

Abstract
目的
探究吉非替尼对非小细胞肺癌A549和H1975细胞死亡形式的影响,并从糖酵解方面探讨其可能机制。
方法
A549细胞加入浓度分别为0、20、30、40 μmol/L的吉非替尼,H1975细胞加入浓度分别为0、20、40、80 μmol/L的吉非替尼。采用MTT法检测吉非替尼对非小细胞肺癌细胞的增殖抑制作用。用乳酸试剂盒检测细胞内乳酸的变化,Western blot法检测细胞内糖酵解相关蛋白(PKM2、HK2)和PI3K-Akt-mTOR信号通路中蛋白的表达水平;2-NBDG检测细胞葡萄糖摄取能力,ATP试剂盒检测胞内ATP水平;JC-1试剂盒检测细胞线粒体膜电位,Annexin V-FITC/PI双染法检测细胞凋亡,Western blot法检测凋亡蛋白(Bax、Bcl-2)以及自噬标志蛋白LC3B的相对表达水平。
结果
MTT结果显示吉非替尼可呈时间剂量依赖性的抑制A549和H1975细胞增殖(P < 0.05),A549细胞24、48、72 h的IC50值分别为48.6、28.6和19.7 μmol/L,H1975细胞24、48、72 h的IC50值分别为321.6、49.1和14.6 μmol/L。乳酸检测结果显示,吉非替尼抑制胞内乳酸水平(P < 0.05)。Western blot结果显示,糖酵解相关蛋白PKM2、HK2表达下调(P < 0.05),PI3K-Akt-mTOR信号通路中相关蛋白表达下调(P < 0.05)。吉非替尼也可以抑制A549和H1975细胞葡萄糖摄取、ATP水平(P < 0.05)。JC-1试剂盒和Annexin V-FITC/PI双染法检测出吉非替尼能够诱导A549和H1975细胞发生凋亡,A549细胞中0、20、30、40 μmol/L的凋亡率分别为(10.77±1.0)%、(14.5±0.4)%、(17.4±0.2)%、(32.1±0.6)%,差异有统计学意义(P < 0.05);而H1975细胞0、20、40、80 μmol/L的凋亡率分别为(10.5±0.6)%、(13.2±0.92)%、(18.9±0.98)%、(35.1± 1.4)%,差异有统计学意义(P < 0.05)。促凋亡蛋白Bax表达增加和抑凋亡蛋白Bcl-2表达下调(P < 0.05)。同时LC3B表达增加证明吉非替尼能够诱导A549和H1975细胞自噬增加(P < 0.05)。
结论
吉非替尼对A549和H1975细胞具有增殖抑制、诱导凋亡和增加自噬作用,凋亡机制可能为吉非替尼影响A549和H1975细胞糖酵解功能和PI3K-Akt-mTOR信号通路。
Keywords: 吉非替尼, 非小细胞肺癌, 糖酵解, 死亡形式
Abstract
Objective
To observe the cell death pattern induced by gefitinib in non-small cell lung cancer A549 and H1975 cells and explore the possible mechanism in light of glycolysis.
Methods
The inhibitory effects of gefitinib at 20, 30, or 40 μmol/L in A549 cells and at 20, 40, or 80 μmol/L in H1975 cells were examined using MTT assay. The changes of lactic acid level in the cells were determined with a lactic acid kit, and the expression levels of glycolysis-related proteins (PKM2 and HK2) and the proteins in PI3K-Akt-mTOR signaling pathway were detected using Western blotting. 2-NBDG was used for detecting glucose uptake capacity of the cells, and ATP kit was used to detect the intracellular ATP level. The mitochondrial membrane potential of the cells was examined with the JC-1 kit, and cell apoptosis was analyzed with Annexin V-FITC/PI double staining. The relative expression levels of the apoptotic proteins Bax and Bcl-2 and the autophagy marker protein LC3B were detected with Western blotting.
Results
MTT assay showed that gefitinib inhibited the proliferation of A549 and H1975 cells in a time- and dose-dependent manner (P < 0.05). The IC50 of gefitinib at 24, 48 and 72 h was 48.6, 28.6 and 19.7 μmol/L in A549 cells and was 321.6, 49.1 and 14.6 μmol/L in H1975 cells, respectively. Gefitinib significantly lowered intracellular lactic acid level of the cells (P < 0.05) and down-regulated the expressions of PKM2 and HK2 proteins (P < 0.05) and PI3K-Akt-mTOR signaling pathway-associated proteins (P < 0.05). Gefitinib obviously inhibited glucose uptake and ATP levels in both A549 and H1975 cells (P < 0.05). Treatment with gefitinib induced obviously enhanced apoptosis in the cells, resulting in apoptosis rates of (10.77± 1.0)%, (14.5±0.4)%, (17.4±0.2)% and (32.1±0.6)% at 0, 20, 30 and 40 μmol/L in A549 cells (P < 0.05) and of (10.5±0.6)%, (13.2± 0.92)%, (18.9±0.98)% and (35.1±1.4)% at 0, 20, 40 and 80 μmol/L in H1975 cells, respectively (P < 0.05). The protein expression of Bax increased and that of Bcl-2 decreased following gefitinib treatment in the cells (P < 0.05). Gefitinib significantly increased autophagy in A549 and H1975 cells as shown by increased LC3B expressions following the treatment (P < 0.05).
Conclusion
Gefitinib can inhibit the proliferation, induce apoptosis and increase autophagy in A549 and H1975 cells. Gefitinib induces apoptosis of the cells possibly by affecting glycolysis and PI3K-Akt-mTOR signaling pathway.
Keywords: gefitinib, non-small cell lung cancer, glycolysis, programmed cell death
肺癌是全世界最常见和致命的恶性肿瘤之一,非小细胞肺癌(NSCLC)占所有肺癌病例的85%[1-2]。由于NSCLC被诊断时通常具有分期晚、病情复杂、并发症多、预后不良等特点,多数患者失去手术机会,化疗是治疗的主要手段[3]。尽管近年手术、放疗和化疗方面均取得了显著进展,但肺癌患者的5年生存率仅为20%[4]。表皮生长因子受体(EGFR)是细胞增殖、存活和代谢的主要调节剂之一[5]。目前,EGFR突变的NSCLC患者的一线治疗选择是表皮生长因子受体酪氨酸激酶抑制剂:包括吉非替尼、厄洛替尼、克唑替尼、阿法替尼、AZD9291等,可抑制癌细胞生长,改善患者生活质量并提高生存期[6-9]。其中吉非替尼是应用于EGFR突变的NSCLC患者的常用一线治疗药物,但因其耐药现象的产生,限制了其临床疗效。尽管已开发出第2或第3代酪氨酸激酶抑制剂,耐药仍然是NSCLC治疗的主要障碍。许多研究者尝试解决该问题,也提出了一些解决方案,但效果并未达到预期[10-13]。这使得我们思考,解决耐药问题是否可以追本溯源,除了从经典的耐药机制入手外,是否可从探寻其他作用机制的方向去研究。
肿瘤细胞的能量代谢一直是热点研究方向,而使用糖代谢抑制剂来降低肿瘤细胞的能量也是其中一个重要的治疗靶点[14]。但耐药NSCLC的分子代谢尚不清楚。虽有研究表明,吉非替尼耐药与线粒体生物能、GLUT1、MCT-1以及Mcl-1等糖酵解相关蛋白的表达有关[15-17],但相关研究相对其他领域而言较少,对照研究则更少,需要更多或更深入的研究,且了解TKI耐药形成过程中不同表型的分子代谢机制也有助于进一步制定治疗策略。故本实验选取A549细胞(EGFR野生型)和H1975细胞EGFR双重突变(L858R/T790M)作为研究对象[18-19],探究吉非替尼对非小细胞肺癌A549和H1975死亡形式的影响,并从糖酵解方面探讨该药物发挥作用以及发生耐药现象的可能机制,并探讨PI3KAkt- mTOR通道与吉非替尼影响非小细胞肺癌的糖酵解是否相关联。
1. 材料和方法
1.1. 实验材料、试剂
人非小细胞肺癌A549和H1975细胞(上海细胞库),DMEM、RIPM 1640培养基、胰酶(Hyclone),胎牛血清(FBS)(浙江天杭生物科技股份有限公司),MTT试剂、JC-1试剂盒、2-NBDG、ATP检测试剂盒、乳酸检测试剂盒、Annexin V-FITC/PI双染试剂盒(上海碧云天公司),PKM2、HK2、PI3K、Akt、mTOR、p-PI3K、p-Akt、Bax、Bcl-2、LC3B、β-actin蛋白抗体(CST),吉非替尼(上海起泰医药科技有限公司)。
1.2. 细胞培养
A549和H1975细胞分别培养在含10%胎牛血清、100 mg/L链霉素、100 U/mL青霉素的DMEM、RIPM 1640培养基中,放置于37 ℃、5% CO2饱和湿度的细胞恒温培养箱中培养。
1.3. MTT测定
A549和H1975分别以1×104/孔细胞接种于6孔板中,A549细胞加入浓度分别为0、20、30、40 μmol/L的吉非替尼的新鲜培养液,H1975细胞加入浓度分别为0、20、40、80 μmol/L的吉非替尼的新鲜培养液,在培养24、48、72 h时加入15 μL/孔MTT溶液,放入培养箱中继续孵育4 h,吸去培养液,加入150 μL/孔DMSO,置于37 ℃培养箱继续孵育30 min待甲臜完全溶解,在490 nm波长处用酶联免疫检测仪测吸光度值A490 nm。计算细胞存活率:细胞存活率(%)=实验组A490 nm/对照组A490 nm× 100%。取均值绘制药物剂量效应曲线,计算药物的半数抑制浓度(IC50)。
1.4. 乳酸的测定
A549和H1975分别以3×105/孔细胞接种于6孔板中,A549细胞组加入浓度分别为0、20、30、40 μmol/L的吉非替尼,H1975细胞组加入浓度分别为0、20、40、80 μmol/L的吉非替尼,给药24 h时,收集细胞,加入适量裂解液,转移至1.5 mL EP管中,在4 ℃下以12 000 r/min离心30 min,取上清液用于后续结果分析,参照乳酸测定试剂盒步骤(Bio Vision)进行测试,并将实验重复3次。
1.5. Western blot法检测蛋白表达水平
A549和H1975分别以3×105/孔细胞接种于6孔板中,A549细胞组加入浓度分别为0、20、30、40 μmol/L的吉非替尼,H1975细胞组加入浓度分别为0、20、40、80 μmol/L的吉非替尼,给药48 h时收集细胞,冰上裂解30 min,12 000 r/min离心30 min,吸取上清液,每组样品取60 μg蛋白进行电泳;蛋白转至聚偏氟乙烯(PVDF)膜;牛奶封闭4 h;孵育一抗过夜;室温下孵育二抗2 h;ECL试剂盒暗室发光显影,凝胶成像系统获取图像。
1.6. 检测细胞葡萄糖摄取
给药24 h时,吸去原培养基,用PBS溶液洗涤2次,换成不含葡萄糖的培养基饥饿处理细胞2 h,每孔加入1 mg/mL的2-NBDG溶液50 μL,37 ℃孵育30 min,用PBS溶液洗涤2次,使用多功能酶标仪检测,并将实验重复3次。
1.7. 细胞内ATP水平检测
药物作用6 h时,离心收集细胞,弃去上清液。加入200 μL/孔裂解液裂解30 min,4 ℃离心机以12 000 r/min将混合物离心5 min,吸取上清液(冰上操作)用于后续分析。每个样中加入100 μL ATP工作液(1:4),并在室温下放置5 min(使反应在黑暗中进行),使背景ATP全部消耗,立即向每孔添加20 μL样品,用酶标仪测量吸光度,并将实验重复3次。
1.8. JC-1染色检测细胞线粒体膜电位差
药物作用12 h时,收集细胞,加入1 mL/孔JC-1(JC-1原液:培养基=1:1000)染色溶液,然后在细胞培养箱中于37 ℃孵育20 min(避光)。1200 r/min离心5 min,弃去上清液,并用PBS溶液洗涤2次,最后用流式细胞仪上机检测,并将实验重复3次。
1.9. Annexin V-FITC/PI双染法检测细胞凋亡
给药24 h时收集细胞,加入500 μL/孔PBS溶液重悬细胞,加入10 μLAnnexin-V-FITC混匀,冰上孵育15 min,之后加15 μL/孔PI染色剂,1 h内检测细胞凋亡情况。
1.10. 统计分析方法
实验数据采用SPSS21.0统计软件进行统计分析,实验数据均以均数±标准差表示,多组比较采用单因素方差分析及两因素方差分析,组间两两比较采用LSDt检验,P < 0.05为差异具有统计学意义。
2. 结果
2.1. 吉非替尼对A549和H1975细胞增殖抑制作用
MTT结果显示,随着吉非替尼浓度增加和作用时间延长,吉非替尼抑制A549和H1975细胞效果增强(图 1,P < 0.05)。A549组24、48、72 h的IC50值分别为48.6、28.6和19.7 μmol/ L,H1975组24、48、72 h的IC50值分别为321.6、49.1和14.6 μmol/L。两株细胞的体外增殖活性明显受到抑制,与对照组相比差异具有统计学意义(P < 0.05)。
1.

吉非替尼对A549和H1975细胞增殖抑制作用
Inhibitory effects of gefitinib on the proliferation of A549 cells and H1975 cells. A: Effects of different concentrations of gefitinib on the cell viability of A549 cells at 24, 48 and 72 h; B: Effects of different concentrations of gefitinib on the cell viability of H1975 cells at 24, 48 and 72 h. **P < 0.01
2.2. 吉非替尼对A549和H1975细胞糖酵解的影响
吉非替尼对A549和H1975细胞糖酵解的最终产物乳酸的抑制有抑制作用(P < 0.05),用Western blot检测吉非替尼对A549和H1975细胞糖酵解的影响,结果显示糖酵解蛋白PKM2和HK2的表达随着吉非替尼浓度的增加而降低(图 2,P < 0.05)。
2.
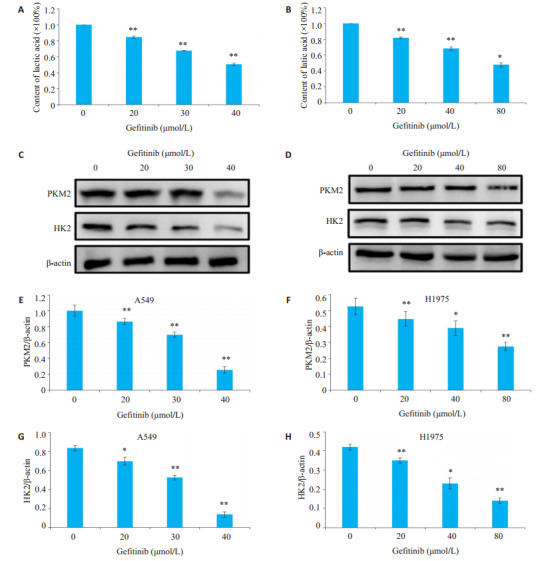
吉非替尼对A549和H1975细胞糖酵解的影响
Effects of gefitinib on glycolysis in A549 and H1975 cells. A: Effects of gefitinib on lactic acid levels in A549 cells; B: Effects of gefitinib on lactic acid levels in H1975 cells; C: Expression of PKM2 and HK2 in A549 cells detected by Western blotting; D: Expression of PKM2 and HK2 in H1975 cells detected by Western blotting; E-H: Quantitative analysis of the results ofWestern blot with Image J grayscale scanning. *P < 0.05, **P < 0.01 vs 0 μmol/L
2.3. 吉非替尼对A549和H1975细胞PI3K-Akt-mTOR信号通路的影响
结果显示,吉非替尼对A549和H1975细胞PI3KAkt- mTOR均有抑制作用(图 3,P < 0.05),但吉非替尼对H1975细胞PI3K-Akt-mTOR抑制作用较小。
3.
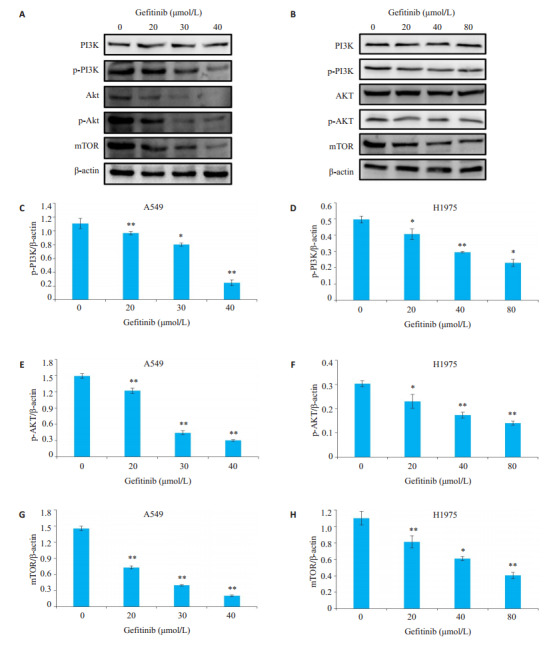
吉非替尼对A549和H1975细胞PI3K-Akt-mTOR信号通路的影响
Effects of gefitinib on PI3K-Akt-mTOR signaling pathway in A549 and H1975 cells. A, B: Expression of PI3K, p-PI3K, Akt, p-Akt and mTOR detected by Western blotting in A549 cells and H1975 cells, respectively; C-H: Quantitative analysis of the results ofWestern blotting with Image J grayscale scanning. *P < 0.05, **P < 0.01 vs 0 μmol/L
2.4. 吉非替尼对A549和H1975细胞葡萄糖摄取和ATP的影响
葡萄糖摄取测定的结果显示,随着吉非替尼浓度的增加,A549和H1975细胞葡萄糖摄取逐渐减少。吉非替尼可抑制A549细胞中的葡萄糖摄取能力和ATP水平(图 4A、C,P < 0.05),同时可抑制H1975细胞中的葡萄糖摄取和ATP水平(图 4B、D,P < 0.05)。
4.

吉非替尼对A549和H1975细胞葡萄糖摄取和ATP水平的影响
Effects of gefitinib on glucose uptake and ATP level in A549 and H1975 cells. A: Effects of gefitinib on glucose uptake of A549 cells; B: Effects of gefitinib on glucose uptake of H1975 cells; C: Effects of gefitinib on ATP level of A549 cells; D: Effects of gefitinib on ATP level of H1975 cells. *P < 0.05, **P < 0.01 vs 0 μmol/L
2.5. 吉非替尼对A549和H1975细胞凋亡的影响
JC-1结果显示,吉非替尼降低了A549和H1975细胞的线粒体膜电位(图 5A、C,P < 0.05)。Annexin VFITC/ PI双染法检验凋亡结果,A549对照组总凋亡率为(10.77±1.0)%,20、30、40 μmol/L吉非替尼组的总凋亡率分别为(14.5±0.4)%、(17.4±0.2)%、(32.1±0.6)%。H1975对照组的总凋亡率为(10.5±0.6)%,20、40、80 μmol/L吉非替尼组的总凋亡率分别为(13.2±0.92)%、(18.9± 0.98)%、(35.1±1.4)%,差异具有统计学意义(图 5E、F,P < 0.05)。为进一步研究吉非替尼诱导的A549和H1975细胞凋亡的可能机制,通过蛋白质印迹检测了凋亡蛋白的表达水平,结果显示随着吉非替尼浓度的增加,A549和H1975细胞促凋亡蛋白Bax的蛋白表达上调(图 5I、J,P < 0.05),而抑凋亡蛋白Bcl-2表达下调(图 5K、L,P < 0.05)。
5.
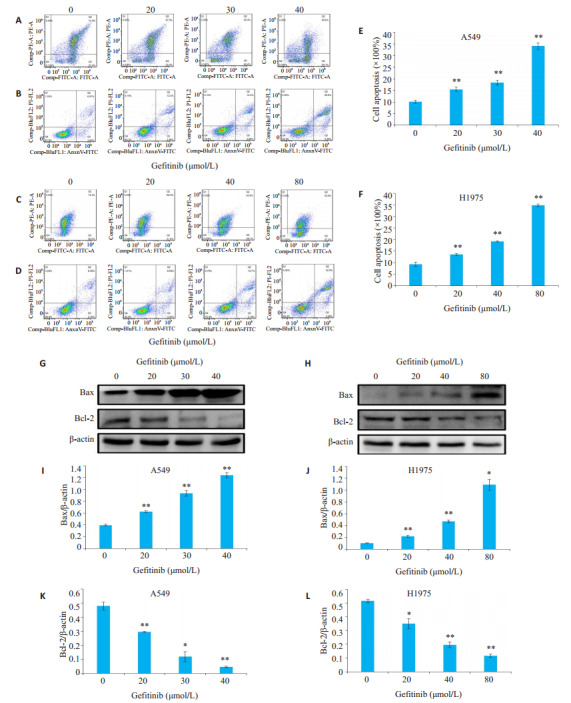
吉非替尼对A549和H1975细胞凋亡的影响
Effects of gefitinib on apoptosis of A549 and H1975 cells. A: Effects of gefitinib on mitochondrial membrane potential in A549 cells; B: Effects of gefitinib on apoptosis in A549 cells determined by flow cytometry; C: Effects of gefitinib on mitochondrial membrane potential in H1975 cells; D: Effects of gefitinib on apoptosis in H1975 cells; E: Quantitative analysis of the cell apoptosis in A549 cells; F: Quantitative analysis of the cell apoptosis in H1975 cells; G: Expression of Bax and Bcl-2 of A549 cells detected by Western blotting; H: Expression of Bax and Bcl-2 of H1975 cells detected by Western blotting; I-L: Quantitative analysis of the results ofWestern blotting with Image J grayscale scanning. *P < 0.05, **P < 0.01 vs 0 μmol/L
2.6. 吉非替尼诱导A549和H1975细胞自噬增加
为研究吉非替尼对A549和H1975细胞除凋亡以外的死亡形式,使用蛋白质印迹方法检测自噬标志蛋白LC3B的表达水平,结果显示,随着吉非替尼浓度的增加,A549和H1975组细胞自噬标志蛋白LC3B的表达增加(图 6,P < 0.05)。
6.
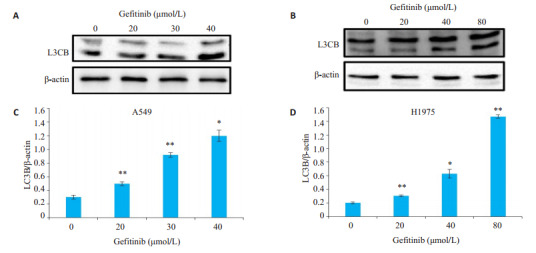
吉非替尼对A549和H1975细胞自噬相关蛋白表达的影响
Effect of gefitinib on the expression of autophagy-related protein in A549 and H1975 cells. A: Expression of L3CB of A549 cells detected by western blotting; B: Expression of L3CB of H 1975 cells detected by Western blotting; C, D: Quantitative analysis of the results ofWestern blotting with Image J grayscale scanning. *P < 0.05, **P < 0.01 vs 0 μmol/L
3. 讨论
糖酵解异常增加是快速增殖癌细胞的共同特征,与正常分化的细胞不同,大多数癌症不管氧气水平如何,细胞都会产生大量的乳酸,这种代谢特性通常称为“有氧糖酵解”[20-21]。癌细胞的新陈代谢重编程是维持细胞增殖并成为癌症的标志[22],己糖激酶(HK)和丙酮酸激酶(PK)是调节糖酵解中两个不可逆步骤的两个关键酶。HK同工酶1-4催化葡萄糖的磷酸化,是糖酵解两个不可逆步骤的第一步,HK2与癌症的促进有关,并已被提议作为潜在的治疗靶标[23]。PK催化磷酸烯醇丙酮酸的去磷酸化为丙酮酸,这是糖酵解过程中最后不可逆的步骤,大多数肿瘤细胞会重新表达PKM2,在有氧糖酵解中,从PKM1到PKM2的转变是公认的代谢重编程事件[24-26],敲低PKM2可以抑制细胞增殖和肿瘤生长[27]。吉非替尼耐药的经典机制是EGFR基因二次突变[28],其他研究发现的机制:如激活IL-6/JAK1/STAT3信号通路[29]、Kras基因突变[30]、激活IGF1R介导的信号通路[31]等,少有人从能量代谢的角度去思考和解决问题。而本实验从糖酵解方面探讨吉非替尼对A549和H1975细胞死亡形式的影响,首先发现吉非替尼能够同时抑制A549和H1975细胞糖酵解相关蛋白PKM2和HK2以及糖酵解最终产物乳酸的生成,但从结果中发现吉非替尼对H1975细胞糖酵解抑制作用较小。
PI3K-Akt-mTOR信号通路是EGFR的主要下游途径之一,EGFR的酪氨酸磷酸化充当PI3K的停靠位点,然后刺激了磷脂酰肌醇3, 4, 5-三磷酸的生成并促进Akt的激活[32],而PI3K-Akt-mTOR通路的过度激活会抑制凋亡并改善肿瘤生长[33-34]。本实验发现吉非替尼对A549和H1975细胞的PI3K-Akt-mTOR通路均有抑制作用,但对H1975细胞PI3K-Akt-mTOR通路的抑制作用较小。Akt可通过将葡萄糖转运蛋白从细胞质转运到质膜来增加葡萄糖的摄取[35]。本实验发现吉非替尼可以通过抑制A549和H1975细胞摄取葡萄糖来降低ATP水平进而诱导细胞调亡。此外,有研究证明Akt调控糖酵解途径涉及改变磷酸果糖激酶II的活性[36],也有研究证明一些药物可通过抑制癌细胞的Akt表达来抑制己糖激酶,从而抑制糖酵解过程[37-38]。而本研究发现吉非替尼对H1975细胞Akt表达的抑制作用较弱,这可能是吉非替尼对H1975的糖酵解抑制水平较弱的作用机制之一。另有研究表明癌细胞可通过开放电压依赖性阴离子通道增加线粒体代谢、提高线粒体膜电位,导致胞质ATP/ADP比率增高,从而抑制糖酵解[39]。本研究发现使用不同浓度的吉非替尼处理细胞后,H1975细胞的线粒体膜电位相对更高,而A549细胞的线粒体膜电位降低较为明显,这亦可能是吉非替尼对H1975的糖酵解抑制水平较弱的作用机制之一。但这些理论中涉及的具体机制有待进一步实验去验证。
mTOR是自噬的主要负调节剂[40],它是mTORC1复合体的一部分,故mTORC1受上游PI3K / Akt途径的调控[41-42]。自噬在癌症中起着双重作用,它既显示出致癌活性又可起到抑制作用[43]。有报道称通过PI3K/Akt/ mTOR途径可促进细胞存活,阻止过度自噬导致的细胞死亡[44]。而本实验中发现吉非替尼能够诱导A549和H1975细胞增加自噬而诱导细胞死亡。
综上所述,吉非替尼可诱导A549和H1975细胞凋亡也可增加细胞自噬,其凋亡机制可能为吉非替尼影响A549和H1975细胞糖酵解蛋白和PI3K-Akt-mTOR信号通路。而在本实验中A549细胞为吉非替尼敏感株,H1975细胞则对吉非替尼不敏感,而两者糖酵解水平的差异可能与Akt的表达以及线粒体膜电位水平变化等机制有关。本研究为糖酵解抑制剂的临床应用提供了实验基础,也为非小细胞肺癌的治疗以及提高其对吉非替尼的敏感性提供了新的方法和思路。
Biography
周巧,本科,E-mail: zhouqiao99@foxmail.com
Funding Statement
国家自然科学基金(81372899);安徽省自然科学基金(1908085QH373);安徽省学术和技术带头人及后备人选学术科研活动资助经费(2019H215);蚌埠医学院科技发展基金(BYKF1816);大学生创新创业项目(201810367005)
Supported by National Natural Science Foundation of China (81372899)
Contributor Information
周 巧 (Qiao ZHOU), Email: zhouqiao99@foxmail.com.
李 姗姗 (Shanshan LI), Email: 393235848@qq.com.
刘 浩 (Hao LIU), Email: liuhao6886@foxmail.com.
References
- 1.Ung MH, MacKenzie TA, Onega TL, et al. Statins associate with improved mortality among patients with certain histological subtypes of lung cancer. Lung Cancer. 2018;126:89–96. doi: 10.1016/j.lungcan.2018.10.022. [Ung MH, MacKenzie TA, Onega TL, et al. Statins associate with improved mortality among patients with certain histological subtypes of lung cancer[J]. Lung Cancer, 2018, 126: 89-96.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Osmani L, Askin F, Gabrielson E, et al. Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): Moving from targeted therapy to immunotherapy. http://cn.bing.com/academic/profile?id=65d297f6b4aaac505e65cb6f0ef2caa4&encoded=0&v=paper_preview&mkt=zh-cn. Semin Cancer Biol. 2018;52(Pt 1):103–9. doi: 10.1016/j.semcancer.2017.11.019. [Osmani L, Askin F, Gabrielson E, et al. Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): Moving from targeted therapy to immunotherapy[J]. Semin Cancer Biol, 2018, 52(Pt 1): 103-9.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smeltzer MP, Faris N, Yu XH, et al. Missed intrapulmonary lymph node metastasis and survival after resection of non-small cell lung cancer. Ann Thorac Surg. 2016;102(2):448–53. doi: 10.1016/j.athoracsur.2016.03.096. [Smeltzer MP, Faris N, Yu XH, et al. Missed intrapulmonary lymph node metastasis and survival after resection of non-small cell lung cancer[J]. Ann Thorac Surg, 2016, 102(2): 448-53.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crabtree T, Puri V, Timmerman R, et al. Treatment of stage I lung cancer in high-risk and inoperable patients: comparison of prospective clinical trials using stereotactic body radiotherapy (RTOG 0236), sublobar resection (ACOSOG Z4032), and radiofrequency ablation (ACOSOG Z4033) J Thorac Cardiovasc Surg. 2013;145(3):692–9. doi: 10.1016/j.jtcvs.2012.10.038. [Crabtree T, Puri V, Timmerman R, et al. Treatment of stage I lung cancer in high-risk and inoperable patients: comparison of prospective clinical trials using stereotactic body radiotherapy (RTOG 0236), sublobar resection (ACOSOG Z4032), and radiofrequency ablation (ACOSOG Z4033)[J]. J Thorac Cardiovasc Surg, 2013, 145(3): 692-9.] [DOI] [PubMed] [Google Scholar]
- 5.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–34. doi: 10.1016/j.cell.2010.06.011. [Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases[J]. Cell, 2010, 141(7): 1117-34.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu HA, Pao W. Targeted therapies: Afatinib: new therapy option for EGFR-mutant lung cancer. Nat Rev Clin Oncol. 2013;10(10):551–2. doi: 10.1038/nrclinonc.2013.154. [Yu HA, Pao W. Targeted therapies: Afatinib: new therapy option for EGFR-mutant lung cancer[J]. Nat Rev Clin Oncol, 2013, 10(10): 551-2.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase Ⅲ trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002. Ann Oncol. 2013;24(1):54–9. doi: 10.1093/annonc/mds214. [Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase Ⅲ trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002[) J]. Ann Oncol, 2013, 24(1): 54-9.] [DOI] [PubMed] [Google Scholar]
- 8.Thongprasert S, Duffield E, Saijo N, et al. Health-related quality-oflife in a randomized phase Ⅲ first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients from Asia with advanced NSCLC (IPASS) J Thorac Oncol. 2011;6(11):1872–80. doi: 10.1097/JTO.0b013e31822adaf7. [Thongprasert S, Duffield E, Saijo N, et al. Health-related quality-oflife in a randomized phase Ⅲ first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients from Asia with advanced NSCLC (IPASS)[J]. J Thorac Oncol, 2011, 6(11): 1872-80.] [DOI] [PubMed] [Google Scholar]
- 9.Yang JC, Hirsh V, Schuler M, et al. Symptom control and quality of life in LUX-Lung 3: a phase Ⅲ study of afatinib or cisplatin/ pemetrexed in patients with advanced lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3342–50. doi: 10.1200/JCO.2012.46.1764. [Yang JC, Hirsh V, Schuler M, et al. Symptom control and quality of life in LUX-Lung 3: a phase Ⅲ study of afatinib or cisplatin/ pemetrexed in patients with advanced lung adenocarcinoma with EGFR mutations[J]. J Clin Oncol, 2013, 31(27): 3342-50.] [DOI] [PubMed] [Google Scholar]
- 10.Wu SG, Shih JY. Management of acquired resistance to EGFR TKItargeted therapy in advanced non-small cell lung cancer. Mol Cancer. 2018;17(1):38–45. doi: 10.1186/s12943-018-0777-1. [Wu SG, Shih JY. Management of acquired resistance to EGFR TKItargeted therapy in advanced non-small cell lung cancer[J]. Mol Cancer, 2018, 17(1): 38-45.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen KL, Cho YT, Yang CW, et al. Olmutinib-induced palmoplantar keratoderma. Br J Dermatol. 2018;178(2):e129–31. doi: 10.1111/bjd.15935. [Chen KL, Cho YT, Yang CW, et al. Olmutinib-induced palmoplantar keratoderma[J]. Br J Dermatol, 2018, 178(2): e129-31.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrews NM, Goss GD. Third-generation epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. http://cn.bing.com/academic/profile?id=0b4c55dc2ffcb4ac63842b25117f2620&encoded=0&v=paper_preview&mkt=zh-cn. Transl Lung Cancer Res. 2019;8(Suppl 3):S247–S264. doi: 10.21037/tlcr.2019.06.01. [Andrews NM, Goss GD. Third-generation epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer[J]. Transl Lung Cancer Res, 2019, 8(Suppl 3): S247-S264.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sullivan I, Planchard D. Next-generation EGFR tyrosine kinase inhibitors for treating EGFR-mutant lung cancer beyond first line. http://cn.bing.com/academic/profile?id=815a6abc65bb6e437ef1e8771cb00d04&encoded=0&v=paper_preview&mkt=zh-cn. Front Med (Lausanne) 2016;3:76–85. doi: 10.3389/fmed.2016.00076. [Sullivan I, Planchard D. Next-generation EGFR tyrosine kinase inhibitors for treating EGFR-mutant lung cancer beyond first line [J]. Front Med (Lausanne), 2016, 3: 76-85.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation [J]. Cell, 2011, 144(5): 646-74.] [DOI] [PubMed] [Google Scholar]
- 15.Kim SM, Yun MR, Hong YK, et al. Glycolysis inhibition sensitizes non-small cell lung cancer with T790M mutation to irreversible EGFR inhibitors via translational suppression of Mcl-1 by AMPK activation. Mol Cancer Ther. 2013;12(10):2145–56. doi: 10.1158/1535-7163.MCT-12-1188. [Kim SM, Yun MR, Hong YK, et al. Glycolysis inhibition sensitizes non-small cell lung cancer with T790M mutation to irreversible EGFR inhibitors via translational suppression of Mcl-1 by AMPK activation[J]. Mol Cancer Ther, 2013, 12(10): 2145-56.] [DOI] [PubMed] [Google Scholar]
- 16.Suzuki S, Okada M, Takeda H, et al. Involvement of GLUT1- mediated glucose transport and metabolism in gefitinib resistance of non-small-cell lung cancer cells. http://cn.bing.com/academic/profile?id=a1e34c57a26ed0c65bad464c59d54b7a&encoded=0&v=paper_preview&mkt=zh-cn. Oncotarget. 2018;9(66):32667–79. doi: 10.18632/oncotarget.25994. [Suzuki S, Okada M, Takeda H, et al. Involvement of GLUT1- mediated glucose transport and metabolism in gefitinib resistance of non-small-cell lung cancer cells[J]. Oncotarget, 2018, 9(66): 32667-79.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang CY, Hsu LH, Chen CY, et al. Inhibition of alternative cancer cell metabolism of EGFR mutated non-small cell lung cancer serves as a potential therapeutic strategy. Cancers (Basel) 2020;12(1):E181–7. doi: 10.3390/cancers12010181. [Huang CY, Hsu LH, Chen CY, et al. Inhibition of alternative cancer cell metabolism of EGFR mutated non-small cell lung cancer serves as a potential therapeutic strategy[J]. Cancers (Basel), 2020, 12(1): E181-7.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73–8. doi: 10.1371/journal.pmed.0020073. [Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain[J]. PLoS Med, 2005, 2 (3): e73-8. DOI:10.1371/journal.pmed.0020073.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer. 2006;118(2):257–62. doi: 10.1002/ijc.21496. [Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers[J]. Int J Cancer, 2006, 118(2): 257-62.] [DOI] [PubMed] [Google Scholar]
- 20.Warbur O. Origin of cancer cells. Oncologia. 1956;9(2):75–83. doi: 10.1159/000223920. [Warbur O. Origin of cancer cells[J]. Oncologia, 1956, 9(2): 75-83.] [DOI] [PubMed] [Google Scholar]
- 21.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. doi: 10.1126/science.1160809. [Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation[J]. Science, 2009, 324(5930): 1029-33.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. [Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate[J]. Cancer Cell, 2012, 21 (3): 297-308.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: cancer's stygian link to the "Warburg Effect" and a pivotal target for effective therapy. Semin Cancer Biol. 2009;19(1):17–24. doi: 10.1016/j.semcancer.2008.11.006. [Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: cancer's stygian link to the "Warburg Effect" and a pivotal target for effective therapy[J]. Semin Cancer Biol, 2009, 19 (1): 17-24.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–3. doi: 10.1038/nature06734. [Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth[J]. Nature, 2008, 452(7184): 230-3.] [DOI] [PubMed] [Google Scholar]
- 25.Mazurek S, Boschek CB, Hugo F, et al. Pyruvate kinase type M2 and its role in tumor growth and spreading.[J]. Semin Cancer Biol, 2005, 15: 300-8.
- 26.David CJ, Chen M, Assanah M, et al. HNRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463(7279):364–8. doi: 10.1038/nature08697. [David CJ, Chen M, Assanah M, et al. HNRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer[J]. Nature, 2010, 463(7279): 364-8.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldberg MS, Sharp PA. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J Exp Med. 2012;209(2):217–24. doi: 10.1084/jem.20111487. [Goldberg MS, Sharp PA. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression[J]. J Exp Med, 2012, 209 (2): 217-24.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin L. Mechanisms of drug resistance to gefitinib and erlotinib. J Int Oncol. 2010;37(10):734–6. [Lin L. Mechanisms of drug resistance to gefitinib and erlotinib[J]. J Int Oncol, 2010, 37(10): 734-6.] [Google Scholar]
- 29.Cao W, Liu Y, Zhang R, et al. Homoharringtonine induces apoptosis and inhibits STAT3 via IL-6/JAK1/STAT3 signal pathway in Gefitinib-resistant lung cancer cells. Sci Rep. 2015;5:8477–85. doi: 10.1038/srep08477. [Cao W, Liu Y, Zhang R, et al. Homoharringtonine induces apoptosis and inhibits STAT3 via IL-6/JAK1/STAT3 signal pathway in Gefitinib-resistant lung cancer cells[J]. Sci Rep, 2015, 5: 8477-85.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen J, Bi H, Hou J, et al. Atorvastatin overcomes gefitinib resistance in KRAS mutant human non-small cell lung carcinoma cells. Cell Death Dis. 2013;4:e814–9. doi: 10.1038/cddis.2013.312. [Chen J, Bi H, Hou J, et al. Atorvastatin overcomes gefitinib resistance in KRAS mutant human non-small cell lung carcinoma cells[J]. Cell Death Dis, 2013, 4: e814-9.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guix M, Faber AC, Wang SE, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGFbinding proteins. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2430495/ J Clin Invest. 2008;118(7):2609–19. doi: 10.1172/JCI34588. [Guix M, Faber AC, Wang SE, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGFbinding proteins[J]. J Clin Invest, 2008, 118(7): 2609-19.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cully M, You H, Levine AJ, et al. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer. 2006;6(3):184–192. doi: 10.1038/nrc1819. [Cully M, You H, Levine AJ, et al. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis[J]. Nat. Rev. Cancer, 2006, 6 (3):184-192.] [DOI] [PubMed] [Google Scholar]
- 33.Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anti Cancer Drugs. 2005;16(8):797–803. doi: 10.1097/01.cad.0000173476.67239.3b. [Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy[J]. Anti Cancer Drugs, 2005, 16(8): 797-803.] [DOI] [PubMed] [Google Scholar]
- 34.Fumarola C, Bonelli MA, Petronini PG, et al. Targeting PI3K/AKT/ mTOR pathway in non small cell lung cancer. Biochem Pharmacol. 2014;90(3):197–207. doi: 10.1016/j.bcp.2014.05.011. [Fumarola C, Bonelli MA, Petronini PG, et al. Targeting PI3K/AKT/ mTOR pathway in non small cell lung cancer[J]. Biochem Pharmacol, 2014, 90(3): 197-207.] [DOI] [PubMed] [Google Scholar]
- 35.Rathmell JC, Fox CJ, Plas DR, et al. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003;23(20):7315–28. doi: 10.1128/MCB.23.20.7315-7328.2003. [Rathmell JC, Fox CJ, Plas DR, et al. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival[J]. Mol Cell Biol, 2003, 23(20): 7315-28.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Novellasdemunt L, Tato I, Navarro-Sabate A, et al. Akt-dependent activation of the heart 6-phosphofructo-2-kinase/fructose-2, 6- bisphosphatase (PFKFB2) isoenzyme by amino acids. J Biol Chem. 2013;288(15):10640–51. doi: 10.1074/jbc.M113.455998. [Novellasdemunt L, Tato I, Navarro-Sabate A, et al. Akt-dependent activation of the heart 6-phosphofructo-2-kinase/fructose-2, 6- bisphosphatase (PFKFB2) isoenzyme by amino acids[J]. J Biol Chem, 2013, 288(15): 10640-51.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang KJ, Zhang MR, Jiang H, et al. Down-regulation of miR-214 inhibits proliferation and glycolysis in non-small-cell lung cancer cells via down-regulating the expression of hexokinase 2 and pyruvate kinase isozyme M2. Biomed Pharmacother. 2018;105:545–52. doi: 10.1016/j.biopha.2018.06.009. [Zhang KJ, Zhang MR, Jiang H, et al. Down-regulation of miR-214 inhibits proliferation and glycolysis in non-small-cell lung cancer cells via down-regulating the expression of hexokinase 2 and pyruvate kinase isozyme M2[J]. Biomed Pharmacother, 2018, 105: 545-52.] [DOI] [PubMed] [Google Scholar]
- 38.Wu J, Zhang XX, Wang YH, et al. Licochalcone A suppresses hexokinase 2-mediated tumor glycolysis in gastric cancer via downregulation of the Akt signaling pathway. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=8d229506cf5b417b5ebba1657becfec3. Oncol Rep. 2018;39(3):1181–90. doi: 10.3892/or.2017.6155. [Wu J, Zhang XX, Wang YH, et al. Licochalcone A suppresses hexokinase 2-mediated tumor glycolysis in gastric cancer via downregulation of the Akt signaling pathway[J]. Oncol Rep, 2018, 39(3): 1181-90.] [DOI] [PubMed] [Google Scholar]
- 39.Fang D, Maldonado EN. VDAC regulation: a mitochondrial target to stop cell proliferation. Adv Cancer Res. 2018;138:41–69. doi: 10.1016/bs.acr.2018.02.002. [Fang D, Maldonado EN. VDAC regulation: a mitochondrial target to stop cell proliferation[J]. Adv Cancer Res, 2018, 138: 41-69.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cuyàs E, Corominas-Faja B, Joven J, et al. Cell cycle regulation by the nutrient-sensing mammalian target of rapamycin (mTOR) pathway. http://cn.bing.com/academic/profile?id=5340ced0aba0cd7c02e24f8390384730&encoded=0&v=paper_preview&mkt=zh-cn. Methods Mol Biol. 2014;1170:113–44. doi: 10.1007/978-1-4939-0888-2_7. [Cuyàs E, Corominas-Faja B, Joven J, et al. Cell cycle regulation by the nutrient-sensing mammalian target of rapamycin (mTOR) pathway[J]. Methods Mol Biol, 2014, 1170: 113-44.] [DOI] [PubMed] [Google Scholar]
- 41.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–74. doi: 10.1016/j.cell.2007.06.009. [Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream [J]. Cell, 2007, 129(7): 1261-74.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8(3):179–83. doi: 10.1016/j.ccr.2005.08.008. [Hay N. The Akt-mTOR tango and its relevance to cancer[J]. Cancer Cell, 2005, 8(3): 179-83.] [DOI] [PubMed] [Google Scholar]
- 43.Kumar A, Singh UK, Chaudhary A. Targeting autophagy to overcome drug resistance in cancer therapy. Future Med Chem. 2015;7(12):1535–42. doi: 10.4155/fmc.15.88. [Kumar A, Singh UK, Chaudhary A. Targeting autophagy to overcome drug resistance in cancer therapy[J]. Future Med Chem, 2015, 7 (12): 1535-42.] [DOI] [PubMed] [Google Scholar]
- 44.Heras-Sandoval D, Pérez-Rojas JM, Hernández-Damián J, et al. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014;26(12):2694–701. doi: 10.1016/j.cellsig.2014.08.019. [Heras-Sandoval D, Pérez-Rojas JM, Hernández-Damián J, et al. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration [J]. Cell Signal, 2014, 26(12): 2694-701.] [DOI] [PubMed] [Google Scholar]