

Abstract
Summary
Most eukaryotic genes produce alternative polyadenylation (APA) isoforms. APA is dynamically regulated under different growth and differentiation conditions. Here, we present a bioinformatics package, named APAlyzer, for examining 3′UTR APA, intronic APA and gene expression changes using RNA-seq data and annotated polyadenylation sites in the PolyA_DB database. Using APAlyzer and data from the GTEx database, we present APA profiles across human tissues.
Availability and implementation
APAlyzer is freely available at https://bioconductor.org/packages/release/bioc/html/APAlyzer.html as an R/Bioconductor package.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Most protein-coding genes in eukaryotes produce alternative polyadenylation (APA) isoforms (Gruber and Zavolan, 2019; Tian and Manley, 2017). APA events in 3′UTRs alter 3′UTR size and content, whereas those in introns additionally change coding sequences. These two types of APA are dynamically regulated across different cell types and under various biological conditions (Ji et al., 2009; Sandberg et al., 2008; Shepard et al., 2011; Singh et al., 2018; Zhang et al., 2005). While sequencing methods focused on the 3′ end provide precise information about the cleavage and polyadenylation site (PAS) of a transcript, RNA-seq data have proven effective in revealing APA changes (Cass and Xiao, 2019; Guvenek and Tian, 2018; Ha et al., 2018; Katz et al., 2010; Singh et al., 2018; Xia et al., 2014). However, poor PAS annotation and intrinsic limitations of de novo PAS prediction are major challenges. Here, we develop a bioinformatics toolkit, named APAlyzer, which utilizes the comprehensive PAS collection in the PolyA_DB database (http://polya-db.org/polya_db/v3/) (Wang et al., 2017, 2018) to examine APA events in all genic regions, including 3′UTRs and introns.
2 Materials and methods
2.1 Definition of APA sites
Reference PASs in genomes, including those in 3′UTRs and introns, are used by APAlyzer for APA analysis (Fig. 1A). The REF4PAS function converts genomic coordinates of 3′UTR and intronic PASs in the PolyA_DB database to genomic references. Conserved 3′UTR APA sites are used as default due to their high usage levels (Ara et al., 2006; Wang et al., 2018). However, all sites could be used for comprehensive analysis, if required. PolyA_DB currently (version 3) contains PAS coordinates in human, mouse, rat and chicken genomes.
Fig. 1.
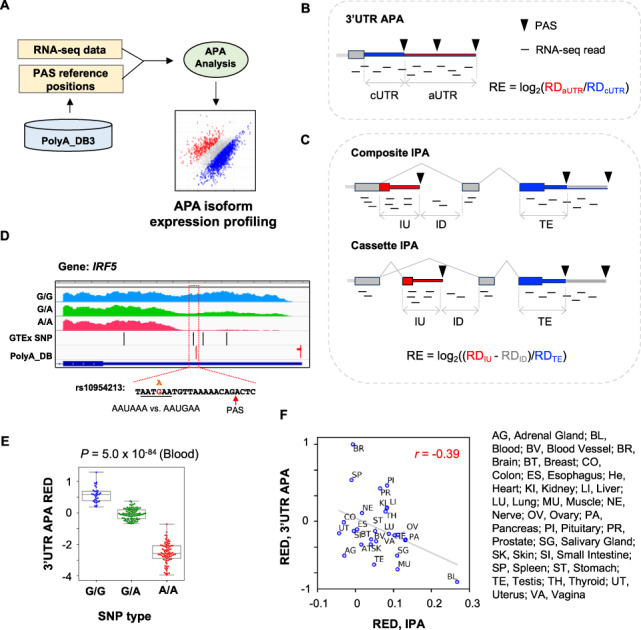
APAlyzer design and examples. (A) Overall design of APAlyzer. (B) Schematic of 3′UTR APA analysis. RE, relative expression between two APA regions; RD, read density. Constitutive regions are in blue, and alternative regions in red. (C) Schematic of IPA analysis. Two IPA types are shown, namely, composite IPA and cassette IPA. Note that only the constitutive region of the 3′ terminal exon (shown in blue) is used for calculation, avoiding complications from 3′UTR APA. (D) An example of 3′UTR APA in human IRF5 gene that is affected by an SNP. RNA-seq data are segregated by three SNP types. SNPs and PASs are indicated. The SNP rs10954213 changes the PAS signal AAUAAA, as indicated. RNA-seq data from blood samples were analyzed. (E) IRF5 3′UTR REDs for three SNP populations. RED was calculated using the median of all samples as reference. P-value was based on the Kolmogorov–Smirnov test between G/G and A/A populations. (F) Scatter plot showing correlation between 3′UTR APA REDs and IPA REDs across human tissues. REDs are REs standardized across all samples. Tissue names are indicated. 3′UTR APA REDs and IPA APA REDs across human tissues were based on the GTEx data. IPA REDs were based on composite and cassette IPA events combined (Supplementary Fig. S1B and C). (Color version of this figure is available at Bioinformatics online.)
2.2 3′UTR APA
For 3′UTR PASs, the PASEXP_3UTR function uses the first and last PASs in the 3′UTR (last exon only) of each gene for APA analysis (Fig. 1B and Supplementary Text S1). The RNA-seq read density (RD, mapped reads divided by region length) for the region between stop codon and the first PAS, also called constitutive 3′UTR or cUTR, is calculated; so is the RD for the region between the first and the last PASs, also called alternative 3′UTR or aUTR (Fig. 1B). 3′UTR APA of each gene is represented by a Relative Expression (RE) score, which is calculated by log2(RDaUTR/RDcUTR) (Fig. 1B). Note that users can also set read count cutoffs using the CUTreads option in the APAdiff function to filter out genes that have a small number of reads mapped in cUTRs or aUTRs. This may be desirable to reduce noise that stems from genes with low read coverages.
2.3 Intronic APA
The PASEXP_IPA function is used to analyze intronic polyadenylation (IPA, Fig. 1C and Supplementary Text S2). An IPA RE is calculated by using log2((RDIU - RDID)/RDTE), where RDIU is read density of intronic upstream region of IPA site, RDID is read density of intronic downstream region of IPA site and RDTE is read density of the constitutive region in 3′ terminal exon (Fig. 1C). Note that RDIU is distinct for the two IPA subtypes, namely, composite IPA and cassette IPA (Fig. 1C). The use of RDID is to address potential influence of intron retention, which is regulated by alternative splicing. An IPA event is discarded if RDIU < RDID. Similar to 3′UTR APA analysis, users can set IU and TE read count cutoffs. Genomic positions for IPA sites, 3′ terminal exons, 5′ splice sites and 3′ splice sites are provided in the package, based on RefSeq and Ensembl databases (including mm9, mm10, hg19 and hg38). Users can also generate PAS information from other sources using the PAS2GTF function in the package.
2.4 Differential APA analysis
The APA difference of a gene between two samples or sample sets is represented by RE difference (RED). The APAdiff function can use different statistical methods depending on the experimental design to assess APA significance. By default, the Fisher’s exact test is used for a nonreplicate design, and a t-test is used for a replicate design. P-value < 0.05 is used to call significance in both cases. To address the multiple testing issue, users can choose Bonferroni or Benjamini and Hochberg method to adjust P-values, using the option p_adjust_methods. Based on REDs and P-values, APAlyzer reports APA regulation as follows: for 3′UTR APA, ‘UP’ indicates 3′UTR lengthening and ‘DN’ 3′UTR shortening; for IPA, ‘UP’ indicates IPA activation and ‘DN’ IPA suppression. ‘NC’ indicates no significant change. Both boxplots (APABox function) and volcano plots (APAVolcano function) can be used to visualize the result.
2.5 Gene expression analysis
APAlyzer uses protein-coding sequence reads to calculate gene expression changes to avoid the confounding situation where the same reads are used for both expression calculation and APA analysis (Supplementary Text S3). The related function is GENEXP_CDS.
3 Examples
3.1 APA of human gene IRF5 in differen SNP populations
As an example, we used APAlyzer to examine APA of human IRF5 in different populations based on the single nucleotide polymorphism (SNP) rs10954213. This SNP was previously shown to affect IRF5 expression through APA (Graham et al., 2007) and, importantly, it is associated with the risk of systemic lupus erythematosus (Graham et al., 2007). Consistent with a previous report (Graham et al., 2007), using the GTEx data of blood samples (phs000424.v7 from dbGaP) (GTEx Consortium et al., 2017), we found that samples from individuals with the genotype A/A showed a much lower RNA-seq read density in the aUTR relative to cUTR as compared to those with G/G or G/A genotypes (Fig. 1D). This is well represented by 3′UTR APA REDs (using RE median of all samples as reference)(Fig. 1E).
3.2 3'UTR and intronic APA profiles across human tissues
We also systematically examined APA across human tissues using 5032 RNA-seq samples from GTEx (GTEx Consortium et al., 2017). The median 3′UTR APA REDs and IPA REDs of each sample were used to represent the global 3′UTR APA and IPA trends for each sample, respectively. Consistent with previous reports (Zhang et al., 2005), brain and blood samples, respectively, showed the highest expression levels of long 3′UTR isoforms and short 3′UTR isoforms, as indicated by their 3′UTR REDs (Fig. 1F and Supplementary Fig. S1A). In addition, we found that composite IPA and cassette IPA events were generally correlated across tissues (r = 0.84, Pearson correlation, Supplementary Fig. S1B–D), indicating similar mechanisms governing both. Notably, consistent with previous reports (Singh et al., 2018; Zhang et al., 2005), blood samples had substantially higher IPA isoform expression for both types than other tissues (Fig. 1F and Supplementary Fig. S1B–C). Using combined IPA profiles (composite IPA + cassette IPA), we observed a modest negative correlation between 3′UTR APA and IPA (r = −0.39, Pearson correlation, Fig. 1F). This result indicates that while 3′UTR APA and IPA in general are related, additional mechanisms, splicing activity for example, may contribute to their distinct regulation in specific tissues. Notably, brain and blood appear to be the most important drivers for the correlation, highlighting their unique APA mechanisms.
4 Conclusions
APAlyzer is a toolbox for APA analysis. In its current version, APAlyzer examines APA by using RNA-seq data based on PASs annotated in the PolyA_DB database. APAlyzer is distinct from other existing tools in many aspects (summarized in Supplementary Table S1), such as analysis of IPA events. We note that because different tools have their own strengths and weaknesses (see comparison of APAlyzer with Roar in Supplementary Fig. S2), users may want to try multiple programs in their research to achieve good sensitivity and specificity.
Supplementary Material
Acknowledgements
The authors thank the members of BT lab for helpful discussions and testing of the program.
Funding
This work was funded by the National Institutes of Health [GM084089 and GM129069 to B.T.].
Conflict of Interest: none declared.
References
- Ara T. et al. (2006) Conservation of alternative polyadenylation patterns in mammalian genes. BMC Genomics, 7, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cass A.A., Xiao X. (2019) mountainClimber identifies alternative transcription start and polyadenylation sites in RNA-seq. Cell Syst., 9, 393–400.e396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R.R. et al. (2007) Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proceedings of the National Academy of Sciences, 104, 6758–6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber A.J., Zavolan M. (2019) Alternative cleavage and polyadenylation in health and disease. Nat. Rev. Genet., 20, 599–614. [DOI] [PubMed] [Google Scholar]
- GTEx Consortium et al. (2017) Genetic effects on gene expression across human tissues. Nature, 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvenek A., Tian B. (2018) Analysis of alternative cleavage and polyadenylation in mature and differentiating neurons using RNA-seq data. Quant. Biol., 6, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha K.C.H. et al. (2018) QAPA: a new method for the systematic analysis of alternative polyadenylation from RNA-seq data. Genome Biol., 19, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z. et al. (2009) Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc. Natl. Acad. Sci. USA, 106, 7028–7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y. et al. (2010) Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods, 7, 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R. et al. (2008) Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science (New York, N.Y.), 320, 1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard P.J. et al. (2011) Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA (New York, N.Y.), 17, 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh I. et al. (2018) Widespread intronic polyadenylation diversifies immune cell transcriptomes. Nat. Commun., 9, 1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B., Manley J.L. (2017) Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol., 18, 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. et al. (2017) PolyA_DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res., 46, D315–D319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. et al. (2018) A compendium of conserved cleavage and polyadenylation events in mammalian genes. Genome Res., 28, 1427–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z. et al. (2014) Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat. Commun., 5, 5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. et al. (2005) Biased alternative polyadenylation in human tissues. Genome Biol., 6, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.