

Abstract
Summary
Fully realizing the promise of personalized medicine will require rapid and accurate classification of pathogenic human variation. Multiplexed assays of variant effect (MAVEs) can experimentally test nearly all possible variants in selected gene targets. Planning a MAVE study involves identifying target genes with clinical impact, and identifying scalable functional assays for that target. Here, we describe MaveQuest, a web-based resource enabling systematic variant effect mapping studies by identifying potential functional assays, disease phenotypes and clinical relevance for nearly all human protein-coding genes.
Availability and implementation
MaveQuest service: https://mavequest.varianteffect.org/. MaveQuest source code: https://github.com/kvnkuang/mavequest-front-end/.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Driven by the advancement of genomic sequencing technologies, and by rapid increases in the number of identified disease-related genes and variants (Brunham and Hayden, 2013), clinical genetic testing is gaining increasingly broad use. An accompanying challenge is the frequent occurrence of (often extremely rare) variants that are difficult to interpret (Blazer et al., 2015). In ClinVar, a popular resource for submitting genetic variants seen in clinical settings, approximately 40% of all variants are missense variants (Landrum et al., 2016). Unfortunately, the majority of missense variants in ClinVar are now classified as ‘variants of uncertain significance’ (VUS) (Starita et al., 2017; Weile and Roth, 2018), which makes any corresponding genetic tests not ‘clinically valid’ (Hoffman-Andrews, 2017), where clinical validity is defined by the extent to which a genetic test reveals a patient’s clinical phenotype or risk (Burke, 2014; Holtzman and Watson, 1999). Many purely computational methods, such as Polyphen-2 (Adzhubei et al., 2010), have been established for predicting the functional effect of given variants. However, experimental functional assays can detect far more disease-associated variants with high confidence than can computational approaches (Sun et al., 2016). Functional evidence is also considered important under the American College of Medical Genetics and Genomics/Association for Molecular Pathology guidelines (Richards et al., 2015), and thus could help shift many VUS variants to more clinically useful categories (e.g. pathogenic or benign). However, conventional functional assays, such as complementation (Osborn and Miller, 2007), are often resource-intensive, and results from such assays are not generally available for rare clinical VUS variants.
Multiplexed assays of variant effect (MAVEs) provide a systematic, experimental approach to study nearly all missense variants in selected gene targets (Starita et al., 2017). Indeed, some variant effect maps have been shown to outperform smaller-scale validated in vitro functional assays in quantitatively predicting disease phenotypes (Sun et al., 2020).
The growing interest in MAVE studies (Weile and Roth, 2018) has presented bioinformatic challenges unique to the early planning stage. For example, to explore the clinical relevance of potential target genes and to identify scalable functional assays for these genes, information must be assembled from multiple database and literature resources. Here, we developed MaveQuest, a web-based service simplifying access to diverse aggregated information about potential functional assays, disease phenotypes and clinical relevance of genes for systematic variant effect mapping.
2 The database
The current version of the MaveQuest database curates literature for information related to 19 200 human genes from the Human Genome Organization’s Gene Nomenclature Committee collection (Braschi et al., 2019). Of these genes, MaveQuest identified cellular phenotypes (each having the potential to enable a scalable functional assay) for 18 979 genes, disease phenotypes for 8460 genes and evidence of clinical relevance for 5203 genes. Figure 1A presents the three categories of data sources that were included in the MaveQuest database.
Fig. 1.
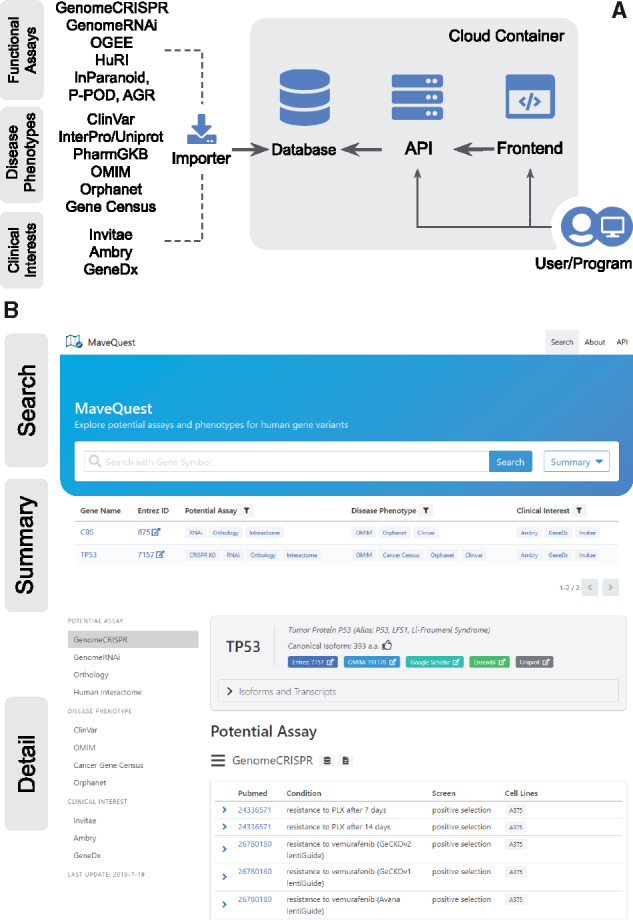
The architecture of MaveQuest. (A) Data from other sources were parsed and imported into the MaveQuest database and are retrieved by the API or the front-end user interface. (B) Three major components of the MaveQuest front-end service
The first data category points the user to potential functional assays. GenomeCRISPR (Rauscher et al., 2017), GenomeRNAi (Schmidt et al., 2013) and the Online Gene Essentiality (Chen et al., 2017) lead for human cell-based phenotypes that could form the basis of a scalable assay. The Human Reference Interactome Mapping project (Luck et al., 2020) provides information on assays to identify variants that ablate specific protein interactions or generally reduce protein folding or stability. Data from InParanoid (Sonnhammer and Östlund, 2015), P-POD (Heinicke et al., 2007) and Alliance of Genome Resources (Howe et al., 2018) databases identify orthologs in non-human species, with links that allow the user to explore whether there are phenotypes associated with disruption of these orthologous genes that might be complemented by human genes to yield a scalable functional assay.
The second category provides data on disease phenotypes with which the query gene has been associated. ClinVar (Landrum et al., 2018) provides clinically-interpreted variants reported for the query gene, which we can visualize to highlight regions enriched for pathogenic or benign variants, together with secondary structures, protein domains and families extracted from InterPro (Mitchell et al., 2019) and Uniprot (UniProt Consortium, 2019) databases. To enable users to further evaluate the clinical significance of query genes, Online Mendelian Inheritance in Man (Hamosh et al., 2005), Orphanet (INSERM, 1997), COSMIC Cancer Gene Census (Sondka et al., 2018) and PharmGKB (Whirl-Carrillo et al., 2012) databases summarize disease- and/or drug-related phenotypes, their mode of inheritance and, in some cases, molecular mechanisms.
The third category contains sequencing panels from three clinical genetic testing providers, Invitae, Ambry and GeneDx, who have each contributed many variant interpretations to ClinVar. The presence of a query gene in clinical genetic sequencing panels from multiple providers suggests clinical interest.
3 The application programming interface
The application programming interface (API) serves as an intermediate between the database and the front-end web application. The API, based on the RESTful standard (Richardson et al., 2007), can be accessed directly using any common programming language. The API currently provides six functions (Supplementary Table S1) that could be further integrated with other MAVE resources as they emerge, e.g. MaveDB (Esposito et al., 2019.)
4 The front-end web application
The front-end interface—enabling queries related to functional assays, disease phenotypes and clinical interests—contains three components (Fig. 1B). The first component, which also serves as the starting page, is a search panel that allows users to look up genes using identifiers. The second component is the gene summary page which lists cell-based phenotypes, disease phenotypes and evidence of clinical interest for each query gene. When the user has searched for partial matches, this information is included for all matching genes. Users can select a specific gene to bring up a detail page. This is the third component, which contains all data in the database associated with that gene. The detail page includes an overview of variants in ClinVar database when available, displaying the distribution of single-nucleotide variants along the protein sequence. Secondary structures, protein domains and families are also visualized for users to identify potential ‘variational hotspots’ (i.e. regions where variants are enriched). This feature is particularly useful for studying large proteins, allowing prioritization of regions harboring more pathogenic or benign variants.
Supplementary Material
Acknowledgements
The authors appreciate the help from members of the Roth Lab and others who tested MaveQuest and provided valuable feedback at various stages.
Funding
This work was supported by the National Human Genome Research Institute of the National Institutes of Health Center of Excellence in Genomic Science [HG004233 and HG010461]; the Canada Excellence Research Chairs Program; a Canadian Institutes of Health Foundation grant and the One Brave Idea Foundation.
Conflict of Interest: none declared.
References
- Adzhubei I.A. et al. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazer K.R. et al. (2015) Next-generation testing for cancer risk: perceptions, experiences, and needs among early adopters in community healthcare settings. Genet. Test. Mol. Biomarkers, 19, 657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braschi B. et al. (2019) Genenames.org: the HGNC and VGNC resources in 2019. Nucleic Acids Res., 47, D786–D792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunham L.R., Hayden M.R. (2013) Hunting human disease genes: lessons from the past, challenges for the future. Hum. Genet., 132, 603–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke W. (2014) Genetic tests: clinical validity and clinical utility. Curr. Protoc. Hum. Genet., 81, 9.15.1–9.15.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.-H. et al. (2017) OGEE v2: an update of the online gene essentiality database with special focus on differentially essential genes in human cancer cell lines. Nucleic Acids Res., 45, D940–D944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito D. et al. (2019) MaveDB: an open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome Biol, 20, 1–11. [DOI] [PMC free article] [PubMed]
- Hamosh A. et al. (2005) Online Mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res., 33, D514–D517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinicke S. et al. (2007) The Princeton protein orthology database (P-POD): a comparative genomics analysis tool for biologists. PLoS One, 2, e766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman-Andrews L. (2017) The known unknown: the challenges of genetic variants of uncertain significance in clinical practice. J. Law Biosci., 4, 648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman N.A., Watson M.S. (1999) Promoting safe and effective genetic testing in the United States. Final report of the task force on genetic testing. J. Child Fam. Nurs., 2, 388–390. [PubMed] [Google Scholar]
- Howe D.G. et al. (2018) Model organism data evolving in support of translational medicine. Lab Anim., 47, 277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INSERM. (1997) Orphanet: An Online Database of Rare Diseases and Orphan Drugs http://www.orpha.net (20 March 2020, date last accessed).
- Landrum M.J. et al. (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res., 44, D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum M.J. et al. (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res., 46, D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck K. et al. (2020) A reference map of the human binary protein interactome. Nature. doi: 10.1038/s41586-020-2188-x. [DOI] [PMC free article] [PubMed]
- Mitchell A.L. et al. (2019) InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res., 47, D351–D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn M.J., Miller J.R. (2007) Rescuing yeast mutants with human genes. Brief. Funct. Genomic. Proteomic, 6, 104–111. [DOI] [PubMed] [Google Scholar]
- Rauscher B. et al. (2017) GenomeCRISPR—a database for high-throughput CRISPR/Cas9 screens. Nucleic Acids Res., 45, D679–D686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S. et al. ; On behalf of the ACMG Laboratory Quality Assurance Committee. (2015), Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med., 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson L. et al. (2007) RESTful Web Services. O’Reilly Media, Inc., Sebastopol, CA.
- Schmidt E.E. et al. (2013) GenomeRNAi: a database for cell-based and in vivo RNAi phenotypes, 2013 update. Nucleic Acids Res., 41(Database issue), D1021–D1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondka Z. et al. (2018) The COSMIC cancer gene census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer, 18, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer E.L.L., Östlund G. (2015) InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res., 43(Database issue), D234–D239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starita L.M. et al. (2017) Variant interpretation: functional assays to the rescue. Am. J. Hum. Genet., 101, 315–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S. et al. (2016) An extended set of yeast-based functional assays accurately identifies human disease mutations. Genome Res., 26, 670–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S. et al. (2020) A proactive genotype-to-patient-phenotype map for cystathionine beta-synthase. Genome Med, 12, 1–18. [DOI] [PMC free article] [PubMed]
- UniProt Consortium. (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res., 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weile J., Roth F.P. (2018) Multiplexed assays of variant effects contribute to a growing genotype-phenotype atlas. Hum. Genet., 137, 665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whirl-Carrillo M. et al. (2012) Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther., 92, 414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.