

Integration of AIEgens with upconversion nanoparticles inhibits tumor recurrence and metastasis.
Abstract
Reactive oxygen species (ROS) are essential for the regulation of antitumor immune responses, where they could induce immunogenic cell death, promote antigen presentation, and activate immune cells. Here, we report the development of near-infrared (NIR)–driven immunostimulants, based on coupling upconversion nanoparticles with aggregation-induced emission luminogens (AIEgens), to integrate the immunological effects of ROS for enhanced adaptive antitumor immune responses. Intratumorally injected AIEgen-upconversion nanoparticles produce high-dose ROS under high-power NIR irradiation, which induces immunogenic cell death and antigen release. These nanoparticles can also capture the released antigens and deliver them to lymph nodes. Upon subsequent low-power NIR treatment of lymph nodes, low-dose ROS are generated to further trigger efficient T cell immune responses through activation of dendritic cells, preventing both local tumor recurrence and distant tumor growth. The utility of dual-mode pumping power on AIEgen-coupled upconversion nanoparticles offers a powerful and controllable platform to activate adaptive immune systems for tumor immunotherapy.
INTRODUCTION
Over the past decade, immunotherapy has emerged as a promising strategy for cancer treatment. However, existing immunotherapeutic strategies usually show ineffectiveness across a wide range of solid tumors, with only a subset of patients showing improved clinical response (1–3). This ineffectiveness can be attributed to the limited presentation of antigens by antigen-presenting cells (APCs) and minimal antitumor response because of the tumor-promoting immunological microenvironment (4–6). In addition, side effects, such as immunotoxicity, notably hinder the clinical application of immunotherapeutic approaches (7, 8). Therefore, a controllable and effective strategy for an enhanced antitumor immune response while reducing side effects is in urgent demand in the field of cancer immunotherapy.
Reactive oxygen species (ROS) are small molecules with oxidation activity, which play an important role in physiological cell functions and immune regulation. Excessive ROS could induce cellular oxidative damage and immunogenic cell death (ICD) of tumors, providing a potential antigenic stimulation for the immune system (9). On the other hand, a low level of ROS can serve as key signaling molecules to promote the maturation of APCs (e.g., dendritic cells) through oxidation of signaling pathway [e.g., nuclear factor κB (NF-κB), mammalian target of rapamycin (mTOR), and extracellular signal–regulated kinase (ERK) signaling] and activation of intracellular calcium channel (10–12). In addition, ROS has proven effective in facilitating a cytosolic antigen delivery within dendritic cells by lysosomal escape and antigen protection, resulting in effective antigen cross-presentation and strong CD8+ T cell response (13, 14). On the basis of these findings, we hypothesize that the combination of these immunological functions of ROS may give rise to more specific, effective, and controllable antitumor immunological response as compared to traditional immunotherapeutic methods.
Photodynamic therapy (PDT) with high spatiotemporal precision and excellent safety has emerged as a promising tool for solid tumor ablation, ICD induction (15). However, these local oxidative damages trigger an insufficient immune response for systemic tumor rejection, owing to the rapid in vivo degradation of tumor antigen and low efficiency of antigen presentation (16, 17). Moreover, it is anticipated that, apart from its direct cytotoxic effects on tumor cells, PDT can be used to modulate intracellular ROS level for activating APCs. However, because of the lack of deep tissue–excitable photosensitive materials and appropriate strategies, the utility of PDT for immune system activation is challenging, especially in in vivo systems.
Here, we report the design and synthesis of a nanosized immunostimulant (Fig. 1A), based on the combination of an aggregation-induced emission (AIE) photosensitizer TPEBTPy with upconversion nanoparticles (UCNPs), to simultaneously realize ICD of tumors and activation of dendritic cells through fine-tuning of ROS generation for enhanced adaptive immune responses against solid tumors. Different from traditional photosensitizers, TPEBTPy with AIE characteristics shows intense fluorescence and strong ROS generation in the aggregated state (18, 19). On the other hand, UCNPs could convert the deep tissue–penetrating near-infrared (NIR) light into visible wavelengths (20), which provides opportunities for applications in bioimaging, optogenetics, and PDT (21–24). To improve light penetration and interaction with TPEBTPy, UCNPs with emission matching the absorption of TPEBTPy were selected as the NIR antennae. Furthermore, the AIE luminogen (AIEgen)–coupled UCNPs (AUNPs) were capable of antigen capture via electrostatic interaction due to their positive z potential. We demonstrated that this hybrid nanomaterial could efficiently eradicate solid tumors, induce ICD, reduce immunosuppressive cells, and capture tumor-associated antigens (TAAs) through high-power NIR light–excited PDT. TAA-loaded AUNPs could be consistently and efficiently taken up by APCs in the draining lymph node (DLN). Subsequent low-power tissue-penetrating NIR irradiation could further enhance T cell stimulation effects of APCs, which caused a better abscopal effect in inhibiting local residual tumor recurrence and controlling distant tumor growth. The underlying mechanisms of ROS-induced adaptive immune response activation were also investigated in this study.
Fig. 1. Design and characterization of AIE-UCNP nano-immunostimulant.
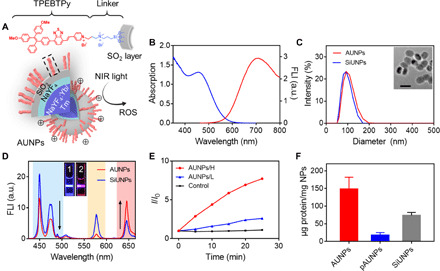
(A) Structure of TPEBTPy and AUNPs. Dashed box indicates a linked TPEBTPy molecule on the AUNPs. (B) Ultraviolet-visible (UV-vis) absorption (blue line) and fluorescence emission (red line) spectra of TPEBTPy. FLI, fluorescence intensity; a.u., arbitrary units. (C) Dynamic light scattering (DLS) measurement of SiUNPs and AUNPs. The inset shows the TEM image of AUNPs. Scale bar, 100 nm. (D) Fluorescence spectra of AUNPs and SiUNPs under NIR irradiation (980 nm). Inset shows fluorescence photographs of SiUNPs (1) and AUNPs (2) in PBS solution under NIR irradiation. (E) The measurement of 1O2 generation of AUNPs in PBS solution under NIR irradiation with high power (H; 0.6 W/cm2) and low power (L; 0.12 W/cm2) by singlet oxygen sensor green (SOSG) in water. Control indicates SOSG alone under high-power NIR irradiation. (F) Quantification of soluble OVA loaded by pAUNPs, AUNPs, and SiUNPs.
RESULTS
Preparation and characterization of AUNPs
TPEBTPy is an AIE photosensitizer designed and synthesized for PDT. Its synthetic route and structural identification data are shown in figs. S1 to S7. The photosensitizer molecule was obtained by introducing electron-donating methoxy groups and two different electron-withdrawing groups, i.e., benzo-2,1,3-thiadiazole and pyridinium, to the tetraphenylethylene unit. In a phosphate-buffered saline (PBS) solution, TPEBTPy exhibited a broad absorption spectrum from 400 to 600 nm with a peak at 460 nm (Fig. 1B). The emission characteristic of TPEBTPy was studied in the mixture of dimethyl sulfoxide (DMSO) and PBS solution. TPEBTPy could dissolve well in pure DMSO and showed weak emission. When the volume fraction of PBS was increased to 70% and above in the DMSO solution, TPEBTPy started to form nanoaggregate and its emission was markedly enhanced with a maximum at 700 nm, exhibiting AIE-active properties (fig. S9A). Upon white light irradiation, TPEBTPy was excited to produce not only singlet oxygen via type II reaction (fig. S9B) but also superoxide radical anion via type I reaction (fig. S9C).
To enable NIR-activated photosensitization, we require the energetic matching between the maximum absorption of TPEBTPy and the emission of UCNPs. NaYF4:Yb/Tm UCNPs with an optically inert shell layer of NaYF4 and SiO2 were selected as an NIR converter. Upon excitation at 980 nm, the core-shell SiO2-UCNPs (SiUNPs) show an emission peak from 425 to 480 nm (Fig. 1D), which matches well with the absorption of TPEBTPy. As shown in figs. S1 and S8, TPEBTPy-Si was further synthesized through modification of TPEBTPy with a trimethoxysilane group, which could be covalently linked onto the surface of SiUNPs via condensation reaction to form AUNPs. In addition, AUNPs modified with polyethylene glycol (PEG)–silane (pAUNPs) via condensation reaction were used to serve as control nanoparticles, which were expected to show minimal absorption for antigen proteins (25). Elemental mapping images (fig. S10A) indicated the successful attachment of TPEBTPy onto the surface of SiUNPs. The hydrodynamic diameter of the AUNPs was around 100 nm, similar to their precursors SiUNPs and pAUNPs (Fig. 1C and fig. S10B). Transmission electron microscopy (TEM) images further confirmed the core-shell (NaYF4:Yb/Tm-SiO2:TPEBTPy) structure of AUNPs (Fig. 1C). A SiO2 layer with an average thickness of 5 nm is beneficial for resonance energy transfer from UCNPs to TPEBTPy. After coating onto the SiUNP surface, the fluorescence intensity of TPEBTPy was significantly increased compared with that of free TPEBTPy molecule in the DMSO solution, indicating activation of the AIE property of TPEBTPy due to the restraint of intramolecular motion (fig. S9D). Upon 980-nm excitation, the emission intensity of AUNPs was markedly decreased in the blue region, while an increase in the red region was observed as compared to that of SiUNPs, which is visible by the naked eye (Fig. 1D), indicating stronger in vivo self-tracking capability than unmodified UCNPs. Meanwhile, AUNPs could generate different amounts of ROS with 980-nm excitation in a laser power–dependent manner, which was mediated by energy transfer (Fig. 1E and fig. S10, C and D). The cytotoxicity of AUNPs was evaluated through use of the MTT [(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)] assay. After incubation with different concentrations of AUNPs for 8 hours, the cell viability of B16F10 cancer cells was not significantly affected, indicating the excellent biocompatibility of AUNPs under dark conditions (fig. S11A). However, AUNPs elicited apparent cytotoxicity under high-power NIR irradiation, with a half-maximal inhibitory concentration of 8.5 μg/ml, indicating their excellent tumor cell ablation ability (fig. S11B). Meanwhile, when pieces of pork tissue were placed between the laser and AUNP-labeled B16F10 cells, the cytotoxicity of AUNPs was modestly impeded by the increased tissue thickness, indicating their potential use for deep tumor therapy (fig. S11C). To enable efficient antigen presentation in vivo, the stability of the antigen needs to be considered as a key factor in the activation of an adaptive immune response. To improve the ability of nanoparticles to capture antigens, apart from pyridinium, we then deliberately added a positively charged quaternary ammonium group between TPEBTPy and trimethoxysilane. The two positive charges change the ζ potential from −26 mV of SiUNPs to +14 mV of AUNPs (fig. S10E), resulting in a significantly enhanced capture of ovalbumin (OVA), a standard antigen model, as compared with those of SiUNPs and pAUNPs (Fig. 1F).
ICD induction and immune cell activation by AUNPs in vitro
It has been demonstrated that PDT might produce ICD by inducing dying tumor cells to release “eat me” immunogenic signals (e.g., CRT and HMGB1), resulting in activation of the immune response (26). To verify whether AUNPs can trigger efficient ICD, B16F10 cells were labeled with AUNPs, followed by treatment with high-power NIR irradiation for 3 min. As shown in Fig. 2 (A and B), AUNPs induce obvious surface-exposed CRT (ecto-CRT) and HSP70 (ecto-HSP70) following NIR treatment, which is much higher than that of Ce6, a commonly used photosensitizer. In addition, enzyme-linked immunosorbent assay also showed that AUNPs plus NIR treatment led to a strong HMGB1 release of tumor cells as compared with those of other control groups (Fig. 2C). All the results indicated that high-power NIR-activated AUNPs could significantly induce the release of damage-associated molecular patterns from B16F10 tumor cells, which could provide potential antigenic stimulation for antitumor immune response.
Fig. 2. ICD induction and immune cell activation by AUNPs in vitro.
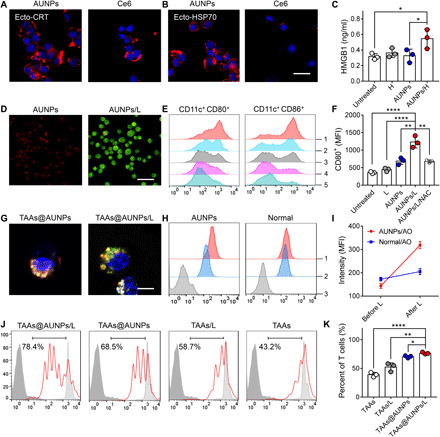
(A and B) Fluorescent imaging of ecto-CRT (A) and ecto-HSP70 (B) expression on the surface of AUNPs or Ce6-labeled B16F10 cells following high-power NIR (980- or 660-nm laser) irradiation. Scale bar, 25 μm. (C) Detection of HMGB1 extracellular release. (D) Detection of ROS level in AUNP (red)–labeled DCs by DCFDA (green), followed by 3-min low-power NIR irradiation (0.12 W/cm2) (AUNPs/L). Scale bar, 50 μm. (E) FACS analysis of CD11c+ CD80+ (left) or CD11c+ CD86+ (right) expression on AUNP-labeled DCs under low-power NIR irradiation (gated on CD11c+). 1, AUNPs/L; 2, AUNPs/L/NAC; 3, AUNPs; 4, L alone; 5, untreated. (F) Quantitative analysis of mean fluorescence intensity (MFI) of AUNP-labeled DCs after different treatments. (G) Fluorescence images of BMDCs stained with LysoTracker (white) after 6 hours of incubation with TAAs-FITC (green) absorbed AUNPs (red), followed by low-power NIR irradiation. Scale bar, 5 μm. (H) FACS analysis and (I) MFI of lysosome rupture of BMDCs stained with acridine orange (AO) after 6 hours of incubation with AUNPs, followed by low-power NIR irradiation (AUNPs, AUNP-labeled DCs; normal, unlabeled DCs). 1, after L; 2, before L; 3, control. (J) FACS analysis of 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE)–labeled CD8+ T cells after 3 days of coculture with BMDCs with different treatments. The light gray histogram indicates nondivided CD8+ T cells in the absence of BMDCs, and dark gray histogram indicates non–CFSE-stained cells. The bar graphs show CFSE MFI of the whole histogram, which is proportional to cell division. (K) Quantitative analysis of the percentages of divided CD8+ T in the total CD8+ T within different groups (n = 3). Statistical significance was assessed using the analysis of variance (ANOVA) test. Data represent means ± SEM. *P < 0.05, **P < 0.01, and ****P < 0.0001.
The immune activation effect of AUNPs to bone marrow dendritic cells (BMDCs) following low-power NIR irradiation was subsequently examined in vitro. Dichlorofluorescein diacetate (DCFDA) staining and MTT assay were used to assess the intracellular ROS level and cytotoxicity, respectively. As shown in fig. S11 (D and F), after incubation with AUNPs, the cell viability of BMDCs was not affected under dark conditions. When these AUNP-loaded BMDCs were stained with DCFDA for 15 min, upon NIR irradiation with 0.3 or 0.6 W/cm2, DCFDA was activated to emit intense green fluorescence, and the AUNPs exhibited apparent cytotoxicity to BMDCs (cell viability, 63 and 29%, respectively) (figs. S11E and S12A). In contrast, lower power of NIR irradiation (0.12 W/cm2) induced modest intracellular green fluorescence in AUNP-loaded BMDCs with minimal effects on their viability (Fig. 2D and fig. S11E), indicating that AUNPs at such a low NIR irradiation dose could generate a mild and safe oxidative stress environment around DCs for further immune activation.
Next, the underlying stimulation effect of AUNPs on BMDCs was tested. As shown in Fig. 2 (E and F) and fig. S12C, BMDCs could take up AUNPs in a time-dependent manner, reach maximum uptake at 12 hours, and exhibit a slight increase in CD86 and CD80 expressions. Upon low-power NIR irradiation for 5 min, BMDCs treated with AUNPs showed a significant increase in the expressions of these surface markers as compared with those of AUNPs alone, NIR irradiated alone (L alone), and untreated control groups. This stimulation effect could be markedly diminished by pretreating DCs with N-acetylcysteine (NAC; a ROS scavenger), indicating that the increased activation was caused by transient ROS surge. Similarly, when the AUNP-labeled BMDCs were covered with pork tissues with increasing thickness, this activation effect was not significantly affected under low-power NIR irradiation (fig. S12B).
ROS-induced lysosomal rupture has been demonstrated as an effective mechanism to facilitate the cytosolic release of antigen and cross-presentation (13, 27). Therefore, we further evaluated the impact of this approach on antigen presentation by pulsing BMDCs with fluorescein isothiocyanate–modified tumor antigens (TAAs-FITC)–loaded AUNPs for 12 hours, followed by LysoTracker 633 staining and low-power NIR irradiation for 3 min. Confocal images showed that TAAs-FITC (green color)–loaded AUNPs (red color) could colocalize precisely in the lysosome (white color) before NIR treatment, whereas low-power NIR treatment significantly eliminated lysosomes and led to TAAs-FITC release within the cytoplasm of BMDCs (Fig. 2G). Besides, the lysosomal integrity of AUNP-loaded BMDCs was checked with acridine orange staining for 30 min. Upon low-power NIR irradiation for 3 min, the green fluorescence of cells was significantly increased as compared to that of nonirradiated cells, as shown by fluorescence-activated cell sorting (FACS) (Fig. 2, H and I), indicating the loss of lysosomal integrity. Lysosomal escape facilitates major histocompatibility complex (MHC) I antigen presentation in DCs. Thus, BMDC-primed CD8+ T cell proliferation was examined to assess the impact of AUNPs on antigen cross-presentation. AUNPs loaded with TAAs (TAAs@AUNPs) and irradiated with low-power NIR significantly increased BMDC-primed CD8+ T cell proliferation over 50% as compared to the free TAAs or other treatments (Fig. 2, J and K). Together, these data suggest that AUNPs could enhance DC activation and antigen cross-presentation through low-power NIR excitation.
AUNP release, uptake, and DC activation in vivo
Before the in vivo application, the biosafety of AUNPs in healthy mice was tested. As shown in fig. S13 (B to D), after subcutaneous injection with AUNPs (0.5 mg/ml, 100 μl, based on TPEBTPy), no obvious difference in body weight and blood biochemistry parameters was observed among the treated and normal groups over 10 days. In addition, histochemistry analysis showed almost no obvious pathological changes in liver and kidney tissues, indicating negligible in vivo toxicity of AUNPs. To examine the proposed immune activation strategy, the in vivo dynamic distribution of AUNPs was monitored by fluorescence imaging under 980-nm excitation. B16F10 tumor–bearing mice were injected intratumorally with AUNPs or pAUNPs (30 μl, 0.5 mg/ml, based on TPEBTPy). After high-power NIR irradiation (0.6 W/cm2) to achieve tumor PDT, the fluorescent signals of DLNs of the mice treated with AUNPs rapidly increased over time and reached the highest intensity at 24 hours after NIR treatment of tumor, which was significantly higher than that of the mouse receiving AUNPs alone, indicating the effective AUNP release from the NIR irradiated tumor. However, pAUNPs showed higher leakage from the tumor, and this led to more intense fluorescence of DLNs in mice with or without high-power NIR irradiation (Fig. 3, D and E).
Fig. 3. AUNP uptake and immune system activation in vivo.
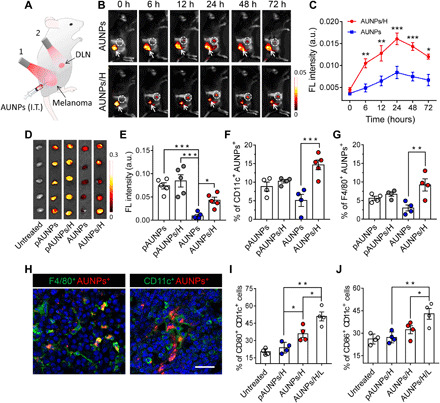
(A) Schematic illustrating of AUNP administration and subsequent NIR-controlled antitumor immune therapy: 1, high-power NIR irradiation (H; 0.6 W/cm2); 2, low-power NIR irradiation (L; 0.12 W/cm2). I.T., intratumoral injection of AUNPs. (B) Time-dependent fluorescence images of B16F10 tumor–bearing mice after intratumoral injection, followed by high-power NIR irradiation (AUNP/H) (excitation source for imaging, 980 nm). The area within the dashed circle indicates a DLN. The white arrow shows the tumor region. (C) Fluorescence intensities of lymph nodes of mice receiving AUNPs/H or AUNPs alone (n = 3). (D) Ex vivo fluorescence images and (E) quantification of fluorescence intensity of harvested lymph nodes after 24 hours of high-power NIR irradiation (excitation source for imaging, 455 nm). (F and G) FACS analysis of the percentages of DCs (CD11c+) (F) and macrophages (F4/80+) (G) with AUNPs or pAUNPs in lymph nodes after 24 hours of high-power NIR irradiation. (H) Representative immunofluorescence images of DLN slices showing CD11c+ AUNPs+ DCs and F4/80+ AUNPs+ macrophages. Scale bar, 100 μm. (I and J) FACS analysis of activated CD11c+ CD80+ (I) and CD11c+ CD86+ (J) DCs in lymph nodes of B16F10 tumor–bearing mice receiving pAUNPs/H, AUNPs/H, and AUNPs/H/L. Statistical significance was assessed using ANOVA test. Data represent means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
High-power NIR irradiation greatly enhanced AUNP uptake by antigen-presenting dendritic cells (CD11c+) and macrophages (F4/80+) in DLNs (Fig. 3, F to H, and fig. S14A). These results, taken together with the low accumulation of pAUNPs in APCs after NIR treatment (Fig. 3, F and G), indicate that the surface property of AUNPs facilitated the capture of TAAs released from damaged tumor and realized more effective APC uptake in DLNs. FACS analysis also showed that the number of activated DCs (CD80+ and CD86+) in the lymph node of mice receiving AUNPs is higher than those of mice receiving pAUNPs. Through exposure to low-power NIR irradiation, the antigen-absorbing AUNPs enabled the highest expression of CD86 and CD80 on DCs in DLNs as compared to those in control groups (Fig. 3, I and J, and fig. S14B), suggesting the successful in vivo DC activation by controlled NIR irradiation. In addition, as shown in fig. S13A, low-power NIR did not lead to a noticeable temperature rise in the skin, indicating the negligible heating effect on immune cell activation. Therefore, we assumed that this in situ nanoparticle-antigen complex could act as an effective tumor vaccine useful for cancer recurrence and metastasis therapy.
Antitumor immunotherapy of AUNP for inhibition of B16F10 tumor growth
On the basis of previous findings, the in vivo antitumor therapeutic effect of this strategy was then evaluated by intratumoral injection of AUNPs into B16F10 tumors in mice. After high-power NIR irradiation for tumor PDT, the DLN region was exposed to low-power NIR for four consecutive days immediately, and tumor growth was monitored by bioluminescent signals (Fig. 4A). As shown in Fig. 4B, the tumor locally injected with AUNPs could be efficiently eradicated by PDT as compared with the untreated tumor and that injected with AUNPs alone. The bioluminescent signals of tumors in mice receiving AUNPs/PDT plus adjuvant low-power NIR irradiation (AUNPs/H/L) gradually decreased over time, showing noticeable residual tumor inhibition effect after PDT. Tumor size measurement showed two-step NIR treatment, which led to approximately ninefold tumor growth inhibition compared with high-power NIR treatment alone on day 19 (Fig. 4C), indicating stronger immune response activation caused by low-power NIR irradiation. Meanwhile, the survival time of mice receiving AUNPs/H/L was much longer than that of control groups (Fig. 4D), which was corroborated with tumor growth analysis.
Fig. 4. Antitumor immunotherapy of AUNP for inhibition of B16F10 tumor growth.
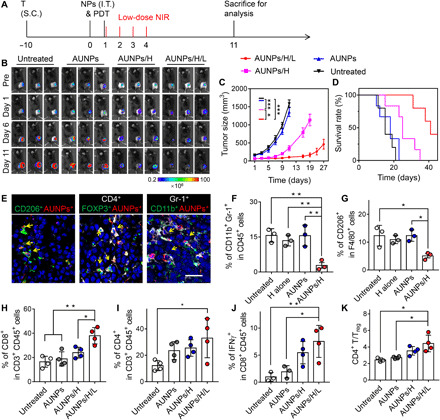
(A) Schematic illustration of the treatment schedule. T, tumor inoculation; S.C., subcutaneous injections. (B) Bioluminescence images of the B16F10 tumor–bearing mice receiving different treatments. (C) Tumor growth curve and (D) survival curve of B16F10 tumor–bearing mice in control and treated groups (n = 5). Statistical significance of tumor size was carried out on day 11. (E) Representative immunofluorescence images of B16F10 tumor tissue slices showing CD206+ AUNPs+ TAMs, CD4+ Foxp3+ AUNPs+ regulatory T cells (Tregs), and CD11b+ Gr-1+ AUNPs+ MDSCs after intratumoral injection of AUNPs. Scale bar, 25 μm. Positive cells were labeled with yellow arrows. (F) FACS analysis of the percentages of CD11b+ Gr-1+ CD45+ MDSCs and (G) CD206+ F4/80+ TAMs in the tumor of B16F10 tumor–bearing mice receiving AUNPs/H. (H to J) FACS analysis of the percentages of CD8+ (H), CD4+ (I), and IFN-γ+ CD8+ (J) T cells in the tumor of B16F10 tumor–bearing mice in control and treated groups. (K) The ratio of the tumor-infiltrating CD4+ T cells to Tregs in the tumor of B16F10 tumor–bearing mice in control and treated groups. Statistical significance was assessed using ANOVA test. Data represent means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
It is well known that reprogrammed immune cells, such as myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and tumor-associated macrophages (TAMs), within the tumor microenvironment participate in tumor progression, metastasis, and recurrence (28,29). Therefore, to understand the underlying mechanism of efficient tumor inhibition, we first assessed the impact of intratumoral administration of AUNPs on the tumor microenvironment. On day 3, tumors receiving AUNPs/H or AUNPs alone were collected and analyzed by immunofluorescence staining and flow cytometry. As shown in Fig. 4E, AUNPs showed high accumulation within CD206+ F4/80+ TAMs, CD4+ Foxp3+ Tregs, and CD11b+ Gr-1+ MDSCs after local injection. Accordingly, after PDT, we observed a significant reduction of all types of immunosuppressive cells in tumors receiving AUNPs. However, there was no obvious effect observed on the untreated tumor or on those receiving a high dose of NIR alone or AUNPs alone (Fig. 4, F and G, and fig. S15). Together, these data indicate that the local delivery and light treatment of AUNPs could efficiently reduce immunosuppressive cells and relieve immune suppression.
Furthermore, FACS analysis of tumor on day 11 showed that mice receiving AUNPs/H/L had remarkable CD8+, CD4+, and interferon-γ (IFN-γ)+ CD8+ T cell infiltration in the tumor compared to that in other groups (Fig. 4, H to J, and fig. S16A). This low-power NIR-assisted immunotherapy also significantly increased the intratumoral effector CD4+ T cells/Tregs ratios, implying ameliorative immunotherapeutic activity within the tumor microenvironment of residual tumor (Fig. 4K). To further test whether this therapeutic strategy could induce systematic immune activation, we used the splenocytes isolated from mice after different treatments to assess the production of antitumor IFN-γ. As shown in fig. S16B, splenocytes from mice receiving AUNPs/H/L exhibited the highest percentage of IFN-γ+ CD8+ T cells when stimulated with TAAs, suggesting its great potential for treating tumor metastasis.
Antitumor immunotherapy of AUNP synergized with αPD-1 for the establishment of systemic immune responses and long-term immune memory
Immune checkpoint blockade therapy is very effective in treating several types of tumors, although its therapeutic effect is always limited by intrinsic immunity (30). Besides, the potential in vivo toxicity due to the breakdown of immune tolerance has also impeded its clinical application (31, 32). In this part of the study, after the demonstration that our strategy could relieve immune suppression and trigger strong systemic immune responses, we found that programmed death-ligand 1 (PD-L1) expression was also enhanced in both primary and distant tumors compared to that in untreated tumors (fig. S17A), which could be caused by systemic distribution of cytokines after treatment. Therefore, we next studied whether this method could be combined with anti–programmed cell death protein 1 (αPD-1), a commonly used immune checkpoint inhibitor, to simultaneously achieve inhibition of residual tumor and distant tumors in a bilateral B16F10 tumor–bearing mouse model. As depicted in Fig. 5A, primary tumors were treated with AUNPs/H/L and injected with αPD-1 at different time points. Bioluminescence imaging and tumor growth profiles showed that αPD-1 therapy alone did not significantly change the primary and distant tumor growth. Expectedly, mice receiving AUNPs/H/L showed significant inhibition of primary tumor recurrence and reduced distant tumor growth, suggesting effective systematic immune responses induced by this strategy (Fig. 5, B to D, and fig. S17B). Encouragingly, the combination of AUNPs/H/L and αPD-1 administration could realize a synergistic effect in inhibiting local tumor recurrence and distant tumor growth, which corresponded with the highest CD3+ CD8+ T cell infiltration within the residual and distant tumors as compared to those in control groups (fig. S17, C to F).
Fig. 5. Antitumor immunotherapy of AUNP synergized with αPD-1 for the establishment of systemic immune responses and long-term immune memory.
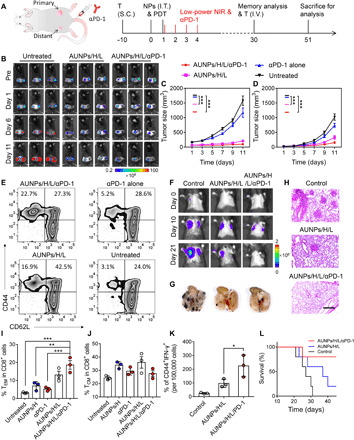
(A) Schematic illustration of synergistic treatment of bilateral B16F10 tumor mouse model (left) and the treatment schedule for immune memory analysis and tumor rechallenge (right). I.V., intravenously. (B) Bioluminescence images of bilateral B16F10 tumor–bearing mice with different treatments. (C) Primary and (D) distant tumor growth curves of the B16F10 tumor–bearing mice with different treatments (n = 5). (E) FACS plots and quantitative analysis of (I) CD8+ CD62− CD44+ effector memory T cells (TEM) and (J) CD8+ CD62+ CD44+ central memory T cells (TCM) in spleen tissue of B16F10 tumor–bearing mice after 30 days of different treatments. (F) Bioluminescence images of mice after rechallenging with intravenous injection of B16F10 tumor cells. (G) Representative lung photographs (21 days) and (H) hematoxylin and eosin (H&E) images of lung tissue slices from control (naïve mice), AUNPs/H/L, and AUNPs/H/L/αPD-1 groups. Scale bar, 100 μm. (K) Number of CD44+ IFN-γ+ per 100,000 cells of lung tissue of mice in control and treated groups. (L) Survival curve of mice in control and treated groups (n = 3). All statistical analyses were assessed using ANOVA test. Data represent means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. Photo credit: Duo Mao, NUS.
Separately, the long-term immune-memory effect was evaluated on day 34 after different treatments (Fig. 5A, right). It was found that the CD8+ CD62+ CD44+ central memory T cells (TCM) were not significantly different among all groups, whereas the amount of CD8+ CD62− CD44+ effector memory T cells (TEM) in the spleens of mice receiving AUNPs/H/L alone or combination therapy was significantly higher than those treated with αPD-1 injection alone or other control groups, indicating the establishment of an antitumor immune-memory based on this strategy (Fig. 5, E, I, and J). Furthermore, when these living mice were rechallenged by intravenous injection of B16F10 tumor cells, the lung tissues of mice receiving combination therapy showed no detectable tumors and the number of tumors only slightly increased in mice receiving AUNPs/H/L, as observed through bioluminescence imaging, photographs of lungs, and histological analysis (Fig. 5, F to H). Impressively, the lung tissues of mice receiving combination therapy showed the most apparent accumulation of CD44+ IFN-γ+ T cells (Fig. 5K), whereas AUNPs/H/L treatment produced a modest number of effector cytotoxic T cells as compared to untreated mice. Besides, the survival rate of mice receiving combination therapy was higher than those of the other two groups (Fig. 5L). Overall, these results demonstrated that our proposed treatment could activate a systematic antitumor immune response for effective inhibition of residual tumors and establish a long-term and effective antitumor immune memory, which could be further synergistically enhanced with αPD-1.
DISCUSSION
We have proposed a new immunostimulant design that combines AIE photosensitizer and UCNPs to achieve effective adaptive immune response through the ROS-dependent immune activation mechanism. This strategy avoids the damaging side effects of traditional immunotherapy and promotes a systemic antitumor immune response and rejection of distant metastases (Fig. 6).
Fig. 6. Proposed scheme of dual-mode ROS-driven tumor immunotherapy.
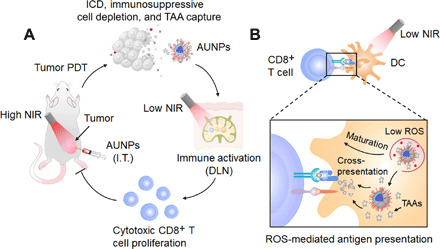
(A) AUNPs are intratumorally injected into the tumor of a mouse. Upon high-power NIR irradiation, AUNPs induce efficient ICD, reduce immunosuppressive cells, and capture TAAs. Subsequently, TAA-loaded AUNPs are specifically taken up by dendritic cells in DLNs. Upon low-power NIR irradiation, AUNPs can produce a low level of ROS to enhance dendritic cell function. This process promotes the expansion of cytotoxic CD8+ T cells and effectively inhibits the growth of residual tumors and distant tumor after PDT. (B) Mechanism underlying the NIR light–driven dendritic cell activation in DLN.
Recent studies have shown that an excessive level of ROS causes ICD of tumor cells and effective antigen release, which could initiate a specific antitumor immune response (10). On the other hand, a low level of ROS has been considered as key signaling molecules for APC activation and subsequent effective T cell priming and expansion. Through our design, we integrate these functions to eradicate solid tumors. We found that our AUNPs could induce ICD more efficiently as compared to a commercial photosensitizer of Ce6. We also verified that the low level of ROS generated by AUNPs was involved in the successful immune response activation, which was consistent with previous conclusions that ROS could regulate the activation of DCs and enhance cross-presentation of antigen (12, 13). We realized local dendritic cell activation and cross-presentation by modulating intracellular ROS level in in vivo system, which could induce stronger CD8+ T cell priming and expansion and further lead to more efficient tumor growth inhibition.
UCNPs allow PDT to be performed through tissue at great depths (33, 34). However, the activation of fluorescent dyes or photosensitizers by UCNPs is generally an inefficient process (35). To enhance the photosensitization process, we designed and synthesized a highly efficient photosensitizer through precise molecular design. In particular, TPEBTPy is a molecule with D (donor)–A′ (auxiliary acceptor)–A (real acceptor) structure. The auxiliary acceptor, benzo-2,1,3-thiadiazole, serves as a spacer to twist the dihedral angle between the donor (methoxy groups) and the acceptor (pyridinium), resulting in HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) separation. This separation leads to a low value of ΔEst (the energy gap between the S1 and T1 states) at 0.39 eV (fig. S1B). The low ΔEst is beneficial for the intersystem crossing process from S1 to T1 state. A high probability of T1 states would promote photosensitization through reaction with nearby oxygen to produce ROS. Moreover, conventional photosensitizers such as Ce6 have conjugated planar structures. Their fluorescence and photosensitization can be easily weakened in the aggregate state due to π-π interactions (36). On the contrary, AIE photosensitizers such as TPEBTPy have twisted three-dimensional molecular structures that weaken intermolecular interactions. These photosensitizers exhibit bright fluorescence and strong photosensitization in the aggregate state when incorporated into nanoparticles (36–39). Therefore, different from other traditional photosensitizer-loaded UCNPs, AUNPs can exhibit strong emission for precise self-tracking and very efficient photosensitization in our therapeutic strategy.
Our data showed that NIR-excited AUNPs have three advantages for cancer PDT and immunotherapy. First, UCNPs are effective NIR antennae for transferring energy from NIR photons to well-matched AIE photosensitizer to efficiently produce ROS in deep tissue. Second, the design of positively charged photosensitizer not only facilitates close contact between oxygen and photosensitizer as well as the rapid diffusion of ROS for effective tumor killing but also ensures the subsequent antigen capturing via electrostatic interaction. Third, through a simple power switching of NIR treatment, the local AUNPs could exhibit potent cytotoxicity and activate the immune system. These results suggest the combination of AIEgens with UCNPs as a feasible noninvasive treatment of choice for many types of tumors.
In closing, we have developed a versatile immunostimulant comprising AUNPs. Our study reveals that a dual-modal ROS activation of these nanoparticles allows high-efficiency antitumor immune activation and antigen processing. Our ROS-activated antitumor strategy could also synergize with immune checkpoint blockade therapy (e.g., αPD-1 therapy) to improve immunological memory and further reduce the risk of cancer recurrence and metastasis. The AIEgen-UCNP coupling is likely to enable the development of an intelligent, noninvasive therapeutic toolbox that could inspire significant research breakthroughs in the field of immunotherapy.
MATERIALS AND METHODS
Materials
All chemicals were purchased from Sigma-Aldrich and used as received without further purification. Deuterated solvents were purchased from Cambridge Isotope Laboratories Inc. The high-performance liquid chromatography (HPLC)–grade solvents methanol and acetonitrile were purchased from Sigma-Aldrich. Tetrahydrofuran (THF) and dichloromethane were dried by distillation using sodium or calcium hydride as the drying agent, respectively. All non-aqueous reactions were carried out under nitrogen atmosphere in oven-dried glassware. RPMI 1640, penicillin, streptomycin, fetal bovine serum (FBS), and trypsin were obtained from Gibco (Thermo Fisher Scientific). Milli-Q water (18.2 megohms) was used to prepare the buffer solutions from 10× PBS stock buffer (1st BASE, Singapore). The murine melanoma cell line B16F10 and B16F10-luc cells were purchased from the American Type Culture Collection and Shanghai Science Light Biology Science and Technology Co. Ltd., respectively. All antibodies for flow cytometry were purchased from BioLegend. All antibodies for immunofluorescence staining were purchased from Abcam. All cytokines for cell culture were purchased from PeproTech Inc. All other biological reagents including 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE) and singlet oxygen sensor green (SOSG) were purchased from Thermo Fisher Scientific.
Preparation of SiUNPs, AUNPs, and pAUNPs
UCNPs with a chemical composition of NaYF4:Yb/Tm were synthesized by a coprecipitation method. In a typical synthesis of blue-emitting UCNPs, 0.278 mmol of Y(CH3CO2)3, 0.12 mmol of Yb(CH3CO2)3, and 0.002 mmol of Tm(CH3CO2)3 lanthanide precursors were mixed with 3 ml of oleic acid and 7 ml of 1-octadecene and heated at 150°C for 1.5 hours to remove any moisture content. After cooling the reaction solution to 50°C, a methanol solution containing 1 mmol of NaOH and 1.6 mmol of NH4F was quickly added and the reaction mixture was further stirred at 50°C for another 0.5 hours. After that, the reaction was heated to 100°C under vacuum for 0.5 hours to remove any low–boiling point solvent. After three times of N2 purge at 100°C, the reaction was heated to 280°C and maintained at this temperature for 2 hours to allow nanoparticle growth. Once the reaction was cooled to room temperature, UCNP products were purified twice by ethanol washing under centrifugation at 6000 rpm for 10 min, followed by ethanol redispersion. Purified UCNPs were dispersed into 4 ml of cyclohexane. For synthesis of UCNPs with a chemical composition of NaYF4:Yb/Er, the experimental procedure was the same as that for NaYF4:Yb/Tm except that 0.272 mmol of Y(CH3CO2)3, 0.12 mmol of Yb(CH3CO2)3, and 0.008 mmol of Er(CH3CO2)3 were used as lanthanide precursors.
To suppress surface quenching of luminescence, an optically inert shell layer of NaYF4 was grown epitaxially onto the as-synthesized UCNPs. Specifically, 0.4 mmol of Y(CH3CO2)3 was mixed with 3 ml of oleic acid and 7 ml of 1-octadecene and heated at 150°C for 1.5 hours. After cooling to 80°C, all UCNP products from the previous step (in 4 ml of cyclohexane) were added to the reaction mixture. The solution was further stirred at 80°C for 1 hour to evaporate the cyclohexane content. Subsequently, a methanol solution containing 1 mmol of NaOH and 1.6 mmol of NH4F was added to the reaction solution at 50°C. After 0.5 hours of continuous stirring at 50°C, the reaction was heated to 100°C under vacuum for 0.5 hours. After three rounds of N2 purging at 100°C, the reaction temperature was ramped to 280°C and maintained at 280°C for 2 hours. Once the reaction was cooled down to room temperature, core-shell UCNP products were washed twice with ethanol and dispersed into 4 ml of cyclohexane.
The as-synthesized UCNPs were transferred into an aqueous phase through an acid-induced ligand removal process. Four milliliters of the cyclohexane was added with 4 ml of ethanol and centrifuged at 14,800 rpm for 5 min. The pellet was redispersed with a mixture of 4 ml of ethanol and 4 ml of HCl aqueous solution (2 M) by sonication. The acid-treated UCNPs were thereafter washed three times with ethanol to remove excess acid content. The ligand-free UCNPs were dispersed into 4 ml of deionized (DI) H2O for subsequent surface modification.
To obtain SiUNPs, 4 ml of the ligand-free UCNPs was slowly added to 18 ml of polyvinylpyrrolidone aqueous solution (50 mg/ml). The solution was sonicated for 20 min and subsequently stirred for 1 hour. After adding 80 ml of ethanol, the reaction mixture was further sonicated for 20 min and stirred for 2 hours. Aqueous ammonia (3.2 ml) was added to adjust the solution pH, followed by 20-min sonication. Subsequently, 80 μl of tetraethyl orthosilicate was added to the solution to initiate silica growth on the surface of UCNPs. The reaction solution was kept stirring for 12 hours, and SiUNPs were purified by three rounds of ethanol washing and dispersed in DI H2O. The final concentration of SiUNPs was 200 mg/ml.
To prepare AUNPs, 50 mg of silica-coated UCNPs was washed with DI water two times and then dispersed in 20 ml of ethanol/water mixture (9:1). The reaction mixture was further sonicated for 20 min and stirred for 2 hours. Ammonium (0.8 ml) was added to adjust the solution pH, followed by 20-min sonication. Subsequently, 200 μl of TPEBTPy-Si in THF (2 mg/ml) was added into the solution to coat on the surface of UCNPs through a condensation reaction. The reaction solution was kept stirring for 1 hour, purified by three rounds of ethanol washing, and dispersed in DI H2O. For further PEG modification, mPEG2000-silane was dissolved in THF (5 mg/ml) and 200 μl of mPEG2000-silane solution was added into the reaction solution containing AUNPs. The reaction solution was continued to stir for 2 hours, and the final products were purified by three rounds of ethanol washing and dispersed in DI H2O.
Characterization of UCNPs and AUNPs
TEM measurements were carried out on a JEOL-JEM 2010F field-emission transmission electron microscope operated at an acceleration voltage of 200 kV. The hydrodynamic sizes of UCNPs were measured by dynamic light scattering (DLS) means on Malvern Nano-ZS Particle Sizer. Upconversion luminescence spectra were recorded at room temperature with an Edinburgh FLS920 fluorescence spectrometer in conjunction with a 980-nm diode laser. To detect the 1O2 generation in solution, 1 ml of AUNP solution (0.1 mg/ml) was mixed with 1 μl of DMSO with SOSG. The solution was kept in the dark and irradiated by a 980-nm laser (0.6 and 0.12 W/cm2) for various times. Then, the solution was collected for fluorescence emission measurements.
To determine the loading efficiency of OVA onto AUNPs, 10 mg of AUNPs and 5 mg of OVA were added into 10 ml of water with stirring for 12 hours to obtain the OVA@AUNPs. Subsequently, the solution was centrifuged at 3000 rpm for 10 min. The supernatant was collected, and the concentration of OVA was quantified using a bicinchoninic acid (BCA) assay. The total OVA uptake by the AUNPs was measured by subtracting the OVA concentration in the supernatant after AUNP capture from the total OVA concentration.
In vivo biodistribution of AUNPs after PDT
To track the in vivo distribution of AUNPs, a Maestro in vivo imaging system (CRi Inc., Woburn, USA) using a 980-nm laser as the excitation source was used to obtain AUNP signals from mice. B16F10 tumor–bearing mice (n = 3) were intravenously injected with 30 μl of AUNPs or pAUNPs (0.5 mg/ml, based on TPEBTPy). At 6 hours after injection, the mice were anesthetized with 2% (v/v) isoflurane and the tumors were irradiated with a 980-nm NIR laser (0.6 W/cm2) for 10 min (with 1-min interval for each minute to avoid heating effect). Subsequently, the mice were immediately imaged with a Maestro in vivo fluorescence imaging system (CRi Inc., Woburn, USA). The autofluorescence was removed by spectral unmixing software, and fluorescence signals were quantified by a Maestro system (emission filter, 850-nm short-pass; acquisition setting, 600 to 900 in 10-nm steps; exposure time, 10 s). In addition, the tumor-bearing mice were also sacrificed at 24 hours after irradiation, and the DLNs in different groups were collected, imaged, and quantified by a Maestro system (excitation filter, 455 nm; emission filter, 672-nm long-pass; acquisition setting, 500 to 900 in 10-nm steps; exposure time, 300 ms).
In vivo antitumor therapy through AUNPs
All animal studies were performed in compliance with the guidelines set by the Tianjin Committee of Use and Care of Laboratory Animals, and the overall project protocols were approved by the Animal Ethics Committee of Nankai University. Six-week-old C57BL/6 mice [obtained from the Laboratory Animal Center of the Academy of Military Medical Sciences (Beijing, China)] were used to establish the B16F10 tumor–bearing mouse model. B16F10 or luciferase-transgenic B16F10 (luc-B16F10) cells (1 × 106 cells per mouse) suspended in 30 μl of saline were injected subcutaneously into the right flank region of the mouse. After 10 days, AUNPs, pAUNPs, or PBS was intratumorally injected into the mice on days 0 and 1 (n = 5, per group) (30 μl, 0.5 mg/ml, based on TPEBTPy). At 6 hours after each injection, the mice were anesthetized with 2% (v/v) isoflurane, and the tumors were irradiated with a 980-nm laser (0.6 W/cm2) for 10 min (with 1-min interval for each minute to avoid heating effect). After PDT, DLNs were imaged and determined by a CRi Maestro in vivo imaging system. DLN regions were irradiated by a 980-nm laser on days 1 to 4 at a power of 0.12 W/cm2 for 10 min (with 1-min interval for each 2 min/four times a day). When using low-power light to treat lymph nodes, the PDT-treated tumor region was covered by a surgical cloth to avoid laser exposure to neighboring tumors.
The abscopal effect of AUNPs was evaluated through the bilateral B16F10 tumor–bearing mouse model. The tumor was built by subcutaneous injection of B16F10 or luc-B16F10 cells (1 × 106 cells per mouse) into the right flank region. Meanwhile, B16F10 or luc-B16F10 cells (4 × 105 cells per mouse) were injected into the left flank region of the same mouse. After 10 days, AUNPs, pAUNPs, or PBS was intratumorally injected into the right tumor of mice on days 0 and 1 (n = 5, per group) (30 μl, 0.5 mg/ml, based on TPEBTPy). At 6 hours after injection, the mice were anesthetized with 2% (v/v) isoflurane, and the tumors were irradiated with a 980-nm NIR laser (0.6 W/cm2). The αPD-1 (clone RMP1-14) blocking antibody (10 mg/kg) was intraperitoneally injected into the animals on days 1 to 4. After PDT, right DLNs of mice were imaged and determined by a CRi Maestro in vivo imaging system and DLN regions were irradiated by a 980-nm laser on days 1 to 4 at a power of 0.12 W/cm2.
To evaluate the therapeutic efficacy, tumor growth was monitored by the IVIS Spectrum Imaging System (PerkinElmer Ltd.). Briefly, d-luciferin (150 mg/kg) was intraperitoneally injected into the mice. The mice were then imaged, and then the bioluminescence signals were quantified in units of maximum photons per second per square centimeter per steradian. The tumor size was measured with a caliper every 2 days. Tumor volumes were calculated as follows: (width2 × length)/2.
To establish a tumor rechallenge model, on day 34 after the first treatment of the unilateral tumor model, mice were rechallenged by intravenous injection of B16F10 cell suspension (100 μl, 5 × 105) into C57BL/6 mouse. Subsequently, tumor growth in the lung was monitored by the IVIS Spectrum Imaging System, and lung tissues were collected for flow cytometry and histological analysis on different time points.
Flow cytometry analysis
At designated time points, DLNs, lungs, spleens, or tumor tissues were collected, ground, and filtered through nylon mesh filters (40 μm) and washed with PBS containing 1% FBS, respectively. The single-cell suspensions were blocked with anti-CD16/32 (BioLegend) for 30 min on ice and then stained with fluorescence-labeled anti-mouse CD45 (30-F11), CD3e (145-2C11), CD4 (GK1.5), CD8 (53-6.7), CD11b (M1/70), Ly-6G/Ly-6C (RB6-8C5), F4/80 (BM8), CD62L (MEL-14), and CD44 (IM7) (all from BioLegend; dilution, 1:100) for 30 min on ice. For DC characterization, DC suspensions were blocked with anti-CD16/32 (BioLegend) for 30 min on ice and then stained with fluorescence-labeled anti-mouse CD11c (N418), CD80 (16-10A1), and CD86 (GL-1) for 30 min on ice. For intracellular protein staining, the samples were blocked with anti-CD16/32 (BioLegend) for 30 min on ice and then stained with anti-mouse CD4 or CD8 for 30 min on ice, respectively. Subsequently, cells were fixed, permeabilized, and stained with anti-mouse Foxp3 (FJK-16s) or IFN-γ (XMG1.2) at room temperature for 30 min, respectively. Last, all samples were washed with PBS containing 1% FBS two times. All the flow cytometric analysis was conducted using the LSRFortessa Cell Analyzer (BD Biosciences), and data analysis was carried out using FlowJo software (www.flowjo.com; Tree Star).
Histological analysis
Mice in different groups were euthanized, and different tissues, including tumors, were excised and fixed in 4% paraformaldehyde. A part of tumors and DLNs were embedded, frozen, and sectioned at a thickness of 8 μm. All tissue section was treated according to the standard manufacturer’s instructions and stained with different primary antibodies: rabbit anti-CD11c (1:200), rat anti-F4/80 (1:200; Abcam), rabbit anti-CD11b (1:1000), rabbit anti–Ly-6G/Ly-6C, rabbit anti-CD4 (1:200), rabbit anti-Foxp3 (1:200), rabbit anti-CD206 (1:200), and rabbit anti–PD-L1 (1:200), followed by staining with fluorescently labeled secondary antibodies [Alexa Fluor 488–conjugated goat anti-rat IgG (H+L) (1:200; Thermo Fisher Scientific) and Alexa Fluor 633–conjugated goat anti-rabbit (H+L) (1:200; Thermo Fisher Scientific)]. The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) containing mounting solution (DAPI Fluoromount-G, SouthernBiotech). All the slices were finally imaged with a confocal microscope (Leica, SP8). The other parts of tumor tissues and other organs were fixed in 4% paraformaldehyde, then embedded into paraffin, and subsequently sliced at a thickness of 5 μm. Slices were stained with hematoxylin and eosin (H&E), imaged by optical microscopy, and assessed by three independent pathologists.
Statistical analysis
Quantitative data were presented as means ± SEM. Analysis of variance (ANOVA) was used for multiple comparisons, and Student’s t test was used for two-group comparisons. The differences in survival in each group were analyzed by the Kaplan-Meier method, and the P value was determined by the log-rank test. All statistical analyses were carried out using GraphPad Prism 5.0. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Supplementary Material
Acknowledgments
Funding: This work was supported by grants from the Singapore NRF Competitive Research Program (R279-000-483-281), NRF Investigatorship (R279-000-444-281), National University of Singapore (R279-000-482-133), the National Science Foundation of China (81830060 and 31771066), and Agency for Science, Technology and Research (A*STAR) (grant nos. A1883c0011 and A1983c0038). Author contributions: D.M., F.H., X.L., and B.L. designed the project. D.M., F.H., and Z.Y. performed the experiments. All authors analyzed and interpreted the data, contributed to the writing of the manuscript, discussed the results and implications, and edited the manuscript at all stages. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/26/eabb2712/DC1
REFERENCES AND NOTES
- 1.Wilt C., Le D. T., Integrating immunotherapy into colorectal cancer care. Oncology 32, 494–498 (2018). [PubMed] [Google Scholar]
- 2.Vonderheide R. H., Domchek S. M., Clark A. S., Immunotherapy for breast cancer: What are we missing? Clin. Cancer Res. 23, 2640–2646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ngwa W., Irabor O. C., Schoenfeld J. D., Hesser J., Demaria S., Formenti S. C., Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 18, 313–322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hughes P. E., Caenepeel S., Wu L. C., Targeted therapy and checkpoint immunotherapy combinations for the treatment of cancer. Trends Immunol. 37, 462–476 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Tang C., Wang X., Soh H., Seyedin S., Cortez M. A., Krishnan S., Massarelli E., Hong D., Naing A., Diab A., Gomez D., Ye H., Heymach J., Komaki R., Allison J. P., Sharma P., Welsh J. W., Combining radiation and immunotherapy: A new systemic therapy for solid tumors? Cancer Immunol. Res. 2, 831–838 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Aloia M. M., Zizzari I. G., Sacchetti B., Pierelli L., Alimandi M., CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 9, 282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinzerling L., Ott P. A., Hodi F. S., Husain A. N., Tajmir-Riahi A., Tawbi H., Pauschinger M., Gajewski T. F., Lipson E. J., Luke J. J., Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J. Immunother. Cancer 4, 50 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuge O., Vasdev N., Allchorne P., Green J. S., Immunotherapy for bladder cancer. Res. Rep. Urol. 7, 65 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galluzzi L., Kepp O., Kroemer G., Enlightening the impact of immunogenic cell death in photodynamic cancer therapy. EMBO J. 31, 1055–1057 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kotsias F., Hoffmann E., Amigorena S., Savina A., Reactive oxygen species production in the phagosome: Impact on antigen presentation in dendritic cells. Antioxid. Redox Signal. 18, 714–729 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Oberkampf M., Guillerey C., Mouriès J., Rosenbaum P., Fayolle C., Bobard A., Savina A., Ogier-Denis E., Enninga J., Amigorena S., Leclerc C., Dadaglio G., Mitochondrial reactive oxygen species regulate the induction of CD8+ T cells by plasmacytoid dendritic cells. Nat. Commun. 9, 2241 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsue H., Edelbaum D., Shalhevet D., Mizumoto N., Yang C., Mummert M. E., Oeda J., Masayasu H., Takashima A., Generation and function of reactive oxygen species in dendritic cells during antigen presentation. J. Immunol. 171, 3010–3018 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Wang C., Li P., Liu L., Pan H., Li H., Cai L., Ma Y., Self-adjuvanted nanovaccine for cancer immunotherapy: Role of lysosomal rupture-induced ROS in MHC class I antigen presentation. Biomaterials 79, 88–100 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Norum O.-J., Selbo P. K., Weyergang A., Giercksky K.-E., Berg K., Photochemical internalization (PCI) in cancer therapy: From bench towards bedside medicine. J. Photochem. Photobiol. B 96, 83–92 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Chen C., Ni X., Jia S., Liang Y., Wu X., Kong D., Ding D., Massively evoking immunogenic cell death by focused mitochondrial oxidative stress using an AIE luminogen with a twisted molecular structure. Adv. Mater. 31, 1904914 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Wachowska M., Gabrysiak M., Muchowicz A., Bednarek W., Barankiewicz J., Rygie T., Boon L., Mroz P., Hambli M. R., Golab J., 5-Aza-2′-deoxycytidine potentiates antitumour immune response induced by photodynamic therapy. Eur. J. Cancer 50, 1370–1381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Firczuk M., Nowis D., Gołąb J., PDT-induced inflammatory and host responses. Photoch. Photobio. Sci. 10, 653–663 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Qian J., Tang B. Z., AIE luminogens for bioimaging and theranostics: From organelles to animals. Chem 3, 56–91 (2017). [Google Scholar]
- 19.Hu F., Xu S., Liu B., Photosensitizers with aggregation-induced emission: Materials and biomedical applications. Adv. Mater. 30, 1801350 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Zhou B., Shi B., Jin D., Liu X., Controlling upconversion nanocrystals for emerging applications. Nat. Nanotechnol. 10, 924 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Zhong Y., Ma Z., Wang F., Wang X., Yang Y., Liu Y., Zhao X., Li J., Du H., Zhang M., Cui Q., Zhu S., Sun Q., Wan H., Tian Y., Liu Q., Wang W., Garcia K. C., Dai H., In vivo molecular imaging for immunotherapy using ultra-bright near-infrared-IIb rare-earth nanoparticles. Nat. Biotechnol. 37, 1322–1331 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen S., Weitemier A. Z., Zeng X., He L., Wang X., Tao Y., Huang A. J. Y., Hashimotodani Y., Kano M., Iwasaki H., Parajuli L. K., Okabe S., Teh D. B. L., All A. H., Tsutsui-Kimura I., Tanaka K. F., Liu X., McHugh T. J., Near-infrared deep brain stimulation via upconversion nanoparticle–mediated optogenetics. Science 359, 679–684 (2018). [DOI] [PubMed] [Google Scholar]
- 23.All A. H., Zeng X., Teh D. B. L., Yi Z., Prasad A., Ishizuka T., Thakor N., Hiromu Y., Liu X., Expanding the toolbox of upconversion nanoparticles for in vivo optogenetics and neuromodulation. Adv. Mater. 31, 1803474 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Idris N. M., Gnanasammandhan M. K., Zhang J., Ho P. C., Mahendran R., Zhang Y., In vivo photodynamic therapy using upconversion nanoparticles as remote-controlled nanotransducers. Nat. Med. 18, 1580–1585 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Jokerst J. V., Lobovkina T., Zare R. N., Gambhir S. S., Nanoparticle PEGylation for imaging and therapy. Nanomedicine 6, 715–728 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galluzzi L., Buqué A., Kepp O., Zitvogel L., Kroemer G., Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Li A. W., Sobral M. C., Badrinath S., Choi Y., Graveline A., Stafford A. G., Weaver J. C., Dellacherie M. O., Shih T., Ali O. A., Kim J., Wucherpfennig K. W., Mooney D. J., A facile approach to enhance antigen response for personalized cancer vaccination. Nat. Mater. 17, 528–534 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biswas S. K., Metabolic reprogramming of immune cells in cancer progression. Immunity 43, 435–449 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Gajewski T. F., Schreiber H., Fu Y. X., Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Topalian S. L., Drake C. G., Pardoll D. M., Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 27, 450–461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Postow M. A., Sidlow R., Hellmann M. D., Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 378, 158–168 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Wang C., Wang J., Zhang X., Yu S., Wen D., Hu Q., Ye Y., Bomba H., Hu X., Liu Z., Dotti G., Gu Z., In situ formed reactive oxygen species–responsive scaffold with gemcitabine and checkpoint inhibitor for combination therapy. Sci. Transl. Med. 10, eaan3682 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Chen G., Qiu H., Prasad P. N., Chen X., Upconversion nanoparticles: Design, nanochemistry, and applications in theranostics. Chem. Rev. 114, 5161–5214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan S., Zeng X., Tang Y., Liu B., Wang Y., Liu X., Activating antitumor immunity and antimetastatic effect through polydopamine-encapsulated core-shell upconversion nanoparticles. Adv. Mater. 31, 1905825 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Wu X., Zhang Y., Takle K., Bilsel O., Li Z., Lee H., Zhang Z., Li D., Fan W., Duan C., Chan E. M., Lois C., Xiang Y., Han G., Dye-sensitized core/active shell upconversion nanoparticles for optogenetics and bioimaging applications. ACS Nano 10, 1060–1066 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu W., Mao D., Xu S., Ji S., Hu F., Ding D., Kong D., Liu B., High-performance photosensitizers with aggregation-induced emission for image-guided photodynamic anticancer therapy. Mater. Horiz. 4, 1110–1114 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Qi J., Chen C., Zhang X., Hu X., Ji S., Kwok R. T., Ding D., Tang B. Z., Light-driven transformable optical agent with adaptive functions for boosting cancer surgery outcomes. Nat. Commun. 9, 1848 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ni X., Zhang X., Duan X., Zheng H.-L., Xue X.-S., Ding D., Near-infrared afterglow luminescent aggregation-induced emission dots with ultrahigh tumor-to-liver signal ratio for promoted image-guided cancer surgery. Nano Lett. 19, 318–330 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Chen C., Ni X., Tian H. W., Liu Q., Guo D.-S., Ding D., Calixarene-based supramolecular AIE dots with highly inhibited nonradiative decay and intersystem crossing for ultrasensitive fluorescence image-guided cancer surgery. Angew. Chem. Int. Ed. (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/26/eabb2712/DC1