

Abstract
Identification of a monogenic etiology is possible in a proportion of patients with childhood-onset nephrolithiasis or nephrocalcinosis. Bartter syndrome (BS), a hereditary tubulopathy characterized by polyuria, hypokalemic alkalosis and growth retardation that rarely presents with isolated nephrocalcinosis. Patients with defect in renal outer medullary potassium channel, encoded by the KCNJ1 gene causing BS type 2, typically present during the neonatal period. We describe a 14-year-old girl with mild late-onset BS type 2 with reported pathogenic compound heterozygous variations in exon 2 of KCNJ1 (c.146G > A and c.657C > G). This patient presented with isolated medullary nephrocalcinosis due to hypercalciuria; absence of hypokalemia and metabolic alkalosis was unique. This case highlights the importance of screening the KCNJ1 gene in patients with hypercalciuria and nephrocalcinosis, even in older children.
Keywords: Bartter syndrome, Nephrocalcinosis, Potassium channel
Introduction
Children with isolated nephrocalcinosis often present a diagnostic dilemma. Identification of a monogenic etiology is possible in 16.7–29.4% of childhood-onset nephrolithiasis or nephrocalcinosis by high throughput sequencing [1–3]. Whole exome sequencing in 65 patients with early-onset nephrocalcinosis or nephrolithiasis identified causative mutations in genes associated with hyperoxaluria (AGXT and GRHPR), familial hypomagnesemia, hypercalciuria and nephrocalcinosis (CLDN16 and CLDN19), Bartter syndrome (BS) type 1 (SLC12A1), infantile hypercalcemia (SLC34A1), distal renal tubular acidosis (ATP6V1B1) and cystinuria (SLC3A1) [2]. While, BS was detected in 3 (4.6%) patients with nephrocalcinosis in the above cohort [2], only one patient was detected among 106 and 143 children with nephrocalcinosis and nephrolithiasis, respectively, in other studies [1, 3]. BS is an inherited salt-losing tubulopathy affecting the thick ascending limb (TAL) and distal convoluted tubule, and characterized by hypokalemic metabolic alkalosis [4]. Five subtypes are distinguished: type 1 (defect in SCL12A1), type 2 (KCNJ1), type 3 (CLCNKB), type 4a (BSND), type 4b (combined CLCNKA and CLCNKB) and type 5 (MAGE-D2) [5, 6]. While all subtypes manifest hypercalciuria and nephrocalcinosis, the age of onset and associated metabolic abnormalities differ. Patients with BS type 1 and 2, resulting from loss of function of the sodium–potassium–chloride cotransporter (NKCC2) and renal outer medullary potassium channel (ROMK), respectively present during early infancy with polyhydramnios, prematurity, polyuria, episodic dehydration and failure to thrive [7]. Later age of onset and heterogenous presentation with isolated nephrocalcinosis is rare in patients with BS type 1 or 2. Patients with BS type 4 and 5 may have an early onset of illness [6]. Clinical heterogeneity is known in BS type 3 that presents antenatally, or during childhood with hypercalciuria and polyuria, or have Gitelman-like presentation with incidentally detected hypokalemia and hypomagnesemia [8]. We describe a patient with late-onset BS type 2 that was diagnosed following evaluation for nephrocalcinosis.
Case report
This 14-year-old developmentally normal girl, born of nonconsanguineous marriage with birth weight of 2.75 kg, was incidentally found to have bilateral medullary nephrocalcinosis on ultrasonography done for abdominal pain (Fig. 1). Family history was non-contributory; there were no siblings. While there was history of maternal polyhydramnios, ultrasound reports for confirmation were not available. There was history of polydipsia and polyuria since 7–8 years of age; urine output was approximately 2.6–3.0 L/m2/day. There was no history of failure to thrive, salt craving, episodic weakness, seizures, tetany, and visual or hearing deficits. Examination, at 14 years, showed weight of 50 kg (0.5 standard deviation score, SDS), height 161 cm (1.08 SDS) and blood pressure 120/86 mmHg. Ambulatory blood pressure monitoring showed normal mean blood pressure (106/67 mmHg) with systolic and diastolic loads of 1.9% and 15.1%, respectively. Venous blood pH (7.35–7.43), pCO2 (43.7–48 mmHg) and bicarbonate levels calculated by Henderson–Hasselbalch equation (23.7–26 mEq/l) were normal while the patient was not receiving any therapy. Blood levels of sodium (140–143 mEq/l), potassium (3.8–4.5 mEq/l), chloride (100–102 mEq/l), urea (18 mg/dl), creatinine (0.6–0.7 mg/dl; estimated GFR 93–110 ml/min/1.73 m2), uric acid (6.3 mg/dl), calcium 8.9–9.2 mg/dl, phosphorus 4.2–4.7 mg/dl and alkaline phosphatase 159–197 U/l were within normal limits.
Fig. 1.
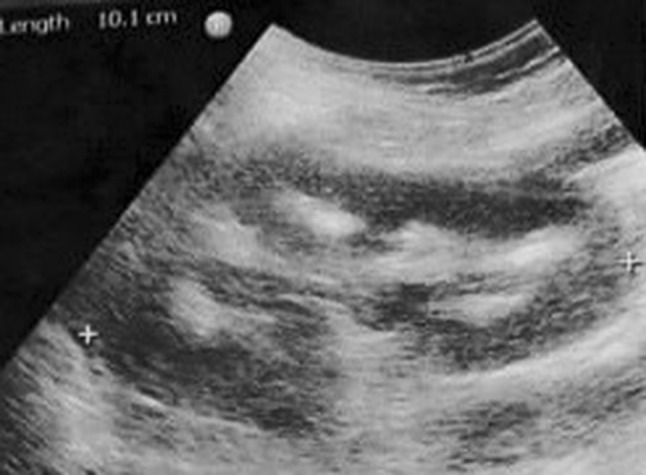
Ultrasonographic appearance of the right kidney showing increased echogenicity of renal medulla suggestive of dense medullary nephrocalcinosis
Blood level of 25-hydroxyvitamin D was normal (70.3 nmol/l) and parathyroid hormone (PTH) was elevated (104–224 pg/ml). Serum magnesium ranged between 1.4 and 2.3 mg/dl (normal 1.7–2.2 mg/dl) and its fractional excretion, assessed on one occasion, was 5.7% (normal < 4%). Early morning fasting urine pH was 5.3, indicating normal distal acidification. Urinalysis confirmed hypercalciuria (calcium 6.9 mg/kg/day; normal < 4 mg/kg/day) and proteinuria (369 mg/day). Urinary β2-microglobin excretion was 130 ng/ml (normal < 300 ng/ml); glucosuria and aminoaciduria were absent. Evaluation of eyes and ears were normal.
At another center, the patient was treated with potassium citrate supplements and hydrochorthiazide (1 mg/kg/day) that resulted in hypokalemia (potassium 2.8–3.1 mEq/l) and alkalosis (bicarbonate 26.9–27.8 mEq/l). Following this therapy, creatinine transiently rose to 0.8 mg/dl (eGFR 83 ml/min/1.73 m2), possibly due to dehydration, and declined to 0.6–0.7 mg/dl (eGFR 95–110 ml/min/1.73 m2) within the next 3–5 months. A clinical diagnosis of familial hypomagnesemia with hypercalciuria was made, based on presence of nephrocalcinosis, high PTH, normal distal acidification and β2-microglobulin excretion, and absence of metabolic acidosis or alkalosis.
Targeted exome sequencing for a panel of genes associated with nephrocalcinosis [2, 9] showed two missense variants on exon 2 of KCNJ1 (c.146G > A and c.657C > G). The former variant led to change from cysteine to tyrosine at position 49, and the latter from serine to arginine at position 219. The variants were confirmed by Sanger sequencing in the parents who were heterozygous carriers (Fig. 2). These variants are not reported in the 1000 genome database; minor allele frequency was 0.005% and 0.0008%, respectively in ExAC database. In silico pathogenicity of both variants was damaging by Mutation Taster 2 and probably damaging by Polyphen-2. Combined annotation dependent depletion (CADD) score was 26.7 and 25.1, respectively (https://cadd.gs.washington.edu/snv) and the reference codons were conserved across species. Both variants caused in vitro functional alteration of ROMK [10] and were classified as pathogenic by the American College of Medical Genetics (ACMG) 2015 criteria [11]. The diagnosis of BS type 2 was made. No variant was detected in other genes associated with Bartter or Gitelman syndromes, CLDN16 and CLDN19.
Fig. 2.

Exon 2 of KCNJ1. Sanger sequence chromatogram and alignment to the reference sequence confirming compound heterozygous variations: a c.657C > G; p.S219R in father of the index patient; b c.146G > A; p.C49T in the mother
Discussion
This 14-year-old girl had a mild and late presentation with isolated nephrocalcinosis without hypokalemia and metabolic alkalosis; compound heterozygous pathogenic variants were present in exon 2 of KCNJ1. Uptake of sodium chloride in the thick ascending limb is mediated by the NKCC2 transporter that transports one Na+, one K+ and two Cl− ions from the lumen into the cell. The basolateral Na-K-ATPase actively exports three Na+ ions out of the cells and imports two K+ ions. The ATP-sensitive inwardly rectifying potassium channel (ROMK) encoded by KCNJ1 recycles K+ into the tubular lumen. While ROMK malfunction indirectly disrupts NKCC2 function, and results in hypokalemia, volume depletion and metabolic alkalosis, these features were absent in the present patient.
Although neonates with ROMK defects might transiently not show hypokalemia [12], the absence of hypokalemia in this patient was unusual. Recycling of potassium by ROMK causes lumen-positive transepithelial potential, which is the main driving force for paracellular uptake of cations, including calcium and magnesium. Thus, hypercalciuria with nephrocalcinosis, and hypermagnesuria are features of BS type 2. While the former were present, serum levels of magnesium were normal in this patient. High PTH levels, have been shown in a large cohort of patients with BS type 2, and attributed to persistent hypercalciuria [13].
Variants in KCNJ1 are chiefly missense or nonsense affecting exon 2 (transcript ID ENST00000392665.6), as was also seen in the present patient [12, 14–17]. The variant c.146G > A has been reported in the compound heterozygous state in a patient with antenatal Bartter syndrome [10]. The c.657C > G variant has been reported both in the compound heterozygous [18] and familial homozygous states in patients with Bartter syndrome [19]. ROMK is gated by intracellular pH with half maximal activation at pH of 6.8 [10]. The gating is driven by protonation of lysine within a triad of arginine-lysine-arginine residues in the transmembrane region [10]. Structural disturbance of the triad alters electrostatic interactions, shifting pKa of the lysine residue to an alkaline pH. Heterologous expression experiments showed shifts in pH gating in vitro due to c.146G > A and c.657C > G to 7.5 and 7.9, respectively [10], inactivating ROMK under physiological conditions. Therefore, both the variants are classified as pathogenic [11].
Antenatal BS, types 1 and 2, typically present early but milder phenotype and later onset has been described in BS type 1 [20–22]. Presentation of BS type 2 beyond childhood is exceptional. In several series of BS type 2, comprising 6–20 patients, prematurity (100%) and polyhydramnios (66–95%) were common manifestations [12, 15–17, 19] and patients were chiefly diagnosed early during infancy (95–100%) [17, 19] or between 1 and 5 years of age (5%) [17].
Table 1 shows features of four reports of late-onset BS type 2. Similar to the present patient, polyhydramnios was present in another patient [23] and most had polyuria during childhood [23–25] without growth failure [23, 24, 26]. While hyperkalemia may occur in the neonatal period in 25–60% patients [15, 19], hypokalemia and metabolic alkalosis are seen between 1-week to 5-years of age [27]. The severity of hypokalemia and metabolic alkalosis is modest compared to those with defects in CLCKNB [16, 19]; in one series only 2 of 12 patients required potassium supplementation [12]. Serum potassium over prolonged follow-up ranged between 3–3.5 mEq/L [15] and 2.3–3.8 mEq/L [27] in six and ten patients, respectively.
Table 1.
Late-onset Bartter syndrome due to defect in the renal outer medullary potassium channel (ROMK)
| Reference | Age at diagnosis, sex | Symptoms in childhood | K+ (mEq/l) | Bicarbonate (mEq/l) |
Growth | Other metabolic abnormalities | Genetic sequencing |
|---|---|---|---|---|---|---|---|
| Li et al. [23] | 32 years, female | Polyhydramnios, polyuria, polydipsia | 2.4–4.0 | 27.5–31.4 | Normal | Nephrocalcinosis, hypercalciuria; high urine chloride, hyperparathyroidism, hyperreninemia |
Compound heterozygous; p.T234I; p.T71M |
| Gollasch et al. [24] | 43 years, female | Polyuria, thirst | 3.0 | 25.7 | Normal | Blood creatinine 1.13 mg/dl, nephrocalcinosis, hypercalciuria; fractional excretion of sodium 2.3% and chloride 2.5%; high transtubular potassium gradient |
Compound heterozygous; p.I66N; p.R292Q |
| Sharma et al. [25] | 9 years, female | Polyuria, polydipsia by 2-year | 2.5 | 32.5 | 5–10th centile | Nephrocalcinosis, hypercalciuria; mild hypercalcemia, high urine chloride; high 1,25(OH)2 vitamin D |
Compound heterozygous; p.G90W; p.I211S |
| Huang et al. [26] | 35 years, male | None | 2.8 | 33 | Normal | Nephrocalcinosis, hypercalciuria | Homozygous; p.L220F |
| Present patient | 12 years, female | Polyhydramnios, polydipsia, polyuria; onset at 6–7 years | 4.2–4.5 | 23–25 | Normal | Nephrocalcinosis, hypercalciuria; high urine chloride; high parathyroid hormone, elevated fractional excretion of magnesium | Compound heterozygous; p.C49T; p.S219R |
While most patients with late onset BS type 2 had hypokalemia and alkalosis (Table 1), these features were not present in our case and was intermittently absent in the case reported by Li et al. [23]. In addition, the case reported by Li et al. also had hyperparathyroidism, similar to our patient. The absence of severe hypokalemia might be because ROMK is also expressed in the collecting duct, where potassium is preserved despite presence of hyperaldosteronism. Presence of variations in the compound heterozygous state might preserve partial channel function accounting for later onset with milder phenotype; similar cases with other compound heterozygous KCNJ1 variants have been reported [23–25].
The present patient requires long term follow-up to determine whether electrolyte abnormalities will manifest during adulthood [23, 24, 26]. Despite milder electrolyte abnormalities, nephrocalcinosis and hypercalciuria is a constant feature in all cases with BS type 2 [16, 19], including all patients with late-onset BS type 2, as was also observed in the present patient. Our finding highlights the need for inclusion of KCNJ1 in the gene panel for exome sequencing for isolated nephrocalcinosis, even in older children.
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
Ethical statement
This article does not contain any interventional studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from the parents of the patients included in this article.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Braun DA, Lawson JA, Gee HY, Halbritter J, Shril S, Tan W, et al. Prevalence of monogenic causes in pediatric patients with nephrolithiasis or nephrocalcinosis. Clin J Am Soc Nephrol. 2016;11(4):664–672. doi: 10.2215/CJN.07540715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daga A, Majmundar AJ, Braun DA, Gee HY, Lawson JA, Shril S, et al. Whole exome sequencing frequently detects a monogenic cause in early onset nephrolithiasis and nephrocalcinosis. Kidney Int. 2018;93(1):204–213. doi: 10.1016/j.kint.2017.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halbritter J, Baum M, Hynes AM, Rice SJ, Thwaites DT, Gucev ZS, et al. Fourteen monogenic genes account for 15% of nephrolithiasis/nephrocalcinosis. J Am Soc Nephrol. 2015;26(3):543–551. doi: 10.1681/ASN.2014040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seyberth HW, Weber S, Komhoff M. Bartter's and Gitelman's syndrome. Curr Opin Pediatr. 2017;29(2):179–186. doi: 10.1097/MOP.0000000000000447. [DOI] [PubMed] [Google Scholar]
- 5.Besouw MTP, Kleta R, Bockenhauer D. Bartter and Gitelman syndromes: questions of class. Pediatr Nephrol. 2019 doi: 10.1007/s00467-019-04371-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laghmani K, Beck BB, Yang SS, Seaayfan E, Wenzel A, Reusch B, et al. Polyhydramnios, transient antenatal Bartter's syndrome, and MAGED2 mutations. N Engl J Med. 2016;374(19):1853–1863. doi: 10.1056/NEJMoa1507629. [DOI] [PubMed] [Google Scholar]
- 7.Seyberth HW. An improved terminology and classification of Bartter-like syndromes. Nat Clin Pract Nephrol. 2008;4(10):560–567. doi: 10.1038/ncpneph0912. [DOI] [PubMed] [Google Scholar]
- 8.Seys E, Andrini O, Keck M, Mansour-Hendili L, Courand PY, Simian C, et al. Clinical and genetic spectrum of Bartter syndrome type 3. J Am Soc Nephrol. 2017;28(8):2540–2552. doi: 10.1681/ASN.2016101057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amar A, Majmundar AJ, Ullah I, Afzal A, Braun DA, Shril S, et al. Gene panel sequencing identifies a likely monogenic cause in 7% of 235 Pakistani families with nephrolithiasis. Hum Genet. 2019;138(3):211–219. doi: 10.1007/s00439-019-01978-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schulte U, Hahn H, Konrad M, Jeck N, Derst C, Wild K, et al. pH gating of ROMK (K(ir)1.1) channels: control by an Arg-Lys-Arg triad disrupted in antenatal Bartter syndrome. Proc Natl Acad Sci U S A. 1999;96(26):15298–15303. doi: 10.1073/pnas.96.26.15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finer G, Shalev H, Birk OS, Galron D, Jeck N, Sinai-Treiman L, et al. Transient neonatal hyperkalemia in the antenatal (ROMK defective) Bartter syndrome. J Pediatr. 2003;142(3):318–323. doi: 10.1067/mpd.2003.100. [DOI] [PubMed] [Google Scholar]
- 13.Landau D, Gurevich E, Sinai-Treiman L, Shalev H. Accentuated hyperparathyroidism in type II Bartter syndrome. Pediatr Nephrol. 2016;31(7):1085–1090. doi: 10.1007/s00467-016-3337-1. [DOI] [PubMed] [Google Scholar]
- 14.Fretzayas A, Gole E, Attilakos A, Daskalaki A, Nicolaidou P, Papadopoulou A. Expanding the spectrum of genetic mutations in antenatal Bartter syndrome type II. Pediatr Int. 2013;55(3):371–373. doi: 10.1111/j.1442-200X.2012.03716.x. [DOI] [PubMed] [Google Scholar]
- 15.Derst C, Konrad M, Kockerling A, Karolyi L, Deschenes G, Daut J, et al. Mutations in the ROMK gene in antenatal Bartter syndrome are associated with impaired K+ channel function. Biochem Biophys Res Commun. 1997;230(3):641–645. doi: 10.1006/bbrc.1996.6024. [DOI] [PubMed] [Google Scholar]
- 16.Brochard K, Boyer O, Blanchard A, Loirat C, Niaudet P, Macher MA, et al. Phenotype–genotype correlation in antenatal and neonatal variants of Bartter syndrome. Nephrol Dial Transpl. 2009;24(5):1455–1464. doi: 10.1093/ndt/gfn689. [DOI] [PubMed] [Google Scholar]
- 17.Peters M, Jeck N, Reinalter S, Leonhardt A, Tonshoff B, Klaus GG, et al. Clinical presentation of genetically defined patients with hypokalemic salt-losing tubulopathies. Am J Med. 2002;112(3):183–190. doi: 10.1016/S0002-9343(01)01086-5. [DOI] [PubMed] [Google Scholar]
- 18.Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet. 1996;13(2):183–188. doi: 10.1038/ng0696-183. [DOI] [PubMed] [Google Scholar]
- 19.Walsh PR, Tse Y, Ashton E, Iancu D, Jenkins L, Bienias M, et al. Clinical and diagnostic features of Bartter and Gitelman syndromes. Clin Kidney J. 2018;11(3):302–309. doi: 10.1093/ckj/sfx118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bettinelli A, Ciarmatori S, Cesareo L, Tedeschi S, Ruffa G, Appiani AC, et al. Phenotypic variability in Bartter syndrome type I. Pediatr Nephrol. 2000;14(10–11):940–945. doi: 10.1007/PL00013418. [DOI] [PubMed] [Google Scholar]
- 21.Madrigal G, Saborio P, Mora F, Rincon G, Guay-Woodford LM. Bartter syndrome in Costa Rica: a description of 20 cases. Pediatr Nephrol. 1997;11(3):296–301. doi: 10.1007/s004670050280. [DOI] [PubMed] [Google Scholar]
- 22.Kurtz CL, Karolyi L, Seyberth HW, Koch MC, Vargas R, Feldmann D, et al. A common NKCC2 mutation in Costa Rican Bartter's syndrome patients: evidence for a founder effect. J Am Soc Nephrol. 1997;8(11):1706–1711. doi: 10.1681/ASN.V8111706. [DOI] [PubMed] [Google Scholar]
- 23.Li J, Hu S, Nie Y, Wang R, Tan M, Li H, et al. A novel compound heterozygous KCNJ1 gene mutation presenting as late-onset Bartter syndrome: case report. Medicine (Baltimore) 2019;98(34):e16738. doi: 10.1097/MD.0000000000016738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gollasch B, Anistan YM, Canaan-Kuhl S, Gollasch M. Late-onset Bartter syndrome type II. Clin Kidney J. 2017;10(5):594–599. doi: 10.1093/ckj/sfx033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma A, Linshaw MA. A novel compound heterozygous ROMK mutation presenting as late onset Bartter syndrome associated with nephrocalcinosis and elevated 1,25(OH)(2) vitamin D levels. Clin Exp Nephrol. 2011;15(4):572–576. doi: 10.1007/s10157-011-0431-3. [DOI] [PubMed] [Google Scholar]
- 26.Huang L, Luiken GP, van Riemsdijk IC, Petrij F, Zandbergen AA, Dees A. Nephrocalcinosis as adult presentation of Bartter syndrome type II. Neth J Med. 2014;72(2):91–93. [PubMed] [Google Scholar]
- 27.Jeck N, Derst C, Wischmeyer E, Ott H, Weber S, Rudin C, et al. Functional heterogeneity of ROMK mutations linked to hyperprostaglandin E syndrome. Kidney Int. 2001;59(5):1803–1811. doi: 10.1046/j.1523-1755.2001.0590051803.x. [DOI] [PubMed] [Google Scholar]