

Whole-brain spatial transcriptomics redefines the neuroanatomical classification of the adult mouse brain.
Abstract
Brain maps are essential for integrating information and interpreting the structure-function relationship of circuits and behavior. We aimed to generate a systematic classification of the adult mouse brain based purely on the unbiased identification of spatially defining features by employing whole-brain spatial transcriptomics. We found that the molecular information was sufficient to deduce the complex and detailed neuroanatomical organization of the brain. The unsupervised (non-expert, data-driven) classification revealed new area- and layer-specific subregions, for example in isocortex and hippocampus, and new subdivisions of striatum. The molecular atlas further supports the characterization of the spatial identity of neurons from their single-cell RNA profile, and provides a resource for annotating the brain using a minimal gene set—a brain palette. In summary, we have established a molecular atlas to formally define the spatial organization of brain regions, including the molecular code for mapping and targeting of discrete neuroanatomical domains.
INTRODUCTION
Mapping the adult brain, in terms of establishing reference maps of subregions and their borders, and determining the diversity of neuron types and their connectivity, is at the core of exploring the structure-function relationship of brain circuits that defines the diversity of animal behaviors (1–4). A central principle in generating brain atlases has, over the past century, been the annotation of tissue landmarks using microscopy. The spatial definitions have, to a large extent, relied on cytoarchitectural features, such as differences in the density and form of cells, as well as chemoarchitectural definitions derived from distribution of key molecules such as neurotransmitters (5, 6). Ongoing collective efforts in the field are now starting to reveal the details of neuron and region connections at the microscale and mesoscale level (4, 7, 8) and gene expression in the human brain (9, 10). These tissue definitions and the resulting mouse brain atlases (11, 12) not only have been essential for establishing the experimental framework to explore brain structure and function relationships but also have resulted in debate and disagreement over the validity of expert-based region annotations (13).
Experimental neuroscience depends on the ability to repeatedly and accurately record and manipulate activity of neuron subtypes in specific brain regions, and genetic targeting approaches have therefore proven extremely valuable in cell type–specific targeting (14, 15). Unexpectedly, spatial classification has primarily been limited to layer-specific gene expression patterns in isocortex (16), not capturing the great diversity of spatially segregated regions, such as the anteroposterior (AP) and mediolateral (ML) specialization in isocortex, or the more complex three-dimensional (3D) organization of hippocampus and other subcortical regions. Only a few brain regions have generally accepted border definitions, raising the importance of spatial definitions to describe nervous system organization from molecular patterns (17). We have applied spatial transcriptomics (ST) on the adult mouse brain to generate detailed anatomic definitions based exclusively on tissue gene expression classification, thereby establishing a molecular brain atlas.
RESULTS
ST at the whole-brain scale
We generated a whole-brain molecular atlas by capturing the spatial patterns of gene expression in the adult mouse brain using ST (18). We hybridized 75 coronal sections from one brain hemisphere that covered the entire AP axis onto ST arrays (Fig. 1A and fig. S1). Using a computational framework designed to generate reference maps (19), we aligned each imaged brain hemisphere section, including the position of the ST spots in the tissue, with the Allen mouse brain reference atlas (ABA; www.brain-map.org) (Fig. 1B and fig. S1) (12). This established a complete brain atlas with information on tissue coordinates, the reference ABA neuroanatomical definitions, and spatially defined gene expression patterns (Fig. 1C). We detected, on average, 4422 genes and 11,210 reads per spot (4422 ± 1164 unique genes, 11,210 ± 5943 reads; fig. S2). The complete dataset after quality control (QC) contained information on the expression of 15,326 unique genes across 34,053 spots (Fig. 1, D and E). As a first demonstration of the spatial expression patterns for well-established regional and cellular markers, we visualized the expression of the cortical marker Rasgrf2, the striatal marker Gpr88, and the thalamic marker Rora in 2D and 3D and compared this to the ABA in situ hybridization (ISH) signal (Fig. 1F) (12). The correspondence between ISH patterns and the gene expression in the molecular atlas supported the possibility of using the complete molecular information in the atlas to extract patterns of spatial gene expression and anatomic definitions without introducing any previous knowledge of spatial boundaries or regions.
Fig. 1. ST to generate a whole-brain molecular atlas.
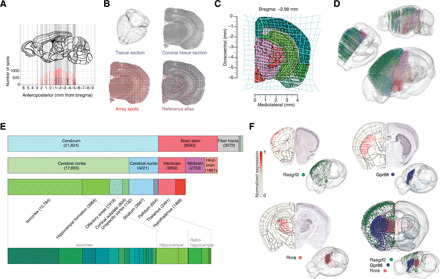
(A) Distribution of coronal sections used to generate the molecular atlas. Adjacent sections shown with light and dark red color. (B) Example of image processing showing the section outline in 3D (top left), the H&E-stained coronal section (top right), alignment with the ST array (bottom left), and alignment with the ABA mouse brain reference atlas (bottom right). (C) Transformation (light blue lines) of section shown in (B) to generate a common reference framework containing ST spot position and ABA neuroanatomical definitions. (D) 3D mapping of all spots color-coded according to main region identity. (E) Distribution of all spots according to main ABA region definitions. (F) Expression of three candidate genes with spatially discrete signals: in isocortex (Rasgrf2, green), striatum (Gpr88, blue), and thalamus (Rora, red). Left side, expression in the molecular atlas; right side, ISH signal from ABA. In 3D, right hemisphere shows spots with gene expression level above the 95th percentile. Left hemisphere shows striatum (light blue) and thalamus (light red) in ABA. Composite 3D image showing expression of all three genes including region definitions. Color scheme in (C) to (E), based on ABA reference color scheme.
Computational approaches to extract new spatial domains from the molecular atlas
To generate a spatial classification of the adult mouse brain based exclusively on molecular patterns, we analyzed the ST gene expression data by performing dimensionality reduction and clustering. We performed dimensionality reduction using the top 50% variable genes by applying an independent component analysis [JADE ICA (20)]. Each independent component (IC) represents a weighted combination of genes (gene loads; fig. S3). We visualized each IC in 2D and 3D in the reference atlas, allowing us to infer whether ICs contained spatial information (biological IC) or likely resulted from technical noise (technical IC) (Fig. 2A and fig. S4).
Fig. 2. Computational approaches to extract new spatial domains from the molecular atlas.
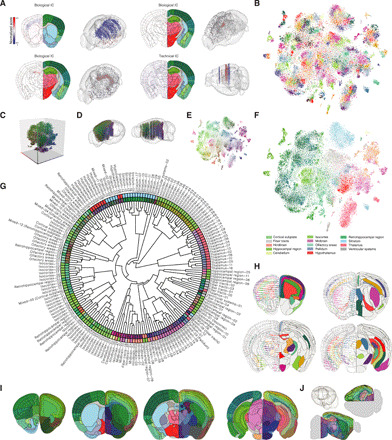
(A) Examples of ICs (biological versus technical). Left side: IC signal (normalized score). Right side: Corresponding ABA reference section. IC scores above the 95th percentile in absolute value are shown in 3D. (B) 2D t-SNE with categorical color scheme to visualize spots in 181 molecular cluster (27 unique colors). (C) 3D t-SNE to visualize molecular similarity of clusters. (D) Mapping of all spots in 3D colored by molecular similarity. (E) 2D t-SNE showing spots according to molecular similarity coloring. (F) 2D t-SNE showing spots according to ABA neuroanatomical regions. (G) Hierarchical clustering of the 181 molecular clusters (fan plot). Molecular clusters annotated with a numerical identifier and a name according to correspondence to ABA subregion. Inner layer is color-coded using molecular similarity. Outer layer colored according to ABA subregions. (H) Example coronal sections from the molecular atlas. Left side: Position and molecular identity of spots for selected clusters (black lines, ABA region borders). Right side: The same clusters shown in the molecular atlas (black lines, molecular cluster borders). (I) Example coronal sections from the molecular atlas (left side, ABA reference atlas; right side, molecular clusters color-coded on the basis of molecular similarity). (J) Virtual sectioning of the molecular atlas (black line shows example of horizontal or a sagittal plane). Molecular similarity colors based on median coordinates of spots per cluster in the 3D t-SNE shown in (C) to (E), (G), and (I).
We selected 45 biological ICs to perform clustering (21), resulting in 181 molecularly defined clusters. We visualized spots with a categorical color scheme according to their cluster identity in a whole-brain reference atlas (movie S1) and in a 2D T-distributed Stochastic Neighbor Embedding (t-SNE) (22) plot (Fig. 2B). We further visualized the molecular relationship between clusters by plotting the spots in a 3D t-SNE or 3D Uniform Manifold Approximation and Projection (UMAP) plot, and applying an RGB (red, green, blue) color scheme corresponding to the x, y, and z coordinates. Plotting based on this color scheme showed the distribution of molecularly similar clusters across the whole brain (Fig. 2, C to E, and figs. S5 to S8). By instead applying a neuroanatomically relevant color code (the ABA reference color scheme) to annotate spots based on their known position in the atlas we revealed the relationship between the molecular classification (derived purely from the gene expression) and the broad spatial fingerprint of the clusters (Fig. 2F). To further map the relationship between clusters, we used hierarchical clustering to generate a dendrogram for the 181 molecular clusters. We then color-coded each molecular cluster based on two definitions in a fan plot: the molecular similarity of clusters based on the 3D t-SNE RGB color code, and the color code denoting neuroanatomical identity (ABA reference color code; Fig. 2G). From these visualizations, we concluded that the molecular clustering respected spatial patterns at the global scale and was aligned with certain aspects of current neuroanatomical definitions.
We next applied machine learning [support vector machine (SVM) algorithm] to transform individual spots and the corresponding molecular clusters into continuous 3D volumes for anatomic and spatial interpretation (Fig. 2H and movie S2). This resulted in a whole-brain atlas with detailed information on the spatial organization of molecular clusters (Fig. 2I and movie S3). The molecular atlas is available for interactive visualization of clusters and gene expression, including manual annotation or modification of subregions and clusters (3D ST Viewer; molecularatlas.org). The 3D molecular atlas further supports virtual sectioning of the brain at any angle (e.g., horizontal and sagittal) to easily visualize the spatial extent of molecular clusters and their borders (Fig. 2J).
To explore the potential of molecular mapping as a road map for new spatial classifications at a finer scale and to compare results from the molecular atlas with available reference maps, we first focused on the spatial organization of the isocortex. We used the neuroanatomical definition of isocortex and nomenclature of the ABA reference, which resulted in the annotation of 55 molecular clusters belonging to isocortex. Visualizing the isocortical clusters in the t-SNE plot showed the underlying molecular-spatial structure, which emerged purely from the gene expression signature of isocortical domains (Fig. 3A). The isocortical clusters showed clear separation by their layer identity, and recapitulated some of the established layer divisions, in terms of position and marker expression (Fig. 3, B to D). More interestingly, we found that the molecular clustering also revealed the global regionalization of isocortical regions, defined by segregation across the AP and ML dimension (Fig. 3E). For example, molecular clusters were clearly segregated into distinct subregions of the frontal cortex (PL, ACA, and ORB), forming borders between primary and secondary somatomotor and somatosensory cortex (MOp, MOs, SSp, and SSs) and delineating spatially the retrosplenial cortex (RSP; Fig. 3, F to I, and movie S4). The molecular atlas did not differentiate between prelimbic and infralimbic parts of prefrontal cortex but established borders between the medial prefrontal and orbital regions, including layer-specific spatial borders. In somatosensory isocortex, we found that the molecular clusters defined eight separate layer-specific clusters, including molecular definitions of layers 5a, 5b, 6a, and 6b. We found that the molecular classification could separate layer 2/3 into two separate clusters with clear spatial organization. This layer 2/3 subdivision was evident in the somatosensory area (primary somatosensory, barrel field), as well as in other isocortical areas such as primary motor area (MOp). The hippocampal region (HIP) was subdivided into 11 molecular clusters, which spatially capture the unique hippocampal organization in 3D and establish dorsoventral (DV) borders for CA1, CA2, and CA3. The dentate gyrus separated into three clear clusters that map onto the granule layer, molecular layer, and polymorph layer (i.e., hilus; fig. S6). The molecular atlas thereby supports detailed spatial classification of isocortex and the hippocampal formation into discrete domains, which capture layer-specific subdivisions (DV axis), gene expression signatures to define region boundaries, and spatial classification of the different isocortical subregion borders across the AP and ML axes (fig. S9).
Fig. 3. New molecular definitions of subregions and layers in isocortex and striatum.
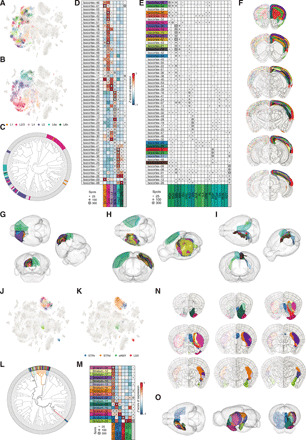
(A) t-SNE showing isocortical clusters (categorical color scheme). (B) t-SNE showing coloring of spots according to layer identity from registration in the ABA. (C) Circular phylogram showing layer identity of isocortical clusters. (D) Visualization of isocortical molecular clusters and gene expression (averaged z scored) for layer markers. (E) Visualization of all isocortical molecular clusters and their correspondence to major isocortical subregions. (F) Visualization of the isocortical clusters in coronal sections (AP axis). Left side: Individual spots belonging to molecular clusters. Right side: The corresponding smoothed clusters in the molecular atlas. (G to I) 3D visualization of molecular clusters found in different isocortical domains. (G) Molecular clusters in anterior isocortex (PL, ILA, ACA, and MO). (H) Molecular clusters in the central part of isocortex (SS, MOp, and PTLp). (I) Molecular clusters in the medial and posterior isocortex (RSP, POST, and PRE). (J) t-SNE showing striatal clusters. (K) t-SNE showing coloring of spots according to ABA reference. STRv, ventral striatum; STRd, dorsal striatum; sAMY, striatum-like amygdala nuclei; LSX, lateral septal complex. (L) Circular phylogram showing ABA reference for striatal clusters. (M) Visualization of the striatal molecular clusters and gene expression (averaged z scored) for reference markers. (N) Visualization of the striatal clusters in coronal sections of the molecular atlas. (O) 3D visualization of the striatal clusters. Left hemisphere, molecular atlas; right hemisphere, ABA striatum regions. Dot surface in (D), (E), and (M) corresponds to the absolute number of spots. Molecular clusters in (A), (E) to (J), and (L) to (O) are shown with the categorical color scheme. Molecular clusters in (B) to (D) show layer identity. Molecular clusters in (K) to (M) show striatal identity.
To further explore how the molecular atlas can reveal spatial organization of subcortical areas, we focused on the striatum (STR), which in the ABA reference is divided into one major dorsal part of striatum (STRd), and the ventral striatum (STRv), including the lateral septal complex (LSX) and the striatum-like amygdalar nuclei (sAMY). In particular, the dorsal striatum has not been molecularly subdivided even if functional mapping suggests several discrete striatal subregions (23). In the molecular atlas, STRd and the other striatal regions can be visualized in the t-SNE plot as 17 discrete clusters, and these molecular clusters reflect spatial information as shown when color-coded with the ABA reference color scheme (Fig. 3, J and K). These molecular clusters establish a new classification of the striatum into spatially restricted subregions, including correlation with discrete marker expression (Fig. 3, L and M). Visualization of the striatal clusters in 2D and 3D supports a spatial organization of dorsal striatum into several discrete molecular domains across the AP and ML axes, in addition to the ventral and septal divisions (Fig. 3, N and O, and movie S5).
In summary, capturing the spatial gene expression signatures of the adult mouse brain allowed us to produce a detailed whole-brain atlas built from only molecular information, thereby establishing a formal system for spatial definitions in neuroanatomy.
Mapping the spatial origin of single cells using the molecular atlas
The molecular profiling of neurons based on single-cell RNA sequencing (scRNA-seq) has been used to establish a molecular code for classification of neuron subtypes. This classification scheme usually lacks information on the spatial origin of the classified neurons. We investigated whether the molecular atlas could serve as a resource to map the spatial origin of single neurons based on their scRNA-seq molecular profiles, thereby providing a spatial dimension to the cell type definitions in large-scale cell classification efforts. To provide proof of principle of the spatial mapping approach, we used available data (24) on scRNA-seq of 23,822 neurons and nonneuronal cells isolated from either primary visual cortex (VISp) or from the frontal cortex [anterior-lateral motor cortex (ALM)].
We trained a neural network classifier with the molecular atlas to develop predictions on the spatial origin of each single cell and quantified spatial mapping onto molecular clusters in ALM and VISp (Fig. 4, A and B). When we used a cutoff in the prediction probability to select 50% of the neurons with the highest confidence in spatial mapping (0.99 probability), we found that 76% of the glutamatergic neurons were accurately classified onto the correct region (VISp versus ALM, 65% without thresholding) and 37% to the correct region and layer (Fig. 4, C and D, and fig. S10). In contrast, we found that GABAergic neurons, as well as most of the nonneuronal cell clusters, contained limited information about spatial origin and could not be accurately mapped onto the molecular atlas (Fig. 4E). In summary, this demonstrates the value of the molecular atlas as a resource to map the spatial origin of neurons using single-cell molecular information.
Fig. 4. Mapping the spatial origin of single cells using the molecular atlas.
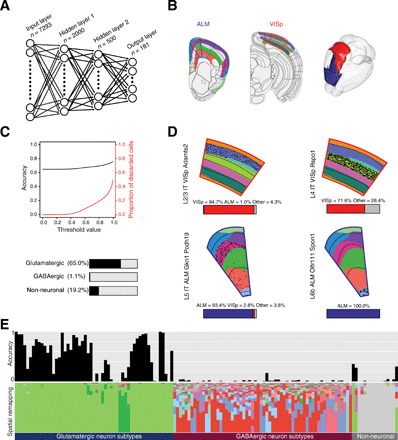
(A) Schematic of the neural network (NN) organization. (B) Molecular atlas clusters that include ALM (blue outline) and VISp (red outline). Black lines show ABA reference. 3D visualization of the molecular clusters. (C) Top: Spatial mapping accuracy for single neurons (glutamatergic) and discarded cells. Bottom: Spatial mapping accuracy for single cells for the three main cell types. (D) Visualization of the spatial mapping of single neurons (scRNA-seq clusters from VISp, red outline; or ALM, blue outline). Black dots show spatial mapping of single neurons onto predicted cluster (clusters shown with categorical color scheme and ABA layers shown with back lines). Quantification of spatial mapping per region is shown for each scRNA-seq cluster with stacked bar plot. (E) Top row shows the accuracy of the spatial mapping of all cells using the NN predictions for each scRNA-seq cluster (133 clusters, 23,822 cells). Bottom row shows the proportion of mapped cells for each scRNA-seq cluster onto the molecular clusters. Color scheme of molecular clusters based on ABA reference colors.
A reduced brain palette captures whole-brain organization
We aimed to determine a reduced gene set that would be sufficient to capture the major brain subdivisions at the global scale. The complete molecular atlas is based on the gene expression patterns of 7663 genes. To establish the genes that best capture the global spatial signals with an unbiased approach, we selected genes either from the ICs or from weights in a SVM model and used the normalized mutual information index (NMI) as a measurement of cluster similarity (Fig. 5A). A panel of 266 unique genes was established on the basis of an NMI 0.6 for clustering using genes from the ICs. We found that the 266 genes—forming a brain palette—when used to define the brain into 181 molecular clusters, established a whole-brain spatial division similar in organization to the clustering found in the full molecular atlas (Fig. 5, B to D). We calculated the cluster similarity between the molecular atlas and the reduced version to visualize the pairwise correlation between spatially conserved subregions (Fig. 5E and fig. S11). We found that the spatial definitions of molecular domains were conserved globally (Fig. 5F), as shown, for example, in a cluster group consisting of several isocortical, retrohippocampal, and olfactory area subregions (Fig. 5G), and in the main hippocampal clusters (Fig. 5H). We concluded from this analysis that, on a whole-brain scale, most subregions can be spatially defined using only the 266 genes found in the reduced brain palette, even if some regions will require a richer gene expression palette for more detailed subdivision (fig. S12). To identify some of the cellular and biological signals that contribute to spatial organization, we used the Gene Ontology (GO) gene annotation (25). We found significant enrichment for 64 of 266 genes in the brain palette, which were assigned to five GO families (combined score > 30, adjusted P value of <1 × 10−3; fig. S13). These genes are associated with cellular processes (dendrite and axon) and biological processes (nervous system development, neurotransmitter transport, and synaptic transmission). The enrichment in genes involved in nervous system development, as well as molecular aspects of dendrite and axon compartments, points to the key biological processes that establish brain regionalization and form and possibly maintain spatial patterns in the adult brain. A large number of the genes that allow spatial clustering (e.g., in the brain palette) are of unknown function, and their role in neurons or glia remains to be determined. In summary, the minimal version of the molecular atlas builds on the brain palette—a list of genes whose expression is sufficient to recapitulate most aspects of the spatial whole-brain subdivisions—revealing some of the biological processes behind spatial identity while also forming an important experimental tool for mapping global brain space with a small number of genes.
Fig. 5. A reduced brain palette captures the whole-brain spatial organization.
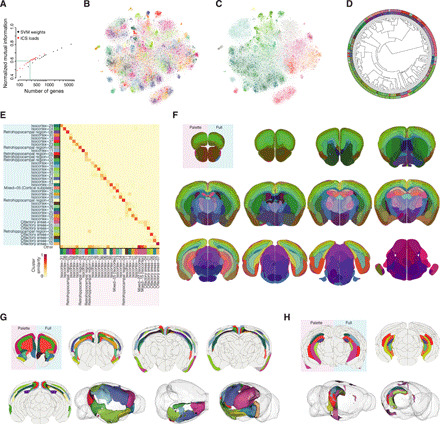
(A) NMI used to select the gene set for a brain palette. NMI computed from either top IC loads (red) or top SVM weights (black). (B) t-SNE showing 181 molecular clusters based on the brain palette (266 genes). Clusters are colored on the basis of the best matching cluster in the full molecular atlas using the same categorical colors. (C) t-SNE showing individual spots colored according to neuroanatomical subregions (ABA reference colors). (D) Circular phylogram showing 181 molecular clusters in the brain palette atlas. The inner layer is color-coded using molecular similarity color scheme. The outer layer is colored according to ABA subregions. (E) Example of the similarity between the full molecular atlas (y axis) and the brain palette molecular atlas (x axis) for selected clusters. (F) Visualization of the brain palette molecular atlas (left side, pink background in the first section) and the full molecular atlas (right side, blue background in the first section) based on molecular similarity color scheme. (G) Visualization of isocortical, retrohippocampal, and olfactory clusters in the brain palette molecular atlas (left side, pink background in the first section) and the full molecular atlas (right side, blue background in the first section). (H) 3D visualization of selected isocortical clusters in the brain palette molecular atlas. Categorical color scheme is used to visualize the same clusters in (B), (E), (G), and (H).
DISCUSSION
We have here established a molecular atlas of the adult mouse brain exclusively on the basis of unsupervised classification of ST patterns on a whole-brain scale. A challenge in neuroscience has for long been the formalized definitions of functionally and molecularly relevant subdivisions of the nervous system. The molecular definition of brain regions is an unbiased approach to map the discrete spatial domains at the whole-brain scale, and also introduces the ability to develop molecular approaches with spatial information to study nervous system organization, the evolutionary conservation of molecular and spatial codes, and their relationship to physiology and pathophysiology.
We found that it is possible to develop a detailed whole-brain spatial annotation based purely on gene expression signatures, for well-studied regions of the brain such as the somatosensory and somatomotor area in isocortex, and the hippocampus, as well as for major regions with poorly defined subdivisions such as the dorsal striatum. In addition, the molecular atlas also establishes the identity of isocortical layers across the entire isocortex, olfactory areas (e.g., piriform area), and the retrohippocampal area (e.g., entorhinal area). The subdivision of these areas into specific layers, and how layer divisions are conserved across different areas, has remained unclear and debated (26). Here, we propose a molecular definition of layers that contains subregional information and, for example, provide evidence for discrete molecular separation of layer 2/3. It remains to be determined whether our inability to identify a more detailed subdivision, for example, of the prefrontal cortex (27), or small amygdalar nuclei, is due to methodological limitations or to the absence of molecular identifiers and instead should be based on another classification level [e.g., connectivity (28)].
Our work furthermore defines a reduced gene set—a brain palette—that is sufficient to capture the spatial complexity of brain subregions on a whole-brain scale. We have demonstrated the power of region classification based on a reduced brain palette, and this approach can be similarly used to define any region of interest by developing local palettes that define subregions and borders with higher spatial resolution. Similar to the classification of neuron subtypes, which is often defined by combinatorial gene expression rather than single markers, the molecular classification of regions and borders will, to some extent, depend on combinatorial expression of some genes. In that sense, the brain palette establishes a reduced set of genes, which are sufficient to map the entire adult brain into relevant subregions. Thereby, the molecular atlas is the basis for assembling palettes for any region of interest that can guide the spatial annotation of subregions and borders using gene expression mapping methods (29–35). For example, focused efforts to map cellular organization in hippocampus and thalamus have demonstrated the capacity to identify spatial distribution of cell types (36, 37). We anticipate that technological advances will lead to increased sequencing depth and spatial resolution (38, 39), potentially leading to a more detailed molecular classification of cell types and brain regions.
We do not know the functional relevance of the molecules that define spatial divisions, and how these reflect biological processes in neurons or glia, but we provide evidence that certain gene ontologies (related to dendrite and axon processes, nervous system development, and glutamatergic neurotransmission) are probably major contributors to the spatial classification signals in the adult brain. We found that the signals that underlie global division of the brain into subregions are enriched in developmentally relevant genes, supporting a role of the developmental axes as important determinants of the adult brain regionalization. Previous work has shown a relationship between gene expression in the adult brain and embryonic development in rodents (40) and humans (41), supporting the role of developmental trajectories in brain maps. However, the mechanisms whereby the gene products contribute to the function or maintenance of regional identity in the adult brain remain unknown. A key question for the future will be to what extent the molecular identifiers of regionalization and spatial domains are evolutionary conserved, similar to conservation of cellular markers across species (for example, whether cortical expansion in the primate brain is a result of an expansion of conserved molecular domains found in other mammalia or is formed by formation of new and discrete spatiomolecular domains) (42). It will therefore be important to map the molecular signatures of regionalization in other species, including humans, and to thereby determine the conserved brain palette.
Classification of the adult brain from a molecular perspective should eventually be fused with large-scale connectivity and activity maps to produce structure-function maps of regions and circuits. Ultimately, generating whole-brain molecular atlases in mouse models of brain disease, and comparing the conserved molecular signatures that define brain regionalization across species, can serve as key points for developing a comprehensive understanding of the role of circuits in normal behavior and provide templates for new strategies to treat brain disorders.
MATERIALS AND METHODS
Experimental part
Brain sectioning
Three male mice (strain C57/BL6, 9 weeks old) were euthanized with an overdose of isoflurane. The brains were rapidly extracted from the skull and immediately submerged in ice-cold artificial cerebrospinal fluid. The brains were then blotted to remove excess liquid, and the hemispheres were separated along the sagittal midline. The left hemisphere was subsequently embedded in Optimal Cutting Temperature compound (OCT) and frozen in isopentane (2-methylbutane, Sigma) on dry ice. The hemisphere was cut into 10-μm sections on a cryostat at −12°C (Cryostar NX70, Thermo Fisher Scientific). All procedures and experiments on animals were performed according to the guidelines of the Stockholm Municipal Committee (approval number N166/15).
Fixation, staining, and imaging
Sections were fixed in 3.6 to 3.8% formaldehyde (Sigma) in phosphate-buffered saline (PBS), washed in PBS, then treated for 1 min with isopropanol, and air-dried. To perform hematoxylin and eosin (H&E) staining of the tissue, sections were incubated in Mayer’s Hematoxylin (Dako) for 7 min, then Bluing buffer for 2 min, and Eosin (Sigma) for 20 s. After drying, the slides were mounted with 85% glycerol and images of sections were taken using Metafer Slide Scanning Platform (MetaSystems). Raw images were stitched together using VSlide software (MetaSystems).
Tissue pre-permeabilization and permeabilization
To pre-permeabilize the tissue, sections of mouse brain were incubated for 20 min at 37°C with collagenase (0.5 U/μl; Thermo Fisher Scientific) in Hanks’ balanced salt solution (HBSS) buffer mixed with bovine serum albumin (0.2 μg/μl; New England Biolabs). Following washing in 0.1× saline sodium citrate (SSC) buffer (Sigma), sections were permeabilized with 0.1% pepsin/HCl (Sigma) at 37°C for 6 min. Then, wells were washed with 0.1× SSC buffer.
Reverse transcription and library preparation
After permeabilization, reverse transcription mix containing SuperScript III reverse transcriptase (Thermo Fisher Scientific) was added to each section and incubated overnight at 42°C as described previously (18). Next, to remove tissue from the slide, sections were incubated for 1 hour at 56°C with Proteinase K Digestion (PKD) buffer (both from Qiagen). Surface probes with bound mRNA/complementary DNA (cDNA) were then cleaved from the slide by USER enzyme (NEB) (18). Released probes were collected from each well and transferred to separate tubes. Next, second-strand synthesis, cDNA purification, in vitro transcription, antisense RNA (aRNA) purification, adapter ligation, post-ligation purification, a second-strand synthesis, and purification were carried out using an automated MBS 8000 system as described previously (43). cDNA was amplified by polymerase chain reaction (PCR) using Illumina indexing primers (18) and purified using carboxylic acid beads on an automated MBS robot system (44). The Agilent Bioanalyzer High Sensitivity DNA Kit was used to analyze the size distribution of the final libraries. The concentration of the libraries was measured with Qubit dsDNA HS (Thermo Fisher Scientific). The libraries were sequenced on the Illumina NextSeq platform using paired-end sequencing. Thirty bases were sequenced on read one to determine the spatial barcode and UMI, and 55 bases were sequenced on read two to cover the genetic region.
Staining of the ST array spots and image alignment
The ST spots were detected by incubation with hybridization mixture containing cyanine-3–labeled oligonucleotides as described previously (18). Fluorescent images were acquired using the same microscope as for the bright-field images.
Imaging and data processing
Image alignment and spot detection
Images of the H&E-stained section and the fluorescent images (Cy3, spots) were aligned by first down-sampling by 40% and then overlaying images (Adobe Photoshop CS6). The alignment was performed using common tissue features visible in both images when applying brightness and contrast filters. Aligned images were cropped to the borders of the array, a mask was created around the Cy3 image outside the tissue area, and the images were then saved for the spot detection.
The spot detection was performed with ImageJ (45), where the spot centroids were detected using the analyze particles feature. The detected spot centroid (inside tissue) pixel coordinates were exported to a file. An R script was used to convert the pixel coordinates to array coordinates and to assign them to an array position by rounding methods.
Data processing
Sequenced raw data were processed using the open source ST Pipeline v1.45 (46) with the genome reference Ensembl GRCm38 v86 and reference Mouse GENCODE vM11 (comprehensive gene annotation). The ST Pipeline was executed with the following settings:
1)Enabled homopolymer filter (A, G, T, C, N) with a length of 10
2)Enabled two-pass mode for the alignment step
3)Removed noncoding RNA [using the latest (v86) noncoding RNA database from Ensembl]
4)Discarded reads whose UMI has more than six low-quality bases
5)Discarded trimmed reads shorter than 20
The matrix of counts (spots by genes) generated by the ST Pipeline was filtered to replace Ensembl IDs by gene names and to keep only protein-coding, long noncoding intergenic, and antisense genes. The matrix of counts underwent another filtering step, where only spots inside the tissue were kept using the file generated in the previous step (image alignment).
Data processing QC
The aligned and cropped H&E image, together with the filtered matrix of counts, was used to perform a QC analysis that consisted of the following:
1)Estimate the average number of reads and genes per spot inside and outside tissue
2)Plot gene expression patterns onto the H&E image (by genes and by reads)
3)Perform a t-SNE followed by k-means clustering of the spots (expression across all genes) and then plot the resulting clustered spot colors onto the H&E image (inside tissue)
4)Plot saturation curves at different sequencing depths
Visual inspection of the generated images and stats was used to discard low-quality sections or to select sections for deeper sequencing.
Registration of brain sections
Both registration and segmentation were performed using the R package wholebrain (19). The microscopy images of H&E-stained tissue were converted into grayscale and registered (fig. S1). Correspondence points were manually adjusted for increased accuracy. The transformation defined during registration was then applied to the spot coordinates extracted during image alignment. Some posterior sections were divided in two or three subregions that were registered separately, as the registration mask could not simultaneously be fitted on the whole section.
Nuclei per spot
Nuclei were segmented from H&E-stained images using an appropriate filter. To account for the variability of spot size (average of 50-μm radius), the Cy3-stained images were segmented using a different filter adapted to spot detection. Segmented spot contours were smoothed to circles and paired with spots detected during alignment (fig. S2G). Segmented nuclei within the radius of every spot were then automatically counted (mean, 6.3 ± 3.2 nuclei/spot).
QC and data postprocessing
Quality control
The assembled atlas consists of 34,103 spots and 23,371 unique genes shared across the spots. The average number of detected genes per spot was 4422, and the average number of reads per spot was 11,210. We tested whether the overall quality of the sections correlated with the sequencing depth and found that the correlation was poor. The number of detected genes per spot is, however, highly correlated with the number of reads per spot.
Discarded genes
We manually discarded six genes detected after visual observation of their spatial expression pattern in 2D and 3D (Ttr, Pmch, Lars2, Prkcd, Enpp2, Nrgn). These genes showed discontinuous pattern of spatial expression that were judged to be replicate dependent and represent a technical artifact. We used a simple heuristic to detect the genes based on computing the sum of absolute difference of mean expression between consecutive sections.
Normalization and batch correction
To account for the variability given by the different batches (mice) and other sources of technical effects, we decided to compute size factors for normalization on each batch using the package Scran (47). Before that, we discarded spots with less than 1000 genes detected (reads > 0), genes with less than 100 spots detected (reads > 0), and the 6 genes mentioned previously. This resulted in a total of 34,053 spots and 15,326 unique genes with an average of 6877 genes per spot. Size factors were computed with the computeSumFactors method providing clusters computed with quickClust using default settings. The pools used for the computation of the size factors ranged from 50 to 100 in intervals of 10. We then used the Scran mmCorrect method to apply a batch correction on the normalized matrices (one per mice) in log space. The batch-corrected matrices were then merged together as a single matrix of counts in log space.
Dimensionality reduction and clustering
Features selection
We used the Seurat (48) package (version 2) built-in function to detect top variable genes. We selected the top 50% genes (7663 genes) with highest dispersion based on variance to mean ratio.
Dimensionality reduction
We performed dimensionality reduction on the normalized batch-corrected expression of the 7663 selected genes. We applied independent component analysis using joint approximate diagonalization of eigen-matrices [ICA JADE (20, 49); R package ica]. We computed the first 80 vectors and classified them as either biological or technical in a similar way to previous studies (50). This classification was performed manually by visualizing the IC scores in 2D, in 3D, and in a 2D t-SNE space computed from the 80 ICs.
Components were classified according to four categories:
1)Artifact. IC showed score that was judged as replicate dependent. No spatial organization could be observed coronally. All spots in some sections had very high scores while having very low scores in other adjacent sections.
2)Outlier. IC was scored high in absolute value in a very small number of spots.
3)Not spatially coherent. IC showed score that was distributed across the brain without spatial continuity.
4)Biological components. IC showed score that was spatially compact and reflecting 3D organization.
Forty-five components were classified as biological (fig. S3), and 35 were considered to be technical (artifact, outlier, or not spatially coherent).
Clustering
We applied the clustering method of the Seurat package on the 45 biological ICs (51). This algorithm first constructs a shared nearest neighbor graph and then computes clusters through modularity optimization. With such an approach, the number of clusters is indirectly defined by the resolution parameter. We tested a broad range of values for this parameter ranging from 0.05 to 50, which grouped spots into 6 to 530 distinct clusters. Several indicators were used to select the final resolution parameter including visual observation of clusters in 2D and 3D, computation of a similarity index (52), and analysis of very small clusters apparition (n < 10 spots) that were considered to be stochastic.
The final clustering was performed using a resolution of 29, while other parameters were set to default value (k = 30 for the nearest neighbor algorithm). Of the 200 generated clusters, 19 of them contained less than 10 spots and were therefore automatically discarded. We confirmed by visualization that the discarded cluster lacked spatial information.
Grouping clusters hierarchically
Hierarchical clustering of the clusters was performed to group them based on their molecular similarity. We started by computing for every cluster the average expression on each of the 45 ICs. The dissimilarity matrix was then generated using pairwise Euclidean distance between clusters in this 45D space. Hierarchical clustering was performed using the hclust function from the R stats package with Ward’s method. Dendrogram and polar phylogram (fan plots) were generated on the basis of functions from the ape package.
Naming clusters
Clusters were named on the basis of the anatomical region (ABA definition) according to main position identity of spots. We used a coarse definition that subdivides the brain into 14 major areas: cerebellum, cortical subplate, fiber tracts, hindbrain, hippocampal region, hypothalamus, isocortex, midbrain, olfactory areas, pallidum, retrohippocampal region, striatum, thalamus, and ventricular systems. Clusters were named after their major region if this area contained more than 40% of the spots. Otherwise, clusters were labeled as Mixed, with the main region specified in parenthesis. An ID was appended to the major region so that each cluster has a unique name. This ID was obtained by sorting clusters by number of spots in decreasing order.
Data visualization
t-SNE
For visualization purposes, spots were projected on the basis of their 45 IC scores onto 2D and 3D spaces using the t-SNE algorithm (22) with a perplexity of 20. No principal components analysis (PCA) initialization step was performed.
UMAP
For visualization purposes, spots were projected on the basis of their 45 IC scores onto 2D and 3D spaces using the UMAP algorithm (53). We used the correlation distance metric with minimal distance set at 0.3 and number of neighbors set at 10.
SVM smoothing
We transformed the molecular clusters defined at the spot level into volumes using an SVM algorithm. We trained a machine learning model to classify a triplet of coordinates ML, DV, and AP into 1 of the 181 clusters. The model was built using the R package e1071 and trained with all spots. We used a radial basis kernel with cost 20 and gamma 0.75 (other parameters were set to default value, stereotaxic coordinates in millimeters). We then used this model to predict the cluster label for every point of a 3D grid inside the brain outline. For visualization of coronal sections, a resolution of 10 μm was used along the ML-DV axis, while AP was a fixed value. For 3D visualizations, the resolution was 20 μm along the ML-DV axis and 42 μm along the AP axis.
3D visualization
For 3D visualization, we converted the voxels (3D pixels from the grid generated during SVM smoothing) into triangular meshes using the R package Rvcg. The 3D grid was first converted into a 3D mesh using the marching cubes algorithm. Then, Laplacian smoothing was applied on the mesh for five iterations. Views of noncoronal cuts were obtained by computing the intersection of smoothed mesh volumes with the desired planes with a custom R script.
Cluster colors
The categorical color scheme was designed to maximize visual contrast and is based on 27 distinct colors. Colors are selected from the Vivid color palette from CARTOColors, with additional colors from ColorBrewer palettes. Colors were automatically selected or manually assigned for some clusters to facilitate visualization.
The molecular similarity color scheme was designed to visualize clusters with similar gene expression profile. The color scheme was automatically generated using a 3D t-SNE projection or a 3D UMAP projection. Each of the three coordinates in the t-SNE space was linearly rescaled between 0 and 1 across all spots. Median coordinates on the three axes were computed per cluster and remapped into the RGB space, thereby defining a unique color for each molecular cluster.
Cluster boundaries
Cluster boundaries were visualized using the ISH signal for single genes provided by the Allen Institute. We selected coronal sections for genes with spatial patterns and registered one hemisphere in the Allen Brain Atlas using the WholeBrain package. This process transforms the image to fit the ABA. We then created a virtual section by symmetrizing the hemisphere, so we could overlay, on one side, the ABA and, on the other side, the molecular clusters.
Allen reference atlas
We used two different versions of the Allen reference atlas. Coronal views display the outlines from the WholeBrain package (19) based on the Allen Institute Common Coordinate Framework (CCF) 2015. 3D plots are based on the CCF 2017.
Video rendering
All videos were rendered using the R package Rvcg. Frames were exported to .png and converted into mp4 videos using the FFmpeg framework.
Interactive plots
Online interactive plots are rendered using the plotly.js library.
Single-cell spatial mapping
Single-cell ground truth
To assess the accuracy of our machine-learning classifier, we used a published single-cell sequencing data, where both the layer of origin and the region (ALM or VISp) were specified for every cell in the dataset (24).
Figure 4 shows prediction on spatial mapping of single cells onto regions. We manually selected clusters to represent the ground truth corresponding to ALM (Isocortex-04, Isocortex-05, Isocortex-08, Isocortex-10, Isocortex-14, Isocortex-09, and Isocortex-33) and to VISp (Isocortex-01, Isocortex-17, Isocortex-19, Isocortex-20, Isocortex-26, Isocortex-28, Isocortex-39, and Isocortex-49). A cell was considered to be correctly mapped if the predicted ST cluster was part of selected clusters for the annotated dissected region.
We further defined a ground truth for layer-specific mapping. We defined the layer identity of the 15 ground truth molecular clusters based on the overlap of at least 30% of the spots to a corresponding ABA layer. We defined spatial origin of single cells based on their cluster identity (region and layer name) in the scRNA-seq data.
Data normalization
We used the ST dataset that had previously been filtered, normalized, and batch-corrected for the unsupervised clustering. We then applied a similar filter to the Single Cell (SC) dataset (24) and filtered both the ST and SC datasets to only use genes that were present in both datasets. This resulted in 7293 shared genes, 34,019 spots in the ST datasets, and 21,852 cells in the SC dataset. We then applied a batch correction on the combined ST and SC dataset using the mnnCorrect method of the Scran package.
ST and SC correlation
To determine the correlation between the gene expression levels of the scRNA-Seq dataset and the gene expression levels in the corresponding molecular clusters, we selected the spots belonging to the 15 different ground truth molecular clusters and averaged the expression across all cells/spots. We performed a gene by gene correlation analysis.
Single-cell spatial mapping method
To map single cells (24) onto the molecular clusters, we performed a supervised learning approach, wherein we trained a classifier with the atlas molecular clusters as labels and then used the classifier to predict a molecular cluster for every single cell. We used as input for the classifier the ST dataset that was normalized as previously described. Our first approach consisted in a multi-label SVMs following the one-versus-rest principle. We used the Support Vector Classification (SVC) class of the Scikit-learn (sklearn) (54) package and trained the model using a tolerance of 0.0001 and 10,000 iterations. The trained model was then used to predict the probabilities of each cluster for each single cell.
Our second approach consisted of a two-layer neural network, where we performed a basic grid search to optimize and select the best hyperparameters. During the grid search, 60% of the ST dataset was used for training, 20% for validation, and 20% for testing. We selected the hyperparameters that gave the best accuracy in the testing set.
The selected hyperparameters were as follows:
1)Hidden layer one 2000 (neurons)
2)Hidden layer two 1000 (neurons)
3)ReLU activation function
4)Cross-entropy loss with stratified balanced weights per cluster
5)Batch normalization to each hidden layer
6)ADAM optimizer with 0.0001 L2 regularization and 0.001 learning rate
7)Batch gradient descent with 500 spots/cells batch size
We used PyTorch to implement the neutral network to take advantage of the fast graphics processing unit (GPU) computation and trained the network using 1000 iterations. The trained network was used to predict for each single cell the probabilities of belonging to each of the 181 molecular clusters. The neural network slightly outperformed the SVM classifier and is therefore the approach presented in Results. Both methods are available as Python tools (https://github.com/jfnavarro/st_analysis).
Visualization of the single-cell spatial mapping
We made a graphical representation to illustrate how the single-cell data were mapped into the molecular clusters. We took the smoothed outlines from ground truth clusters at Bregma = 1.42 mm for ALM and Bregma = −3.58 mm for VISp. We manually adjusted contours of these clusters to best capture the spatial organization along the AP axis with a single coronal cut. We did not manage to display all the eight VISp clusters due to color conflicts; therefore, Isocortex-49 was not shown on the schematic illustration (17/21,852 cells predicted). The prediction for a cell was represented by a black dot positioned randomly inside the outline of the predicted ST molecular cluster.
Reduced brain palette
Gene selection
We applied two methods to select genes with maximum contribution to the molecular profile of clusters. We first used a multi-label SVM classifier following a one-versus-rest approach. We trained the model using the filtered, normalized, and batch-corrected dataset. We used again the sklearn class SVC with a tolerance of 0.0001 and 10,000 iterations. The trained model provides the weights distributions of the genes for each cluster, which we then filtered and sorted by absolute value. Top k genes were taken for each cluster and merged together for several k values. The second approach consisted in selecting the top k genes loads in absolute values from the 45 biological ICs.
Establishing a brain palette
We ran the ICA dimensionality reduction with technical-biological classification of components followed by clustering for multiple reduced sets of genes. Because of the need to reiterate the analysis several times, we made a fast version of the pipeline: We used the icafast algorithm from the R package ica and automatized the classification of ICs for each gene palette. We computed the first 80 ICs, performed a cross-correlation at zero lag with the 45 original components, selected the correlation from the best match in absolute value, and kept components above a cutoff set at 0.4 correlation. We then performed clustering with these new components as input and adjusted the resolution parameter, so 181 clusters are retained after discarding clusters with less than 10 spots.
To evaluate the similarity between the original and the new clusters, we calculated the NMI using the NMI R package for all the reduced palettes of genes (Fig. 5A). On the basis of this indicator, we decided to focus on the first set of genes to exceed a 0.6 NMI value (266 genes selected from IC loads) as we believed it to be a good trade-off between the number of genes and the ability to retrieve initial clusters. After deciding to work with this specific set of genes, we ran the full pipeline previously described: dimensionality reduction with ICA JADE, manual classification of components as either technical or biological, clustering with Seurat, and visualization of the resulting clusters with 2D coronal views, 3D plots, 2D t-SNE, and fan plot.
Enrichment analysis
We performed an enrichment analysis using the brain palette (266 genes) with the enrichR R package. We queried the GO Biological terms and the GO Cellular terms databases (2018) (25).
Supplementary Material
Acknowledgments
We thank J. Frisén, D. Fürth, A. Mollbrink, E. Wärnberg, P. Le Merre, H. Brünner, K. Ampatzis, and M. Carlén for providing advice and technical help. Funding: This study was supported by the Swedish Research Council (VR 2017-01457 to K.M.), the Swedish Brain Foundation (Hjärnfonden grant to K.M.), the Swedish Foundation for Strategic Research (SSF grant FFL12-0006 to K.M.), the Knut and Alice Wallenberg Foundation (KAW 2015.0296 Spatial Brain grant to J.L.), the Swedish Foundation for Strategic Research (SSF grant SB16-0014, K.M. and J.L.), the Olav Thon Foundation (doctoral and postdoctoral funding for J.F.N. and A.J., respectively), EU JPND INSTALZ (doctoral and postdoctoral funding for J.F.N. and A.J., respectively), and Karolinska Institutet (senior research fellow grant to K.M., doctoral funding KID grant for A.M.). Author contributions: C.O. and J.F.N. analyzed and visualized data. A.J. and A.M. performed experiments and collected data. J.L. conceived and supervised the project. K.M. conceived and supervised the project and wrote the manuscript with input from all authors. Competing interests: J.L. is scientific consultant for 10X Genomics Inc. holding the IP for the barcoded slides. The remaining authors declare that they have no competing interests. Data and materials availability: Raw sequencing data are available using GEO accession number GSE147747. The 3D ST Viewer can be used for exploration of the data (https://github.com/jfnavarro/st_viewer). Data and visualization of the atlas (e.g., vectorized molecular atlas plates, interactive version) are available at http://molecularatlas.org. The code that supports analyses and findings of this study is available at https://github.com/cantin-ortiz/molecular-atlas.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/26/eabb3446/DC1
REFERENCES AND NOTES
- 1.Crick F., Jones E., Backwardness of human neuroanatomy. Nature 361, 109–110 (1993). [DOI] [PubMed] [Google Scholar]
- 2.Denk W., Briggman K. L., Helmstaedter M., Structural neurobiology: Missing link to a mechanistic understanding of neural computation. Nat. Rev. Neurosci. 13, 351–358 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Swanson L. W., Lichtman J. W., From Cajal to Connectome and beyond. Annu. Rev. Neurosci. 39, 197–216 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Zeng H., Mesoscale connectomics. Curr. Opin. Neurobiol. 50, 154–162 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.L. W. Swanson, A history of neuroanatomical mapping, in Brain Mapping: The Systems, A. W. Toga, J. C. Mazziotta, Eds. (Academic Press, 2000), pp. 77–109. [Google Scholar]
- 6.Zilles K., Amunts K., Centenary of Brodmann’s map—Conception and fate. Nat. Rev. Neurosci. 11, 139–145 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Oh S. W., Harris J. A., Ng L., Winslow B., Cain N., Mihalas S., Wang Q., Lau C., Kuan L., Henry A. M., Mortrud M. T., Ouellette B., Nguyen T. N., Sorensen S. A., Slaughterbeck C. R., Wakeman W., Li Y., Feng D., Ho A., Nicholas E., Hirokawa K. E., Bohn P., Joines K. M., Peng H., Hawrylycz M. J., Phillips J. W., Hohmann J. G., Wohnoutka P., Gerfen C. R., Koch C., Bernard A., Dang C., Jones A. R., Zeng H., A mesoscale connectome of the mouse brain. Nature 508, 207–214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmidt H., Gour A., Straehle J., Boergens K. M., Brecht M., Helmstaedter M., Axonal synapse sorting in medial entorhinal cortex. Nature 549, 469–475 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Hawrylycz M. J., Lein E. S., Guillozet-Bongaarts A. L., Shen E. H., Ng L., Miller J. A., van de Lagemaat L. N., Smith K. A., Ebbert A., Riley Z. L., Abajian C., Beckmann C. F., Bernard A., Bertagnolli D., Boe A. F., Cartagena P. M., Chakravarty M. M., Chapin M., Chong J., Dalley R. A., Daly B. D., Dang C., Datta S., Dee N., Dolbeare T. A., Faber V., Feng D., Fowler D. R., Goldy J., Gregor B. W., Haradon Z., Haynor D. R., Hohmann J. G., Horvath S., Howard R. E., Jeromin A., Jochim J. M., Kinnunen M., Lau C., Lazarz E. T., Lee C., Lemon T. A., Li L., Li Y., Morris J. A., Overly C. C., Parker P. D., Parry S. E., Reding M., Royall J. J., Schulkin J., Sequeira P. A., Slaughterbeck C. R., Smith S. C., Sodt A. J., Sunkin S. M., Swanson B. E., Vawter M. P., Williams D., Wohnoutka P., Zielke H. R., Geschwind D. H., Hof P. R., Smith S. M., Koch C., Grant S. G. N., Jones A. R., An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gandal M. J., Zhang P., Hadjimichael E., Walker R. L., Chen C., Liu S., Won H., van Bakel H., Varghese M., Wang Y., Shieh A. W., Haney J., Parhami S., Belmont J., Kim M., Losada P. M., Khan Z., Mleczko J., Xia Y., Dai R., Wang D., Yang Y. T., Xu M., Fish K., Hof P. R., Warrell J., Fitzgerald D., White K., Jaffe A. E.; Psych ENCODE Consortium, Peters M. A., Gerstein M., Liu C., Iakoucheva L. M., Pinto D., Geschwind D. H., Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.G. Paxinos, K. B. J. Franklin, The Mouse Brain in Stereotaxic Coordinates (Gulf Professional Publishing, 2004). [Google Scholar]
- 12.Lein E. S., Hawrylycz M. J., Ao N., Ayres M., Bensinger A., Bernard A., Boe A. F., Boguski M. S., Brockway K. S., Byrnes E. J., Chen L., Chen L., Chen T.-M., Chin M. C., Chong J., Crook B. E., Czaplinska A., Dang C. N., Datta S., Dee N. R., Desaki A. L., Desta T., Diep E., Dolbeare T. A., Donelan M. J., Dong H.-W., Dougherty J. G., Duncan B. J., Ebbert A. J., Eichele G., Estin L. K., Faber C., Facer B. A., Fields R., Fischer S. R., Fliss T. P., Frensley C., Gates S. N., Glattfelder K. J., Halverson K. R., Hart M. R., Hohmann J. G., Howell M. P., Jeung D. P., Johnson R. A., Karr P. T., Kawal R., Kidney J. M., Knapik R. H., Kuan C. L., Lake J. H., Laramee A. R., Larsen K. D., Lau C., Lemon T. A., Liang A. J., Liu Y., Luong L. T., Michaels J., Morgan J. J., Morgan R. J., Mortrud M. T., Mosqueda N. F., Ng L. L., Ng R., Orta G. J., Overly C. C., Pak T. H., Parry S. E., Pathak S. D., Pearson O. C., Puchalski R. B., Riley Z. L., Rockett H. R., Rowland S. A., Royall J. J., Ruiz M. J., Sarno N. R., Schaffnit K., Shapovalova N. V., Sivisay T., Slaughterbeck C. R., Smith S. C., Smith K. A., Smith B. I., Sodt A. J., Stewart N. N., Stumpf K.-R., Sunkin S. M., Sutram M., Tam A., Teemer C. D., Thaller C., Thompson C. L., Varnam L. R., Visel A., Whitlock R. M., Wohnoutka P. E., Wolkey C. K., Wong V. Y., Wood M., Yaylaoglu M. B., Young R. C., Youngstrom B. L., Yuan X. F., Zhang B., Zwingman T. A., Jones A. R., Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Bota M., Dong H.-W., Swanson L. W., From gene networks to brain networks. Nat. Neurosci. 6, 795–799 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Kim C. K., Adhikari A., Deisseroth K., Integration of optogenetics with complementary methodologies in systems neuroscience. Nat. Rev. Neurosci. 18, 222–235 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo L., Callaway E. M., Svoboda K., Genetic dissection of neural circuits: A decade of progress. Neuron 98, 256–281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belgard T. G., Marques A. C., Oliver P. L., Abaan H. O., Sirey T. M., Hoerder-Suabedissen A., García-Moreno F., Molnár Z., Margulies E. H., Ponting C. P., A transcriptomic atlas of mouse neocortical layers. Neuron 71, 605–616 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lein E., Borm L. E., Linnarsson S., The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science 358, 64–69 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Ståhl P. L., Salmén F., Vickovic S., Lundmark A., Navarro J. F., Magnusson J., Giacomello S., Asp M., Westholm J. O., Huss M., Mollbrink A., Linnarsson S., Codeluppi S., Borg Å., Pontén F., Costea P. I., Sahlén P., Mulder J., Bergmann O., Lundeberg J., Frisén J., Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Fürth D., Vaissière T., Tzortzi O., Xuan Y., Märtin A., Lazaridis I., Spigolon G., Fisone G., Tomer R., Deisseroth K., Carlén M., Miller C. A., Rumbaugh G., Meletis K., An interactive framework for whole-brain maps at cellular resolution. Nat. Neurosci. 21, 139–149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cardoso J. F., Souloumiac A., Blind beamforming for non-Gaussian signals. IEE Proc. F 140, 362–370 (1993). [Google Scholar]
- 21.Satija R., Farrell J. A., Gennert D., Schier A. F., Regev A., Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Maaten L., Hinton G., Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008). [Google Scholar]
- 23.Voorn P., Vanderschuren L. J. M. J., Groenewegen H. J., Robbins T. W., Pennartz C. M. A., Putting a spin on the dorsal–ventral divide of the striatum. Trends Neurosci. 27, 468–474 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Tasic B., Yao Z., Graybuck L. T., Smith K. A., Nguyen T. N., Bertagnolli D., Goldy J., Garren E., Economo M. N., Viswanathan S., Penn O., Bakken T., Menon V., Miller J., Fong O., Hirokawa K. E., Lathia K., Rimorin C., Tieu M., Larsen R., Casper T., Barkan E., Kroll M., Parry S., Shapovalova N. V., Hirschstein D., Pendergraft J., Sullivan H. A., Kim T. K., Szafer A., Dee N., Groblewski P., Wickersham I., Cetin A., Harris J. A., Levi B. P., Sunkin S. M., Madisen L., Daigle T. L., Looger L., Bernard A., Phillips J., Lein E., Hawrylycz M., Svoboda K., Jones A. R., Koch C., Zeng H., Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., Sherlock G., Gene Ontology: Tool for the unification of biology. Nat. Genet. 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Narayanan R. T., Udvary D., Oberlaender M., Narayanan R. T., Udvary D., Oberlaender M., Cell type-specific structural organization of the six layers in rat barrel cortex. Front. Neuroanat. 11, 91 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carlén M., What constitutes the prefrontal cortex? Science 358, 478–482 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Harris J. A., Mihalas S., Hirokawa K. E., Whitesell J. D., Choi H., Bernard A., Bohn P., Caldejon S., Casal L., Cho A., Feiner A., Feng D., Gaudreault N., Gerfen C. R., Graddis N., Groblewski P. A., Henry A. M., Ho A., Howard R., Knox J. E., Kuan L., Kuang X., Lecoq J., Lesnar P., Li Y., Luviano J., Conoughey S. M., Mortrud M. T., Naeemi M., Ng L., Oh S. W., Ouellette B., Shen E., Sorensen S. A., Wakeman W., Wang Q., Wang Y., Williford A., Phillips J. W., Jones A. R., Koch C., Zeng H., Hierarchical organization of cortical and thalamic connectivity. Nature 575, 195–202 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sylwestrak E. L., Rajasethupathy P., Wright M. A., Jaffe A., Deisseroth K., Multiplexed intact-tissue transcriptional analysis at cellular resolution. Cell 164, 792–804 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen F., Wassie A. T., Cote A. J., Sinha A., Alon S., Asano S., Daugharthy E. R., Chang J.-B., Marblestone A., Church G. M., Raj A., Boyden E. S., Nanoscale imaging of RNA with expansion microscopy. Nat. Methods 13, 679–684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ke R., Mignardi M., Pacureanu A., Svedlund J., Botling J., Wählby C., Nilsson M., In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 10, 857–860 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Lee J. H., Daugharthy E. R., Scheiman J., Kalhor R., Yang J. L., Ferrante T. C., Terry R., Jeanty S. S. F., Li C., Amamoto R., Peters D. T., Turczyk B. M., Marblestone A. H., Inverso S. A., Bernard A., Mali P., Rios X., Aach J., Church G. M., Highly multiplexed subcellular RNA sequencing in situ. Science 343, 1360–1363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lubeck E., Coskun A. F., Zhiyentayev T., Ahmad M., Cai L., Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen K. H., Boettiger A. N., Moffitt J. R., Wang S., Zhuang X., Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eng C.-H. L., Lawson M., Zhu Q., Dries R., Koulena N., Takei Y., Yun J., Cronin C., Karp C., Yuan G.-C., Cai L., Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shah S., Lubeck E., Zhou W., Cai L., In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron 92, 342–357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phillips J. W., Schulmann A., Hara E., Winnubst J., Liu C., Valakh V., Wang L., Shields B. C., Korff W., Chandrashekar J., Lemire A. L., Mensh B., Dudman J. T., Nelson S. B., Hantman A. W., A repeated molecular architecture across thalamic pathways. Nat. Neurosci. 22, 1925–1935 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vickovic S., Eraslan G., Salmén F., Klughammer J., Stenbeck L., Schapiro D., Äijö T., Bonneau R., Bergenstråhle L., Navarro J. F., Gould J., Griffin G. K., Borg Å., Ronaghi M., Frisén J., Lundeberg J., Regev A., Ståhl P. L., High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 16, 987–990 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodriques S. G., Stickels R. R., Goeva A., Martin C. A., Murray E., Vanderburg C. R., Welch J., Chen L. M., Chen F., Macosko E. Z., Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zapala M. A., Hovatta I., Ellison J. A., Wodicka L., Del Rio J. A., Tennant R., Tynan W., Broide R. S., Helton R., Stoveken B. S., Winrow C., Lockhart D. J., Reilly J. F., Young W. G., Bloom F. E., Lockhart D. J., Barlow C., Adult mouse brain gene expression patterns bear an embryologic imprint. Proc. Natl. Acad. Sci. U.S.A. 102, 10357–10362 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nowakowski T. J., Bhaduri A., Pollen A. A., Alvarado B., Mostajo-Radji M. A., Lullo E. D., Haeussler M., Sandoval-Espinosa C., Liu S. J., Velmeshev D., Ounadjela J. R., Shuga J., Wang X., Lim D. A., West J. A., Leyrat A. A., Kent W. J., Kriegstein A. R., Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hodge R. D., Bakken T. E., Miller J. A., Smith K. A., Barkan E. R., Graybuck L. T., Close J. L., Long B., Johansen N., Penn O., Yao Z., Eggermont J., Höllt T., Levi B. P., Shehata S. I., Aevermann B., Beller A., Bertagnolli D., Brouner K., Casper T., Cobbs C., Dalley R., Dee N., Ding S.-L., Ellenbogen R. G., Fong O., Garren E., Goldy J., Gwinn R. P., Hirschstein D., Keene C. D., Keshk M., Ko A. L., Lathia K., Mahfouz A., Maltzer Z., Graw M. M., Nguyen T. N., Nyhus J., Ojemann J. G., Oldre A., Parry S., Reynolds S., Rimorin C., Shapovalova N. V., Somasundaram S., Szafer A., Thomsen E. R., Tieu M., Quon G., Scheuermann R. H., Yuste R., Sunkin S. M., Lelieveldt B., Feng D., Ng L., Bernard A., Hawrylycz M., Phillips J. W., Tasic B., Zeng H., Jones A. R., Koch C., Lein E. S., Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jemt A., Salmén F., Lundmark A., Mollbrink A., Navarro J. F., Ståhl P. L., Yucel-Lindberg T., Lundeberg J., An automated approach to prepare tissue-derived spatially barcoded RNA-sequencing libraries. Sci. Rep. 6, 37137 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lundin S., Stranneheim H., Pettersson E., Klevebring D., Lundeberg J., Increased throughput by parallelization of library preparation for massive sequencing. PLOS ONE 5, e10029 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schindelin J., Rueden C. T., Hiner M. C., Eliceiri K. W., The ImageJ ecosystem: An open platform for biomedical image analysis. Mol. Reprod. Dev. 82, 518–529 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Navarro J. F., Sjöstrand J., Salmén F., Lundeberg J., Ståhl P. L., ST Pipeline: An automated pipeline for spatial mapping of unique transcripts. Bioinformatics 33, 2591–2593 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Lun A. T. L., McCarthy D. J., Marioni J. C., A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res. 5, 2122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R., Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cardoso J.-F., Souloumiac A., Jacobi angles for simultaneous diagonalization. SIAM J. Matrix Anal. Appl. 17, 161–164 (1996). [Google Scholar]
- 50.Saunders A., Macosko E. Z., Wysoker A., Goldman M., Krienen F. M., de Rivera H., Bien E., Baum M., Bortolin L., Wang S., Goeva A., Nemesh J., Kamitaki N., Brumbaugh S., Kulp D., McCarroll S. A., Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waltman L., van Eck N. J., A smart local moving algorithm for large-scale modularity-based community detection. Eur. Phys. J. B 86, 471 (2013). [Google Scholar]
- 52.Bohland J. W., Bokil H., Pathak S. D., Lee C.-K., Ng L., Lau C., Kuan C., Hawrylycz M., Mitra P. P., Clustering of spatial gene expression patterns in the mouse brain and comparison with classical neuroanatomy. Methods 50, 105–112 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Becht E., Innes L. M., Healy J., Dutertre C.-A., Kwok I. W. H., Ng L. G., Ginhoux F., Newell E. W., Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–44 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., Vanderplas J., Passos A., Cournapeau D., Brucher M., Perrot M., Duchesnay É., Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/26/eabb3446/DC1