

Abstract
Using PROteolysis TArgeting Chimeras (PROTACs) to degrade proteins that are important for tumorigenesis has emerged as a potential therapeutic strategy for cancer. PROTACs are heterobifunctional molecules consisting of one ligand for binding to a protein of interest (POI) and another to an E3 ubiquitin (E3) ligase, connected via a linker. PROTACs recruit the E3 ligase to the POI and cause proximity-induced ubiquitination and degradation of the POI by the ubiquitin proteasome system (UPS). PROTACs have been developed to degrade a variety of cancer targets with unprecedented efficacy against a multitude of tumor types. To date, most of the PROTACs developed have utilized ligands to recruit E3 ligases that are ubiquitously expressed in both tumor and normal tissues. These PROTACs can cause on-target toxicities if the POIs are not tumor-specific. Therefore, identifying and recruiting the E3 ligases that are enriched in tumors with minimal expression in normal tissues holds the potential to develop tumor-specific/selective PROTACs. In this review, we will discuss the potential of PROTACs to become anticancer therapeutics, chemical and bioinformatics approaches for PROTAC design, and safety concerns with a special focus on the development of tumor-specific/selective PROTACs. In addition, the identification of tumor types in terms of solid versus hematological malignancies that can be best targeted with PROTAC approach will be briefly discussed.
1. Introduction
PROTACs are heterobifunctional molecules consisting of two different ligands, of which one is for binding to a POI and another to an E3 ligase. These two ligands are connected via a linker. PROTACs hijack the UPS to induce ubiquitination and degradation of the POI by bringing it to close proximity to the E3 ligase (Fig. 1). PROTACs are potentially advantageous as compared to traditional small molecule inhibitors (SMIs). First, pertaining to its unique mechanism of action i.e. an event-driven pharmacology, a PROTAC molecule is capable of catalyzing the degradation of multiple POI molecules. Due to this catalytic mode of action, PROTACs are required at significantly lower concentrations than SMIs to elicit a desired pharmacological effect, which may reduce the toxicities of SMIs. Another advantage of PROTACs over SMIs is that PROTACs can target undruggable proteins such as transcription factors (TFs). For example, the PROTACs targeting STAT3, a largely undruggable TF, have been recently reported [1, 2]. Additionally, PROTACs can be used to overcome drug resistance resulting from mutations of a POI. For example, PROTACs targeting mutated forms of proteins such as the mutants of BCR-ABL [3], receptor tyrosine kinases (RTKs) [4], estrogen receptor alpha (ERα) [5] and Bruton’s tyrosine kinase (BTK) [6] have been reported. Furthermore, PROTACs can overcome resistance to SMIs resulting from target upregulation by degrading the target. For example, androgen receptor (AR) degraders have been shown to overcome the resistance developed to AR antagonist enzalutamide during prostate cancer treatment [7].
Fig. 1. Mechanisms of PROTAC-mediated protein degradation.
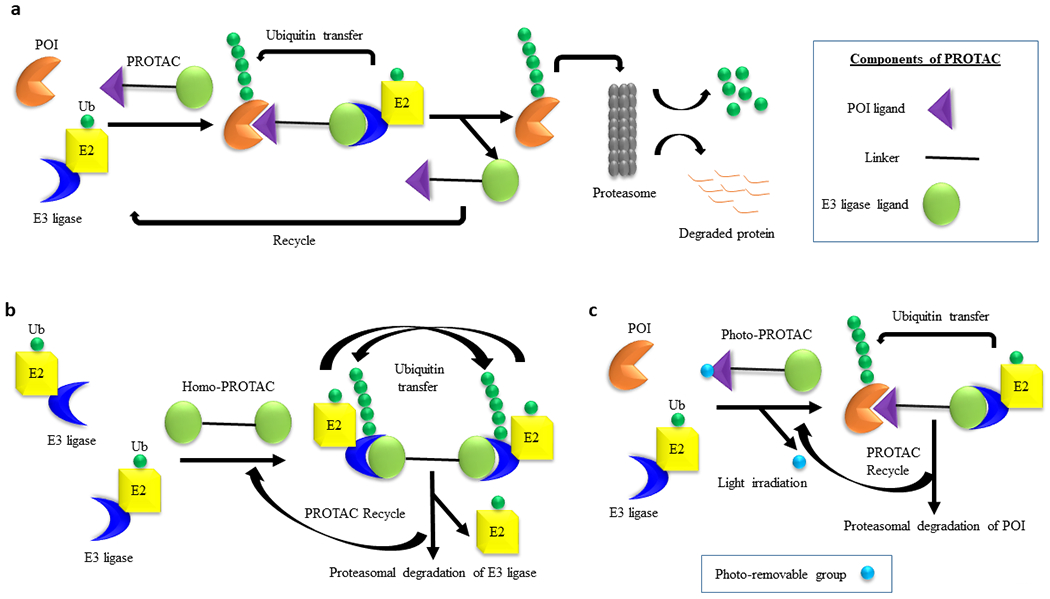
a, Schematic representation of the general mechanism of PROTAC-induced degradation of POI. A PROTAC molecule recruits an E3 ligase to a POI followed by polyubiquitin of the latter by E2 conjugating enzyme. The polyubiquitinated POI is recognized by the proteasome for its degradation. The PROTAC molecule is recycled to induce next round of degradation. The three different structural components of a PROTAC are depicted in the box. b, Mechanism of the self-degradation of an E3 ligase with homo-PROTACs. A homo-PROTAC recruits an E3 ligase molecule (e.g., CRBN or VHL) to another E3 ligase molecule followed by bidirectional polyubiquitination of E3 ligase molecules and their subsequent degradation by the proteasome. c, Mechanism of POI degradation by Photo-PROTACs. In a photo-PROTAC, a photo-removable group is attached to the POI ligand. Upon irradiation with external light, the photo-removable group is detached from the photo-PROTAC converting it to an active PROTAC for the degradation of POI.
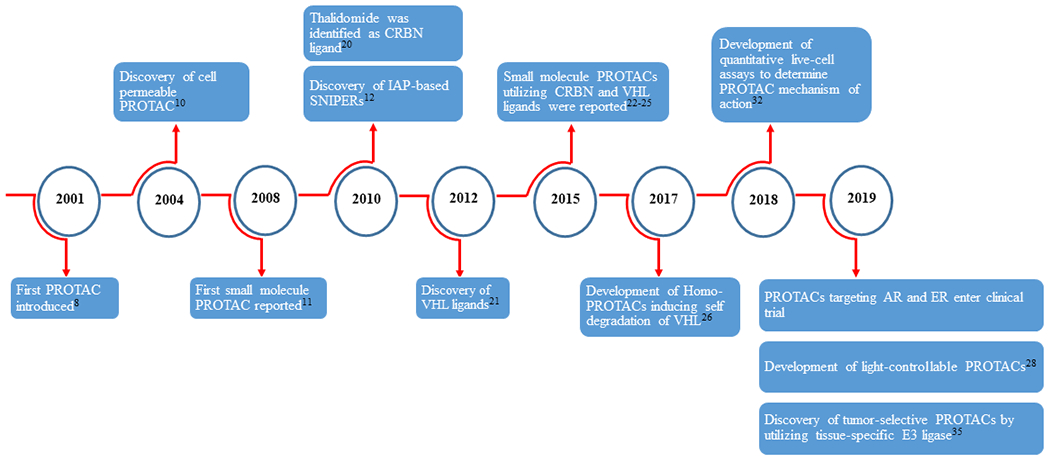
The field of PROTAC research is still relatively new but has been witnessing rapid developments (Fig. 2). The first PROTAC, which was developed by Sakamoto et al. [8] to target methionine aminopeptidase-2 (MetAP-2), was based on a phosphopeptide, but not a small molecule. Two years later, PROTACs targeting ER and AR in breast cancer and prostate cancer, respectively, were reported by Craig Crews’ group [9]. This was followed by the development of the first cell permeable PROTAC consisting of a hydroxypeptide moiety of hypoxia-inducible factor (HIF)-1α targeting FKBP12 and AR [10]. It took another four years before the first small molecule-based PROTAC, which utilizes Nutlin-3a as mouse double minute 2 homolog (MDM2) E3 ligase ligand to recruit MDM2 to AR, was developed [11]. Soon after, Hashimoto’s and Naito’s groups discovered degraders of cellular retinoic acid binding proteins (CRABPs) 1 and 2 that are based on inhibitor of apoptosis proteins (IAPs) E3 ligases, which they named specific and non-genetic IAP-dependent protein erasers (SNIPERs) [12]. This was followed by the discovery of several other potent SNIPERs against different targets [13, 14, 15, 16, 17, 18, 19]. However, the PROTAC field witnessed a transformation after the discoveries of thalidomide, an immunomodulatory imide drug (IMiD), as a ligand of the cereblon (CRBN) E3 ligase in 2010 [ref 20], and development of the small molecule von-Hippel Lindau (VHL) ligands by Crews’ group in 2012 [ref 21]. These ligands have been widely and successfully used to develop CRBN- and VHL-based PROTACs since four key research articles on PROTACs were published by the research groups of Bradner, Ciulli and Crews [22, 23, 24, 25].
Fig. 2. Milestones in PROTAC discovery.
Numbers in superscript indicate reference number.
The researchers have employed several creative approaches to advance the PROTAC field in the recent past. This includes the development of Homo-PROTACs that can induce E3 ligase self-degradation (e.g., the Homo-PROTACs targeting VHL or CRBN E3 ligases) (Fig. 1b) [26, 27], and light-controllable PROTACs, termed as photo-PROTACs or opto-PROTACs or simply PHOTACs, whose activity can be controlled by UV or visible light (Fig. 1c) [28, 29, 30, 31]. Researchers from Promega Corporation have developed some powerful biophysical technologies to help understand unprecedented modes of PROTAC action [32, 33]. Recently, we reported the first use of PROTAC technology to selectively target anti-apoptotic BCL-XL protein in tumor cells with reduced platelet toxicity by employing VHL or CRBN E3 ligases that are minimally expressed in platelets [34, 35, 36]. More recently, the macrocyclic PROTACs to enhance the selectivity between homologous proteins, e.g., the bromodomain proteins, have been reported by Ciulli’s group [37]. In this review, we will discuss the advantages and limitations of currently used E3 ligase ligands for PROTAC development and safety considerations. In particular, we will discuss the possibility of developing tumor-specific/selective PROTACs by utilizing tissue-specific and tumor-selective E3 ligases. Finally, we will touch on the relative efficacy of PROTACs in solid versus hematological cancers and the future of PROTACs as promising cancer therapeutics.
2. E3 ligases exploited to design PROTACs
More than 600 E3 ligases have been discovered in the human genome of which only few have been exploited for the PROTAC design. Mainly ligands of CRBN, VHL, MDM2, IAPs, DCAF15, DCAF16, RNF4 and RNF114 E3 ligases have been used for PROTAC development against various cancer targets (Table 1). PROTACs based on each of these E3 ligases have their advantages and disadvantages. As discussed above, the first generation of PROTACs utilized a peptide moiety against an E3 ligase and subsequently small-molecule-based PROTACs were developed. Here we will discuss small-molecule-based PROTACs or protein degraders generated with some of these widely used E3 ligase ligands.
Table 1.
Key cancer targets that have been successfully targeted with small molecular PROTACs
| E3 ligase | Target | Cancer/cell type | Study type | Refs |
|---|---|---|---|---|
| MDM2 | AR | Hela cells | In vitro | [11] |
| BRD4 | Colon cancer | In vitro | [38] | |
| PARP1 | Breast cancer | In vitro | [39] | |
| IAP | CRABP-2 | Fibrosarcoma | In vitro | [41] |
| ERα | Breast cancer | In vitro/In vivo | [13, 14] | |
| BCR-ABL | CML | In vitro | [15, 16] | |
| AR | Prostate cancer | In vitro | [17] | |
| TACC3 | Fibrosarcoma | In vitro | [19] | |
| VHL | AR | CRPC | In vitro/In vivo | [50, 54] |
| ERRα | Breast cancer | In vitro/In vivo | [24] | |
| RIPK2 | Acute monocytic leukemia | In vitro | [24] | |
| BRD4 | Cervical cancer | In vitro | [23, 43] | |
| BCR-ABL | CML | In vitro | [44] | |
| TBK1 | KRAS-mutant NSCLC | In vitro | [45] | |
| TRIM24 | AML | In vitro | [46] | |
| FLT-3 | AML | In vitro | [47] | |
| ALK | NSCLC; ALCL | In vitro/In vivo | [48] | |
| FAK | Breast cancer; Prostate cancer | In vitro | [49] | |
| P38 | Breast cancer | In vitro | [51] | |
| BCL-XL | T-ALL; SCLC; Breast cancer | In vitro/In vivo | [35, 36] | |
| CRBN | BETs | AML | In vitro | [25] |
| BRD4 | Burkitt’s lymphoma | In vitro | [22] | |
| PCAF/GCN5 | Acute monocytic leukemia | In vitro | [65] | |
| ALK | NSCLC; ALCL; Neuroblastoma | In vitro | [105] | |
| BTK | Burkitt’s lymphoma | In vitro | [60, 61] | |
| CDK9 | Colon cancer; Breast cancer | In vitro | [57] | |
| CDK6 | Multiple myeloma; AML | In vitro | [58, 59] | |
| BCL-XL | T-ALL; SCLC | In vitro | [34] | |
| STAT3 | AML, ALCL | In vitro/In vivo | [1, 2] | |
| MDM2 | B-ALL | In vitro/In vivo | [40] | |
| DCAF16 | FKBP12 | HEK293 Cells | In vitro | [70] |
| RNF4 | BRD4 | Hela cells | In vitro | [71] |
| RNF114 | BRD4 | Breast cancer | In vitro | [72] |
Note: CML, Chronic myelogenous leukemia; CRPC, Castration resistant prostate cancer; NSCLC, Non-small cell lung cancer; ALCL, Anaplastic large cell lymphoma; AML, Acute myeloid leukemia; T-ALL, T-cell acute lymphoblastic leukemia; SCLC, Small-cell lung cancer; B-ALL, B-cell acute lymphoblastic leukemia
MDM2-based PROTACs:
The first MDM2-based PROTAC utilizing Nutlin-3a as an MDM2 ligand and non-steroidal selective androgen receptor modulator (SARM) as the ligand for AR was developed to degrade AR [11]. PROTACs based on MDM2 are advantageous because MDM2 acts as an oncogene by suppressing the activity of the tumor suppressor p53. The binding of a PROTAC to MDM2 can stabilize the p53 protein in p53-wild type tumors, at the same time inducing the degradation of its target. Recently, Crews’ group has demonstrated that a MDM2-based PROTAC targeting bromodomain-containing protein 4 (BRD4), named A1874, shows superior efficacy versus the corresponding VHL-based PROTAC utilizing the same ligand for the POI in the wild-type p53-harboring colon cancer HCT116 cell line. This is because A1874 simultaneously stabilizes the p53 protein and targets BRD4 for degradation [38]. Another MDM2-targeted PROTAC based on a niraparib derivative (PARP1 ligand) and a nutlin-3 derivative has been reported, which induces poly (ADP) ribose polymerase 1 (PARP1) degradation leading to apoptosis induction in MDA-MB-231 human breast cancer cells [39]. Moreover, the PROTACs targeting MDM2 for degradation have also been reported [40]. These PROTACs have been shown to be highly efficient in degrading MDM2, leading to potent growth inhibition of human B-cell acute lymphoblastic leukemia (B-ALL) RS4;11 cells both in culture and in mice [40].
IAP-based protein degraders:
The IAP family of proteins are negative regulators of apoptosis and are comprised of eight different members. Among them cIAP1, cIAP2 and XIAP are the best known. Due to the E3 ligase activity of IAP proteins, their inhibitors have been exploited to develop protein degraders. Hashimoto’s and Naito’s groups discovered the first IAP-based protein degraders or SNIPERs using methyl bestatin (MeBS) as an IAP ligand targeting CRABP-1/2. However, in addition to degrading its target protein, these SNIPERs also induced auto-ubiquitination and subsequent degradation of cIAP1 [ref 12]. To overcome this auto-ubiquitination, they replaced the ester group with amide group at the linker attachment site of MeBS, resulting in the generation of SNIPERs that degraded CRABP-2 only without any effect on cIAP1 [41]. Subsequently, Naito’s group reported several other SNIPERs against different targets such as ERα [13, 14], BCR-ABL [15, 16], AR [17], BRD4 [ref 18] and TACC3 [ref 19]. The SNIPERs/IAP-based PROTACs could be advantageous in the sense that they can exert more potent antitumor effects by simultaneously degrading IAPs and target proteins because some tumor cells upregulate IAPs to evade apoptosis.
VHL-based PROTACs:
All small molecule VHL-based PROTACs came into existence after the discovery of the VHL ligands by Crews’ group in 2012 [refs 21, 42]. Thereafter, the Crews group reported the first small molecule VHL-based PROTACs targeting estrogen receptor-related receptor α (ERRα) and receptor-interacting serine/threonine protein kinase 2 (RIPK2) [ref 24]. The VHL-based BRD4 PROTAC, named MZ1, developed by Ciulli’s group utilized JQ1 as BRD4 ligand. Though JQ1 inhibits BRD2, 3 and 4, after being converted to a PROTAC, MZ1 became a selective BRD4 degrader [23]. Later, Ciulli’s group reported a more potent VHL-based BRD4 degrader, named AT1 [ref 43]. Subsequently, PROTACs targeting various proteins in tumor cells have been designed based on the VHL ligands [4, 44, 45, 46, 47, 48, 49, 50, 51, 52]. Ciulli’s group reported the first proof-of-concept of Homo-PROTACs that dimerize VHL molecules leading to its self-degradation [26]. Recently, we converted a dual inhibitor of BCL-XL and BCL-2, named ABT263 (or navitoclax), into selective VHL-based BCL-XL PROTACs, which provides further evidence that promiscuous inhibitors can be converted into target-specific PROTACs [35, 36]. A major advantage of VHL-based PROTACs is their target-specificity [44, 53]. Moreover, the VHL-based PROTACs may have better tumor-selectivity as compared to PROTACs based on other E3 ligases like IAP for certain targets. For example, we have shown that VHL is differentially expressed between tumor cells and platelets, which helped us to design platelet-sparing BCL-XL PROTACs [35, 36]. Recently, Wang’s group reported the development of highly potent AR degraders (DC50 = 0.2-1 nM in prostate cancer cells) by utilizing weak affinity VHL ligands (binding affinity in the micromolar range) [54]. This study suggests that the binding affinity of VHL ligands does not need to be very high to develop potent PROTACs. This is because a PROTAC employing a weak binding ligand to a VHL protein and high binding ligand to a target protein is capable to forming ternary complex with the target protein and VHL E3 ligase. On the other hand, VHL-based PROTACs also have some drawbacks. For example, VHL is a tumor suppressor protein, which is frequently mutated in several tumor cells such as clear cell renal cell carcinomas or kidney cancer [55, 56]. Therefore, VHL-based PROTACs cannot be used to treat kidney cancer with mutation or deletion in VHL gene. Also, careful optimization of dosing is needed to avoid tumor suppressor function of VHL when using VHL-based PROTACs. Another drawback associated with the VHL-based PROTACs is their high molecular weights, which makes it challenging to develop orally bioavailable PROTACs.
CRBN-based PROTACs:
Since the discovery of CRBN as the target of IMiD drugs i.e., thalidomide, lenalidomide and pomalidomide [20], many CRBN-based PROTACs against numerous targets have been developed. The first CRBN-based PROTACs targeting the bromodomain and extra-terminal (BET) proteins or FK506 binding protein 12 (FKBP12) were reported by Bradner’s group in 2015 [ref 25]. The BET-targeting PROTAC, named dBET1, utilizing JQ1 as the BRD ligand, induced degradation of BRD2, BRD3 and BRD4. In the same year, another CRBN-based BET targeting PROTAC, named ARV-825, was made by Crews’ group, which selectively induced BRD4 degradation in Burkitt’s lymphoma cells [22]. Subsequently, several CRBN-based PROTACs targeting cyclin-dependent kinases (CDKs) [57, 58, 59], BTK [60, 61], epigenetic erasers (SIRT2 and HDAC6) [refs 62, 63, 64], epigenetic writers (PCAF/GCN5) [ref 65], BCL6 [ref 66] and STAT3 [refs 1, 2] have been reported by different groups. Moreover, the Homo-PROTACs designed by linking two molecules of pomalidomide where CRBN acts as both an E3 ligase and a target [27, 67], and CRBN-VHL hetero-dimerizing PROTACs [68] targeting CRBN have been reported. Interestingly, these hetero-dimerizing PROTACs preferentially degrade CRBN and could only induce mild degradation of VHL [68]. In general, CRBN-based PROTACs may degrade a broader range of targets compared with VHL-based PROTACs. For example, using the same BCR-ABL ligands, the CRBN-based PROTACs degraded both BCR-ABL and c-ABL, whereas VHL-based PROTACs could only degrade c-ABL [44]. Similarly, using the same promiscuous kinase inhibitor as a warhead, the CRBN-based PROTAC degrades more kinases than the VHL-based PROTAC [53]. This may be because the protein-binding surface of CRBN is much larger than that of VHL, so CRBN is expected to contribute more in forming the ternary complex than is necessary for triggering ubiquitination, which results in broader target adaptation of CRBN compared with VHL. Since CRBN is ubiquitously expressed in tumor cells and normal cells, therefore the PROTACs based on CRBN have a lesser chance of being tissue-selective. The advantage with CRBN-based PROTACs is their relatively lower molecular weight than VHL-based PROTACs, and thus it is more feasible to develop orally bioavailable PROTACs using a CRBN ligand.
PROTACs based on other E3 ligases:
In addition to the above four E3 ligases that have been commonly exploited for PROTAC design, other E3 ligases belonging to the DCAF family (DCAF15 and DCAF16) and RNF family (RNF4 and RNF114) have the potential to be used for protein degradation because of the recent discoveries of ligands for these E3 ligases. For example, indisulam, a sulfonamide class of drug, exerts its cytotoxicity in numerous leukemia and lymphoma cell lines by recruiting DCAF15 to degrade the RNA binding motif protein 39 (RBM39) [ref 69]. Although indisulam acts as molecular glue to degrade RBM39, it has the potential to be used as a DCAF15 ligand to generate PROTACs for other POIs. In addition, a PROTAC that targets nuclear FKBP12 for degradation by engaging DCAF16 has been reported [70]. Furthermore, covalent ligands for RNF4 and RNF114 have been identified recently and have been used to generate PROTACs targeting BRD4 [refs 71, 72]. The other E3 ligases that can potentially be recruited to degrade a target may include β-TRCP and KEAP1 E3 ligases. In fact, the first PROTAC design was based on a β-TRCP E3 ligase utilizing a phosphopeptide moiety as a β-TRCP binder [8]. A peptide-based PROTAC which recruits the KEAP1 E3 ligase to degrade Tau protein demonstrates the potential of PROTAC technology to treat neurodegenerative disorders such as Alzheimer’s disease [73].
3. Considerations for PROTAC optimization
PROTACs are bifunctional small molecules that bind both E3 ligases and POIs. Despite the rapid progress in this field, the discovery and optimization of PROTACs are still laborious. For example, it is quite challenging to predict which E3 ligase is the best suitable to target a specific POI. A common practice is to exhaust all the E3 ligases that could possibly be used to develop PROTACs. However, in some cases, preliminary experimental data may suggest that a certain E3 ligase-target POI pair is incompatible. However, further investigation proves contrary to the previous conclusion [74]. In addition, high binary binding affinities are not necessarily needed to result in efficient degradation of a target protein [53], indicating the difficulty in predicting the best ligand of a POI to be used for PROTAC construction without preliminary experimental data. Furthermore, the selection of a linker tethering site on the ligands of a POI can be challenging, even when co-crystal structures of a specific ligand with the POI are available. It has been well documented that PROTACs derived from different ligand tethering sites could have significantly different potency and selectivity profiles [74, 75, 76].
Several key components can be fine-tuned to improve PROTAC-mediated protein degradation. Among them, linkerology probably plays the most critical role in both the biological and physicochemical properties of PROTACs. First, the linker length needs to be optimized. If a linker is too short, it may result in steric clash to disrupt ternary complex formation and reduce the ability of a PROTAC to degrade its target. If a linker is too long, it can increase the relative motility of the two heads of a PROTAC and decrease the association constant of E3 ligase binding to POI, thus reducing the stability of the ternary complex. The latter can also increase the molecular weight and potentially reduce its cell permeability. There are several frequently used linker types in PROTAC design. The initial explorations are usually started with flexible linkers such as polyethylene glycol (PEG) or polymethylene chains. The introduction of a hydrophilic PEG-containing linker can improve water solubility. Replacement of PEG linkers with other oxygen-containing chains might further alter the protein degradation profile. Potentially, the oxygen atom(s) may interact with residues at the ternary binding interface thus increasing positive cooperativity [43]. Polymethylene linkers are also frequently used to generate PROTACs for a variety of targets and several of them have achieved respectful in vivo activities, indicating appropriate pharmacokinetic properties can be achieved with these linkers [1, 35]. Besides, ‘click chemistry’ based on copper-catalyzed azide-alkyne cycloaddition (CuAAC) and the Diels-Alder (DA) reaction have been applied in PROTAC preparation [76]. The resulting PROTACs with heterocycle-containing linkers allow fast validation of degradation capability. This technique also enables protein degradation by self-assembly of active PROTACs in live cells [77].
Increasing the rigidity of the linker unit is an effective way to improve the pharmacokinetic properties and oral bioavailability of PROTACs. Proper rigidity in the linker unit can also constrain a PROTAC in its bioactive conformation, which may lead to improved protein degradation [50, 54]. Aromatic, heterocyclic, and macrocyclic linker structures have been reported to minimize unfavorable conformations [37, 78]. Hydrophilic motifs such as piperazine hold the potential to improve water solubility and balance lipophilicity [50]. However, the discovery of an optimal rigid linker can be very challenging without the co-crystal structure of ternary complex. With the VCB: PROTAC: SMARCA2BD co-crystal structure in hand, Farnaby et al. gained insights into the PPI interface, which guided their linker design and facilitated their PROTAC optimization. For example, they replaced the PEG unit of a lead PROTAC targeting SMARCA2 with a phenyl ring, which recapitulated the geometry of the more flexible linker and formed an additional T-stack interaction to Y98 of VHL [78]. Testa et al. analyzed the crystal structure of the BRD4: MZ1: VHL ternary complex and rationally designed a macrocyclic-containing PROTAC with a decent BRD4 degradation activity [37].
The linker tethering sites on both the POI ligand and E3 ligase ligand could significantly affect binary binding affinity, ternary complex conformation, as well as the physiochemical and pharmacokinetic properties of the PROTACs. Similar effects can be derived from modifying the linkages, the structural moieties that connect the POI ligand and E3 ligase ligand to the linker unit. Amide bonds, ether bonds, alkylamines, carbon-carbon single bonds, and carbon-carbon triple bonds are frequently used as linkage in various PROTACs. Given a fine-tuned linker, minor changes on the POI warhead or E3 ligand portion may further improve the protein degradation efficacy. Again, a better binary binding cannot secure improved protein degradation. For example, a PROTAC with weak binding affinity against VHL resulted in the highly potent and efficient degradation of AR [54]. Indeed, PROTAC-mediated cellular activities are regulated by multiple factors including E3 activity, target capability, binary and ternary binding affinity, cooperativity, cell permeability, compound solubility, and stability.
4. Pros and Cons of PROTACs
As discussed above, PROTACs have several advantages over conventional SMIs pertaining to their unique mechanism of action. The main advantages are their catalytic mode of action, high selectivity, potential to target undruggable proteins, and the capacity to overcome resistance from SMIs by targeting mutated proteins. Moreover, it is possible to achieve tumor-specific/selective degradation of a target protein with PROTACs by using ligands for tissue-specific and/or tumor-selective E3 ligases. Due to these properties, PROTACs avoid many side effects and limitations of SMIs. However, there are still some safety concerns associated with PROTACs, which need to be taken into consideration before advocating for their clinical translation. These Pros and Cons of PROTACs in comparison to SMIs are summarized in Table 2. The safety challenges associated with PROTACs have recently been reviewed in greater detail by Moreau and colleagues [79]. In general, the toxicities associated with PROTACs can be broadly categorized into on-target and off-target toxicities. We will discuss these two types of toxicities associated with PROTACs and potential strategies to overcome these toxicities in the following paragraphs.
Table 2.
Pros and Cons of PROTACs in comparison to SMIs
| Pros/Cons | PROTACs | SMIs |
|---|---|---|
| Pros | 1. More potent and longer lasting effects reduce dosing, dosing frequency and toxicity due to event-driven pharmacology and catalytic nature 2. Added layer of selectivity due to the requirement for the formation of a cooperative ternary complex 3. Capable of targeting undruggable proteome and mutated proteins 4. Possible to achieve tumor-selectivity by utilizing tissue- and/or tumor-specific E3 ligase ligands |
1. Due to low molecular weight, generally cell permeability and/or tissue penetration are not challenging and easy to achieve oral bioavailability 2. Partial and transient inhibition of a target protein important for normal cell function by a SMI may be more tolerable than complete depletion of the protein for a longer duration by a PROTAC |
| Cons | 1. High molecular weight may reduce cell permeability and tissue penetration and thus poses some challenges for oral administration 2. Complete degradation of certain proteins and degradation of off-target proteins in a complex or in close proximity to E3 ligase could be detrimental 3. Accumulation of natural substrates of E3 ligase may have undesirable toxicities |
1. Occupancy-driven pharmacology requires higher and more frequent dosing to achieve a therapeutic effect, and thus can potentially produce more toxicities 2. Efficacy and specificity depend on the binding affinity of SMIs to targets, thus inability to target undruggable proteins without an active binding site or mutated proteins |
The on-target toxicities induced by PROTACs are mainly due to the following three reasons. First, some proteins such as kinases have not only enzymatic functions but also scaffold functions. The latter may be essential for normal cellular functions in certain cells. In contrary to SMIs which only block enzymatic function, complete protein degradation with PROTACs can block both enzymatic activity as well as also remove the scaffolding function of such proteins and the latter may sometimes have undesirable consequences. However, removing the scaffolding function of some proteins with a PROTAC can be beneficial in certain conditions. For example, an interleukin-1 receptor-associated kinase 4 (IRAK4) PROTAC has been shown to be more therapeutically relevant compared to IRAK4 inhibitors in part by removing the scaffolding signaling mediated by IRAK4 [52]. Similarly, a FAK degrader was proven to be beneficial by inhibiting its non-enzymatic function [49]. Second, SMIs rarely completely inhibit the functions of their targets while potent PROTACs can nearly completely deplete their targets. An incomplete inhibition of certain proteins with SMIs may be tolerable, while complete depletion of the same proteins with a PROTAC can be detrimental if the proteins carry some vital functions. For example, the inhibition of BET proteins with SMIs is relatively well tolerated than their degradation with PROTAC dBET1 [25]. Third, a SMI may cause the transient inhibition of a protein function while a PROTAC causes prolonged depletion of the protein mainly because of the catalytic nature of PROTACs and the time needed to restore a depleted protein via new synthesis. However, this prolonged degradation of a target protein can be either beneficial or harmful depending on the target and cellular/tissue context. This property of PROTACs can cause on-target toxicities especially when a POI is indispensable for normal cells/tissue function. However, if a POI has redundant function in normal tissues, then its prolonged degradation could prove to be beneficial. For example, in the case of our BCL-XL PROTACs the prolonged degradation of BCL-XL may be advantageous because BCL-XL exhibits functional redundancy in normal tissues except platelets, and therefore its prolonged depletion accounts for better therapeutic efficacy [35, 80, 81]. Another on-target toxicity of PROTACs may result from the accumulation of the natural substrates of an E3 ligase especially when the tissue E3 ligase expression is low. These toxicities could be avoided by utilizing ligands against tissue-specific and tumor-selective E3 ligases for designing tissue/tumor-specific/selective PROTACs [82].
PROTACs do not need to have a high binding affinity to target proteins like SMIs in order to induce target degradation [75]. Off-target toxicities often result from the degradation of proteins to which the binding affinity of a PROTAC is lower to otherwise inhibit their activity by inhibitors. The degradation of proteins that are not directly bound to a PROTAC can occur when the protein is a part of the same complex as the target protein or is in close vicinity [83]. Since PROTACs need to form a ternary complex with the target protein and E3 ligase, the formation of these complexes is inhibited at higher concentrations with most PROTACs. This phenomenon is known as the “Hook effect” which results from the binary bindings of a PROTAC with either the target protein or E3 ligase, and lead to lesser degradation of the target protein at higher concentrations [75]. The generation of PROTAC-E3 ligase binary complexes resulting from the Hook effect at high PROTAC concentrations can lead to degradation of non-targeted proteins via recruitment of these binary complexes to lower-affinity targets [79]. The ligands of some E3 ligases such as CRBN ligands (thalidomide, pomalidomide and lenalidomide) or DCAF15 ligand (e.g., indisulam) can induce the degradation of neosubstrates by recruiting them to CRBN or DCAF15, respectively [69, 84, 85]. Moreover, some CRBN-based PROTACs have been shown to degrade the natural substrates of CRBN such as IKZF1 and IKZF3 [86, 87]. This may lead to undesirable toxicities, since some of these substrates are important for normal homeostasis [88]. Moreover, the accumulation of off-target ubiquitinated proteins can dysregulate the normal proteostasis by saturating the UPS. Most of these PROTAC on- and off-target toxicities can potentially be reduced by generating tumor-specific/selective PROTACs as discussed in the next section.
5. Tumor-specific/selective PROTACs
Although numerous PROTACs have been generated in the last a few years, only few of them are tumor-selective. In 2013, Crews’ group reported the development of two phospho-PROTACs, which induced specific degradation of fibroblast growth factor receptor substrate 2α (FRS2α) or phosphatidylinositol-3-kinase (PI3K) after they were phosphorylated by tropomyosin receptor kinase A (TrkA) or erythroblastosis oncogene B3 (ErbB3), respectively [89]. However, these PROTACs utilized peptide moieties as ligands for the target proteins and thus are not considered tissue/tumor-specific. Recently, the light-controllable photo-PROTACs, whose action can be controlled under visible or UVA light to direct tumor-specific degradation of targeted proteins have been reported by several groups [28, 29, 30, 31]. Photo-PROTACs require an external light source to cause tumor-specific degradation of target proteins. This can only be accomplished in a clinical setting by the use of photodynamic therapy (PDT) for limited types of cancer. We were the first to report tumor-selective and platelet-sparing BCL-XL PROTACs by taking advantage of the E3 ligases that are barely expressed in platelets [34, 35, 36]. Since platelets depend on BCL-XL for survival, BCL-XL inhibition by the dual BCL-XL/BCL-2 inhibitor ABT263 or monoselective BCL-XL inhibitors (A1155463 or A1331852) causes severe platelet toxicity [81, 90], which limits the clinical development of these inhibitors as safer anticancer therapeutics. Through surveying the RNA sequencing data and by performing immunoblot analysis, we identified the VHL E3 ligase as minimally expressed in platelets compared to numerous tumor cells. Our VHL-based BCL-XL PROTACs with ABT263 as a warhead potently degraded BCL-XL in tumor cells both in vitro and in vivo, but not in platelets. The first lead VHL-based BCL-XL PROTAC, named DT2216, potently inhibited the tumor growth in BCL-XL-dependent mouse and patient-derived xenografts and synergistically worked with other BCL-2 family inhibitors and standard-of-care chemotherapy. DT2216 was found to be a BCL-XL selective PROTAC without any degradation of BCL-2. Our biochemical analyses revealed that although DT2216 could form a ternary complex with either BCL-XL and BCL-2 in a cell free system as analyzed by AlphaLISA assay, it could only form a stable ternary complex with BCL-XL in cells as analyzed by a recently developed nanoBRET assay. This partly explained why DT2216 could not degrade BCL-2. Moreover, DT2216 induced BCL-XL degradation in a lysine 87-dependent manner, and since BCL-2 lacks the corresponding lysine, this provides further justification for the inability of DT2216 to not degrade BCL-2.
These findings are in agreement with Craig Crews’ recent suggestion that it is advantageous to develop tumor-specific/selective PROTACs to reduce on-target toxicities where a POI is important for normal tissue function [82]. The tumor-specific/selective degradation of tumor-associated POIs by PROTACs can be achieved by several strategies. If a POI is tumor specific, one can generate a tumor-specific PROTAC by targeting this POI to any available E3 ligases in the tumor tissues. For example, Crews’ group reported the generation of PROTACs for BCR-ABL1, an oncogenic fusion protein specifically expressed in chronic myelogenous leukemia (CML) cells, using the VHL ligand [91]. Alternatively, if a POI is not tumor-specific but specific for a tumor-derived tissue, one can still generate tumor-selective PROTACs by targeting the POI to any available E3 ligases in the tissue, providing the POI is dispensable for normal tissue function or the normal tissue is unessential. For example, several PROTACs targeting BTK, a protein that is important for normal B cell development and development of B cell lymphoma, to CRBN have been generated and exhibit the potential to treat ibrutinib-resistant non-Hodgkin’s lymphomas [6, 92]. Furthermore, if a POI is not tissue/tumor-specific, one can generate a tumor-selective PROTAC by targeting this POI to a tissue-specific/tumor-selective E3 ligase to limit its on-target toxicity. As discussed above, our BCL-XL PROTAC DT2216 is an example of this approach. In addition, several tissue-specific/selective E3 ligases have been identified by Crews’ group, including the pancreas-selective E3 ligase ASB9, which can be potentially used to generate pancreatic cancer selective PROTACs [82], if the targeted POI is not essential for normal pancreatic functions.
However, many tumor-associated POIs are widely expressed in both tumor and normal tissues and are also important for normal cell functions, even though they are frequently upregulated to promote tumorigenesis. Therefore, a PROTAC targeting a tumor-associated POI to a tissue-specific E3 ligase may not be sufficient to reduce the toxicity to normal tissues. Comparatively, using E3 ligases that are highly expressed in tumors (tumor-specific E3 ligases) but are lowly or not expressed in normal tissues, to target the tumor-associated POI has an increased advantage of achieving selective tumor cell killing while minimizing on-target toxicity to normal tissues. The development of tumor-specific/selective PROTACs primarily relies on the identification of tissue-specific and tumor-selective E3 ligases. In the following subsections we will discuss some preliminary analysis regarding the identification of tissue-specific and tumor-selective E3 ligases that can be potentially exploited for generating tumor-specific/selective PROTACs.
Profiling tissue expression of E3 ligases
Accumulating public omics data make it possible to identify tissue-selective E3 ligases, which can be potentially used to design PROTACs to specifically target POIs in a tissue-selective manner. We extracted the RNA-sequencing (RNA-seq) data produced by the Genotype-Tissue Expression (GTEx) Program and analyzed the E3 ligase gene expression profiles in various normal tissues [93, 94]. The samples were collected from more than 50 non-diseased tissues across nearly 1000 individuals. The data of 611 E3 ligases were successfully extracted and the E3 ligases were classified by the criteria described in the caption of Table 3. As shown in Fig. 3, we found that some E3 ligases show significant cluster in some tissues according to their expression levels, for example, in the brain, muscle, and testis. Among the E3 ligases, we found that 3% of E3 ligases cannot be detected, 9% of E3 ligases are lowly expressed, and 4% are highly expressed in almost all normal tissues (Fig. 4a; Table 3). About 9% of E3 ligases are specifically expressed in one tissue, and 12% of E3 ligases are enriched in 2-7 tissue types (group enriched) (Fig. 4a). The E3 ligase expression pattern in many E3 ligases can be validated by the immunohistochemistry (IHC) data from the Human Protein Atlas (THPA) (https://www.proteinatlas.org/) or the human tissue proteomic data [95]. For example, PRPF19 is highly expressed, while KCTD21 is lowly expressed in most of the normal tissues; KLHL41 is specifically expressed in skeletal muscles; RNF112 is mainly expressed in brain tissues, and TRIM69 is specifically expressed in testis (Fig. 4b). Additional tissue-selective E3 ligases including ASB9, FBXL16, HERC5, RAPSN and TRIM46 were also recently reported by Crews’ group [82]. These findings suggest that some E3 ligases exhibit a tissue-selective expression pattern in normal human tissues. These E3 ligases can potentially be employed to design tissue-specific or -selective PROTACs.
Table 3.
Identification of tissue- and tumor-selective E3 ligases
| Highly expressed E3 ligases in normal tissues | Lowly expressed E3 ligases in normal tissues | E3 ligases highly expressed in tumors but lowly expressed in most normal tissues |
|---|---|---|
| BCL6 | ARMC5 | ASB2 |
| BTBD2 | ASB7 | ASB4 |
| CPSF1 | ATRX | ASB5 |
| FBXL5 | CBLL1 | ASB9 |
| FBXW5 | CNOT4 | ASB11 |
| PJA2 | FBXL4 | ASB15 |
| PRPF19 | FBXO3 | ASB16 |
| RBCK1 | FBXO22 | BIRC3 |
| RING1 | FBXO33 | BIRC7 |
| RNF5 | FBXO42 | DPF1 |
| RNF10 | HERC4 | ENC1 |
| RNF11 | KBTBD4 | FBXL13 |
| RNF114 | KBTBD6 | FBXL16 |
| RNF126 | KCTD21 | FBXO2 |
| RNF167 | KDM2B | FBXO32 |
| RNF181 | KLHL8 | FBXO40 |
| RNF187 | KLHL9 | FBXO41 |
| SPSB3 | KLHL15 | HECW1 |
| STUB1 | KLHL18 | HERC5 |
| TRIM8 | KLHL20 | KCNA1 |
| TRIM28 | KLHL26 | KCNA7 |
| WSB1 | LTN1 | KCNB2 |
| ZFAND3 | MDM2 | KCNC1 |
| ZFAND5 | MIB1 | KCNC2 |
| MYNN | KCNC3 | |
| PCGF1 | KCND2 | |
| PEX2 | KCNG1 | |
| RC3H1 | KCNS1 | |
| RC3H2 | KCNV1 | |
| RCHY1 | KCTD4 | |
| RNF2 | KCTD16 | |
| RNF111 | KLHL1 | |
| RNF121 | KLHL6 | |
| SMURF2 | KLHL30 | |
| SOCS4 | KLHL31 | |
| SPOPL | KLHL34 | |
| TRAF3 | MARCH4’ | |
| TRAF6 | MARCH10’ | |
| TRIM23 | MARCH11’ | |
| TRIM68 | MKRN3 | |
| UBE3B | NEURL3 | |
| UBOX5 | OTUD7A | |
| UBR1 | RAPSN | |
| VPS8 | RFPL4B | |
| WDSUB1 | RNF17 | |
| XIAP | RNF39 | |
| ZBTB2 | RNF157 | |
| ZBTB5 | RNF175 | |
| ZBTB11 | RNF182 | |
| ZBTB14 | RNF183 | |
| ZBTB25 | RNF186 | |
| ZBTB33 | SKP2 | |
| ZBTB45 | SOCS7 | |
| ZBTB49 | SPSB4 | |
| ZNF131 | TRIM7 | |
| TRIM9 | ||
| TRIM10 | ||
| TRIM15 | ||
| TRIM17 | ||
| TRIM31 | ||
| TRIM36 | ||
| TRIM46 | ||
| TRIM50 | ||
| TRIM54 | ||
| TRIM63 | ||
| TRIM67 | ||
| TRIM72 | ||
| TRIML1 | ||
| ZBTB32 |
Note: The original RNA-sequencing (RNA-seq) data of normal tissues used for the analyses were obtained from the GTEx Portal (https://www.gtexportal.org) in 2017. RNA-seq data of the tissues with same origin and similar gene expression pattern were averaged to represent one tissue. For example, there are 11 brain tissues as shown in Figure 3, we calculated the average of gene expression level in the 11 brain tissues to represent the brain tissue. In the final analysis, 31 tissues were used to identify the normal tissue specific/selective E3 ligases. The criteria of classification for E3 ligases are: 1) Not expressed in normal tissues – RPKM (Reads Per Kilobase of exon model per Million mapped reads) <1 in all 31 tissues; 2) Tissue specific – RPKM is 5-fold higher in one tissue than all other tissues; 3) Tissue enriched – RPKM is 2.5-fold higher in one tissue than in all other tissues; 4) Group enriched – RPKM is 2.5-fold higher in a group of 2-7 tissues than all other tissues; 5) Expressed low in all tissues – RPKM ≥1 and <10 in all 31 tissues and; 6) Expressed high in all tissues – RPKM ≥10 in all 31 tissues; and 7) Mixed – The remaining genes detected in 1–31 tissues with RPKM>1 and are not in the above categories. In the table, only type 5) Expressed low in all tissues (left column) and 6) Expressed high in all tissues (top column) were listed. Tumor-selective/specific E3 ligases (right column) were identified by overlapping the E3 ligases highly expressed in tumors with those of E3 ligases not or lowly expressed in all normal tissues or selectively expressed in limited normal tissues. The RNA-seq data of 12 cancer types were downloaded from the Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov). The criteria for determination of tumor-specific/selective E3 ligases are: 1) Log fold change of tumor-RPKM/normal tissue- RPKM> 1.5; 2) Statistical significance of p<0.05 when comparing the gene expression in tumors to their corresponding normal tissues. RPKM, Fragments Per Kilobase of exon model per Million mapped fragments.
Fig. 3. Profile of E3 ligase expression in normal tissues.

The original RNA-sequencing (RNA-seq) data used for the analyses were obtained from the GTEx Portal (https://www.gtexportal.org). Heatmap was drawn based on their gene expression levels (RPKM). Red circle indicates selective E3 cluster in tissues.
Fig. 4. Distribution of E3 ligases in normal tissues.
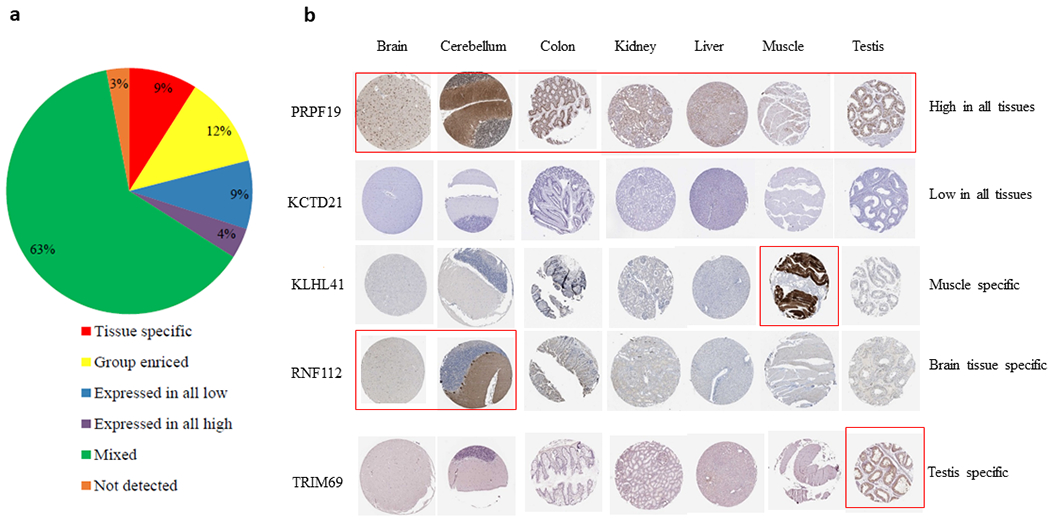
a, Percentage of E3 ligases in normal tissues according to the classification criteria shown in Table 3. b, Immunohistochemistry (IHC) staining of representative E3 ligases in 7 normal tissues. The figures were acquired from the Human Protein Atlas (THPA) (https://www.proteinatlas.org). Red rectangle indicates high expression of E3 ligases in the tissues.
Identification of tumor-specific/selective E3 ligases
To identify the E3 ligases that are upregulated in tumors and can be considered tumor-specific/selective, we extracted the RNA-seq data of 12 cancer types from the Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov) database and analyzed the E3 ligase expression profiles across the tumor types. Using a cutoff point of Logfold change > 1.5 and statistical significance of p <0.05, we identified 114 E3 ligases that were highly expressed in at least one cancer type (Fig. 5a). Many of these E3 ligases are shared by multiple tumors and can be considered tumor-selective because they are also more or less expressed in normal tissues. We are most interested in identifying tumor-specific/selective E3 ligases that are highly expressed in tumors but are not expressed or lowly expressed in all normal tissues, or if not all, at least are lowly expressed in most of the normal tissues. These tumor-specific/selective E3 ligases can be used to design tumor-specific/selective PROTACs to degrade tumor-associated POIs while sparing normal tissues. To find such E3 ligases, we overlapped the tumor E3 ligases with the E3 ligases that are not detected or lowly expressed in normal tissues as identified above. As shown in Table 3, 69 E3 ligases meet this criterion. For example, Birc7, a member of the IAP family, is highly and specifically expressed in melanoma [96] and many other tumors [97], which however is lowly or almost not expressed in normal tissues (Fig. 5b). Moreover, it is an effective E3 ligase to mediate the degradation of Smac [98]. Therefore, Birc7 can be potentially targeted to generate melanoma-specific PROTACs. Among these tumor-specific E3 ligases identified, many have been validated to have E3 activities to degrade their substrates through the UPS, such as TRIM31 [99, 100], TRIM67 [101], FBXO2 [102], TRIM50 [103] etc., which have great potential to be used to design tumor-specific/selective PROTACs. However, there are still several challenges to use tissue- and/or tumor-selective E3 ligase to target tumor cells. First, only a fraction of E3 ligases among more than 600 E3 ligases have been reported to have E3 ligase activity to induce protein ubiquitination and degradation. In addition, protein structures for the majority of the E3 ligases have yet to be solved. Furthermore, development of small molecule ligands of E3 ligases is challenging and time consuming but is urgently needed to design PROTACs. Because space limitations, a discussion on how to develop new E3 ligase ligands will be addressed in a separate review in the future.
Fig. 5. Identification of tumor-specific/selective E3 ligases.
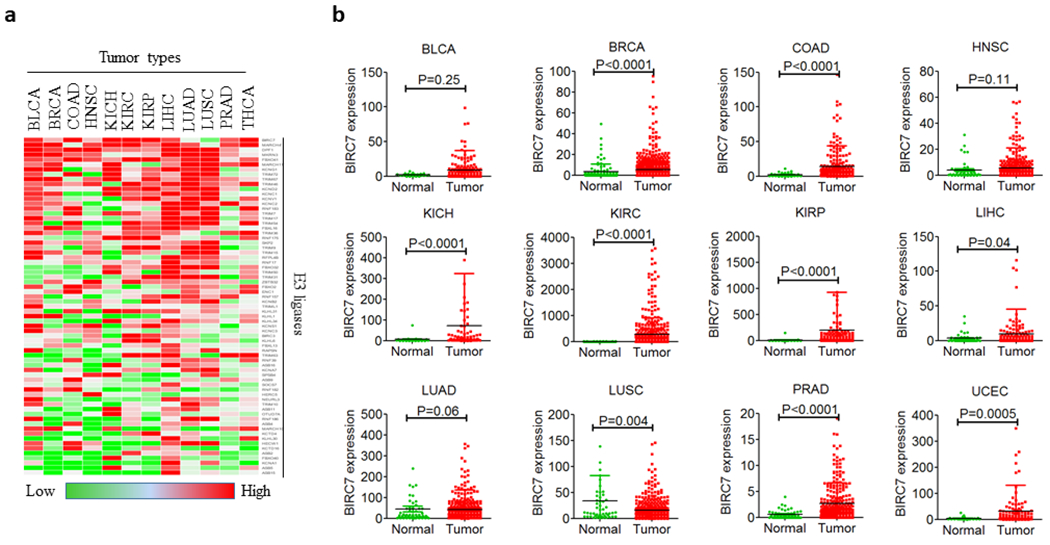
a, Tumor-specific/selective E3 ligases in 12 tumor tissues. BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; COAD, colon adenocarcinoma; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; PRAD, prostate adenocarcinoma; THCA, thyroid carcinoma. b, BIRC7 expression (RPKM) in 12 tumor and corresponding normal tissues. Comparisons were made by two-tailed Student’s t-test.
6. Efficacy of PROTACs in hematological vs solid cancers
PROTACs have been developed against different POIs for solid tumors and hematological malignancies and exhibit a high potency against some tumor cells in a target-dependent manner. For example, PROTACs against BRD4, BTK, BCR-ABL and CDK-6 etc. have shown efficacy in leukemia [22, 44, 58, 60], whereas AR, ER, FAK, P38 PROTACs have been developed against different solid tumors [13, 49, 51, 104] (Table 1). Two PROTACs against AR and ER, named ARV-110 and ARV-471, respectively, are the first to be tested in clinic (Identifier: NCT03888612; NCT04072952). Some of the PROTACs, such as PROTACs targeting BCL-XL and ALK [35, 105], have broad-spectrum antitumor activities, which can effectively kill leukemia and solid tumor cells in vitro and in xenograft models. However, it is important to assess the relative efficacy of such PROTACs in solid versus hematological malignancies and to identify the tumor types that could be best targeted using PROTAC strategy. This is important because unlike SMIs, PROTACs are larger in size, therefore tissue penetration and cell permeability remain major challenges. Due to their high molecular weight and tissue penetration constraints, some PROTACs can only be administered by the intravenous route and thus may be more efficient against hematological malignancies compared to solid tumors, or they may be required at higher doses to efficiently kill solid tumor cells than leukemia/lymphoma cells in vivo.
7. Future of PROTACs as cancer therapeutics
Two decades since the first PROTAC was discovered, we are now entering into a new era of PROTAC research, as the first PROTACs targeting AR and ER have entered clinic. They have the potential to become the first PROTAC therapeutics against cancer if the clinical studies prove AR- and ER-PROTACs are effective and superior to AR- and ER-SMIs. In addition, there are several other PROTAC drug candidates with promising preclinical data that can potentially be tested in clinical trials in the near future [1, 35, 40]. So far, the research has mainly focused on the antitumor potency rather than the safety of such molecules. However, researchers have adopted innovative strategies to guide tumor-specific actions of PROTACs. For example, photo-PROTACs that can be used to induce target degradation in desired tissues with the help of an external source of light have been discovered. In the future, research should be directed towards identifying more strategies to develop safer PROTACs with a greater potential to be successful in a clinical setting. The development of PROTACs based on tissue-specific and tumor-selective E3 ligases could be one such potential strategy that can revolutionize the field of PROTACs as cancer therapeutics, as we recently demonstrated [35, 36]. In addition, the strategies that can enhance the antitumor immunity to develop newer anticancer therapeutics have gained much attention in the last few years. Basically, the compounds that can increase antitumor immunity by neo-antigen presentation or by depleting regulatory T-cells could be potential cancer drug candidates. Of note are the PROTACs that can potentially enhance the anti-tumor immunity by inducing the presentation of peptides derived from target degradation to antigen-presenting cells [106, 107]. Another strategy could be exploiting E3 ligases that have a tumor suppressor natural substrate. This is because the engagement of the E3 ligase with a PROTAC could stabilize the tumor suppressor protein leading to enhanced antitumor activity, as in the case of MDM2-based BRD4 PROTAC [38]. Recently, researchers have developed strategies similar to PROTACs to induce small-molecule targeted degradation of RNAs (e.g. oncogenic micro-RNAs) by recruiting nucleases. These compounds are known as Ribonuclease Targeting Chimeras (RIBOTACs), which also hold potential to become future anticancer therapeutics [108, 109]. Overall, PROTACs and similar compounds could represent a new class of drugs adding to the armament of cancer therapeutics which currently consists of chemotherapeutics, SMIs, monoclonal antibodies and cell-based therapy.
ACKNOWLEDGMENTS:
The authors would like to thank the members of the Zheng and Zhou laboratories for thoughtful discussion and assistance. We greatly acknowledge help from Ms. Janet S. Wiegand for grammatical corrections. This study was supported by US National Institutes of Health (NIH) grants R01CA211963 (D.Z.), R01CA219836 (D.Z.), R01CA242003 (D.Z. and G.Z.) and R21CA223371 (G.Z.).
Footnotes
CONFLICT OF INTEREST: S.K., Y.H., X.Z, G.Z., and D.Z. are inventors of two pending patent applications for use of Bcl-xL PROTACs as senolytic and antitumor agents. G.Z., and D.Z. are co-founders of and have equity in Dialectic Therapeutics, which develops Bcl-xL PROTACs to treat cancer. The remaining authors declare no competing financial interests.
References
- 1.Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy K, McEachern D et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019; 36: 498–511 e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou H, Bai L, Xu R, Zhao Y, Chen J, McEachern D et al. Structure-Based Discovery of SD-36 as a Potent, Selective, and Efficacious PROTAC Degrader of STAT3 Protein. J Med Chem 2019; 62: 11280–11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao Q, Ren C, Liu L, Chen J, Shao Y, Sun N et al. Discovery of SIAIS178 as an Effective BCR-ABL Degrader by Recruiting Von Hippel-Lindau (VHL) E3 Ubiquitin Ligase. J Med Chem 2019; 62: 9281–9298. [DOI] [PubMed] [Google Scholar]
- 4.Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem Biol 2018; 25: 67–77.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez TL, Hancock M, Sun S, Gersch CL, Larios JM, David W et al. Targeted degradation of activating estrogen receptor α ligand-binding domain mutations in human breast cancer. Breast Cancer Res Treat 2020; 180: 611–622. [DOI] [PubMed] [Google Scholar]
- 6.Buhimschi AD, Armstrong HA, Toure M, Jaime-Figueroa S, Chen TL, Lehman AM et al. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 2018; 57: 3564–3575. [DOI] [PubMed] [Google Scholar]
- 7.Kregel S, Wang C, Han X, Xiao L, Fernandez-Salas E, Bawa P et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia 2020; 22: 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A 2001; 98: 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics 2003; 2: 1350–1358. [DOI] [PubMed] [Google Scholar]
- 10.Schneekloth JS Jr., Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K et al. Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc 2004; 126: 3748–3754. [DOI] [PubMed] [Google Scholar]
- 11.Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett 2008; 18: 5904–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Itoh Y, Ishikawa M, Naito M, Hashimoto Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J Am Chem Soc 2010; 132: 5820–5826. [DOI] [PubMed] [Google Scholar]
- 13.Okuhira K, Demizu Y, Hattori T, Ohoka N, Shibata N, Nishimaki-Mogami T et al. Development of hybrid small molecules that induce degradation of estrogen receptor-alpha and necrotic cell death in breast cancer cells. Cancer Sci 2013; 104: 1492–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohoka N, Morita Y, Nagai K, Shimokawa K, Ujikawa O, Fujimori I et al. Derivatization of inhibitor of apoptosis protein (IAP) ligands yields improved inducers of estrogen receptor alpha degradation. J Biol Chem 2018; 293: 6776–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demizu Y, Shibata N, Hattori T, Ohoka N, Motoi H, Misawa T et al. Development of BCR-ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorg Med Chem Lett 2016; 26: 4865–4869. [DOI] [PubMed] [Google Scholar]
- 16.Shibata N, Miyamoto N, Nagai K, Shimokawa K, Sameshima T, Ohoka N et al. Development of protein degradation inducers of oncogenic BCR-ABL protein by conjugation of ABL kinase inhibitors and IAP ligands. Cancer Sci 2017; 108: 1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shibata N, Nagai K, Morita Y, Ujikawa O, Ohoka N, Hattori T et al. Development of Protein Degradation Inducers of Androgen Receptor by Conjugation of Androgen Receptor Ligands and Inhibitor of Apoptosis Protein Ligands. J Med Chem 2018; 61: 543–575. [DOI] [PubMed] [Google Scholar]
- 18.Ohoka N, Ujikawa O, Shimokawa K, Sameshima T, Shibata N, Hattori T et al. Different Degradation Mechanisms of Inhibitor of Apoptosis Proteins (IAPs) by the Specific and Nongenetic IAP-Dependent Protein Eraser (SNIPER). Chem Pharm Bull (Tokyo) 2019; 67: 203–209. [DOI] [PubMed] [Google Scholar]
- 19.Ohoka N, Nagai K, Hattori T, Okuhira K, Shibata N, Cho N et al. Cancer cell death induced by novel small molecules degrading the TACC3 protein via the ubiquitin-proteasome pathway. Cell Death Dis 2014; 5: e1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y et al. Identification of a primary target of thalidomide teratogenicity. Science 2010; 327: 1345–1350. [DOI] [PubMed] [Google Scholar]
- 21.Buckley DL, Gustafson JL, Van Molle I, Roth AG, Tae HS, Gareiss PC et al. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1alpha. Angew Chem Int Ed Engl 2012; 51: 11463–11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol 2015; 22: 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zengerle M, Chan KH, Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol 2015; 10: 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol 2015; 11: 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015; 348: 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maniaci C, Hughes SJ, Testa A, Chen W, Lamont DJ, Rocha S et al. Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat Commun 2017; 8: 830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steinebach C, Lindner S, Udeshi ND, Mani DC, Kehm H, Kopff S et al. Homo-PROTACs for the Chemical Knockdown of Cereblon. ACS Chem Biol 2018; 13: 2771–2782. [DOI] [PubMed] [Google Scholar]
- 28.Xue G, Wang K, Zhou D, Zhong H, Pan Z. Light-Induced Protein Degradation with Photocaged PROTACs. J Am Chem Soc 2019; 141: 18370–18374. [DOI] [PubMed] [Google Scholar]
- 29.Pfaff P, Samarasinghe KTG, Crews CM, Carreira EM. Reversible Spatiotemporal Control of Induced Protein Degradation by Bistable PhotoPROTACs. ACS Cent Sci 2019; 5: 1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynders M, Matsuura BS, Berouti M, Simoneschi D, Marzio A, Pagano M et al. PHOTACs enable optical control of protein degradation. Sci Adv 2020; 6: eaay5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu J, Chen H, Ma L, He Z, Wang D, Liu Y et al. Light-induced control of protein destruction by opto-PROTAC. Sci Adv 2020; 6: eaay5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riching KM, Mahan S, Corona CR, McDougall M, Vasta JD, Robers MB et al. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem Biol 2018; 13: 2758–2770. [DOI] [PubMed] [Google Scholar]
- 33.Daniels DL, Riching KM, Urh M. Monitoring and deciphering protein degradation pathways inside cells. Drug Discov Today Technol 2019; 31: 61–68. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X, Thummuri D, He Y, Liu X, Zhang P, Zhou D et al. Utilizing PROTAC technology to address the on-target platelet toxicity associated with inhibition of BCL-XL. Chem Commun (Camb) 2019; 55: 14765–14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khan S, Zhang X, Lv D, Zhang Q, He Y, Zhang P et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat Med 2019; 25: 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, Thummuri D, Liu X, Hu W, Zhang P, Khan S et al. Discovery of PROTAC BCL-XL degraders as potent anticancer agents with low on-target platelet toxicity. Eur J Med Chem 2020; 192: 112186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Testa A, Hughes SJ, Lucas X, Wright JE, Ciulli A. Structure-Based Design of a Macrocyclic PROTAC. Angew Chem Int Ed Engl 2020; 59: 1727–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hines J, Lartigue S, Dong H, Qian Y, Crews CM. MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of p53. Cancer Res 2019; 79: 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao Q, Lan T, Su S, Rao Y. Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chem Commun (Camb) 2019; 55: 369–372. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Yang J, Aguilar A, McEachern D, Przybranowski S, Liu L et al. Discovery of MD-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J Med Chem 2019; 62: 448–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Itoh Y, Ishikawa M, Kitaguchi R, Sato S, Naito M, Hashimoto Y. Development of target protein-selective degradation inducer for protein knockdown. Bioorg Med Chem 2011; 19: 3229–3241. [DOI] [PubMed] [Google Scholar]
- 42.Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ et al. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J Am Chem Soc 2012; 134: 4465–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol 2017; 13: 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E et al. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem Int Ed Engl 2016; 55: 807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crew AP, Raina K, Dong H, Qian Y, Wang J, Vigil D et al. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J Med Chem 2018; 61: 583–598. [DOI] [PubMed] [Google Scholar]
- 46.Gechijian LN, Buckley DL, Lawlor MA, Reyes JM, Paulk J, Ott CJ et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat Chem Biol 2018; 14: 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burslem GM, Song J, Chen X, Hines J, Crews CM. Enhancing Antiproliferative Activity and Selectivity of a FLT-3 Inhibitor by Proteolysis Targeting Chimera Conversion. J Am Chem Soc 2018; 140: 16428–16432. [DOI] [PubMed] [Google Scholar]
- 48.Kang CH, Lee DH, Lee CO, Du Ha J, Park CH, Hwang JY. Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC). Biochem Biophys Res Commun 2018; 505: 542–547. [DOI] [PubMed] [Google Scholar]
- 49.Cromm PM, Samarasinghe KTG, Hines J, Crews CM. Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J Am Chem Soc 2018; 140: 17019–17026. [DOI] [PubMed] [Google Scholar]
- 50.Han X, Wang C, Qin C, Xiang W, Fernandez-Salas E, Yang CY et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J Med Chem 2019; 62: 941–964. [DOI] [PubMed] [Google Scholar]
- 51.Smith BE, Wang SL, Jaime-Figueroa S, Harbin A, Wang J, Hamman BD et al. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat Commun 2019; 10: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nunes J, McGonagle GA, Eden J, Kiritharan G, Touzet M, Lewell X et al. Targeting IRAK4 for Degradation with PROTACs. ACS Med Chem Lett 2019; 10: 1081–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S et al. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem Biol 2018; 25: 78–87 e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han X, Zhao L, Xiang W, Qin C, Miao B, Xu T et al. Discovery of Highly Potent and Efficient PROTAC Degraders of Androgen Receptor (AR) by Employing Weak Binding Affinity VHL E3 Ligase Ligands. J Med Chem 2019; 62: 11218–11231. [DOI] [PubMed] [Google Scholar]
- 55.Clifford SC, Prowse AH, Affara NA, Buys CH, Maher ER. Inactivation of the von Hippel-Lindau (VHL) tumour suppressor gene and allelic losses at chromosome arm 3p in primary renal cell carcinoma: evidence for a VHL-independent pathway in clear cell renal tumourigenesis. Genes Chromosomes Cancer 1998; 22: 200–209. [DOI] [PubMed] [Google Scholar]
- 56.Clifford SC, Walsh S, Hewson K, Green EK, Brinke A, Green PM et al. Genomic organization and chromosomal localization of the human CUL2 gene and the role of von Hippel-Lindau tumor suppressor-binding protein (CUL2 and VBP1) mutation and loss in renal-cell carcinoma development. Genes Chromosomes Cancer 1999; 26: 20–28. [PubMed] [Google Scholar]
- 57.Robb CM, Contreras JI, Kour S, Taylor MA, Abid M, Sonawane YA et al. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem Commun (Camb) 2017; 53: 7577–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Su S, Yang Z, Gao H, Yang H, Zhu S, An Z et al. Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J Med Chem 2019; 62: 7575–7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brand M, Jiang B, Bauer S, Donovan KA, Liang Y, Wang ES et al. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem Biol 2019; 26: 300–306 e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zorba A, Nguyen C, Xu Y, Starr J, Borzilleri K, Smith J et al. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc Natl Acad Sci U S A 2018; 115: E7285–E7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jaime-Figueroa S, Buhimschi AD, Toure M, Hines J, Crews CM. Design, synthesis and biological evaluation of Proteolysis Targeting Chimeras (PROTACs) as a BTK degraders with improved pharmacokinetic properties. Bioorg Med Chem Lett 2020; 30: 126877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schiedel M, Herp D, Hammelmann S, Swyter S, Lehotzky A, Robaa D et al. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J Med Chem 2018; 61: 482–491. [DOI] [PubMed] [Google Scholar]
- 63.Yang K, Song Y, Xie H, Wu H, Wu YT, Leisten ED et al. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg Med Chem Lett 2018; 28: 2493–2497. [DOI] [PubMed] [Google Scholar]
- 64.Yang H, Lv W, He M, Deng H, Li H, Wu W et al. Plasticity in Designing PROTACs for Selective and Potent Degradation of HDAC6. Chem Commun (Camb) 2019; 55: 14848–14851. [DOI] [PubMed] [Google Scholar]
- 65.Bassi ZI, Fillmore MC, Miah AH, Chapman TD, Maller C, Roberts EJ et al. Modulating PCAF/GCN5 Immune Cell Function through a PROTAC Approach. ACS Chem Biol 2018; 13: 2862–2867. [DOI] [PubMed] [Google Scholar]
- 66.McCoull W, Cheung T, Anderson E, Barton P, Burgess J, Byth K et al. Development of a Novel B-Cell Lymphoma 6 (BCL6) PROTAC To Provide Insight into Small Molecule Targeting of BCL6. ACS Chem Biol 2018; 13: 3131–3141. [DOI] [PubMed] [Google Scholar]
- 67.Lindner S, Steinebach C, Kehm H, Mangold M, Gutschow M, Kronke J. Chemical Inactivation of the E3 Ubiquitin Ligase Cereblon by Pomalidomide-based Homo-PROTACs. J Vis Exp 2019. [DOI] [PubMed] [Google Scholar]
- 68.Girardini M, Maniaci C, Hughes SJ, Testa A, Ciulli A. Cereblon versus VHL: Hijacking E3 ligases against each other using PROTACs. Bioorg Med Chem 2019; 27: 2466–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017; 356. [DOI] [PubMed] [Google Scholar]
- 70.Zhang X, Crowley VM, Wucherpfennig TG, Dix MM, Cravatt BF. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat Chem Biol 2019; 15: 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ward CC, Kleinman JI, Brittain SM, Lee PS, Chung CYS, Kim K et al. Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem Biol 2019; 14: 2430–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Spradlin JN, Hu X, Ward CC, Brittain SM, Jones MD, Ou L et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat Chem Biol 2019; 15: 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lu M, Liu T, Jiao Q, Ji J, Tao M, Liu Y et al. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur J Med Chem 2018; 146: 251–259. [DOI] [PubMed] [Google Scholar]
- 74.Zoppi V, Hughes SJ, Maniaci C, Testa A, Gmaschitz T, Wieshofer C et al. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel–Lindau (VHL) based dual degrader probe of BRD9 and BRD7. Journal of medicinal chemistry 2018; 62: 699–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pettersson M, Crews CM. PROteolysis TArgeting Chimeras (PROTACs) - Past, present and future. Drug Discov Today Technol 2019; 31: 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Y, Jiang X, Feng F, Liu W, Sun H. Degradation of proteins by PROTACs and other strategies. Acta Pharm Sin B 2020; 10: 207–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lebraud H, Wright DJ, Johnson CN, Heightman TD. Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS central science 2016; 2: 927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Farnaby W, Koegl M, Roy MJ, Whitworth C, Diers E, Trainor N et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat Chem Biol 2019; 15: 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moreau K, Coen M, Zhang AX, Pachl F, Castaldi MP, Dahl G et al. Proteolysis-targeting chimeras in drug development: A safety perspective. Br J Pharmacol 2020; 177: 1709–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eichhorn JM, Alford SE, Sakurikar N, Chambers TC. Molecular analysis of functional redundancy among anti-apoptotic Bcl-2 proteins and its role in cancer cell survival. Exp Cell Res 2014; 322: 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S et al. Programmed anuclear cell death delimits platelet life span. Cell 2007; 128: 1173–1186. [DOI] [PubMed] [Google Scholar]
- 82.Schapira M, Calabrese MF, Bullock AN, Crews CM. Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov 2019; 18: 949–963. [DOI] [PubMed] [Google Scholar]
- 83.Hsu JH, Rasmusson T, Robinson J, Pachl F, Read J, Kawatkar S et al. EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex. Cell Chem Biol 2020; 27: 41–46 e17. [DOI] [PubMed] [Google Scholar]
- 84.Donovan KA, An J, Nowak RP, Yuan JC, Fink EC, Berry BC et al. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. Elife 2018; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ishoey M, Chorn S, Singh N, Jaeger MG, Brand M, Paulk J et al. Translation Termination Factor GSPT1 Is a Phenotypically Relevant Off-Target of Heterobifunctional Phthalimide Degraders. ACS Chem Biol 2018; 13: 553–560. [DOI] [PubMed] [Google Scholar]
- 86.Dobrovolsky D, Wang ES, Morrow S, Leahy C, Faust T, Nowak RP et al. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood 2019; 133: 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jiang B, Wang ES, Donovan KA, Liang Y, Fischer ES, Zhang T et al. Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew Chem Int Ed Engl 2019; 58: 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schmitt C, Tonnelle C, Dalloul A, Chabannon C, Debre P, Rebollo A. Aiolos and Ikaros: regulators of lymphocyte development, homeostasis and lymphoproliferation. Apoptosis 2002; 7: 277–284. [DOI] [PubMed] [Google Scholar]
- 89.Hines J, Gough JD, Corson TW, Crews CM. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc Natl Acad Sci U S A 2013; 110: 8942–8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood 2011; 118: 1663–1674. [DOI] [PubMed] [Google Scholar]
- 91.Burslem GM, Schultz AR, Bondeson DP, Eide CA, Savage Stevens SL, Druker BJ et al. Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Res 2019; 79: 4744–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sun Y, Ding N, Song Y, Yang Z, Liu W, Zhu J et al. Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia 2019; 33: 2105–2110. [DOI] [PubMed] [Google Scholar]
- 93.Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015; 348: 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M et al. Human genomics. The human transcriptome across tissues and individuals. Science 2015; 348: 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R et al. A draft map of the human proteome. Nature 2014; 509: 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vucic D, Stennicke HR, Pisabarro MT, Salvesen GS, Dixit VM. ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr Biol 2000; 10: 1359–1366. [DOI] [PubMed] [Google Scholar]
- 97.Liu B, Han M, Wen JK, Wang L. Livin/ML-IAP as a new target for cancer treatment. Cancer Lett 2007; 250: 168–176. [DOI] [PubMed] [Google Scholar]
- 98.Ma L, Huang Y, Song Z, Feng S, Tian X, Du W et al. Livin promotes Smac/DIABLO degradation by ubiquitin-proteasome pathway. Cell Death Differ 2006; 13: 2079–2088. [DOI] [PubMed] [Google Scholar]
- 99.Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat Immunol 2017; 18: 214–224. [DOI] [PubMed] [Google Scholar]
- 100.Song H, Liu B, Huai W, Yu Z, Wang W, Zhao J et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun 2016; 7: 13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yaguchi H, Okumura F, Takahashi H, Kano T, Kameda H, Uchigashima M et al. TRIM67 protein negatively regulates Ras activity through degradation of 80K-H and induces neuritogenesis. J Biol Chem 2012; 287: 12050–12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu B, Lu H, Li D, Xiong X, Gao L, Wu Z et al. Aberrant Expression of FBXO2 Disrupts Glucose Homeostasis Through Ubiquitin-Mediated Degradation of Insulin Receptor in Obese Mice. Diabetes 2017; 66: 689–698. [DOI] [PubMed] [Google Scholar]
- 103.Ma X, Ma X, Qiu Y, Zhu L, Lin Y, You Y et al. TRIM50 suppressed hepatocarcinoma progression through directly targeting SNAIL for ubiquitous degradation. Cell Death Dis 2018; 9: 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Salami J, Alabi S, Willard RR, Vitale NJ, Wang J, Dong H et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun Biol 2018; 1: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Powell CE, Gao Y, Tan L, Donovan KA, Nowak RP, Loehr A et al. Chemically Induced Degradation of Anaplastic Lymphoma Kinase (ALK). J Med Chem 2018; 61: 4249–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Moser SC, Voerman JSA, Buckley DL, Winter GE, Schliehe C. Acute Pharmacologic Degradation of a Stable Antigen Enhances Its Direct Presentation on MHC Class I Molecules. Front Immunol 2017; 8: 1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jensen SM, Potts GK, Ready DB, Patterson MJ. Specific MHC-I Peptides Are Induced Using PROTACs. Front Immunol 2018; 9: 2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Costales MG, Aikawa H, Li Y, Childs-Disney JL, Abegg D, Hoch DG et al. Small-molecule Targeted Recruitment of a Nuclease to Cleave an Oncogenic RNA in a Mouse Model of Metastatic Cancer. Proc Natl Acad Sci U S A 2020; 117: 2406–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Costales MG, Childs-Disney JL, Haniff HS, Disney MD. How We Think About Targeting RNA With Small Molecules. J Med Chem 2020. Online ahead of print ( 10.1021/acs.jmedchem.9b0192). [DOI] [PMC free article] [PubMed] [Google Scholar]