

Abstract
Many genetic disorders are detectable in the prenatal period, and the capacity to identify them has increased remarkably as molecular genetic testing techniques continue to improve and become incorporated into clinical practice. The indications for prenatal genetic testing vary widely, including follow-up of an anomaly found by routine ultrasound or maternal aneuploidy screening, a family history of genetic disease, advanced maternal or paternal age, or evaluation of a low-risk pregnancy due to parental concern. The interpretation of genetic variants identified in the prenatal period poses unique challenges due to the lack of ability for deep phenotyping as well as the option to make critical decisions regarding pregnancy continuation and perinatal management. In this review, we address the various modalities currently available and commonly used for genetic testing, including pre-implantation genetic testing of embryos, cell-free DNA testing, and diagnostic procedures such as chorionic villous sampling, amniocentesis, or percutaneous umbilical blood sampling, from which samples may be sent for a wide variety of genetic tests. We discuss the difference between these modalities for the genetic diagnosis of a fetus, their strengths and weaknesses, and strategies for their optimal use in order to direct perinatal care.
Introduction
Recent advances in genetic and genomic technologies have allowed for the identification of many new disease-causing genes and for the molecular diagnosis of thousands of individuals affected by rare genetic disorders 1, 2, 3. Exome sequencing (ES) and genome sequencing (GS), in particular, have truly broadened the horizons for the diagnosis not only of children and young adults suspected to have genetic conditions 4, but also for neonates and infants 5, 6, 7, in whom recognition of a genetic syndrome by clinical presentation alone may be challenging. Even more challenging is the recognition of a genetic syndrome in the fetus, as physical assessment is limited to imaging techniques such as ultrasound or, in some centers, fetal MRI. Until recently, prenatal screening for genetic disorders was limited to ultrasound findings with the addition of serum markers such as alpha fetoprotein (AFP) and human chorionic gonadotropin (hCG). The rise of cell-free DNA (cfDNA) analysis from a maternal blood sample has resulted in an increased ability to detect common aneuploidy syndromes such as Down syndrome, Edward syndrome, and Patau syndrome in which an extra copy of chromosome 21, 18, or 13 is detected, respectively. The use of cfDNA continues to evolve, with testing for microdeletion syndromes such as 22q11 deletion syndrome now routinely offered, and limited testing available for single gene (monogenic) disorders 8.
Options for diagnostic testing, in which fetal samples are collected for analysis, have also expanded beyond the karyotype, where chromosomes are directly visualized by light microscopy to evaluate for aneuploidy (abnormalities in chromosome number) or large chromosomal losses, gains, or rearrangements. Chromosomal microarray analysis (CMA), by which submicroscopic copy number changes can be detected, is now recommended as the first-tier test in the evaluation of a fetus with one or more congenital anomalies 9, 10, and fetal ES/GS is also likely to become more commonplace. The evaluation after stillbirth has also evolved; though the underlying etiology for stillbirth may or may not be genetic 11, CMA 12 and now ES/GS 13 are commonly employed with a high diagnostic yield.
Identification of a genetic disorder in the fetus can lead to life-saving interventions initiated either prenatally or early in the post-natal period. Equally important is identifying which fetuses are not at risk for a familial disorder to potentially spare an unaffected newborn any invasive and costly unnecessary treatments (as in the case of a male neonate born to a family with a history of ornithine transcarbamylase deficiency, a urea cycle disorder which may necessitate empiric protein restriction and a central line for parenteral nutrition to prevent a metabolic crisis in the newborn period until the diagnosis is confirmed or refuted). In this review, we discuss various strategies available for fetal genetic diagnosis, beginning with pre-conception testing and prenatal screening for genetic disorders, followed by diagnostic testing options (Figure 1).
Figure 1.
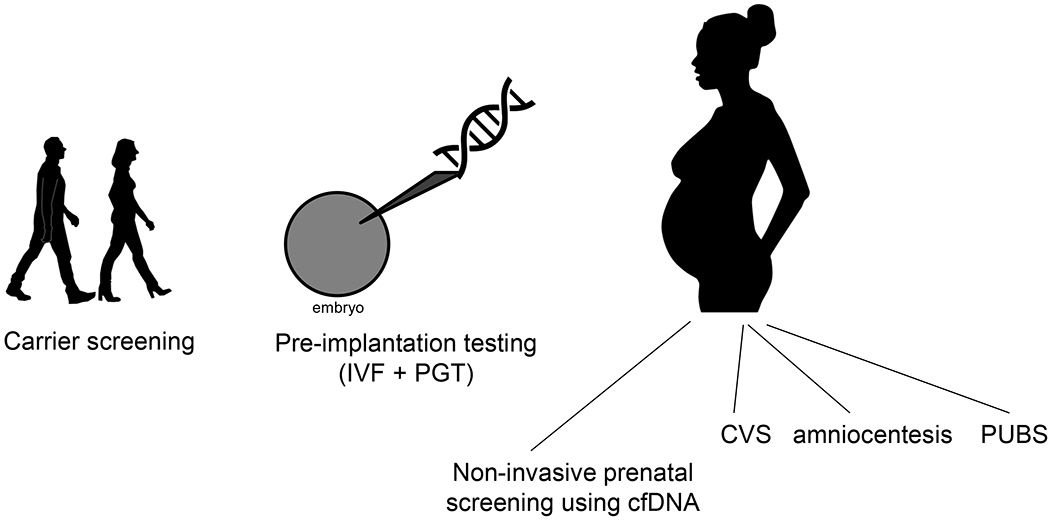
Opportunities for prenatal genetic evaluation. IVF, in vitro fertilization; PGT, pre-implantation genetic testing; CVS, chorionic villous sampling; PUBS, percutaneous umbilical blood sampling; cfDNA, cell-free DNA
Pre-conception Testing and Prenatal Screening
Genetic information relevant to fetal diagnosis may be generated prior to conception, and routine prenatal screening of the parents or fetus may offer important clues that direct the fetal diagnostic genetic evaluation. These screening tests are commonly mistaken for diagnostic tests for the fetus and therefore an understanding of their use and limitations is important for those pursuing a prenatal genetic evaluation.
Pre-conception or pre-implantation genetic testing
The genetic diagnostic evaluation may begin even before the pregnancy. The American College of Obstetricians and Gynecologists (ACOG) recommends universal carrier screening for spinal muscular atrophy, cystic fibrosis, and hemoglobinopathies 14. Other conditions are known to have high carrier frequencies within particular populations, and screening for these conditions is recommended amongst specific ethnic groups, such as screening for Tay-Sachs disease in the Ashkenazi Jewish, Cajun, and French Canadian populations 14. However, “expanded carrier screening” panels are commercially available that evaluate for the presence of pathogenic variants in hundreds of severe, early-onset disorders in which an unaffected parent may harbor a pathogenic variant 15. A recent study utilizing a database containing over 100,000 individuals has identified carrier rates for 415 severe, recessive genetic disorders ranging from 32.6% to 62.9% depending on ethnic group, and calculated that a screening panel using these 415 genes would identify 0.17-2.52% of reproductive partners as being at risk for having an affected pregnancy and that 4-63 per 10,000 fetuses would be affected 16. While these tests are ideally obtained prior to conception 17, many couples at risk for a genetic disorder in the fetus (e.g. due to ancestry or family history) undergo carrier screening after a pregnancy has occurred. One disadvantage of carrier screening post-conception is that the option for the use of assisted reproductive technologies (ART) to select for an unaffected fetus is lost. In pregnancy, if both parents are found to be carriers of an autosomal recessive condition or a mother is found to be a carrier of an X-linked recessive condition and carries a male fetus, targeted diagnostic genetic testing may proceed as described in a subsequent section of this review. In practice, at-risk couples may decline diagnostic testing or only one parent may be tested for carrier status and found to have a pathogenic variant. Complete genetic risk assessment is therefore important for providers evaluating the neonate to determine the optimal strategy for diagnosis and treatment.
As previously mentioned, if a couple is found prior to conception to be at high risk to have a fetus affected by a genetic disorder, as in the situation where both parents are carriers for pathogenic variants in the same disease gene (resulting in a 25% risk of disease in the fetus), ART may be useful. The genotype of the parents must first be determined, after which in vitro fertilization is performed. Cells from the embryo (trophoectoderm cells which will form the placenta) are harvested on day five of development and tested, a process called pre-implantation genetic testing for monogenic disorders (PGT-M). Importantly, unique markers around the gene of interest that carries the pathogenic variant are used to screen for the presence of the pathogenic variant itself (karyomapping) rather than sequencing of the gene. Embryos that either carry no copies of the affected allele or only one copy (indicative of carrier status) can then be implanted 18. Distinct from PGT-M is pre-implantation genetic testing for aneuploidy, or PGT-A. During the process of PGT-M, the embryonic genome can be screened for aneuploidy to improve the chances of implantation and live birth (embryos conceived via IVF with PGT have high rates of aneuploidy resulting in pregnancy loss) 19, 20. Historically, this was done via fluorescence in situ hybridization (FISH) with direct visualization of chromosome number, though current techniques such as array comparative genomic hybridization (CGH), single nucleotide polymorphism (SNP) arrays, or massively-parallel (“next-generation”) sequencing are now used to evaluate for imbalances in chromosome number 19. Importantly, neither PGT-M nor PGT-A can guarantee that the fetus does not have a genetic disorder. PGT may not be successful, particularly if “allele dropout” occurs during the embryo testing process, which relies on amplification of genetic material to detect affected alleles. If an allele from one parent that harbors a pathogenic variant fails to amplify, the embryo’s testing will result in a false negative result. Errors in the genetic analysis – from human error to problems with the diagnostic technology – may also lead to misdiagnosis of the embryo 21. Additionally, PGT typically screens only for complete chromosomal aneuploidy or large segment aneuploidy and will generally not evaluate for microdeletions or duplications or monogenic disorders. Furthermore, as trophoectoderm cells are sampled for PGT, which will develop into the placenta, it is possible that these cells are euploid while the inner cell mass of the embryo, which develops into the fetus, is aneuploid. This placental/fetal discordance can complicate any prenatal genetic testing. Thus, in the presence of imaging findings or a postnatal evaluation concerning for a genetic disorder, having had PGT should not preclude a thorough diagnostic genetic evaluation. This concept is crucial for parents and to avoid false reassurance by “negative” PGT.
Prenatal Screening
Traditional serum screening has been used for decades to identify pregnancies at risk for aneuploidy in addition to other conditions. This may be undertaken during any trimester of pregnancy, where comparison of maternal serum levels of various analytes to population mean values allows for identification of high risk pregnancies. Second trimester ultrasonography may also reveal features suggestive of aneuploidy, including “soft markers” such as choroid plexus cysts, an echogenic intracardiac focus, or echogenic bowel, in addition to major congenital anomalies that are more predictive of a possible genetic disorder. In the first trimester, levels of human chorionic gonadotropin (hCG) and pregnancy-associated plasma protein A (PAPP-A) plus measurement of the fetal nuchal translucency are used along with maternal factors to calculate a risk of aneuploidy. Quadruple or “quad” screening, performed in the second trimester, involves maternal serum measurements of hCG, AFP, inhibin A, and unconjugated estriol while the “triple” screen does not include inhibin A. Patterns of abnormalities seen in the concentrations of these analytes may indicate a higher risk of a particular aneuploidy syndrome – for example, pregnancies affected by Down syndrome have lower levels of AFP and unconjugated estriol and higher levels of hCG and inhibin-A while pregnancies with trisomy 18 have diminished values for all analytes. In addition to estimating a risk of aneuploidy, elevated levels of AFP may indicate the presence of an open neural tube defect and low levels of estriol can be suggestive of fetal Smith-Lemli-Opitz syndrome, a defect in cholesterol metabolism. Various methods of combined first and second trimester screens exist as well, and are summarized in a recent ACOG practice bulletin 22. Advantages of maternal serum screening include the low cost and ability to identify high risk pregnancies early in gestation. The main disadvantage is the relatively low sensitivity and specificity compared to analysis of cfDNA.
Previously referred to as non-invasive prenatal testing (NIPT) or screening (NIPS), cfDNA testing entered clinical practice in 2011. It is now widely used as a screening test for aneuploidy syndromes such as Down syndrome, Edward syndrome, and Patau syndrome, most commonly caused by trisomies of chromosomes 21, 18, and 13 due to nondisjunction during gametogenesis 23, although they may also occur by other mechanisms such as chromosomal translocation or via mitotic errors leading to mosaicism in the fetus. Fetal sex and sex chromosome abnormalities such as Turner syndrome (monosomy X) and Klinefelter syndrome (47,XXY) can also be detected. This test relies on the presence of segments of fetal DNA in the maternal bloodstream, which allows for an analysis of a maternal blood sample to determine relative proportions of certain chromosomes in the fetus. A variety of techniques may be used, commonly involving either massively-parallel sequencing with estimation of relative quantities of chromosomal material or SNP analysis to evaluate the relative distribution of maternal and fetal alleles 24. Importantly, cfDNA testing does not replace the need for early ultrasound and serum marker assessment, as these techniques can identify other high risk fetal conditions such as anencephaly and gastroschisis. In addition, the isolated genetic material for cfDNA is from placental cytotrophoblast cells 25, similar to the trophoectoderm sampled for PGT (Figure 2). Thus, it is possible for false negatives or positives to occur due to lack of concordance between the placenta and fetus, where placental cells are aneuploid with a euploid fetus, or vice versa. Regardless, cfDNA screening has been shown to have a higher specificity and positive predictive value for the detection of trisomies 18 and 21 in both high and low risk women 26. CfDNA is also thought to be more sensitive for the detection of trisomy 21, though the low number of affected fetuses in recent studies limits this conclusion. The false negative rate has been reported at 4-19% for trisomy 21 using standard screening 26 compared to a sensitivity of nearly 100% using cfDNA to detect trisomy 21; test performance is worse for trisomies 18 and 13 27.
Figure 2.

Overview of embryonic development and its relevance to prenatal genetic testing. Placental cell lines are demonstrated in italicized font. Testing modalities are encompassed within the text boxes. PGT, pre-implantation genetic testing; cfDNA, cell-free DNA; CVS, chorionic villous sampling. Adapted from [42].
Both false positive and false negative results on cfDNA screening, however, represent interesting clinical scenarios and have led to important biological insights. A “positive” result on cfDNA screening with normal fetal karyotype may represent maternal malignancy (particularly when multiple abnormalities are present) 28, 29 or could be the result of various other maternal conditions 29. Sex chromosome discordance has been found when the mother has received a solid organ or bone marrow transplant from a male donor, resulting in a 46,XY finding on cfDNA with a female fetus. The mother herself may be mosaic for a condition such as Turner syndrome, as somatic mosaicism with loss of one copy of the X chromosome occurs with age 29. Maternal mosaicism for autosomal aneuploidy is more rare, but has also been reported 29. Additionally, the pregnant woman may have an undiagnosed chromosomal microdeletion or duplication leading to “false positive” cfDNA results, as the chromosomal imbalance present in the mother is misinterpreted as fetal in origin 30, 31. Other false positives for aneuploidy may be due to a prior co-twin demise, confined placental mosaicism, fetal mosaicism, or may be related to testing technique. A recent systematic review found that one-third of false positive cases ended up having an explanation, most commonly maternal copy number variation (48%), followed by confined placental mosaicism (32%), and maternal cancer (15%) 25. A high risk cfDNA result with a normal fetal karyotype may also indicate that monosomy or trisomy rescue has occurred to produce a euploid fetus, which confers a risk of uniparental disomy that could lead to autosomal recessive or imprinting disorders in the fetus 32. Due to the low positive predictive value of cfDNA (in part because the conditions tested for are very rare), a high risk result on cfDNA screening should prompt diagnostic testing. False negative results were explained in 54% of cases in a systematic review and often reflected fetal mosaicism, which occurred in 92% of cases 25. Particularly, isochromosome 21q resulting in Down syndrome is commonly associated with a false negative cfNDA test as this translocation usually occurs de novo in the fetus and may not be present in placental cytotrophoblast cells 33. Low fetal fraction, or a decreased proportion of fetal cfDNA relative to maternal DNA in the sample, may also result in a “no call” result. This is an important consideration as many obese women will have a low fetal fraction and up to 23% of “no call” results have been found to have aneuploidy 34. These potential limitations of cfDNA testing are important to bear in mind for perinatal care providers.
In the future, additional tests based on cfDNA are likely to be more commonly offered, such as evaluation for other rare autosomal trisomies (RATs), in which positive findings may confer a risk of miscarriage in addition to fetal mosaicisim or uniparental disomy 32; for chromosomal copy number variants (CNVs) such as microdeletions or duplications; or for single nucleotide variants (SNVs) found by sequencing single genes, panels of genes, or the entire exome or genome. It is recognized that pregnant women are interested in learning about the possibility of severe genetic disorders in the fetus, particularly if a procedure such as an amniocentesis is not needed 35. CNV analysis using cfDNA is currently most commonly offered for 22q11 deletion syndrome, although it is available for other conditions as well, albeit with a lower positive predictive value (high false positive rate) 24. Though an earlier study demonstrated a high sensitivity of a SNP-based approach to detect CNVs in artificial samples 36 a more recent study of 349 “positive” cfDNA results in pregnant women showed that only 32 had a microdeletion confirmed by invasive testing and an overall positive predictive value of 9.2% 24. The positive predictive value is likely to improve as deeper sequencing techniques evolve, as was demonstrated in a recent analysis of over 94,000 pregnancies 37. However, routine use of cfDNA to screen for microdeletion syndromes is not currently recommended by professional societies and further research in this area is needed 34.
Non-invasive Prenatal Screening and Diagnosis of Monogenic Disorders
The next frontier of cfDNA testing will likely involve the non-invasive detection of monogenic disorders. CfDNA has been used to identify pathogenic variants causing achondroplasia and thanatophoric dysplasia in fetuses at risk for these conditions based upon family history or ultrasound findings, with 96% accuracy 38. A recent study using cfDNA to evaluate for de novo or paternally-inherited variants in a panel of 30 genes associated with autosomal dominant disorders was successful in identifying these conditions prenatally in 32 of 422 pregnancies (with a diagnostic yield of 19% for fetuses presenting with abnormal ultrasound results), without false positive or false negative findings, though clinical follow-up was not available for all pregnancies 8. Importantly, these studies and commercially-available non-invasive gene panel testing choose to evaluate for de novo or paternally-inherited variants as these are technically easier to detect. Variants that the fetus may have inherited from the mother are harder to identify, as it is difficult to distinguish between the fetal and maternal alleles in a maternal blood sample, particularly across multiple genetic loci. This and other technical hurdles limit current widespread implementation of the use of cfDNA to screen for fetal monogenic disorders 39. However, there have been recent successful attempts at sequencing the fetal genome using cfDNA from a maternal blood sample as early as the first trimester 40, 41, and challenges such as accurate identification of the fetal alleles are likely to be overcome as genomic sequencing technologies continue to improve. As with the prior techniques discussed, interpretations of test performance are limited by the rarity of the genetic disorders involved. The detection of monogenic disorders via cfDNA in a maternal sample therefore represents an exciting emerging development in fetal genetic diagnosis, though it incurs similar ethical and practical challenges in interpreting fetal genomic data 39, 40 as presented in the section to follow.
Diagnostic Fetal Genetic Testing
We will now discuss the various methods used to identify or confirm a genetic diagnosis in the fetus. Multiple methods for obtaining fetal cells exist, though not all methods sample the same tissue of origin and therefore may yield discordant results (Figure 2). Once fetal cells are obtained, the same cytogenetic or molecular genetic tests that are available in the postnatal period for diagnosis may be performed prenatally.
Sample Acquisition
Fetal samples are commonly obtained either by chorionic villous sampling (CVS) between 10-14 weeks of gestation or by amniocentesis after 16 weeks of gestation. Percutaneous umbilical blood sampling (PUBS) is less commonly used and involves taking a sample of the fetal blood directly from the umbilical cord.
The choice of sample depends on several factors such as timing, risk to the fetus, and the cell line sampled in each type of test – an issue particular to fetal genetic testing. CVS involves sampling the fetal side of the placenta, composed of the cytotrophoblast and extraembryonic mesoderm 42. Therefore, genetic testing of a sample obtained by CVS may yield a false positive or false negative result if the fetus and placenta are genetically discordant, as in confined placental mosaicism (CPM) which is thought to occur in 1-2% of CVS samples 42, 43. This is similar to the issue that arises in PGT and in cfDNA testing, but does not apply to amniocentesis, which directly samples fetal cells that have been shed into the amniotic fluid (Figure 2). Furthermore, genetic testing after CVS may proceed using a direct preparation of the sample obtained, or on a sample cultured to grow additional cells and augment the yield of DNA. The direct preparation of a CVS sample favors analysis of the cytotrophoblast cells, which are rapidly dividing, whereas the mesenchymal cells may grow in a cultured sample. Thus, the direct preparation may reveal a chromosomal abnormality though the cultured sample and fetus are both normal, due to nondisjunction in the cytotrophoblast as it rapidly divides 42. Tetraploidy, for example, is typically seen in direct preparation CVS samples in combination with a normal fetus 44. Other patterns of discordance have also been seen, such as the direct preparation and fetus being normal but the cultured cells being abnormal, representing a nondisjunction event limited to the chorionic mesoderm 42. A situation in which both the direct preparation and cultured samples are abnormal but the fetus is normal can also be seen in the setting of monosomy or disomy rescue, in which meiotic nondisjunction leads to a trisomic or monosomic placenta but the fetus “corrects” the incorrect chromosomal number. In these situations, the involved chromosome pair may therefore be of single parental origin, which can cause imprinting disorders if certain chromosomes (6, 7, 11, 14, 15, or 20) with imprinted regions are affected 43. Interestingly, true false negatives from a CVS samples (normal CVS but an abnormal fetus) are rarely seen 42, and most false positive rare autosomal trisomy results on CVS (97% of trisomies other than 21, 18, or 13) represent CPM 43, 44, which may be associated with adverse outcomes such as fetal growth restriction, though this risk has recently been shown to be low except in the case of CPM for trisomy 16 43.
As previously mentioned, amniocentesis avoids this issue as placental cells are not sampled, thus amniocentesis may be preferred as the follow-up test after a high risk cfDNA result, particularly if no fetal anomalies are seen on ultrasound and a false positive is therefore suspected. An amniocentesis may also be more useful for diagnosis of certain conditions that are typically mosaic in the fetus, with disease-causing variants that are absent from the blood but present in other tissues. Pallister-Killian syndrome, tetrasomy 12p, is an example of such a disorder; postnatal diagnosis of this condition typically requires a skin biopsy rather than blood test for karyotype. As Pallister-Killian syndrome typically manifests with congenital diaphragmatic hernia (CDH), an amniocentesis to evaluate for this condition when CDH is detected on prenatal imaging may be particularly helpful.
Testing of Fetal Samples
Once a sample has been obtained, choosing the optimal test takes into account multiple factors, including the results and limitations of prenatal screening or parental carrier screening. Although the testing modalities described below are also available in the postnatal period, the turnaround time is faster prenatally and clinical interpretation of identified variants differs in the prenatal period given the decreased ability to resolve variants of uncertain significance based on fetal phenotype and the implication that these results may have on parental stress and pregnancy management. In practice, fetal CMA is often used as a first-tier test and performed directly on an amniotic fluid or CVS sample; leftover sample is then cultured and can be used for another test (such as gene panel testing) if the CMA is negative – although proceeding in a tiered approach can add weeks to the turnaround time. Maternal samples are usually analyzed along with fetal samples to evaluate for maternal cell contamination.
Biochemical or Enzyme Testing
Biochemical analyses or measurements of enzyme activity can be performed on the amniotic fluid or fetal cells from CVS or amniocentesis and may have the advantage of a more rapid turnaround time than molecular genetic testing. This can be used to evaluate for inborn errors of metabolism and other conditions such as Smith-Lemli-Opitz syndrome, in which elevated levels of 7-dehydrocholesterol are seen in the amniotic fluid 45. However, gene sequencing approaches to diagnose inborn errors of metabolism are becoming more commonplace than enzyme activity or other biochemical analysis of fetal samples, particularly as the gene or even variant(s) of interest may be known due to family history.
Karyotype and Chromosomal Microarray
Historically, genetic testing of samples obtained via amniocentesis or CVS was limited to karyotype or FISH testing. A karyotype is usually performed on cultured (dividing) cells and typically returns within 7-10 days; this test will detect aneuploidy such as Down syndrome, large chromosomal rearrangements, and triploidy. FISH can be used to detect aneuploidy or certain targeted CNVs (such as the recurrent 22q11 deletion) on non-cultured or cultured cells and results can be obtained in as rapidly as 1-2 days if performed on a direct sample. CMA, in which submicroscopic chromosomal CNVs may be detected, can be performed on either a direct or cultured sample and is recommended over karyotype as the first tier test in the evaluation of an anomalous fetus, with an approximately 6% diagnostic yield 9, 10. Low-pass genome sequencing of a fetal sample has recently been shown to be highly sensitive and perhaps more successful at identifying disease-causing CNVs with a lower minimum DNA requirement for testing46 in addition to the ability to detect balanced rearrangement and potentially lower cost47. Appropriate use of CMA or other technologies for CNV detection in low-risk pregnancies, particularly in those in which no anomalies are detected on fetal ultrasound, is currently under debate, as it has been found that a proportion of these pregnancies (1/71 pregnancies with a normal ultrasound and 1/131 “low risk” pregnancies in a recent study 48) also carry clinically significant CNVs – estimated at 0.86% in a recent meta-analysis 49. As many of these studies utilized oligonucleotide-based array CGH, which are unable to detect uniparental disomy or triploidy as SNP arrays can, the true incidence of abnormal results may be underestimated 49. Assessing the clinical significance of submicroscopic CNVs, which may be associated with varying degrees of developmental delay or intellectual disability, particularly in a structurally-normal fetus, can be challenging and leave parents feeling conflicted with regard to decision-making during the pregnancy. As previously mentioned, SNVs, small insertions/deletions (indels) or other structural variants (variants > 50 base pairs in size and including inversions, complex rearrangements, or copy number events) affecting single genes are generally not detected by prenatal CMA. If suspicion exists for a monogenic disorder, therefore, testing of single genes or panels of genes should be considered.
Single Gene Testing
Single gene testing is typically indicated if the fetal presentation is highly suspicious for a specific condition, based upon available information such as serum screening results, fetal ultrasound findings, or family history. In the setting of a known familial disorder, for example, if a sibling is affected or if both parents are known to be carriers of an autosomal recessive condition, targeted evaluation of the fetal DNA for the known pathogenic variants can be undertaken. In such scenarios where the specific disease-causing variant is known, targeted variant testing is preferred to sequencing of the entire gene as it eliminates the possibility of variants of uncertain significance, aids in the interpretation of a positive result, and is more cost efficient. In other cases, such as a constellation of anomalies pathognomonic for a particular syndrome, sequencing of the entire gene may be the preferred approach. Of note, for certain monogenic conditions that are not caused by SNVs, such as Fragile X syndrome or congenital myotonic dystrophy – both caused by short tandem repeats (STRs) of triplet nucleotides – specific molecular genetic tests other than sequencing may be indicated 14.
Gene Panel Testing
Gene panel testing involves sequencing multiple genes in parallel from a single sample. This testing modality is often used for fetal diagnosis as many presentations are nonspecific. Examples include fetal skeletal dysplasia and fetal akinesia sequence, in which lack of fetal movement leads to multiple anomalies such as arthrygryposis, club feet, and polyhydramios, or skeletal dysplasias. An advantage to gene panel testing is this ability to investigate multiple genetic hypotheses at once in order to increase the chance of arriving at a fetal diagnosis in the shortest possible time (2-3 weeks). Limitations to this modality include the cost (typically hundreds of dollars) in addition to the challenge of selecting the correct list of genes to sequence that will ultimately yield the diagnosis. As previously mentioned, deep phenotyping is difficult in the prenatal period and thousands of genes are known to cause Mendelian disorders. Many labs have developed panels specifically for prenatal use, focusing on common indications for fetal testing with a rapid turnaround time. These panels generally include only severe conditions known to present in utero that correlate with the fetal phenotype to minimize parental and provider confusion and anxiety. As this modality is also sequencing-based, consideration of variants that are difficult to detect by massively-parallel sequencing, such as STRs or structural variants, is important in interpreting a negative test result.
Exome or Genome Sequencing
Even broader than gene panel testing is exome sequencing (ES) or genome sequencing (GS), where ES involves sequencing the 2% of the genome that is protein-coding 50 and GS includes non-coding regions. Though genome sequencing includes more base pairs and has greater ability to detect structural variants, copy number variants, non-coding variants, and even some SNVs and indels due to more even coverage across the genome 51, it has not been demonstrated in the (postnatal) clinical setting to have a substantially increased diagnostic yield despite a substantially higher cost 52. One potential advantage for the fetal period is speed of testing, as it does not require the capture step used to target the protein-coding region of the genome for sequencing. As GS incurs high cost and introduces increased complexity to the data analysis, ES has been used more commonly in large prenatal diagnosis studies. ES, first reported in the prenatal setting for diagnosis of fetal akinesia sequence/non-immune hydrops in a family with recurrent fetal losses53 has subsequently been shown to be high yield for fetuses with multiple congenital anomalies or otherwise highly suspected to have a genetic disorder, with yields of approximately 25-80% depending upon the population analyzed 54, 55, 56, 57, 58, 59. The information gleaned from prenatal exome has been shown to aid in parental decision-making and perinatal management, even when non-diagnostic 56, 57. More recently, two large studies of ES for fetuses with any structural anomaly, including increased nuchal translucency, and negative prior testing for aneuploidy or CNVs found diagnostic yields of 10-12.5% 60, 61. The highest-yield diagnostic rates were found in fetuses with skeletal anomalies or multiple congenital anomalies 60, 61. For both studies, only pathogenic or likely pathogenic variants thought to explain the fetal presentation were returned to the family after committee review. Thus, the analysis did not proceed in the same way as a postnatal exome in that variants responsible for developmental disorders that could not be validated by the fetal phenotype were not returned 61. The differences in variant interpretation between pre- and postnatal exome is important to bear in mind in the case where the prenatal ES is non-diagnostic, as postnatal re-analysis may later be diagnostic. Prenatal ES may also be non-diagnostic if the fetus carries potentially disease-causing variants in a gene that is not linked with human disease, as more than 80% of the 20,000 human genes are yet to be associated with a Mendelian condition 62. Further, establishing a diagnosis in a gene of uncertain significance is particularly challenging in the prenatal period with limited phenotype information and time, complicating parental decision-making. ES has also proven high yield 13 and valuable for diagnosis after fetal demise or after termination of pregnancy 61, which aids in recurrence risk counseling for families 63, 64. Even if fetal DNA cannot be obtained, an autosomal recessive condition may be identified by duo ES of the parents 13, 65.
The limitations of fetal ES include 1) the turnaround time, as women may be attempting to use the information to decide whether or not to continue a pregnancy and commonly more accessible under 24 weeks gestation, 2) the cost, with ES generally costing thousands of dollars, compared to hundreds of dollars for single gene or gene panel testing, and 3) interpretation, as determination of disease-causing variants is made more challenging with the incomplete phenotype information inherent to a fetal presentation 54. Notably, similar challenges present in the neonatal period as well and are increasingly being overcome. Ultra-rapid ES with a turnaround time of 24-48 hours has been transformative for neonatal care7 and can be used for prenatal diagnosis as well, as this is also a population that stands to benefit from prompt diagnosis65. Given that this information can be incredibly helpful in prenatal and perinatal management, parents have expressed a desire to have this option for testing, particularly to learn about conditions which may be treatable 66. The American College of Medical Genetics and Genomics has recently released a “points to consider” document describing the use of fetal ES and considering its use for the diagnosis of anomalous fetuses when prior methods have failed67.
Conclusion
As genomic sequencing technology and the interpretation of genomic data generated by these approaches continue to evolve, opportunities for fetal genetic diagnosis are expanding at a rapid rate. An understanding of the options for screening and diagnosis of genetic disorders in the fetus is important to any perinatal care provider. Identifying a genetic diagnosis in the fetus (or demonstrating that a fetus has not inherited a pathogenic variant of concern due to family history) is valuable to aid in pregnancy management decisions and can be critical for medical management of the newborn. Future directions include advancing opportunities for fetal intervention and therapy in addition to neonatal treatment to take full advantage of a genetic diagnosis found in the prenatal period.
Acknowledgments:
MHW is supported by UM1HG008900. The authors thank the patients and families that we care for at our respective institutions and at the Maternal Fetal Care Center at Boston Children’s Hospital, who continue to teach and inspire us.
Footnotes
Conflicts of Interest: The authors have no competing financial interests relevant to this article.
References
- 1.Posey JE, O’Donnell-Luria AH, Chong JX, Harel T, Jhangiani SN, Coban Akdemir ZH, et al. Insights into genetics, human biology and disease gleaned from family based genomic studies. Genet Med 2019, 21(4):798–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bamshad MJ, Nickerson DA, Chong JX. Mendelian Gene Discovery: Fast and Furious with No End in Sight. Am J Hum Genet 2019, 105(3): 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boycott KM, Hartley T, Biesecker LG, Gibbs RA, Innes AM, Riess O, et al. A Diagnosis for All Rare Genetic Diseases: The Horizon and the Next Frontiers. Cell 2019, 177(1): 32–37. [DOI] [PubMed] [Google Scholar]
- 4.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013, 369(16): 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saunders CJ, Miller NA, Soden SE, Dinwiddie DL, Noll A, Alnadi NA, et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med 2012, 4(154): 154ra135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith LD, Willig LK, Kingsmore SF. Whole-Exome Sequencing and Whole-Genome Sequencing in Critically Ill Neonates Suspected to Have Single-Gene Disorders. Cold Spring Harb Perspect Med 2015, 6(2): a023168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willig LK, Petrikin JE, Smith LD, Saunders CJ, Thiffault I, Miller NA, et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med 2015, 3(5): 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Li J, Saucier JB, Feng Y, Jiang Y, Sinson J, et al. Non-invasive prenatal sequencing for multiple Mendelian monogenic disorders using circulating cell-free fetal DNA. Nat Med 2019, 25(3): 439–447. [DOI] [PubMed] [Google Scholar]
- 9.Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, Zachary JM, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. The New England journal of medicine 2012, 367(23): 2175–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Committee Opinion No.682: Microarrays and Next-Generation Sequencing Technology: The Use of Advanced Genetic Diagnostic Tools in Obstetrics and Gynecology. Obstet Gynecol 2016, 128(6): e262–e268. [DOI] [PubMed] [Google Scholar]
- 11.McPherson E, Nestoridi E, Heinke D, Roberts DJ, Fretts R, Yazdy MM, et al. Alternatives to Autopsy for Fetal and Early Neonatal (Perinatal) Deaths: Insights from the Wisconsin Stillbirth Service Program. Birth Defects Res 2017, 109(18):1430–1441 [DOI] [PubMed] [Google Scholar]
- 12.Reddy UM, Page GP, Saade GR, Silver RM, Thorsten VR, Parker CB, et al. Karyotype versus microarray testing for genetic abnormalities after stillbirth. New Eng J Med 2012, 367(23): 2185–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shamseldin HE, Kurdi W, Almusafri F, Alnemer M, Alkaff A, Babay Z, et al. Molecular autopsy in maternal-fetal medicine. Genet Med 2018, 20(4):420–427 [DOI] [PubMed] [Google Scholar]
- 14.Genetics Co. Committee Opinion No. 691: Carrier Screening for Genetic Conditions. Obstet Gynecol 2017, 129(3): e41–e55. [DOI] [PubMed] [Google Scholar]
- 15.Genetics Co. Committee Opinion No. 690: Carrier Screening in the Age of Genomic Medicine. Obstet Gynecol 2017, 129(3): e35–e40. [DOI] [PubMed] [Google Scholar]
- 16.Guo MH, Gregg AR. Estimating yields of prenatal carrier screening and implications for design of expanded carrier screening panels. Genet Med 2019, 21(9):1940–1947 [DOI] [PubMed] [Google Scholar]
- 17.Gregg AR, Edwards JG. Prenatal genetic carrier screening in the genomic age. Semin Perinatol 2018, 42(5): 303–306. [DOI] [PubMed] [Google Scholar]
- 18.Dolan SM, Goldwaser TH, Jindal SK. Preimplantation Genetic Diagnosis for Mendelian Conditions. JAMA 2017, 318(9): 859–860. [DOI] [PubMed] [Google Scholar]
- 19.Rubio C, Bellver J, Rodrigo L, Castillón G, Guillén A, Vidal C, et al. In vitro fertilization with preimplantation genetic diagnosis for aneuploidies in advanced maternal age: a randomized, controlled study. Fertil Steril 2017, 107(5): 1122–1129. [DOI] [PubMed] [Google Scholar]
- 20.Rechitsky S, Pakhalchuk T, San Ramos G, Goodman A, Zlatopolsky Z, Kuliev A. First systematic experience of preimplantation genetic diagnosis for single-gene disorders, and/or preimplantation human leukocyte antigen typing, combined with 24-chromosome aneuploidy testing. Fertil Steril 2015, 103(2): 503–512. [DOI] [PubMed] [Google Scholar]
- 21.Brezina PR, Kutteh WH. Clinical applications of preimplantation genetic testing. BMJ 2015, 350: g7611. [DOI] [PubMed] [Google Scholar]
- 22.Committee on Practice Bulletins—Obstetrics CmoG, and the Society for Maternal-Fetal Medicine. Practice Bulletin No. 163: Screening for Fetal Aneuploidy. Obstet Gynecol 2016, 127(5): e123–137. [DOI] [PubMed] [Google Scholar]
- 23.Fisher JM, Harvey JF, Morton NE, Jacobs PA. Trisomy 18: studies of the parent and cell division of origin and the effect of aberrant recombination on nondisjunction. Am J Hum Genet 1995, 56(3): 669–675. [PMC free article] [PubMed] [Google Scholar]
- 24.Schwartz S, Kohan M, Pasion R, Papenhausen PR, Platt LD. Clinical experience of laboratory follow-up with noninvasive prenatal testing using cell-free DNA and positive microdeletion results in 349 cases. Prenat Diagn 2018, 38(3): 210–218. [DOI] [PubMed] [Google Scholar]
- 25.Hartwig TS, Ambye L, Sørensen S, Jørgensen FS. Discordant non-invasive prenatal testing (NIPT) - a systematic review. Prenat Diagn 2017, 37(6): 527–539. [DOI] [PubMed] [Google Scholar]
- 26.Bianchi DW, Parker RL, Wentworth J, Madankumar R, Saffer C, Das AF, et al. DNA sequencing versus standard prenatal aneuploidy screening. N Engl J Med 2014, 370(9): 799–808. [DOI] [PubMed] [Google Scholar]
- 27.Gil MM, Quezada MS, Revello R, Akolekar R, Nicolaides KH. Analysis of cell-free DNA in maternal blood in screening for fetal aneuploidies: updated meta-analysis. Ultrasound Obstet Gynecol 2015, 45(3): 249–266. [DOI] [PubMed] [Google Scholar]
- 28.Bianchi DW, Chudova D, Sehnert AJ, Bhatt S, Murray K, Prosen TL, et al. Noninvasive Prenatal Testing and Incidental Detection of Occult Maternal Malignancies. JAMA 2015, 314(2): 162–169. [DOI] [PubMed] [Google Scholar]
- 29.Bianchi DW. Cherchez la femme: maternal incidental findings can explain discordant prenatal cell-free DNA sequencing results. Genet Med 2018, 20(9): 910–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou X, Sui L, Xu Y, Song Y, Qi Q, Zhang J, et al. Contribution of maternal copy number variations to false-positive fetal trisomies detected by noninvasive prenatal testing. Prenat Diagn 2017, 37(4): 318–322. [DOI] [PubMed] [Google Scholar]
- 31.Snyder MW, Gammill HS, Shendure J. Copy-Number Variation and False Positive Results of Prenatal Screening. N Engl J Med 2015, 373(26): 2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pertile MD, Halks-Miller M, Flowers N, Barbacioru C, Kinnings SL, Vavrek D, et al. Rare autosomal trisomies, revealed by maternal plasma DNA sequencing, suggest increased risk of feto-placental disease. Sci Transl Med 2017, 9(405). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huijsdens-van Amsterdam K, Page-Christiaens L, Flowers N, Bonifacio MD, Ellis KMB, Vogel I, et al. Isochromosome 21q is overrepresented among false-negative cell-free DNA prenatal screening results involving Down syndrome. Eur J Hum Genet 2018, 26(10): 1490–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Committee Opinion No. 640: Cell-Free DNA Screening For Fetal Aneuploidy. Obstet Gynecol 2015, 126(3): e31–37. [DOI] [PubMed] [Google Scholar]
- 35.Sullivan HK, Bayefsky M, Wakim PG, Huddleston K, Biesecker BB, Hull SC, et al. Noninvasive Prenatal Whole Genome Sequencing: Pregnant Women’s Views and Preferences. Obstet Gynecol 2019, 133(3): 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wapner RJ, Babiarz JE, Levy B, Stosic M, Zimmermann B, Sigurjonsson S, et al. Expanding the scope of noninvasive prenatal testing: detection of fetal microdeletion syndromes. Am J Obstet Gynecol 2015, 212(3): 332, e331–339. [DOI] [PubMed] [Google Scholar]
- 37.Liang D, Cram DS, Tan H, Linpeng S, Liu Y, Sun H, et al. Clinical utility of noninvasive prenatal screening for expanded chromosome disease syndromes. Genet Med 2019, 21(9): 1998–2006. [DOI] [PubMed] [Google Scholar]
- 38.Chitty LS, Mason S, Barrett AN, McKay F, Lench N, Daley R, et al. Non-invasive prenatal diagnosis of achondroplasia and thanatophoric dysplasia: next-generation sequencing allows for a safer, more accurate, and comprehensive approach. Prenat Diagn 2015, 35(7): 656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jenkins LA, Deans ZC, Lewis C, Allen S. Delivering an accredited non-invasive prenatal diagnosis service for monogenic disorders and recommendations for best practice. Prenat Diagn 2018, 38(1): 44–51. [DOI] [PubMed] [Google Scholar]
- 40.Rabinowitz T, Polsky A, Golan D, Danilevsky A, Shapira G, Raff C, et al. Bayesian-based noninvasive prenatal diagnosis of single-gene disorders. Genome Res 2019, 29(3): 428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kitzman JO, Snyder MW, Ventura M, Lewis AP, Qiu R, Simmons LE, et al. Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med 2012, 4(137): 137ra176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bianchi DW, Wilkins-Haug LE, Enders AC, Hay ED. Origin of extraembryonic mesoderm in experimental animals: relevance to chorionic mosaicism in humans. Am J Med Genet 1993, 46(5): 542–550. [DOI] [PubMed] [Google Scholar]
- 43.Grati FR, Ferreira J, Benn P, Izzi C, Verdi F, Vercellotti E, et al. Outcomes in pregnancies with a confined placental mosaicism and implications for prenatal screening using cell-free DNA. Genet Med 2019. doi: 10.1038/s41436-019-0630-y [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 44.Malvestiti F, Agrati C, Grimi B, Pompilii E, Izzi C, Martinoni L, et al. Interpreting mosaicism in chorionic villi: results of a monocentric series of 1001 mosaics in chorionic villi with follow-up amniocentesis. Prenat Diagn 2015, 35(11): 1117–1127. [DOI] [PubMed] [Google Scholar]
- 45.Haas D, Haege G, Hoffmann GF, Burgard P. Prenatal presentation and diagnostic evaluation of suspected Smith-Lemli-Opitz (RSH) syndrome. Am J Med Genet A 2013, 161A(5): 1008–1011. [DOI] [PubMed] [Google Scholar]
- 46.Wang H, Dong Z, Zhang R, Chau MHK, Yang Z, Tsang KYC, et al. Low-pass genome sequencing versus chromosomal microarray analysis: implementation in prenatal diagnosis. Genet Med 2019. doi: 10.1038/s41436-019-0634-7 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong Z, Ye L, Yang Z, Chen H, Yuan J, Wang H, et al. Balanced Chromosomal Rearrangement Detection by Low-Pass Whole-Genome Sequencing. Curr Protoc Hum Genet 2018, 96: 8.18.11–18.18.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sagi-Dain L, Cohen Vig L, Kahana S, Yacobson S, Tenne T, Agmon-Fishman I, et al. Chromosomal microarray vs. NIPS: analysis of 5541 low-risk pregnancies. Genet Med 2019, 21(11): 2462–2467. [DOI] [PubMed] [Google Scholar]
- 49.Srebniak MI, Joosten M, Knapen MFCM, Arends LR, Polak M, van Veen S, et al. Frequency of submicroscopic chromosomal aberrations in pregnancies without increased risk for structural chromosomal aberrations: systematic review and meta-analysis. Ultrasound Obstet Gynecol 2018, 51(4): 445–452. [DOI] [PubMed] [Google Scholar]
- 50.O’Donnell-Luria AH, Miller DT. A Clinician’s perspective on clinical exome sequencing. Human genetics 2016, 135(6): 643–654. [DOI] [PubMed] [Google Scholar]
- 51.Talkowski ME, Ordulu Z, Pillalamarri V, Benson CB, Blumenthal I, Connolly S, et al. Clinical diagnosis by whole-genome sequencing of a prenatal sample. N Engl J Med 2012, 367(23): 2226–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alfares A, Aloraini T, Subaie LA, Alissa A, Qudsi AA, Alahmad A, et al. Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet Med 2018, 20(11): 1328–1333. [DOI] [PubMed] [Google Scholar]
- 53.Shamseldin HE, Swaid A, Alkuraya FS. Lifting the lid on unborn lethal Mendelian phenotypes through exome sequencing. Genet Med 2013, 15(4): 307–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drury S, Williams H, Trump N, Boustred C, Lench N, Scott RH, et al. Exome sequencing for prenatal diagnosis of fetuses with sonographic abnormalities. Prenat Diagn 2015, 35(10): 1010–1017. [DOI] [PubMed] [Google Scholar]
- 55.Greenbaum L, Pode-Shakked B, Eisenberg-Barzilai S, Dicastro-Keidar M, Bar-Ziv A, Goldstein N, et al. Evaluation of Diagnostic Yield in Fetal Whole-Exome Sequencing: A Report on 45 Consecutive Families. Front Genet 2019, 10: 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Koning MA, Haak MC, Adama van Scheltema PN, Peeters-Scholte CMPC, Koopmann TT, Nibbeling EAR, et al. From diagnostic yield to clinical impact: a pilot study on the implementation of prenatal exome sequencing in routine care. Genet Med 2019, 21(10): 2303–2310. [DOI] [PubMed] [Google Scholar]
- 57.Chandler N, Best S, Hayward J, Faravelli F, Mansour S, Kivuva E, et al. Rapid prenatal diagnosis using targeted exome sequencing: a cohort study to assess feasibility and potential impact on prenatal counseling and pregnancy management. Genet Med 2018, 20(11): 1430–1437. [DOI] [PubMed] [Google Scholar]
- 58.Vora NL, Powell B, Brandt A, Strande N, Hardisty E, Gilmore K, et al. Prenatal exome sequencing in anomalous fetuses: new opportunities and challenges. Genet Med 2017, 19(11): 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Best S, Wou K, Vora N, Van der Veyver IB, Wapner R, Chitty LS. Promises, pitfalls and practicalities of prenatal whole exome sequencing. Prenat Diagn 2018, 38(1): 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petrovski S, Aggarwal V, Giordano JL, Stosic M, Wou K, Bier L, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet 2019, 393(10173): 758–767. [DOI] [PubMed] [Google Scholar]
- 61.Lord J, McMullan DJ, Eberhardt RY, Rinck G, Hamilton SJ, Quinlan-Jones E, et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet 2019, 393(10173): 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Posey JE, O’Donnell-Luria AH, Chong JX, Harel T, Jhangiani SN, Coban Akdemir ZH, et al. Insights into genetics, human biology and disease gleaned from family based genomic studies. Genet Med 2019, 21(4): 798–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Filges I, Friedman JM. Exome sequencing for gene discovery in lethal fetal disorders--harnessing the value of extreme phenotypes. Prenatal diagnosis 2015, 35(10): 1005–1009. [DOI] [PubMed] [Google Scholar]
- 64.Alamillo CL, Powis Z, Farwell K, Shahmirzadi L, Weltmer EC, Turocy J, et al. Exome sequencing positively identified relevant alterations in more than half of cases with an indication of prenatal ultrasound anomalies. Prenat Diagn 2015, 35(11): 1073–1078. [DOI] [PubMed] [Google Scholar]
- 65.Monies D, Abouelhoda M, Assoum M, Moghrabi N, Rafiullah R, Almontashiri N, et al. Lessons Learned from Large-Scale, First-Tier Clinical Exome Sequencing in a Highly Consanguineous Population. Am J Hum Genet 2019, 104(6): 1182–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kalynchuk EJ, Althouse A, Parker LS, Saller DN, Rajkovic A. Prenatal whole-exome sequencing: parental attitudes. Prenat Diagn 2015, 35(10): 1030–1036. [DOI] [PubMed] [Google Scholar]
- 67.Monaghan KG, Leach NT, Pekarek D, Prasad P, Rose NC, Committee APPaG. The use of fetal exome sequencing in prenatal diagnosis: a points to consider document of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2020. doi: 10.1038/s41436-019-0731-7 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]