

Abstract
Vascular endothelial growth factor receptor 2 (VEGFR-2) binds to VEGFR-A, VEGFR-C and VEGFR-D and participates in the formation of tumor blood vessels, mediates the proliferation of endothelial cells, enhances microvascular permeability, and blocks apoptosis. Blocking or downregulating the signal transduction of VEGFR is the main way to discover new drugs for many human angiogenesis-dependent malignancies. Mesenchymal epithelial transfer factor tyrosine kinase (c-Met) is a high affinity receptor for hepatocyte growth factor (HGF). Abnormal c-Met signaling plays an important role in the formation, invasion and metastasis of human tumors. Therefore, the HGF/c-Met signaling pathway has become a significant target for cancer treatment. Related studies have shown that the conduction of the VEGFR and c-Met signaling pathways has a synergistic effect in inducing angiogenesis and inhibiting tumor growth. In recent years, multi-target small molecule inhibitors have become a research hotspot, among which the research of VEGFR and c-Met dual-target small molecule inhibitors has become more and more extensive. In this review, we comprehensively summarize the chemical structures and biological characteristics of novel VEGFR/c-Met dual-target small-molecule inhibitors in the past five years.
Keywords: dual VEGFR/c-Met inhibitors, anticancer agents, research progress
1. Introduction
The occurrence of tumor is a multi-stage and complex process which seriously endangers human life and health [1]. Tumor metastasis, growth and survival depend on cell differentiation, proliferation, angiogenesis and apoptosis, which are regulated by a variety of signal transduction pathways and protein kinases [2,3]. At present, cancer therapy that interferes with a single biomolecule or pathway has been successfully applied [4]. However, the problem of drug resistance often arises in the research of single target drugs and combination drugs [4,5]. It is found that multi-target drugs may overcome drug resistance and achieve higher efficacy than single target drugs, which makes the molecules of multi-target drugs widely studied [4,5,6].
The vascular endothelial growth factor (VEGF) pathway is one of the most essential active regulators of angiogenesis. It can promote the proliferation and migration of vascular endothelial cells and induce the formation of blood vessels [7,8]. There are three main vascular endothelial growth factor receptors (VEGFR-1, VEGFR-2 and VEGFR-3), which are the key intermediate products of tumor angiogenesis and new blood vessels and provide nutrition and oxygen for tumor growth [9]. The binding of VEGF with the receptor leads to the homologous or heterogenous dimerization of the receptor, phosphorylation of the kinase area in the cell, activation of many major signaling pathways, and production of many physiological effects [10]. Vascular endothelial growth factor receptor-2 (VEGFR-2) is the main effector of VEGF/VEGFR signal transduction in promoting tumor angiogenesis. It is expressed on the surface of blood vessels and plays a key role in tumor angiogenesis [11,12]. The phosphorylation of VEGFR-2 activates the Raf-1/MAPK/ERK signaling pathway, which will eventually lead to angiogenesis, enhanced vascular permeability, tumor proliferation and tumor migration [13]. Therefore, inhibition of the VEGFR-2/VEGF signaling pathway is considered to be one of the most eventful and valuable pathways in the development of tumor chemotherapy [10]. At present, a number of VEGFR-2 inhibitors approved by the Food and Drug Administration (FDA) are used as chemotherapy drugs in clinical cancer treatment [14]. However, drug resistance leads to decreased efficacy and increased toxicity, resulting in unnecessary side effects [15]. Therefore, the treatment of tumors with VEGFR inhibitors alone was limited.
Mesenchymal epithelial transfer factor tyrosine kinase (c-Met) is a crucial member of the receptor tyrosine kinases (RTKs) family [16,17,18]. In normal cells, c-Met is activated by extracellular binding to its natural ligand, hepatocyte growth factor/scatter factor (HGF/SF) [19]. Many human cancers involve abnormal expression of HGF/SF or c-Met or activation of c-Met kinase mutations. The aberrant expression of c-Met/HGF signaling arises from c-Met mutations, c-Met/HGF overexpression or c-Met genomic amplification, which can promote the proliferation, migration, invasion and genesis of tumors [18,20,21]. Blocking the abnormal activation of c-Met activity is a promising method for the treatment of cancer caused by c-Met activity. Therefore, the HGF/c-Met signaling pathway has become an attractive target for tumor therapy [22,23]. At present, most small molecule inhibitors that interfere with the active site of the kinase domain have been found to be competitive inhibitors of ATP, which could block the transmission of the c-Met signaling pathway by blocking the phosphorylation of tyrosine [24]. According to their structures and binding modes with the c-Met kinase domain, small molecule inhibitors can be roughly divided into type I and type II [18,25]. Studies have shown that type I c-Met inhibitors are more selective than type II c-Met inhibitors, but type II inhibitors may be more effective than type I inhibitors since most type II inhibitors are multi-kinase inhibitors, which also have strong inhibition on VEGFR and other homologous kinases [26,27]. There is an obvious structural feature of type II c-Met inhibitors of ’5-atom regulation‘ [28,29,30]. Therefore, receptor tyrosine kinase c-Met is considered to be an essential target for the discovery of small molecule anticancer inhibitors [31,32]. The biochemical pathways of various cancers can be inhibited by drug combinations or single chemical entities with different mechanisms, which can regulate multiple targets of multifactor diseases. Multiple target inhibitors can block the transduction of multiple signal pathways, which may have a satisfactory effect on tumor therapy [33,34,35,36,37]. Thus, multi-target inhibition combined with other therapies will become a favorable approach for future clinical antitumor treatment. Among them, antitumor therapy, which is based on dual-target VEGFR and c-Met tyrosine kinase inhibitors, is a promising approach for tumor treatment [36,37,38,39,40]. The synergistic collaboration of VEGFR and c-Met promotes the development of angiogenesis and the progression of various human cancers. Thus, dual-target VEGFR/c-Met tyrosine kinase inhibitors may have more extensive advantages than VEGFR or c-Met selective inhibitors alone [37,40]. A large number of multi-kinase inhibitors have been found and applied to cancer treatment or scientific research. As shown in Table 1, these compounds, such as Foretinib, Golvatinib and Dovitinib, can act on multiple targets and have high cytotoxic activity and kinase selectivity. At present, most of the compounds have entered into clinical trials to further evaluate their pharmacodynamic (PD) and pharmacokinetic (PK) properties, in order to explore their mechanism of action and evaluate their efficacy in vivo, side effects and drug resistance. These compounds exhibited slight inhibitory effects against VEGFR and c-Met kinases, so their active skeletons are worthy of further study and may have a positive effect on the development of small anticancer inhibitors of dual-target VEGFR/c-Met kinase. Studies show that VEGFR and c-Met targeting pathways exhibit synergistic effects in inducing angiogenesis and the development of various human cancers (Figure 1). It is crucial to inhibit multiple signaling pathways, including VEGF and c-Met, to improve antitumor efficacy and overcome drug resistance [41,42]. Therefore, dual-target c-Met/VEGFR inhibitors are considered to be a promising tumor treatment method, which may be superior to c-Met selective or VEGFR selective single target inhibitors alone [43,44]. The purpose of this review is to summarize the chemical structures and biological characteristics of VEGFR/c-Met dual-target inhibitors reported in the past five years, which will help researchers to design new dual-target VEGFR/c-Met dual small molecule inhibitors.
Table 1.
Multi-target vascular endothelial growth factor receptor/mesenchymal epithelial transfer factor tyrosine kinase (VEGFR/c-Met) inhibitors in clinical trials.
| Compounds | Structural Formula | Target | Indication | Phase | Signaling Pathway | Research and Development Company |
|---|---|---|---|---|---|---|
| Foretinib [45] |
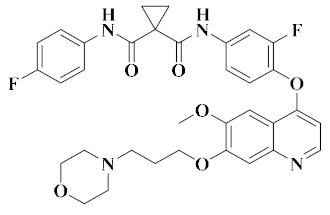
|
c-Met, VEGFR-2 (KDR), Tie-2, VEGFR-3/FLT4 | Gastric cancer and head/neck cancer | II | Protein tyrosine kinase | Exelixis |
| Golvatinib [46] |
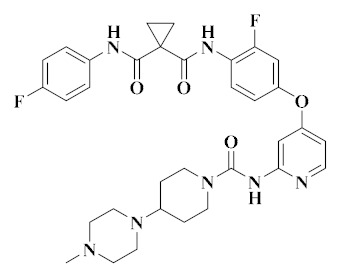
|
c-Met, VEGFR-2 | Head and neck cancer, liver cancer | II | Angiogenesis; protein tyrosine kinase | Eisai |
| Dovitinib [47] |
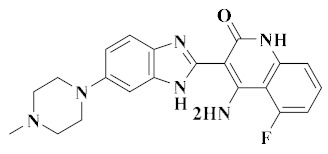
|
FLT3, c-Kit, FGFR-1/3, VEGFR1-4, EGFR, c-Met | Solid tumors | IV | Angiogenesis | Novartis |
| Tivozanib [48] |
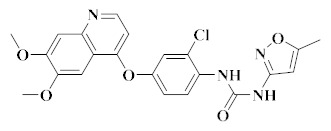
|
VEGFR-1, VEGFR-2, VEGFR-3, c-Met, PDGFR, c-Kit | Advanced renal cell carcinoma | III | Angiogenesis | Aevo |
| BMS-794833 [49] |
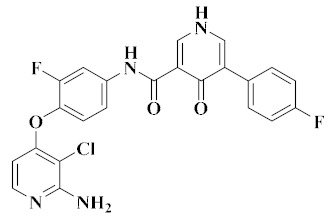
|
c-Met, VEGFR-2, Ron, Axl, FLT3 | Gastric cancer | I | Angiogenesis; protein tyrosine kinase | Bristol Myers Squibb |
| BMS-777607 [50] |
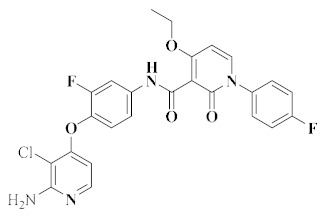
|
c-Met, Axl, Ron, VEGFR-2, lck | Advanced solid tumors | II | Protein tyrosine kinase | Bristol Myers Squibb |
| MGCD-265 [51] |
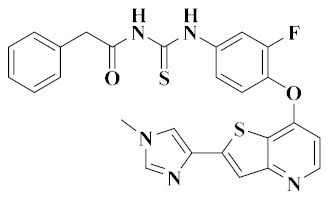
|
c-Met, Ron, VEGFR-1, VEGFR-2 | Non-small cell lung cancer | II | Angiogenesis; protein tyrosine kinase | MethylGene |
| AC480 [52] |
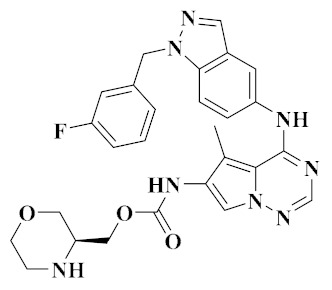
|
HER1, HER2, HER4, VEGFR-2, c-Kit, Lck, MET | Advanced solid tumors | I | Angiogenesis | Ambit Biosciences |
| CP-724714 [53] |
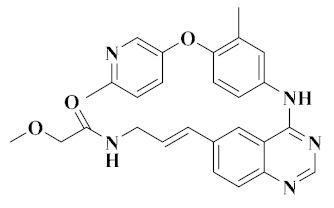
|
HER2/ErbB2, EGFR, VEGFR-2, c-Met | Advanced solid tumors | II | Protein tyrosine kinase | Pfizer |
| AMG-458 [54] |
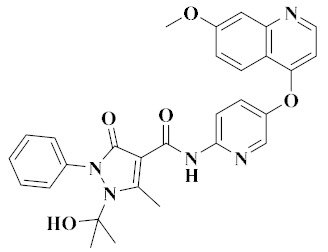
|
c-Met, VEGFR-2 | Solid tumors | Non-medicinal | Protein tyrosine kinase | Amgen |
Figure 1.

The crosstalk between VEGFR and c-Met (GAB1: GRB2-associated-binding protein 1; GRB2: Growth factor receptor-bound protein 2; SOS: Son of sevenless; RAS: Rat sarcoma viral oncogene homolog; RAF: Root abundant factor; FAK:Focal adhesion kinase; PI3K/AKT: Phosphoinositide 3-kinase/protein kinase B; mTOR:Mammalian target of rapamycin/mechanistic target of rapamycin.).
2. Chemical Design Strategies of Dual-Target VEGFR/c-Met Kinase Anticancer Inhibitors
Dual-target VEGFR/c-Met kinase inhibitors are designed by combining the pharmacophores or fragments of the leading compounds acting on VEGFR and c-Met kinase targets into a molecule and modifying their skeletons. In this way, the resulting compounds have active pharmacophores or fragments for targeting VEGFR and c-Met kinases, and may have high inhibitory activity towards them [44,55]. As shown in Figure 2, the pharmacophores of VEGFR inhibitors and the pharmacophores of c-Met inhibitors are combined to form new dual-target VEGFR/c-Met inhibitors through the principles of skeleton and bioelectronic exclusion principle. They contain the main structural features of two kinase drugs, which bind to the protein sequence of VEGFR and c-Met kinases or the binding site of ligands respectively, thus blocking the signaling and bypassing pathways of VEGFR and c-Met [41,56,57]. Therefore, the development of novel dual-target VEGFR/c-Met kinase inhibitors is of great significance for future cancer treatment.
Figure 2.

Design of dual-target VEGFR/c-Met kinase inhibitors (PDB code: 3U6J, 3LQ8).
Most of the active pharmacophores of VEGFR are heterocycles containing nitrogen and sulfur atoms, such as quinolines, pyridines, thiophenes and pyridines, which are great structural fragments for hydrogen bonding with the amino acid residues of the kinase [43,58]. The active fragment of c-Met meets the ‘5-atom regulation’ of type II c-Met, which can be a flexible chain or rigid ring structure containing one or more hydrogen bond donors or receptors, so as to facilitate the formation of a hydrogen bond with the amino acid residues of the kinase [59,60]. Most of the dual-target VEGFR/c-Met inhibitors have the active pharmacophore of selective VEGFR inhibitors and the ‘5-atom’ active fragment of c-Met inhibitors so that the dual-target VEGFR/c-Met inhibitors can penetrate the ATP binding vesicles of VEGFR and c-Met receptors (PDB code: 3U6J, 3LQ8), and occupy the ATP pocket of VEGFR and c-Met binding through hydrogen bonding [57,60]. The skeleton modification of the obtained double target kinase inhibitors may be an effective way to obtain small molecular compounds, reduce toxicity and improve metabolic stability and water solubility. According to the structure of the parent cores, 11 derivatives are summarized, such as pyridines, quinolones, pyrrolopyridines, benzimidazoles and thienopyrimidines.
3. Dual VEGFR and c-Met Small Molecule Inhibitors
3.1. Pyridine Derivatives
Pyridine skeletons are widely used in VEGFR inhibitors and c-Met inhibitors. In particular, the pyridine nucleus in receptor tyrosine kinase inhibitors can extend into the ATP binding pocket of VEGFR-2 and c-Met receptors, and the nitrogen atom of the pyridine ring can interact with the amino acid residues in the hinge region. The pyridine skeleton plays a key role in the activity of small molecule antitumor inhibitors.
In 2016, Hao et al. introduced a series of aminopyrimidine derivatives and assayed their enzymatic activities against VEGFR-2 and c-Met kinases. Most target compounds have inhibition potency both on VEGFR-2 and c-Met with IC50 values in the nanomolar range, especially compound 1 (Figure 3). The substitution of compound 1 (VEGFR-2: IC50 = 0.170 ± 0.055 µM, c-Met: IC50 = 0.210 ± 0.030 µM) with a dimethylamino group was better than that by pyrrolidine and morpholine. As shown in Figure 4, it was found that the pyrimidine of 4-aminopyrimidine-5-formoxime could bind to the pocket of ATP in VEGFR-2 and c-Met receptor proteins. CYS-919 of the VEGFR-2 hinge region and MET-1160 of the c-Met hinge region can form hydrogen bonds with the nitrogen atoms of pyrimidine [36]. The results indicated that compound 1 was a dual inhibitor of VEGFR-2 and c-Met that holds promising potential.
Figure 3.

Pyridine derivatives of dual VEGFR/c-Met inhibitors.
Figure 4.

(A) The model of compound 1 binding to VEGFR-2 (PDB code: 3U6J); (B) The model of compound 1 binding to c-Met (PDB code: 3LQ8).
A series of pyrimidine derivatives have been reported and half of the target compounds have been evaluated to exhibit moderate to potent c-Met inhibitory activities. Among these, it was noteworthy that compound 2 (Figure 3) showed most potent c-Met (IC50 = 17 nM) inhibitory potency. In particular, compound 2 significantly decreased the selectivity for VEGFR-2 (IC50 = 55 nM) kinase, which may provide an opportunity to investigate the further selectivity and reduce VEGFR-2 related side effects [19]. The results suggested that compound 2 could be a potentially interesting lead compound for the development of anticancer agents. Compared with compound 1, the substitution of halogen chloride in the side chain of the pyridine ring has more significant kinase inhibitory activity than the long chain of compound 2.
In our previous studies, Wang et al. screened out four series of Sorafenib derivatives bearing pyrazole scaffolds and evaluated them for cytotoxicity, and some compounds were further evaluated for kinase activities. In particular, the most promising compound, compound 3 (Figure 3), exhibited the best activity against A549, HepG2 and MCF-7 cell lines. The IC50 value of compound 3 on VEGFR-2 kinase was 0.56 µM. Compound 3 showed moderate to no activity against CRAF, c-Met, EGFR and FLT3 kinases. Through molecular docking simulation studies, it was found that compound 3 formed hydrogen bonds with VEGFR-2 amino acid residues CYS-919, ASP-1046 and GLU-885 [61]. Similarly, new hydrogen bonds also played a central role in improving the inhibition of pyrazole derivatives on c-Met kinase. As a potential inhibitor of VEGFR-2/c-Met kinases, further study is needed to determine its mechanism.
A series of pyridine derivatives with pyrazolone skeletons were evaluated. The cell proliferation assay in vitro showed that most of the target compounds had inhibition potency towards both VEGFR-2 and c-Met. Compound 4 (Figure 3) showed the greatest inhibitory activities against BaF3-TPR-Met and HUVEC cancer cell lines with IC50 values of 0.30 µM and 0.13 µM, respectively. A triazole ring formed by the cyclization of pyridylamide can improve the anti-proliferation activity of the compound, and the tertiary amine substituent shows better anti-proliferation activity, indicating that the tertiary amine has good tolerance in maintaining strong inhibitory activity. This compound also exhibited the most potent inhibitory activity against VEGFR-2 and c-Met with IC50 values of 0.19 µM and 0.11 µM, respectively. Molecular docking of compound 4 (Figure 5) into the ATP binding sites of c-Met and VEGFR-2 was performed and the result suggested that compound 4 could bind well with the active site of c-Met (MET-1160 and ASP-1222) and VEGFR-2 (CYS-919 and ASP-1046) [5]. These results indicated that compound 4 has become a promising candidate for further development of more potent dual-target c-Met/VEGFR-2 kinase inhibitors.
Figure 5.

Docking of compound 4 with VEGFR-2 (PDB code: 3U6J) and c-Met (PDB code: 3LQ8) (Most dual-target VEGFR/c-Met inhibitors can be divided into four parts: A, B, C and D. Part A: The main structural skeleton, mainly nitrogen-containing heterocycles. Part B: Substituted benzene ring; Part C: Meet the ‘5-atom regulations’. Part D; Benzene ring).
Pyridine derivatives bearing 1,6-naphthyridine scaffolds were scrutinized by Wang et al. Based on the scaffold-hopping strategy of heteroatom migration, the optimal compound 5 (MET: IC50 = 9.8 nM, VEGFR-2: IC50 = 8.8 nM) was designed. Further molecular docking showed that the cyclopropane carbonyl from compound 5 formed a good hydrophobic interaction with c-Met kinase amino acid residues TRY-1159 and LSY-1611. For VEGFR-2 kinase, the smaller steric cyclopropane carbonyl of compound 5 can be well tolerated and has a favorable pharmacokinetic profile (F = 63%, CL = 0.12 L/h/kg, and AUC0–∞ = 42.2 h µg/mL) [62]. The results showed that a 1,6-naphthidine scaffold could be used as a new scaffold for kinase inhibitors.
In study by Zeidan, two series of picolinamide derivatives bearing (thio)urea and dithiocarbamate were reported. All compounds were evaluated for their cytotoxic activity against A549 cancer cells. Compound 6 (Figure 3) showed enhanced potency towards VEGFR-2 (IC50 = 0.027 µM), c-Met (IC50 = 8.300 µM), EGFR (IC50 = 0.084 µM), HER-2 (IC50 = 0.104 µM), and MER (IC50 = 3.950 µM) kinases. Cell cycle analysis of A549 cells treated with compound 6 showed cell cycle arrest at the G2/M phase and pro-apoptotic activity [63]. Therefore, compound 6 deserves further study.
With the development of related research, new antiangiogenic drugs have been found. Biological results showed that compound 7 (Figure 3) showed significant inhibition of VEGFR-2 (IC50 = 2.35 nM), Tie-2 and EphB4 kinases. Compound 7 showed good selectivity for VEGFR-1 (IC50 = 25.00 nM), VEGFR-3 (IC50 = 45.00 nM) and c-Met (IC50 = 57.80 nM) kinases (Table 2) and was found to be a multi-target inhibitor. Biological evaluation and molecular docking showed that N-(pyridin-2-yl)acrylamide was beneficial to the potency of these triple inhibitors [64]. Compound 7 is a multi-kinase inhibitor with good kinase selectivity, and its role in the research of anticancer drugs is of great significance.
Table 2.
RTKs selectivity profile of compound 7.
| Compound | IC50 (nM) | |||||||
|---|---|---|---|---|---|---|---|---|
| VEGFR-2 | c-Met | VEGFR-1 | VEGFR-3 | EGFR | IGF1-R | B-Raf | c-Kit | |
| 7 | 2.35 | 33.12 | 25.00 | 45.00 | 8.70 | 40.00 | 32.00 | 68.90 |
3.2. Quinoline Derivatives
Liu and co-workers used Foretinib as the lead compound and introduced 1,2,4-triazolone into the ‘5-atom’ part to synthesize a series of new 4-phenoxyquinoline derivatives. All compounds showed moderate to excellent cytotoxic activity against different cancer cells. Compound 8 (Figure 6) showed significant antitumor activity against all tested cell lines (HT-29, H460, A549, and MKN-45), especially against the HT-29 cell line (IC50 = 0.08 µM). In addition, compound 8 exhibited a high inhibitory effect on c-Met kinase (IC50 = 1.57 nM), and also showed a strong inhibitory effect on c-Kit (IC50 = 3.38 nM) and FLT3 (IC50 = 8.19 nM). In addition, compound 8 showed moderate selectivity for PDGFRα (IC50 = 95.23 nM), Ron (IC50 = 140.72 nM), and VEGFR-2 (IC50 = 480.47 nM) kinases (Table 3) [65]. The results indicated that compound 8 provided a new scaffold for further selectivity.
Figure 6.

Quinoline derivatives of dual VEGFR/c-Met inhibitors.
Table 3.
Inhibition of tyrosine kinases by compounds 8, 9 and 10.
| Compound | IC50 (nM) | |||||||
|---|---|---|---|---|---|---|---|---|
| c-Met | c-Kit | FLT3 | PDGFRα | Ron | VEGFR | EGFR | ALK | |
| 8 | 1.57 | 3.38 | 8.19 | 95.23 | 140.47 | 480.47 | >10000 | >10000 |
| 9 | 0.09 | 2.45 | 268.81 | 19.13 | 82.56 | 151.52 | 980.83 | 2840.72 |
| 10 | 1.98 | 380 | 400 | 242 | 375 | 689 | >10000 | >10000 |
In another effort, Liu and co-workers also reported a series of novel 4-phenoxyquinoline derivatives containing 3-oxo-3,4-dihydroquinoxaline moieties. The most promising compound, compound 9 (Figure 6), has significant cytotoxicity against HT-29, H460, A54 9, MKN-45 and U87MG cell lines, with IC50 values of 0.06 µM, 0.05µM, 0.18µM, 0.023µM, and 0.66µM, respectively. Preliminary research also showed that compound 9 also showed a high inhibitory effect on c-Met (IC50 = 0.90 nM), c-Kit (IC50 = 2.45 nM) and PDGFRα (IC50 = 19.13 nM). In addition, compound 9 has moderate to excellent selectivity for Ron, VEGFR-2, FLT3, EGFR, and ALK kinases (Table 3) [22]. So, compound 9 remains a multi-target inhibitor of tyrosine kinase. Compared with compound 8, the selectivity of VEGFR-2 kinase was improved when the ‘5-atom’ moiety of compound 9 was 3-oxo-3,4-dihydroquinoline.
During the discovery of potent VEGFR/c-Met kinase inhibitors, Nan et al. reported a series of 4-phenoxyquinoline derivatives containing sulfonylurea and evaluated them for their c-Met kinase inhibition and cytotoxicity against four cell lines. Pharmacological data indicated that most compounds showed moderate to significant potency, with the most promising compound, compound 10 (Figure 6, c-Met: IC50 = 1.98 nM), showing better selectivity over other tyrosine kinases (Table 3). Compound 10 had significant cytotoxicity to HT460, MKN-45, HT-29 and MDA-MB-231 cell lines, with IC50 values of 0.055 μM, 0.064 μM, 0.16 μM and 0.49 μM, respectively. Docking studies show that the introduction of sulfonylurea into the structure maintains the strong cytotoxicity of the compounds [18].
Compound 11 (Figure 6) had good pharmacokinetic properties in vivo, had no obvious AO-mediated metabolite in a xenotransplantation model of H1993 and SNU-5 with high expression of c-Met, and has good antitumor effect. The selectivity of compound 11 was studied with human tyrosine kinases. The results showed that compound 11 had high selectivity to c-Met, and the IC50 values of the other tested tyrosine kinases were higher than 700 nM [66].
3.3. Pyrrolopyridine Derivatives
In our previous studies, Wang et al. screened two series of aromatic hydrazone derivatives bearing a 1H-pyrrolo[2,3-b]pyridine moiety and evaluated them for cytotoxicity; some compounds were further evaluated for kinase activities. Most of the compounds showed moderate to extremely good cytotoxic activity towards four cancer cell lines (A549, HepG2, MCF-7 and PC-3 cells). Among them, the inhibitory activities of compounds 12 (Figure 7) and 13 (Figure 7) were better than those of positive control (Foretinib). Compounds 12 and 13 showed moderate inhibition on c-Met kinase, and the activity of compound 12 (IC50 = 0.506 μM) was slightly better than that of compound 13 (IC50 = 0.907 μM), similar to the cytotoxicity results. The activity data showed that compounds 12 and 13 had selective inhibition on c-Met (IC50 = 0.5 μM, and 0.9 μM) compared with FLT3 (IC50 = 2.6 μM, and 3.8 μM), VEGFR-2 (IC50 = 8.9 μM, and 14.4 μM) and EGFR (IC50 > 100 μM, and IC50 = 45.0 μM) kinases. Therefore, compounds 12 and 13 can be considered as multi-target small molecule inhibitors [67].
Figure 7.

Pyrrolopyridine derivatives of dual VEGFR/c-Met inhibitors.
Wang et al. used Foretinib as the lead compound and a pyrrolo[2,3-b]pyridine moiety as the core, and then introduced 4-oxopyridazinone into the ‘5-atom’ moiety to limit the conformation. Finally, the optimal compound 14 was found. It had significant cytotoxicity against A549 (IC50 = 2.190 μM), HepG2 (IC50 = 1.320 μM), MCF-7 (IC50 = 6.270 μM) and PC-3 (IC50 = 4.630 μM) cell lines in vitro (Table 4), and it showed good kinase selectivity for FLT3 (IC50 = 0.770 μM), VEGFR-2 (IC50 = 2.400 μM), c-Kit (IC50 = 2.200 μM) and EGFR (IC50 > 10.000 μM) kinases (Table 5). Therefore, compound 14 can also be considered as a multi-target small molecule inhibitor [68].
Table 4.
Inhibition of cells by compounds 12, 13 and 14.
| Compound | IC50 (μM) | |||
|---|---|---|---|---|
| A549 | HepG2 | MCF-7 | PC-3 | |
| 12 | 0.82 | 1.00 | 0.93 | 0.92 |
| 13 | 1.30 | 1.42 | 4.53 | 4.29 |
| 14 | 2.19 | 1.32 | 6.27 | 4.63 |
Table 5.
Inhibition of kinases by compounds 12, 13 and 14.
| Compound | IC50 (μM) | ||||
|---|---|---|---|---|---|
| c-Met | VEGFR-2 | c-Kit | FLT3 | EGFR | |
| 12 | 0.5 | 8.9 | 2.6 | >100 | |
| 13 | 0.9 | 14.4 | 3.8 | 45.0 | |
| 14 | 0.073 | 2.4 | 2.2 | 0.77 | >10 |
3.4. Benzimidazole Derivatives
Benzimidazole is a privileged lead nucleus which exists in many anticancer drugs. Moreover, some reports showed that the anticancer activities of some benzimidazole derivatives were mediated by targeting VEGFR-2. Some benzimidazole derivatives, such as benzimidazolyl amino quinoline derivatives, exhibited dual inhibition of VEGFR-2 and c-Met kinases.
Three series of benzimidazole derivatives which were hybridized with piperazine, oxadiazole and triazolo-thiadiazole, respectively, were synthesized. Compound 15 (Figure 8) had good cytotoxic activity on HCT-116 (IC50 = 2.19 ± 0.09 µM) and A549 (IC50 = 10.97 ± 0.09 µM) cells (Table 5). Compound 15 exhibited the best activity against VEGFR-2 and c-Met kinases, with an inhibition percent of 29.22% for VEGFR-2 and 71.66% for c-Met, which were close to those of the reference inhibitor AG-1478 (VEGFR-2: 73.73%, c-Met: 33.12%). The selectivity for c-Met kinase was stronger than that for VEGFR-2 kinase. Molecular simulation studies showed that the thiadiazole ring of compound 15 interacted with the amino acid residue CYS-919 of VEGFR-2 kinase to form a hydrogen bond (Figure 9). The sulfur atom of thiadiazole in compound 15 formed a hydrogen bond interaction with the amino acid residue ASP-1222 of c-Met kinase. The docking results indicated that the addition of triazolothiazole fragments enhanced the binding of compound 15 to the receptor, which may enhance the binding affinity and enhance the antitumor activity of the compound [69]. Therefore, compound 15 may be a good dual-target VEGFR-2/c-Met small molecule inhibitor.
Figure 8.

Benzimidazole derivatives of dual VEGFR/c-Met inhibitors.
Figure 9.

(A,B) The model of compounds 15 (magenta), 16 (green) and 17 (orange) bound to VEGFR-2 (PDB code: 2QU5); (C,D). The model of compounds 15, 16 and 17 bound to c-Met (PDB code: 3CD8).
Ibrahim et al. also reported in the same year that the docking results of compound 16 (Figure 8) were related to its moderate enzyme inhibitory activity on VEGFR-2 and its good inhibitory activity on c-Met (35.88% and 88.48%, respectively), as shown in Table 5. The inhibitory rates of compound 17 (Figure 8) on NCI-H522 and SK-MEL-2 were 48.70% and 42.62%, respectively, and the inhibitory activities on VEGFR-2 and c-Met kinases were 35.88% and 82.48% (Table 6), respectively. The binding mode of compound 16 with VEGFR-2 and c-Met was related to the data from the kinase assay, which indicated that compound 17 is a good dual-target VEGFR-2/c-Met small molecule inhibitor [70].
Table 6.
Inhibition percent of compounds 15, 16 and 17 for VEGFR-2/c-Met kinases.
| Compound | % Inhibition at 10 µM | |
|---|---|---|
| VEGFR-2 | c-Met | |
| 15 | 29.22 | 71.66 |
| 16 | 35.88 | 82.48 |
| 17 | 21.07 | 76.01 |
| AG-1478 | 73.73 | 33.12 |
3.5. Thienopyrimidine Derivatives
The thienopyrimidine skeleton had been widely used in FGFR1 inhibitors, B-Raf inhibitors, FLT3 inhibitors and EGFR kinase inhibitors. In 2017, Li et al. identified a series of thienopyrimidine derivatives as novel dual-target VEGFR-2/c-Met kinase inhibitors. The inhibition rates of compound 18 (Figure 10) on HUVEC and BaF3-TPR-Met cell lines were 79.52% and 95.97%, respectively. Cell analysis showed that the introduction of amides in the solvent area would improve the compounds’ cell cytostatic activity, and that long chain polar substituents made the compounds more tolerant. Therefore, the introduction of polar flexible chains in the solvent area are conducive to improving the inhibition rate of the compounds on tumor cells. Further evaluation of the kinase activity of the compounds towards VEGFR-2 and c-Met was carried out using ELISA. The result showed that most of the target compounds had inhibition potency towards VEGFR-2 and c-Met with IC50 values in the nanomolar range. Among the test compounds, compound 18 (which was found to be the most potent compound) could inhibit the kinase activities of VEGFR-2 and c-Met with IC50 values of 0.048 ± 0.006 µM and 0.025 ± 0.003 µM, respectively. This study provided us with clear SARs, which would help design more effective VEGFR-2/c-Met inhibitors [71].
Figure 10.

Thienopyrimidine and pyrrolotriazine derivatives of dual VEGFR/c-Met inhibitors.
3.6. Pyrrolotriazine Derivatives
Pyrrole [1,2-f] [1,2,4] triazine has been widely used in kinase domains. In 2018, Wei et al. designed and synthesized a series of novel pyrrolotriazine double c-Met/VEGFR-2 kinase derivatives. Compound 18 showed strong antiproliferative activity in BaF3-TPR-Met cells (IC50 = 0.710 ± 0.160 nM) and HUVEC cells (IC50 = 3.740 ± 0.311 nM), and inhibited the phosphorylation of VEGFR-2 and c-Met and the corresponding downstream signaling pathways (MAPK and PI3K). Compound 19 (Figure 10) has great selectivity to c-Met and VEGFR-2, and potent inhibitory activity against them (IC50 = 2.300 ± 0.100 nM and 5.000 ± 0.500 nM). Western blotting was used to study the inhibitory effect of compound 19 on VEGFR-2 activation and the MAPK/PI3K signaling pathway in HUVEC cells. Flow cytometry analysis showed that different concentrations of compound 19 (1 nM and 10 nM) significantly inhibited the proliferation of BaF3-TPR-Met cells, and the percentage of cells in the G0/G1 phase increased in a concentration dependent manner, which may be achieved by periodically blocking and inducing apoptosis in the G0/G1 phase. Compound 19 has good pharmacodynamics and physicochemical properties. In addition, docking experiments showed that compound 19 occupied the ATP pocket combined with VEGFR-2 (CYS-919 and ASP-1046) and c-Met (MET-1160, ASP-1222, LYS-1161 and LYS-1110) kinases through hydrogen bonding [56]. The evaluation experiments indicated that compound 19 was a potential dual-target c-Met/VEGFR-2 kinase inhibitor for cancer therapy deserving further study.
3.7. Quinazoline Derivatives
Quinazoline drugs can inhibit a variety of tyrosine kinases, including c-kit, c-Met, VEGFR, EGFR, PDGFR, FGFR, and so on. Some quinazoline derivatives are highly effective antitumor drugs, such as Cabozantinib and Fortinib. Therefore, quinazoline derivatives are promising antitumor drugs, especially for targeting c-Met/VEGFR-2 kinases.
Recently, [1,4]dioxa[2,3-f]quinazoline derivatives were scrutinized by Deng et al. in order to explore their potential as VEGFR-2/c-Met kinase inhibitors. Compounds 20 and 21 (Figure 11) exhibited potent inhibitory activity against VEGFR-2 (IC50 = 4.8 nM and 3.5 nM) and c-Met (IC50 = 5.8 nM and 7.3 nM) kinases. Importantly, compound 20 showed excellent antiproliferative activities on MHCC97H and HUVEC cells with IC50 values of 15.7 nM and 0.8 nM, respectively. As shown in Figure 12A, compounds 20 and 21 formed hydrogen bonds with the ASP-1064 residue on the binding site of VEGFR-2 kinase, and the second nitrogen atom of quinazoline formed hydrogen bonds with the CYS-919 residue. Figure 12B showed that compounds 20 and 21 were linked to the binding site of c-Met kinase, and quinazoline formed hydrogen bond interactions with MET-1160 [72]. All results indicated that compounds 20 and 21 were dual-target VEGFR-2/c-Met kinase inhibitors that held promising potential in cancer therapy.
Figure 11.

Quinazoline and diazepine derivatives of dual-target VEGFR/c-Met inhibitors.
Figure 12.

(A) The model of compounds 20 (green) and 21 (orange) bound to VEGFR-2 (PDB code: 3U6J); (B) The model of compounds 20 and 21 bound to c-Met (PDB code: 3LQ8).
3.8. Diazepine Derivatives
In recent years, a series of 6,11-dihydro-5H-benzo[e]pyrimido-[5,4-b][1,4]diazepine derivatives have been evaluated for their inhibition of c-Met kinase. In addition to the kinase activity of c-Met (IC50 = 24.4 nM), compound 22 (Figure 11) also has strong inhibition on other kinases such as VEGFR-2 (IC50 = 62.5 nM), EGFR (IC50 = 267.6 nM), RET (IC50 = 162.8 nM), c-Kit (IC50 = 258.7 nM) and FLT3 (IC50 = 851.8 nM). These results indicated that compound 22 was a promising multi-target inhibitor of tyrosine kinase. It also showed good pharmacokinetic characteristics in rats, had acceptable safety in preclinical research, and had significant antitumor activity in a Caki-1 tumor xenotransplantation model [73]. Compound 22 provided a new scaffold for further optimization.
3.9. Pyrazolopyrimidine Derivatives
A series of 1H-pyrazolo[3,4-d]pyrimidine derivatives were reported and evaluated for their BRAFV600E and VEGFR-2 inhibitory activity as well as their anti-proliferative activity. Among them, compound 23 (Figure 13) had high inhibition towards BRAFV600E (IC50 = 0.171 μM) and VEGFR-2 (IC50 = 0.779 μM), and a good anti-proliferation effect on A375, HT-29 and HUVEC cell lines. Moreover, compound 23 blocked A375 and HUVEC cells mainly in the G0/G1 phase. Compound 23 showed moderate inhibition on VEGFR-1 and VEGFR-3 kinases, but no significant inhibition on c-Met kinase [74]. The results showed that compound 23 had good selectivity and was a multi-kinase inhibitor.
Figure 13.

Structures of dual VEGFR/c-Met inhibitors.
3.10. Naphthyridinone Derivatives
Zhuo et al. developed a series of 2,7-naphthyridone-based derivatives of BMS-777607 as new MET kinase inhibitors. Among them, compound 24 (Figure 13) had significant c-Met (IC50 = 12.6 nM) and VEGFR-2 (IC50 = 17.3 nM) kinase inhibitory activities [2]. It is shown that 2,7-naphthyridone can be used as a novel dual-target antitumor drug scaffold.
3.11. Triazine Derivatives
Tetrahydrobenzo[b]thiophene derivatives have been found in the study of multi-target antitumor drugs. The most promising compound complexes 25 (Figure 13) and 26 (Figure 13) had significant c-Met kinase (IC50 = 0.42 nM and 0.49 nM, respectively) inhibitory activity. They also had inhibitory activity towards c-Kit (IC50 = 0.62 nM and 0.16 nM, respectively), FLT3 (IC50 = 0.49 nM and 0.24 nM, respectively), VEGFR-2 (IC50 = 0.26 nM and 0. 24 nM, respectively), EGFR (IC50 = 0.38 nM and 0.49 nM, respectively) and PDGFα (IC50 = 0.41 nM and 0.22 nM, respectively) kinases [75]. Therefore, compounds 25 and 26 are potential multi-target antitumor inhibitors.
4. Structure–Activity Relationship
The structural characteristics of most dual-target VEGFR/c-Met inhibitors are shown in Figure 14, and they can be divided into four parts: A, B, C and D. In part A, different nitrogen-containing aromatic heterocycles can be introduced, and substituents or heterocycles containing other heteroatoms such as nitrogen and sulfur atoms can be added to form substituted pyridine, pyrrolidine, quinazoline and other structures. When the side chain of the A-ring of pyridine derivatives is replaced by an amide chain, they show better activity. The side chains of quinoline and quinazoline derivatives are mostly substituted by alkyl oxygen groups, which can change the water solubility of the compounds and have little effect on the activity. If the side chains of the pyrrolidine derivative ring are replaced by a lipophilic group, its activity may be improved. Part A is a major structural fragment which formed a hydrogen bond with the amino acid residues of VEGFR (CYS-919) and c-Met (MET-1160) kinases (Figure 15). Part B is mainly composed of a pyridine ring and a benzene ring with various substitutions or not. Part C usually meets the ‘5-atom regulation’. In part C, a flexible chain or rigid ring structure such as pyrazolone, naphthyridine or triazolo-thiadiazole can be introduced. Urea structures contain hydrogen bond donor or acceptor atoms for forming hydrogen bonds with residues of VEGFR (LYS-868, ASP-1046, etc.) and c-Met (ASP1220, LYS1110, LEU1245, etc.) kinases (Figure 15). A new five atom connection bridge is also formed so that part D can be fully embedded in the hydrophobic pocket (Figure 15). When part D is mainly composed of benzyl or fluorophenyl, the activity of the compound is better. Among other compounds which do not conform to the five atom rule, tetrahydrobenzothiophene derivatives with a triazine structure have better activity.
Figure 14.

General structural formula of dual-target VEGFR/c-Met kinase inhibitors (Most dual-target VEGFR/c-Met inhibitors can be divided into four parts: A, B, C and D. Part A: The main structural skeleton, mainly nitrogen-containing heterocycles. Part B: Benzene ring or substituted benzene ring; Part C: Usually meet the ‘5-atom regulations’. Part D; Mostly benzene ring, substituted benzene ring or substituted pyridine ring).
Figure 15.

(A) The model of dual-target VEGFR/c-Met inhibitors bound to VEGFR. (B) The model of dual-target VEGFR/c-Met inhibitors bound to c-Met.
5. Conclusion and Perspectives
So far, dozens of small molecule kinase inhibitors have been approved for the treatment of different types of human cancer, but the emergence of drug resistance and toxicity caused by single-agent treatment or combinations of drugs are the main challenges faced by cancer patients. Studies have shown that multi-target inhibitors can combine with different targets simultaneously, regulate multiple signaling pathways and control the development of diseases to increase the therapeutic effect. Therefore, the development of dual-target inhibitors is of great significance for cancer treatment in the future.
In addition, this review not only summarizes the dual-target VEGFR/c-Met small molecule antitumor inhibitors with significant cytotoxicity and kinase inhibitory activity published in recent years, but also shows some active skeletons and fragments. These active skeletons and fragments may play an role in enhancing the inhibitory activity of antitumor derivatives, reducing toxicity or improving the stability and water solubility of compounds. It can be clearly seen from the above that the research and exploration of dual-target kinase compounds have a positive effect on the development of drugs for the treatment of various cancers. Other types of dual-target drugs are also emerging in current cancer treatments. Multi-target inhibition combined with other therapies will become a considerable future clinical antitumor treatment, and more small molecule antitumor inhibitors with higher potency and selectivity will appear.
Author Contributions
Q.Z. and W.Z. drafted the first version of the manuscript. P.Z. supervised and edited the paper. All authors contributed to the elaboration of further versions and the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
We gratefully acknowledge the generous support provided by the National Natural Science Foundation of China (21662014), Natural Science Foundation of Jiangxi, China (20181ACB20025, 20181BBG70003), Youth Top Talent Support Program of Jiangxi Science & Technology Normal University (2019QNBJRC008), Science and Technology Project Founded by the Education Department of Jiangxi Province, China (GJJ190583) and Nanchang Key Laboratory of Molecular Targeted Anticancer Drug Design and Evaluation (2019-NCZDSY-007).
Conflicts of Interest
There are no conflicts to declare.
References
- 1.Dokla E.M., Fang C.-S., Abouzid K.A., Chen C.S. 1,2,4-Oxadiazole derivatives targeting EGFR and c-Met degradation in TKI resistant NSCLC. Eur. J. Med. Chem. 2019;182:111607. doi: 10.1016/j.ejmech.2019.111607. [DOI] [PubMed] [Google Scholar]
- 2.Zhuo L.-S., Xu H., Wang M.-S., Zhao X.-E., Ming Z.-H., Zhu X.-L., Huang W., Yang G.-F. 2,7-naphthyridinone-based MET kinase inhibitors: A promising novel scaffold for antitumor drug development. Eur. J. Med. Chem. 2019;178:705–714. doi: 10.1016/j.ejmech.2019.06.033. [DOI] [PubMed] [Google Scholar]
- 3.Robinson D.R., Wu Y.-M., Lin S.-F. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19:5548–5557. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- 4.Nulgumnalli M.R., Divya P., Sundeep K., Akhila M. Dual or Multi-Targeting Inhibitors: The Next Generation Anticancer Agents. Euro J. Med. Chem. 2018;143:1277–1300. doi: 10.1016/j.ejmech.2017.10.021. [DOI] [PubMed] [Google Scholar]
- 5.Gu W., Dai Y., Qiang H., Shi W., Liao C., Zhao F., Huang W., Qian H. Discovery of novel 2-substituted-4-(2-fluorophenoxy) pyridine derivatives possessing pyrazolone and triazole moieties as dual c-Met/VEGFR-2 receptor tyrosine kinase inhibitors. Bioorganic Chem. 2017;72:116–122. doi: 10.1016/j.bioorg.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Arpita D., Eric J.S. Treatment of Advanced Renal Cell Carcinoma Patients with Cabozantinib, an Oral Multityrosine Kinase Inhibitor of MET, AXL and VEGF Receptors. Future Oncol. 2019;15:2337–2348. doi: 10.2217/fon-2019-0021. [DOI] [PubMed] [Google Scholar]
- 7.Sun W., Hu S., Fang S., Yan H. Design, synthesis and biological evaluation of pyrimidine-based derivatives as VEGFR-2 tyrosine kinase inhibitors. Bioorganic Chem. 2018;78:393–405. doi: 10.1016/j.bioorg.2018.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Masabumi S. VEGF-VEGFR System as a Target for Suppressing Inflammation and Other Diseases. Endocr. Metab. Immune Disord. Drug Targets. 2015;2:135–144. doi: 10.2174/1871530315666150316121956. [DOI] [PubMed] [Google Scholar]
- 9.Yuan X., Yang Q., Liu T., Li K., Liu Y., Zhu C., Zhang Z., Li L., Zhang C., Xie M., et al. Design, synthesis and in vitro evaluation of 6-amide-2-aryl benzoxazole/benzimidazole derivatives against tumor cells by inhibiting VEGFR-2 kinase. Eur. J. Med. Chem. 2019;179:147–165. doi: 10.1016/j.ejmech.2019.06.054. [DOI] [PubMed] [Google Scholar]
- 10.Reddy V.G., Reddy T.S., Jadala C., Reddy M.S., Sultana F., Akunuri R., Bhargava S.K., Wlodkowic D., Srihari P., Kamal A. Pyrazolo-benzothiazole hybrids: Synthesis, anticancer properties and evaluation of antiangiogenic activity using in vitro VEGFR-2 kinase and in vivo transgenic zebrafish model. Eur. J. Med. Chem. 2019;182:111609. doi: 10.1016/j.ejmech.2019.111609. [DOI] [PubMed] [Google Scholar]
- 11.Modi S.J., Kulkarni V.M. Vascular Endothelial Growth Factor Receptor (VEGFR-2)/KDR Inhibitors: Medicinal Chemistry Perspective. Med. Drug Discov. 2019;2:100009. doi: 10.1016/j.medidd.2019.100009. [DOI] [Google Scholar]
- 12.Ferrara N., Adamis A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016;15:385–403. doi: 10.1038/nrd.2015.17. [DOI] [PubMed] [Google Scholar]
- 13.Abdel-Mohsen H.T., Omar M.A., El Kerdawy A.M., Mahmoud A.E., Ali M.M., El Diwani H.I. Novel potent substituted 4-amino-2-thiopyrimidines as dual VEGFR-2 and BRAF kinase inhibitors. Eur. J. Med. Chem. 2019;179:707–722. doi: 10.1016/j.ejmech.2019.06.063. [DOI] [PubMed] [Google Scholar]
- 14.Peng F.-W., Liu D.-K., Zhang Q.-W., Xu Y.-G., Shi L. VEGFR-2 inhibitors and the therapeutic applications thereof: a patent review (2012–2016) Expert Opin. Ther. Patents. 2017;27:987–1004. doi: 10.1080/13543776.2017.1344215. [DOI] [PubMed] [Google Scholar]
- 15.Fan H., Wei D., Zheng K., Qin X., Yang L., Yang Y., Duan Y., Xu Y., Hu L. Discovery of Dioxino[2,3-f]quinazoline derivative VEGFR-2 inhibitors exerting significant antipro-liferative activity in HUVECs and mice. Eur. J. Med. Chem. 2019;175:349–356. doi: 10.1016/j.ejmech.2019.04.015. [DOI] [PubMed] [Google Scholar]
- 16.Lu D., Yan J., Wang L., Liu H., Zeng L., Zhang M., Duan W., Ji Y., Cao J., Geng M., et al. Design, Synthesis, and Biological Evaluation of the First c-Met/HDAC Inhibitors Based on Pyridazinone Derivatives. ACS Med. Chem. Lett. 2017;8:830–834. doi: 10.1021/acsmedchemlett.7b00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lemmon M.A., Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nan X., Jiang Y.-F., Li H.-J., Wang J.-H., Wu Y.-C. Design, synthesis and evaluation of sulfonylurea-containing 4-phenoxyquinolines as highly selective c-Met kinase inhibitors. Bioorganic Med. Chem. 2019;27:2801–2812. doi: 10.1016/j.bmc.2019.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Zhao Y., Zhang J., Zhuang R., He R., Xi J., Pan X., Shao Y., Pan J., Sun J., Cai Z., et al. Synthesis and evaluation of a series of pyridine and pyrimidine derivatives as type II c-Met inhibitors. Bioorganic Med. Chem. 2017;25:3195–3205. doi: 10.1016/j.bmc.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Zhai X., Bao G., Wang L., Cheng M., Zhao M., Zhao S., Zhou H., Gong P. Design, synthesis and biological evaluation of novel 4-phenoxy-6,7-disubstituted quinolines possessing (thio)semicarbazones as c-Met kinase inhibitors. Bioorganic Med. Chem. 2016;24:1331–1345. doi: 10.1016/j.bmc.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 21.Maria B., Jonathan Z., Moshe E., Marina W., Vadim E.F. c-Met as a New Marker of Cellular Senescence. Aging. 2019;9:2889–2897. doi: 10.18632/aging.101961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J., Yang D., Yang X., Nie M., Wu G., Wang Z., Li W., Liu Y., Gong P. Design, synthesis and biological evaluation of novel 4-phenoxyquinoline derivatives containing 3-oxo-3,4-dihydroquinoxaline moiety as c-Met kinase inhibitors. Bioorganic Med. Chem. 2017;25:4475–4486. doi: 10.1016/j.bmc.2017.06.037. [DOI] [PubMed] [Google Scholar]
- 23.Anestis A., Zoi I., Karamouzis M.V. Current advances of targeting HGF/c-Met pathway in gastric cancer. Ann. Transl. Med. 2018;6:247. doi: 10.21037/atm.2018.04.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh P.K., Silakari O. Molecular dynamics guided development of indole based dual inhibitors of EGFR (T790M) and c-MET. Bioorganic Chem. 2018;79:163–170. doi: 10.1016/j.bioorg.2018.04.001. [DOI] [PubMed] [Google Scholar]
- 25.Fakhrudin N., Waltenberger B., Cabaravdic M., Atanasov A., Heiss E., Grzywacz A., Mihaly-Bison J., Breuss J., Rollinger J., Bochkov V., et al. Discovery of Plumericin as a novel potent NF-?B inhibitor from Himatanthus sucuuba. Planta Medica. 2013;79:2328–2342. doi: 10.1055/s-0033-1352060. [DOI] [Google Scholar]
- 26.Yang Y., Li Y., Hou Y., Qin M., Gong P., Liu J., Zhao Y. Design, synthesis, and biological evaluation of 4-phenoxyquinoline derivatives as potent c-Met kinase inhibitor. Bioorganic Med. Chem. Lett. 2019 doi: 10.1016/j.bmcl.2019.126666. [DOI] [PubMed] [Google Scholar]
- 27.Dorsch D., Schadt O., Stieber F., Meyring M., Grädler U., Bladt F., Friese-Hamim M., Knühl C., Pehl U., Blaukat A. Identification and optimization of pyridazinones as potent and selective c-Met kinase inhibitors. Bioorganic Med. Chem. Lett. 2015;25:1597–1602. doi: 10.1016/j.bmcl.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 28.Tang Q., Wang L., Tu Y., Zhu W., Luo R., Tu Q., Wang P., Wu C., Gong P., Zheng P. Discovery of novel pyrrolo[2,3-b]pyridine derivatives bearing 1,2,3-triazole moiety as c-Met kinase inhibitors. Bioorganic Med. Chem. Lett. 2016;26:1680–1684. doi: 10.1016/j.bmcl.2016.02.059. [DOI] [PubMed] [Google Scholar]
- 29.Xu D., Wang T.-L., Sun L.-P., You Q.-D. Recent progress of small molecular VEGFR inhibitors as anticancer agents. Mini-Rev. Med. Chem. 2011;11:18–31. doi: 10.2174/138955711793564015. [DOI] [PubMed] [Google Scholar]
- 30.Hojjat-Farsangi M. Small-Molecule Inhibitors of the Receptor Tyrosine Kinases: Promising Tools for Targeted Cancer Therapies. Int. J. Mol. Sci. 2014;15:13768–13801. doi: 10.3390/ijms150813768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmed E.M., Khalil N.A., Taher A.T., Refaey R.H., Nissan Y.M. Triazolopyridazine derivatives: Synthesis, cytotoxic evaluation, c-Met kinase activity and molecular docking. Bioorganic Chem. 2019;92:103272. doi: 10.1016/j.bioorg.2019.103272. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J., Shan Y., Pan X., He L. Recent advances in antiangiogenic agents with VEGFR as target. Mini-Rev. Med. Chem. 2011;11:920–946. doi: 10.2174/138955711797068355. [DOI] [PubMed] [Google Scholar]
- 33.Dejuan S., Yuqian Z. Dual-target kinase drug design: Current strategies and future directions in cancer therapy. Euro J. Med. Chem. 2020 doi: 10.1016/j.ejmech.2019.112025Get. [DOI] [PubMed] [Google Scholar]
- 34.Li W., Man X.-Y., Chen J.-Q., Zhou J., Cai S.-Q., Zheng M. Targeting VEGF/VEGFR in the treatment of psoriasis. Discov. Med. 2014;18:97–104. [PubMed] [Google Scholar]
- 35.Hu C.-T., Wu J.-R., Cheng C.-C., Wu W.-S. The Therapeutic Targeting of HGF/c-Met Signaling in Hepatocellular Carcinoma: Alternative Approaches. Cancers. 2017;9:58. doi: 10.3390/cancers9060058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qiang H., Gu W., Huang W., Shi W., Qiu Q., Dai Y., Huang W., Qian H. Design, synthesis and biological evaluation of 4-aminopyrimidine-5-cabaldehyde oximes as dual inhibitors of c-Met and VEGFR-2. Bioorganic Med. Chem. 2016;24:3353–3358. doi: 10.1016/j.bmc.2016.03.061. [DOI] [PubMed] [Google Scholar]
- 37.Karaman S., Leppänen V.-M., Alitalo K. Vascular endothelial growth factor signaling in development and disease. Development. 2018 doi: 10.1242/dev.151019. [DOI] [PubMed] [Google Scholar]
- 38.Rothenberger N.J., Stabile L.P. Hepatocyte Growth Factor/c-Met Signaling in Head and Neck Cancer and Implications for Treatment. Cancers. 2017;9:39. doi: 10.3390/cancers9040039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zillhardt M., Park S.M., Romero I.L., Sawada K., Montag A. Foretinib (GSK1363089), an orally available multi-kinase inhibitor of c-Met and VEGFR-2, blocks proliferation, induces anoikis, and impairs ovarian cancer metastasis. Clin. Cancer Res. 2011;17:4042–4051. doi: 10.1158/1078-0432.CCR-10-3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi L., Wu T.-T., Wang Z., Xue J.-Y., Xu Y.-G. Discovery of quinazolin-4-amines bearing benzimidazole fragments as dual inhibitors of c-Met and VEGFR-2. Bioorganic Med. Chem. 2014;22:4735–4744. doi: 10.1016/j.bmc.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J., Jiang X., Jiang Y., Guo M., Zhang S., Li J., He J., Liu J., Wang J., Ouyang L. Recent advances in the development of dual VEGFR and c-Met small molecule inhibitors as anticancer drugs. Eur. J. Med. Chem. 2016;108:495–504. doi: 10.1016/j.ejmech.2015.12.016. [DOI] [PubMed] [Google Scholar]
- 42.Kwon Y., Smith B.D., Zhou Y., Kaufman M.D., Godwin A.K. Effective inhibition of c-MET-mediated signaling, growth and migration of ovarian cancer cells is influenced by the ovarian tissue microenvironment. Oncogene. 2013;34:144–153. doi: 10.1038/onc.2013.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Falcon B.L., Chintharlapalli S., Uhlik M.T., Pytowski B. Antagonist antibodies to vascular endothelial growth factor receptor 2 (VEGFR-2) as anti-angiogenic agents. Pharmacol. Ther. 2016;164:204–225. doi: 10.1016/j.pharmthera.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 44.Kulkarni A., Vijaykumar V.E., Natarajan S.K., Sengupta S., Sabbisetti V.S. Sustained inhibition of cMET-VEGFR2 signaling using liposome-mediated delivery increases efficacy and reduces toxicity in kidney cancer. Nanomedicine. 2016;12:1853–1861. doi: 10.1016/j.nano.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 45.Goltsov A.A., Fang B., Pandita T.K., Maru D., Swisher S.G., Hofstetter W.L. HER2 Confers Resistance to Foretinib Inhibition of MET-Amplified Esophageal Adenocarcinoma Cells. Ann. Thorac. Surg. 2018;105:363–370. doi: 10.1016/j.athoracsur.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 46.IS6-4 - Results of Phase 1 Studies of Golvatinib (E7050), A c-Met and EPH Receptor-Targeted Multi-Kinase Inhibitor. Ann. Oncol. 2012 doi: 10.1016/S0923-7534(20)31988-8. [DOI] [Google Scholar]
- 47.Semrad T.J., Kim E.J., Tanaka M.S., Sands J., Roberts C., Burich R.A., Li Y., Gandara D.R., Lara P., Mack P.C. Phase II Study of Dovitinib in Patients Progressing on Anti-Vascular Endothelial Growth Factor Therapy. Cancer Treat. Res. Commun. 2017;10:21–26. doi: 10.1016/j.ctarc.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Molina A.M., Hutson T.E., A Nosov D., Tomczak P., Lipatov O.N., Sternberg C.N., Motzer R.J., Eisen T. Efficacy of tivozanib treatment after sorafenib in patients with advanced renal cell carcinoma: crossover of a phase 3 study. Eur. J. Cancer. 2018;94:87–94. doi: 10.1016/j.ejca.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fargnoli J., Henley B., Wautlet B., Borzilleri R. 106 Preclinical studies and characterization of BMS-794833, a small molecule inhibitor of Met and VEGFR-2 kinases. Eur. J. Cancer Suppl. 2010 doi: 10.1016/S1359-6349(10)71811-5. [DOI] [Google Scholar]
- 50.Zhang W., Ai J., Shi D., Peng X., Ji Y., Liu J., Geng M.-Y., Li Y. Discovery of novel type II c-Met inhibitors based on BMS-777607. Eur. J. Med. Chem. 2014;80:254–266. doi: 10.1016/j.ejmech.2014.04.056. [DOI] [PubMed] [Google Scholar]
- 51.Wang Q., Quan H., Zhao J., Xie C.-Y., Wang L., Lou L.-G. RON confers lapatinib resistance in HER2-positive breast cancer cells. Cancer Lett. 2013;340:43–50. doi: 10.1016/j.canlet.2013.06.022. [DOI] [PubMed] [Google Scholar]
- 52.Soria J.-C., Cortes J., Massard C., Armand J.-P., De Andreis D., Ropert S., Lopez E., Catteau A., James J., Marier J.-F., et al. Phase I safety, pharmacokinetic and pharmacodynamic trial of BMS-599626 (AC480), an oral pan-HER receptor tyrosine kinase inhibitor, in patients with advanced solid tumors. Ann. Oncol. 2012;23:463–471. doi: 10.1093/annonc/mdr137. [DOI] [PubMed] [Google Scholar]
- 53.Jitesh P.J., Richard S.F., Mary C., Kevin G.C., Richard D.C., Nicolas C., Erling O.E. Discovery and Pharmacologic Characterization of CP-724,714, a Selective ErbB2 Tyrosine Kinase Inhibitor. Cancer Res. 2007;67:9887–9893. doi: 10.1158/0008-5472.CAN-06-3559. [DOI] [PubMed] [Google Scholar]
- 54.Li B., Torossian A., Sun Y., Du R., Dicker A., Lu B. Higher Levels of c-Met Expression and Phosphorylation Identify Cell Lines with Increased Sensitivity to AMG-458, a Novel Selective c-Met Inhibitor with Radiosensitizing Effects. Int. J. Radiat. Oncol. 2012;84:e525–e531. doi: 10.1016/j.ijrobp.2012.06.025. [DOI] [PubMed] [Google Scholar]
- 55.Zhan Z., Ai J., Liu Q., Ji Y., Chen T., Xu Y., Geng M., Duan W. Discovery of Anilinopyrimidines as Dual Inhibitors of c-Met and VEGFR-2: Synthesis, SAR, and Cellular Activity. ACS Med. Chem. Lett. 2014;5:673–678. doi: 10.1021/ml500066m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi W., Qiang H., Huang W., Bi X., Huang W., Qian H. Exploration of novel pyrrolo[2,1-f][1,2,4]triazine derivatives with improved anticancer efficacy as dual inhibitors of c-Met/VEGFR-2. Eur. J. Med. Chem. 2018;158:814–831. doi: 10.1016/j.ejmech.2018.09.050. [DOI] [PubMed] [Google Scholar]
- 57.Cui J.J. Targeting Receptor Tyrosine Kinase MET in Cancer: Small Molecule Inhibitors and Clinical Progress. J. Med. Chem. 2013;57:4427–4453. doi: 10.1021/jm401427c. [DOI] [PubMed] [Google Scholar]
- 58.Kaya M., Cokakli M., Berk A.T., Yaman A., Yesilirmak D. Associations of VEGF/VEGF-receptor and HGF/c-Met promoter poly-morphisms with progression/regression of retinopathy of prematurity. Curr. Eye Res. 2013;38:137–142. doi: 10.3109/02713683.2012.731550. [DOI] [PubMed] [Google Scholar]
- 59.Yang Y., Zhang Y., Yang L., Zhao L., Si L., Zhang H., Liu Q., Zhou J. Discovery of imidazopyridine derivatives as novel c-Met kinase inhibitors: Synthesis, SAR study, and biological activity. Bioorganic Chem. 2017;70:126–132. doi: 10.1016/j.bioorg.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 60.Lai S., Chen J., Huang H., Zhang X., Jiang H., Li W., Wang P., Wang J., Liu F.-N. Structure activity relationships of chrysoeriol and analogs as dual c-Met and VEGFR2 tyrosine kinase inhibitors. Oncol. Rep. 2018;40:405–422. doi: 10.3892/or.2018.6542. [DOI] [PubMed] [Google Scholar]
- 61.Wang M., Xu S., Lei H., Wang C., Xiao Z., Jia S., Zhi J., Zheng P., Zhu W. Design, synthesis and antitumor activity of Novel Sorafenib derivatives bearing pyrazole scaffold. Bioorganic Med. Chem. 2017;25:5754–5763. doi: 10.1016/j.bmc.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 62.Wang M.-S., Zhuo L.-S., Yang F.-P., Wang W.-J., Huang W., Yang G.-F. Synthesis and biological evaluation of new MET inhibitors with 1,6-naphthyridinone scaffold. Eur. J. Med. Chem. 2019;185:111803. doi: 10.1016/j.ejmech.2019.111803. [DOI] [PubMed] [Google Scholar]
- 63.Zeidan M.A., Mostafa A.S., Gomaa R.M., Abou-Zeid L.A., El-Mesery M., El-Sayed M.A.-A., Selim K.B. Design, synthesis and docking study of novel picolinamide derivatives as anticancer agents and VEGFR-2 inhibitors. Eur. J. Med. Chem. 2019;168:315–329. doi: 10.1016/j.ejmech.2019.02.050. [DOI] [PubMed] [Google Scholar]
- 64.Li C., Shan Y., Sun Y., Si R., Liang L., Pan X., Wang B.-H., Zhang J. Discovery of novel anti-angiogenesis agents. Part 7: Multitarget inhibitors of VEGFR-2, TIE-2 and EphB4. Eur. J. Med. Chem. 2017;141:506–518. doi: 10.1016/j.ejmech.2017.10.030. [DOI] [PubMed] [Google Scholar]
- 65.Liu J., Nie M., Wang Y., Hu J., Zhang F., Gao Y., Liu Y., Gong P. Design, synthesis and structure-activity relationships of novel 4-phenoxyquinoline derivatives containing 1,2,4-triazolone moiety as c-Met kinase inhibitors. Eur. J. Med. Chem. 2016;123:431–446. doi: 10.1016/j.ejmech.2016.07.059. [DOI] [PubMed] [Google Scholar]
- 66.Zhao F., Zhang L.-D., Hao Y., Chen N., Bai R., Wang Y.-J., Zhang C.-C., Li G.-S., Hao L., Shi C., et al. Identification of 3-substituted-6-(1-(1H-[1,2,3]triazolo[4,5-b]pyrazin-1-yl)ethyl)quinoline derivatives as highly potent and selective mesenchymal-epithelial transition factor (c-Met) inhibitors via metabolite profiling-based structural optimization. Eur. J. Med. Chem. 2017;134:147–158. doi: 10.1016/j.ejmech.2017.03.085. [DOI] [PubMed] [Google Scholar]
- 67.Wang W., Xu S., Duan Y., Liu X., Zhang H., Wang C., Zhao B., Zheng P., Zhu W. Synthesis and bioevaluation and doking study of 1H-pyrrolo[2,3-b]pyridine derivatives bearing aromatic hydrazone moiety as c-Met inhibitors. Eur. J. Med. Chem. 2017;145:315–327. doi: 10.1016/j.ejmech.2017.12.078. [DOI] [PubMed] [Google Scholar]
- 68.Wang L.X., Liu X., Xu S., Tang Q., Duan Y., Xiao Z., Zhi J., Jiang L., Zheng P., Zhu W. Discovery of novel pyrrolo-pyridine/pyrimidine derivatives bearing pyridazinone moiety as c-Met kinase inhibitors. Eur. J. Med. Chem. 2017;141:538–551. doi: 10.1016/j.ejmech.2017.10.027. [DOI] [PubMed] [Google Scholar]
- 69.Ibrahim H.A., Awadallah F.M., Refaat H.M., Amin K.M. Molecular docking simulation, synthesis and 3D pharmacophore studies of novel 2-substituted-5-nitro-benzimidazole derivatives as anticancer agents targeting VEGFR-2 and c-Met. Bioorganic Chem. 2018;77:457–470. doi: 10.1016/j.bioorg.2018.01.014. [DOI] [PubMed] [Google Scholar]
- 70.A Ibrahim H., Awadallah F.M., Refaat H.M., Amin K.M. Design, synthesis and molecular modeling study for some new 2-substituted benzimidazoles as dual inhibitors for VEGFR-2 and c-Met. Futur. Med. Chem. 2018;10:493–509. doi: 10.4155/fmc-2017-0174. [DOI] [PubMed] [Google Scholar]
- 71.Li J., Gu W., Bi X., Li H., Liao C., Liu C., Huang W., Qian H. Design, synthesis, and biological evaluation of thieno[2,3-d]pyrimidine derivatives as novel dual c-Met and VEGFR-2 kinase inhibitors. Bioorganic Med. Chem. 2017;25:6674–6679. doi: 10.1016/j.bmc.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 72.Wei D., Fan H., Zheng K., Qin X., Yang L., Yang Y., Duan Y., Zhang Q., Zeng C., Hu L. Synthesis and anti-tumor activity of[1,4]dioxino[2,3-f]quinazoline derivatives as dual inhibitors of c-Met and VEGFR-2. Bioorganic Chem. 2019;88:102916. doi: 10.1016/j.bioorg.2019.04.010. [DOI] [PubMed] [Google Scholar]
- 73.Huang D., Huang L., Zhang Q.-W., Li J. Synthesis and biological evaluation of novel 6,11-dihydro-5H-benzo[e]pyrimido-[5,4-b][1,4]diazepine derivatives as potential c-Met inhibitors. Eur. J. Med. Chem. 2017;140:212–228. doi: 10.1016/j.ejmech.2017.08.060. [DOI] [PubMed] [Google Scholar]
- 74.Wang Y., Wan S., Li Z., Fu Y., Wang G., Zhang J., Wu X. Design, synthesis, biological evaluation and molecular modeling of novel 1H-pyrazolo[3,4-d]pyrimidine derivatives as BRAFV600E and VEGFR-2 dual inhibitors. Eur. J. Med. Chem. 2018;155:210–228. doi: 10.1016/j.ejmech.2018.05.054. [DOI] [PubMed] [Google Scholar]
- 75.Abdo N.Y.M., Mohareb R.M., Halim P.A. Uses of cyclohexane-1,3-dione for the synthesis of 1,2,4-triazine derivatives as anti-proliferative agents and tyrosine kinases inhibitors. Bioorganic Chem. 2020;97:103667. doi: 10.1016/j.bioorg.2020.103667. [DOI] [PubMed] [Google Scholar]