

Abstract
Summary
We introduce shallowHRD, a software tool to evaluate tumor homologous recombination deficiency (HRD) based on whole genome sequencing (WGS) at low coverage (shallow WGS or sWGS; ∼1X coverage). The tool, based on mining copy number alterations profile, implements a fast and straightforward procedure that shows 87.5% sensitivity and 90.5% specificity for HRD detection. shallowHRD could be instrumental in predicting response to poly(ADP-ribose) polymerase inhibitors, to which HRD tumors are selectively sensitive. shallowHRD displays efficiency comparable to most state-of-art approaches, is cost-effective, generates low-storable outputs and is also suitable for fixed-formalin paraffin embedded tissues.
Availability and implementation
shallowHRD R script and documentation are available at https://github.com/aeeckhou/shallowHRD.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Aggressive subtypes of breast and ovarian cancers are frequently associated with homologous recombination deficiency (HRD) making these tumors sensitive to poly(ADP-ribose) polymerase inhibitors (Coleman et al., 2019). HRD arises upon inactivation of BRCA1/2, RAD51C or PALB2 and is characterized by specific tumor genome instability (Nik-Zainal et al., 2016; Staaf et al., 2019). Even though HRD genes are mostly known, exhaustive testing of their inactivation is difficult. This motivates developing surrogate genomic markers of HRD. Recent developments based on high throughput sequencing, HRDetect, Signature 3, SigMA, scarHRD, achieved excellent capacity to evaluate HRD (Davies et al., 2017; Gulhan et al., 2019; Polak et al., 2017; Sztupinszki et al., 2018). However, these methods are technically complex, time- and data-storage consuming, often need a matched normal sample and can be costly.
We introduce shallowHRD, a software for HRD testing based on the number of large-scale genomic alterations (LGA) obtained from whole genome sequencing (WGS) at low coverage (shallow WGS or sWGS; ∼1X). sWGS robustly detect copy number alterations (CNAs), even in fixed-formalin paraffin embedded (FFPE) samples and liquid biopsies (Van Roy et al., 2017) at low cost and with easy-storable outputs. The concept of LGAs follows single-nucleotide polymorphism (SNP) array approaches, exploiting an increased number of large-scale intra-chromosomal CNAs characteristic of HRD (Abkevich et al., 2012; Birkbak et al., 2012; Popova et al., 2012).
2 Materials and methods
2.1 Data
In-house sWGS of breast and ovarian cancers (26 primary tumors, 39 patient-derived xenografts from frozen blocks and 4 primary tumors FFPE) and down-sampled to ∼1X WGS (108 normal tissues, 79 primary tumors from the TCGA breast cancer) were processed by Control-FREEC (v11.5) (Boeva et al., 2012) (Supplementary Material).
2.2 shallowHRD
The tool takes as input ‘sample_name.bam_ratio.txt’, which includes CNA profile where x is normalized read counts in a sliding window, is genomic coordinate and the profile segmentation with Si, Zi segment median and size (in megabases, Mb).
2.2.1 Workflow
is detected and the profile segmentation is optimized as follows: Segments are defined as ‘large’ if , where are quartiles of Zi (Zi > 3 Mb) distribution. M is detected as the first local minimum of density, where i, j are large segments (Supplementary Fig. S1). . Adjacent segments are merged if ; starting from the largest segment.
LGAs, defined as intra-chromosome arm CNA breaks with adjacent segments Mb, are counted after removing segments <3 Mb.
The sample is annotated as ‘non-HRD’ (LGA < 15), ‘borderline’ (15 ≤ LGA ≤ 19) or ‘HRD’ (LGA > 19).
Sample quality is defined by M and cMAD, , where corresponds to the segment enclosing x, before optimization: ‘bad’ (cMAD > 0.5 | cMAD > 0.14 and M > 0.45), ‘average’ (cMAD > 0.14 and M < 0.45 | cMAD < 0.14 and M > 0.45) or ‘normal or highly contaminated’ (M < 0.025) (Supplementary Material and Fig. S2).
CCNE1 amplification is called if , where c is the segment enclosing the gene (4 was set arbitrarily).
shallowHRD output contains: (A) Tumor genome profile. (B) Density plot for . (C) CNA segmentation summary. (D) Sample quality and HRD diagnostics (Supplementary Fig. S3).
3 Results
In-house sWGS and down-sampled WGS of normal samples (TCGA) were employed to develop the sWGS methodology similar to the large-scale state transitions (LST) in SNP-arrays (Popova et al., 2012) (Section 2). LGAs inferred from sWGS corresponded well to the LSTs with identical HRD calls for 8 primary tumors tested (76–97% match in segments 10 Mb) (Supplementary Fig. S4). sWGS coverage >0.3X provide adequate quality, also for FFPE (Supplementary Figs. S2 and 5).
Validation by down-sampled WGS (TCGA) showed LGA to be coherent to SNP-arrays LST (r = 0.92; slope = 0.88; P < 2.2e–16, Pearson) with increased discrepancy in average quality samples (n = 13), and HRD diagnostics discordant in three and borderline in four cases (Fig. 1A;Supplementary Material, Supplementary Figs. S6 and 7, Supplementary Table S1). CCNE1 amplification was found in four non-HRD cases, in-line with previous observations of almost mutual exclusivity with HRD (Goundiam et al., 2015). Thus, sWGS LGAs is suitable to take over the SNP-array LSTs, which is a clinically validated method for HRD detection.
Fig. 1.
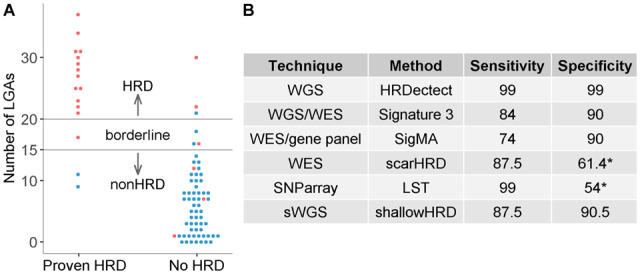
shallowHRD validation in down-sampled WGS of the TCGA (A) and performance (B). Proven/No HRD: cases with/without inactivation of BRCA1/2, RAD51C or PALB2 (Supplementary Material); HRD (red) and non-HRD (blue) cases in SNP-arrays; LGAs: large-scale genomic alterations; WES: whole exome sequencing. aLow specificity could be due to non-complete annotation of HRD
Tumor content for sWGS limits to >0.3 as estimated from the TCGA and in silico dilution series (Supplementary Material, Supplementary Figs. S8 and 9).
Fifteen and 20 LGAs represent soft and stringent cut-offs with sensitivity of 87.5% and 81.25% (16 cases HRD) and specificity of 90.5% and 95.2% (63 non-HRD cases), respectively, which is compatible with other state-of-the-art approaches (Fig. 1B).
To conclude, shallowHRD implements a fast and straightforward evaluation of tumor HRD in breast, ovarian and other cancers such as pancreatic or prostatic, performing similar to most state-of-the-art approaches, the technique is cheap and suitable for all type of samples.
Supplementary Material
Acknowledgements
The results here are in part-based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. The authors thank The Seven Bridges Cancer Genomics Cloud for computational facilities.
Funding
This work was supported by the Ligue Nationale Contre le Cancer (to A.E.).
Conflict of Interest:
E. Manié, T. Popova and M.-H. Stern are co-inventors of the LST method (US20170260588, US20150140122 and exclusive Licence to Myriad Genetics).
References
- Abkevich V. et al. (2012) Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer, 107, 1776–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkbak N.J. et al. (2012) Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov., 2, 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeva V. et al. (2012) Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics, 28, 423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman R.L. et al. (2019) Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N. Engl. J. Med., 381, 2403–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H. et al. (2017) HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med., 23, 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goundiam O. et al. (2015) Histo-genomic stratification reveals the frequent amplification/overexpression of CCNE1 and BRD4 genes in non-BRCAness high grade ovarian carcinoma. Int. J. Cancer, 137, 1890–1900. [DOI] [PubMed] [Google Scholar]
- Gulhan D.C. et al. (2019) Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet., 51, 912–919. [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S. et al. (2016) Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature, 534, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P. et al. (2017) A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet., 49, 1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popova T. et al. (2012) Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res., 72, 5454–5462. [DOI] [PubMed] [Google Scholar]
- Staaf J. et al. (2019) Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study. Nat. Med., 25, 1526–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztupinszki Z. et al. (2018) Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer, 4, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Roy N. et al. (2017) Shallow whole genome sequencing on circulating cell-free DNA allows reliable noninvasive copy-number profiling in neuroblastoma patients. Clin. Cancer Res., 23, 6305–6314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.