitai G., Shemesh A., Sitbon E., Shklar M., Netanely D., Venger I., Pietrokovski S.. Network analysis of protein structures identifies functional residues. J. Mol. Biol. 2004; 344:1135–1146. [DOI] [PubMed] [Google Scholar]
- 3. Brinda K.V., Vishveshwara S.. A network representation of protein structures: implications for protein stability. Biophys. J. 2005; 89:4159–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. del Sol A., Fujihashi H., Amoros D., Nussinov R.. Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol. Syst. Biol. 2006; 2:2006.0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bode C., Kovacs I.A., Szalay M.S., Palotai R., Korcsmaros T., Csermely P.. Network analysis of protein dynamics. FEBS Lett. 2007; 581:2776–2782. [DOI] [PubMed] [Google Scholar]
- 6. Eyal E., Chennubhotla C., Yang L.W., Bahar I.. Anisotropic fluctuations of amino acids in protein structures: insights from X-ray crystallography and elastic network models. Bioinformatics. 2007; 23:i175–184. [DOI] [PubMed] [Google Scholar]
- 7. Ghosh A., Vishveshwara S.. A study of communication pathways in methionyl- tRNA synthetase by molecular dynamics simulations and structure network analysis. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:15711–15716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chennubhotla C., Bahar I.. Signal propagation in proteins and relation to equilibrium fluctuations. PLoS Comput. Biol. 2007; 3:1716–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chennubhotla C., Yang Z., Bahar I.. Coupling between global dynamics and signal transduction pathways: a mechanism of allostery for chaperonin GroEL. Mol. Biosyst. 2008; 4:287–292. [DOI] [PubMed] [Google Scholar]
- 10. Sethi A., Eargle J., Black A.A., Luthey-Schulten Z.. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:6620–6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vishveshwara S., Ghosh A., Hansia P.. Intra and inter-molecular communications through protein structure network. Curr. Protein Pept. Sci. 2009; 10:146–160. [DOI] [PubMed] [Google Scholar]
- 12. Bhattacharyya M., Ghosh A., Hansia P., Vishveshwara S.. Allostery and conformational free energy changes in human tryptophanyl-tRNA synthetase from essential dynamics and structure networks. Proteins. 2010; 78:506–517. [DOI] [PubMed] [Google Scholar]
- 13. Fanelli F., Seeber M.. Structural insights into retinitis pigmentosa from unfolding simulations of rhodopsin mutants. FASEB J. 2010; 24:3196–3209. [DOI] [PubMed] [Google Scholar]
- 14. Doncheva N.T., Klein K., Domingues F.S., Albrecht M.. Analyzing and visualizing residue networks of protein structures. Trends Biochem. Sci. 2011; 36:179–182. [DOI] [PubMed] [Google Scholar]
- 15. Pandini A., Fornili A., Fraternali F., Kleinjung J.. Detection of allosteric signal transmission by information-theoretic analysis of protein dynamics. FASEB J. 2012; 26:868–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Papaleo E., Lindorff-Larsen K., De Gioia L.. Paths of long-range communication in the E2 enzymes of family 3: a molecular dynamics investigation. Phys. Chem. Chem. Phys. 2012; 14:12515–12525. [DOI] [PubMed] [Google Scholar]
- 17. Venkatakrishnan A.J., Deupi X., Lebon G., Tate C.G., Schertler G.F., Babu M.M.. Molecular signatures of G-protein-coupled receptors. Nature. 2013; 494:185–194. [DOI] [PubMed] [Google Scholar]
- 18. Sethi A., Tian J., Derdeyn C.A., Korber B., Gnanakaran S.. A mechanistic understanding of allosteric immune escape pathways in the HIV-1 envelope glycoprotein. PLoS Comput. Biol. 2013; 9:e1003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tse A., Verkhivker G.M.. Molecular dynamics simulations and structural network analysis of c-Abl and c-Src kinase core proteins: capturing allosteric mechanisms and communication pathways from residue centrality. J. Chem. Inf. Model. 2015; 55:1645–1662. [DOI] [PubMed] [Google Scholar]
- 20. Bhattacharyya M., Ghosh S., Vishveshwara S.. Protein structure and function: looking through the network of side-chain interactions. Curr. Protein Pept. Sci. 2016; 17:4–25. [DOI] [PubMed] [Google Scholar]
- 21. Felline A., Mariani S., Raimondi F., Bellucci L., Fanelli F.. Structural determinants of constitutive activation of gα proteins: transducin as a paradigm. J. Chem. Theory Comput. 2017; 13:886–899. [DOI] [PubMed] [Google Scholar]
- 22. Felline A., Ghitti M., Musco G., Fanelli F.. Dissecting intrinsic and ligand-induced structural communication in the beta3 headpiece of integrins. Biochim. Biophys. Acta. 2017; 1861:2367–2381. [DOI] [PubMed] [Google Scholar]
- 23. Salamanca Viloria J., Allega M.F., Lambrughi M., Papaleo E.. An optimal distance cutoff for contact-based protein structure networks using side-chain centers of mass. Sci. Rep. 2017; 7:2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Behnen P., Felline A., Comitato A., Di Salvo M.T., Raimondi F., Gulati S., Kahremany S., Palczewski K., Marigo V., Fanelli F.. A small chaperone improves folding and routing of rhodopsin mutants linked to inherited blindness. iScience. 2018; 4:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Felline A., Belmonte L., Raimondi F., Bellucci L., Fanelli F.. Interconnecting flexibility, structural communication, and function in RhoGEF oncoproteins. J. Chem. Inf. Model. 2019; 59:4300–4313. [DOI] [PubMed] [Google Scholar]
- 26. Vijayabaskar M.S., Vishveshwara S.. Interaction energy based protein structure networks. Biophys. J. 2010; 99:3704–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sladek V., Tokiwa H., Shimano H., Shigeta Y.. Protein residue networks from energetic and geometric data: are they identical. J. Chem. Theory Comput. 2018; 14:6623–6631. [DOI] [PubMed] [Google Scholar]
- 28. Doncheva N.T., Assenov Y., Domingues F.S., Albrecht M.. Topological analysis and interactive visualization of biological networks and protein structures. Nat. Protoc. 2012; 7:670–685. [DOI] [PubMed] [Google Scholar]
- 29. Martin A.J., Vidotto M., Boscariol F., Di Domenico T., Walsh I., Tosatto S.C.. RING: networking interacting residues, evolutionary information and energetics in protein structures. Bioinformatics. 2011; 27:2003–2005. [DOI] [PubMed] [Google Scholar]
- 30. Piovesan D., Minervini G., Tosatto S.C.. The RING 2.0 web server for high quality residue interaction networks. Nucleic Acids Res. 2016; 44:W367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T.. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kayikci M., Venkatakrishnan A.J., Scott-Brown J., Ravarani C.N.J., Flock T., Babu M.M.. Visualization and analysis of non-covalent contacts using the Protein Contacts Atlas. Nat. Struct. Mol. Biol. 2018; 25:185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vanwart A.T., Eargle J., Luthey-Schulten Z., Amaro R.E.. Exploring residue component contributions to dynamical network models of allostery. J. Chem. Theory Comput. 2012; 8:2949–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feher V.A., Durrant J.D., Van Wart A.T., Amaro R.E.. Computational approaches to mapping allosteric pathways. Curr. Opin. Struct. Biol. 2014; 25:98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Van Wart A.T., Durrant J., Votapka L., Amaro R.E.. Weighted Implementation of Suboptimal Paths (WISP): An optimized algorithm and tool for dynamical network analysis. J. Chem. Theory Comput. 2014; 10:511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seeber M., Felline A., Raimondi F., Muff S., Friedman R., Rao F., Caflisch A., Fanelli F.. Wordom: a user-friendly program for the analysis of molecular structures, trajectories, and free energy surfaces. J. Comput. Chem. 2011; 32:1183–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bhattacharyya M., Bhat C.R., Vishveshwara S.. An automated approach to network features of protein structure ensembles. Protein Sci. 2013; 22:1399–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pasi M., Tiberti M., Arrigoni A., Papaleo E.. xPyder: a PyMOL plugin to analyze coupled residues and their networks in protein structures. J. Chem. Inf. Model. 2012; 52:1865–1874. [DOI] [PubMed] [Google Scholar]
- 39. Brown D.K., Penkler D.L., Sheik Amamuddy O., Ross C., Atilgan A.R., Atilgan C., Tastan Bishop O.. MD-TASK: a software suite for analyzing molecular dynamics trajectories. Bioinformatics. 2017; 33:2768–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tiberti M., Invernizzi G., Lambrughi M., Inbar Y., Schreiber G., Papaleo E.. PyInteraph: a framework for the analysis of interaction networks in structural ensembles of proteins. J. Chem. Inf. Model. 2014; 54:1537–1551. [DOI] [PubMed] [Google Scholar]
- 41. Sercinoglu O., Ozbek P.. gRINN: a tool for calculation of residue interaction energies and protein energy network analysis of molecular dynamics simulations. Nucleic Acids Res. 2018; 46:W554–W562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ribeiro A.A., Ortiz V.. MDN: a web portal for network analysis of molecular dynamics simulations. Biophys. J. 2015; 109:1110–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chakrabarty B., Naganathan V., Garg K., Agarwal Y., Parekh N.. NAPS update: network analysis of molecular dynamics data and protein-nucleic acid complexes. Nucleic Acids Res. 2019; 47:W462–W470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bahar I., Lezon T.R., Bakan A., Shrivastava I.H.. Normal mode analysis of biomolecular structures: functional mechanisms of membrane proteins. Chem. Rev. 2010; 110:1463–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tirion M.M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996; 77:1905–1908. [DOI] [PubMed] [Google Scholar]
- 46. Atilgan C., Okan O.B., Atilgan A.R.. Network-based models as tools hinting at nonevident protein functionality. Annu. Rev. Biophys. 2012; 41:205–225. [DOI] [PubMed] [Google Scholar]
- 47. Raimondi F., Felline A., Seeber M., Mariani S., Fanelli F.. A mixed protein structure network and elastic network model approach to predict the structural communication in biomolecular Systems: The PDZ2 domain from tyrosine phosphatase 1E as a case study. J. Chem. Theory Comput. 2013; 9:2504–2518. [DOI] [PubMed] [Google Scholar]
- 48. Seeber M., Felline A., Raimondi F., Mariani S., Fanelli F.. WebPSN: a web server for high-throughput investigation of structural communication in biomacromolecules. Bioinformatics. 2015; 31:779–781. [DOI] [PubMed] [Google Scholar]
- 49. Aydinkal R.M., Sercinoglu O., Ozbek P.. ProSNEx: a web-based application for exploration and analysis of protein structures using network formalism. Nucleic Acids Res. 2019; 47:W471–W476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chakrabarty B., Parekh N.. NAPS: network analysis of protein structures. Nucleic Acids Res. 2016; 44:W375–W382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T., Ben-Tal N.. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016; 44:W344–W350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang Z., Zhu L., Cao Y., Wu G., Liu X., Chen Y., Wang Q., Shi T., Zhao Y., Wang Y. et al.. ASD: a comprehensive database of allosteric proteins and modulators. Nucleic Acids Res. 2011; 39:D663–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Doyle D.A., Lee A., Lewis J., Kim E., Sheng M., MacKinnon R.. Crystal structures of a complexed and peptide-free membrane protein-binding domain: Molecular basis of peptide recognition by PDZ. Cell. 1996; 85:1067–1076. [DOI] [PubMed] [Google Scholar]
- 54. Lockless S.W., Ranganathan R.. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999; 286:295–299. [DOI] [PubMed] [Google Scholar]
- 55. Shulman A.I., Larson C., Mangelsdorf D.J., Ranganathan R.. Structural determinants of allosteric ligand activation in RXR heterodimers. Cell. 2004; 116:417–429. [DOI] [PubMed] [Google Scholar]
- 56. Tocchini-Valentini G., Rochel N., Wurtz J.M., Mitschler A., Moras D.. Crystal structures of the vitamin D receptor complexed to superagonist 20-epi ligands. PNAS. 2001; 98:5491–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yamamoto K., Abe D., Yoshimoto N., Choi M., Yamagishi K., Tokiwa H., Shimizu M., Makishima M., Yamada S.. Vitamin D receptor: ligand recognition and allosteric network. J. Med. Chem. 2006; 49:1313–1324. [DOI] [PubMed] [Google Scholar]
- 58. Yadav P.K., Xie P., Banerjee R.. Allosteric communication between the pyridoxal 5′-phosphate (PLP) and heme sites in the H2S generator human cystathionine beta-synthase. J. Biol. Chem. 2012; 287:37611–37620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Joseph R.E., Xie Q., Andreotti A.H.. Identification of an allosteric signaling network within Tec family kinases. J. Mol. Biol. 2010; 403:231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Datta D., Scheer J.M., Romanowski M.J., Wells J.A.. An allosteric circuit in caspase-1. J. Mol. Biol. 2008; 381:1157–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Youden W.J. Index for rating diagnostic tests. Cancer. 1950; 3:32–35. [DOI] [PubMed] [Google Scholar]
- 62. Le C.T. A solution for the most basic optimization problem associated with an ROC curve. Stat. Methods Med. Res. 2006; 15:571–584. [DOI] [PubMed] [Google Scholar]
- 63. Bohning D., Bohning W., Holling H.. Revisiting Youden's index as a useful measure of the misclassification error in meta-analysis of diagnostic studies. Stat. Methods Med. Res. 2008; 17:543–554. [DOI] [PubMed] [Google Scholar]
- 64. Gurevich V.V., Gurevich E.V.. The molecular acrobatics of arrestin activation. Trends Pharmacol. Sci. 2004; 25:105–111. [DOI] [PubMed] [Google Scholar]
- 65. DeWire S.M., Ahn S., Lefkowitz R.J., Shenoy S.K.. beta-arrestins and cell signaling. Annu. Rev. Physiol. 2007; 69:483–510. [DOI] [PubMed] [Google Scholar]
- 66. Scheerer P., Sommer M.E.. Structural mechanism of arrestin activation. Curr. Opin. Struct. Biol. 2017; 45:160–169. [DOI] [PubMed] [Google Scholar]
- 67. Hirsch J.A., Schubert C., Gurevich V.V., Sigler P.B.. The 2.8 A crystal structure of visual arrestin: a model for arrestin's regulation. Cell. 1999; 97:257–269. [DOI] [PubMed] [Google Scholar]
- 68. Han M., Gurevich V.V., Vishnivetskiy S.A., Sigler P.B., Schubert C.. Crystal structure of beta-arrestin at 1.9 angstrom: Possible mechanism of receptor binding and membrane translocation. Structure. 2001; 9:869–880. [DOI] [PubMed] [Google Scholar]
- 69. Zhan X., Gimenez L.E., Gurevich V.V., Spiller B.W.. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J. Mol. Biol. 2011; 406:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhou X.E., He Y.Z., de Waal P.W., Gao X., Kang Y.Y., Van Eps N., Yin Y.T., Pal K., Goswami D., White T.A. et al.. Identification of phosphorylation codes for arrestin recruitment by G protein-coupled receptors. Cell. 2017; 170:457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sekine S., Nureki O., Dubois D.Y., Bernier S., Chenevert R., Lapointe J., Vassylyev D.G., Yokoyama S.. ATP binding by glutamyl-tRNA synthetase is switched to the productive mode by tRNA binding. EMBO J. 2003; 22:676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Van Wynsberghe A.W., Cui Q.. Conservation and variation of structural flexibility in protein families. Structure. 2010; 18:281–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The webPSN server is freely available at http://webpsn.hpc.unimore.it