

Abstract
Myotonic dystrophy type 1 is the most common type of adult-onset muscular dystrophy. This is an autosomal dominant disorder and caused by the expansion of the CTG repeat in the 3′ untranslated region of the dystrophia myotonica protein kinase (DMPK) gene. Messenger RNAs containing these expanded repeats form aggregates as nuclear RNA foci. Then, RNA binding proteins, including muscleblind-like 1, are sequestered to the RNA foci, leading to systemic abnormal RNA splicing. In this study, we used CRISPR-Cas9 genome editing to excise this CTG repeat. Dual cleavage at the 5′ and 3′ regions of the repeat using a conventional Cas9 nuclease and a double nicking with Cas9 nickase successfully excised the CTG repeat. Subsequently, the formation of the RNA foci was markedly reduced in patient-derived fibroblasts. However, contrary to expectations, a considerable amount of off-target digestions and on-target genomic rearrangements were observed using high-throughput genome-wide translocation sequencing. Finally, the suppression of DMPK transcripts using CRISPR interference significantly decreased the intensity of RNA foci. Our results indicate that close attention should be paid to the unintended mutations when double-strand breaks are generated by CRISPR-Cas9 for therapeutic purposes. Alternative approaches independent of double-strand breaks, including CRISPR interference, may be considered.
Keywords: CRISPR-Cas9, genome editing, myotonic dystrophy, DMPK, RNA foci, off-target, CRISPR interference
Graphical Abstract
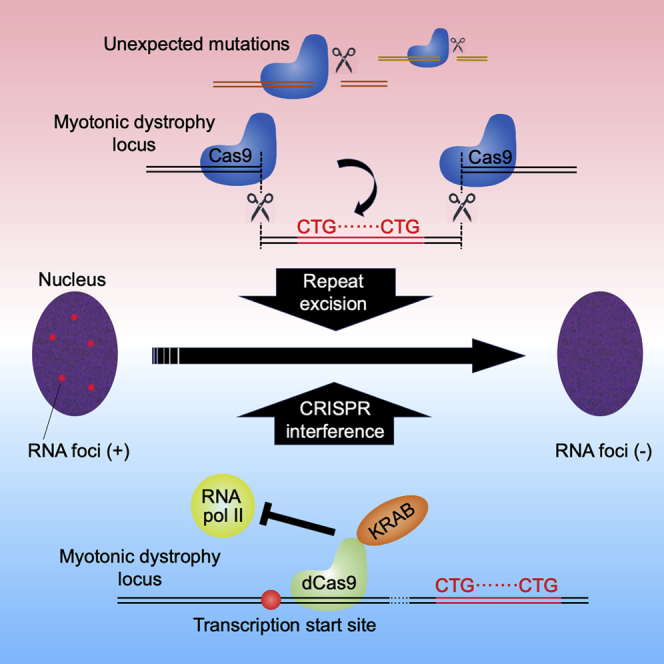
Ikeda et al. report unexpected mutations caused by excision of the CTG repeat in the myotonic dystrophy locus using CRISPR-Cas9 and double nicking strategy. They further show that CRISPR interference significantly suppresses the RNA foci, proposing that the alternative approach independent of double-strand breaks may be considered for therapeutic purposes.
Introduction
Myotonic dystrophy type 1 (DM1) is the most common type of adult-onset muscular dystrophy, affecting 1 in 8,000 individuals.1 DM1 is an autosomal dominant disorder that is characterized by systemic symptoms, including progressive muscular atrophy, muscular weakness, myotonia, cardiac arrhythmia, insulin resistance, gastrointestinal dysfunctions, cataract, and cognitive impairment.1 It is caused by the expansion of the CTG repeat in the 3′ untranslated region (UTR) of the dystrophia myotonica protein kinase (DMPK) gene.2,3 Healthy subjects have 5–37 CTG repeats, whereas DM1 symptoms are identified in individuals with more than 50 CTG repeats.2, 3, 4 The larger repeat sizes tend to be associated with worse clinical manifestations, and several thousand repeats are typically observed in severe congenital DM1 patients.5,6 The expanded CTG repeats exhibit somatic and intergenerational instability with a bias toward expansion.7,8
The expanded CTG repeats in the 3′ UTR of DMPK are transcribed to mRNA as CUG repeats. It was previously demonstrated that the expanded CUG repeat forms stable hairpin structures that aggregate as RNA foci.9, 10, 11 The intranuclear RNA foci sequester RNA binding proteins, including muscleblind-like 1 (MBNL1), a known splicing regulator.12, 13, 14 This in turn leads to the depletion of soluble MBNL1 with normal regulatory function.15 In addition, the RNA foci upregulate the activity of another splicing regulator, CUGBP Elav-like family member 1 (CELF1), by activating the protein kinase C pathway and suppressing the expression of specific microRNAs for CELF1.16,17 The altered function of these splicing regulators results in the abnormal splicing of many genes, including CLCN1, BIN1, or IRα, which account for some aspects of the systemic features of DM1.18, 19, 20, 21
The CRISPR-Cas9 system was first discovered as a microbial adaptive immune system.22 It has since been successfully applied in the genome editing of eukaryotic cells and in a variety of research fields.23, 24, 25 The most basic CRISPR-Cas9 system uses Cas9 nuclease derived from Streptococcus pyogenes and a single-guide RNA (sgRNA) with a complementary sequence to the target region of interest. These two components form a complex that is able to induce double-strand breaks (DSBs) at the target site. After cleavage, the DSBs are repaired by one of the two major repair pathways, that is, non-homologous end joining (NHEJ) or homology-directed repair (HDR).26 This powerful tool has been adapted for medical therapeutics, including DM1. Thus far, several groups have successfully excised the CTG repeat of the DMPK gene using the conventional Cas9 nuclease system in cultured cells and model mice.27, 28, 29, 30
Although CRISPR-Cas9 is an innovative technology, care must be taken to avoid causing undesirable mutations when used for therapeutic purposes.31 One way to avoid this lies in the use of the double nicking strategy.32 In this system, Cas9 nickase, a D10A mutant of Cas9, is utilized with a pair of offset sgRNAs complementary to opposite strands of the target site. The nicks of both of the DNA strands lead to a DSB with a 5′ overhang. A large reduction in off-target cutting is expected due to the need for two sgRNAs, since it is unlikely that two off-target nicks will be generated by chance in close proximity.32,33 Importantly, by dual DSBs, the region encompassed by up to several Mb can be removed and the 5′ and 3′ cut ends can be rejoined using the NHEJ or HDR repair systems.34 Another candidate is CRISPR interference (CRISPRi), a strategy in which the transcription of any gene is downregulated without inducing DSBs.35,36 This strategy utilizes catalytically inactive Cas9 (dCas9) fused with a transcription suppressor, KRAB, and sgRNA designed at the vicinity of transcription start sites (TSSs). This DSB-free method is expected to be much safer than DSB-dependent genome editing.
In the present study, we demonstrated that both a conventional Cas9 nuclease and a double nicking strategy using Cas9 nickase successfully excised the CTG repeat tract by designing sgRNAs at the 5′ and 3′ flanking regions. Using these procedures, the formation of RNA foci was markedly inhibited. However, the unbiased detection of genomic alterations using linear amplification-mediated high-throughput genome-wide translocation sequencing (LAM-HTGTS)37,38 revealed unexpected on- and off-target mutations as a result of using these procedures. Lastly, we showed that the downregulation of DMPK transcription by CRISPRi significantly suppressed the formation of RNA foci. Based on these observations, we propose that approaches that are independent of a DSB formation, such as CRISPRi, should be considered when applying the CRISPR-Cas9 technologies for therapeutic purposes in the future.
Results
Excision of DMPK CTG Repeat by Cas9 Nuclease
First, we tested whether the conventional CRISPR-Cas9 system using Cas9 nuclease and a pair of sgRNAs designed at the 5′ and 3′ region of the CTG repeat could be used to remove the repeat sequence in HEK293 cells. We confirmed by Sanger sequencing that the strain of HEK293 cells we used contained five CTG repeats (data not shown). As shown in Figure 1A, three sgRNAs were designed on both the 5′ and the 3′ regions of the CTG repeats. Each sgRNA was expressed with Cas9 nuclease in HEK293 cells and showed similar indel frequencies, according to the result of a T7 endonuclease 1 assay (Figure S1). Then, Cas9 nuclease and two sgRNAs, one targeting the 5′ region and another targeting the 3′ region of the repeat, were co-transfected. Three days after transfection, genomic PCR was performed using primers designed at the outer sites of the sgRNA targeting loci (Figure 1A). Upon these co-expressions, lower molecular weight bands were observed (Figure 1B), suggesting the excision of the CTG repeats.
Figure 1.

Excision of DMPK CTG Repeat Using Cas9 Nuclease
(A) Schematic representation of 3′ UTR of the DMPK gene is shown. The CTG repeat sequence is indicated in red characters. Three guide RNAs were designed at the 5′ and 3′ region of the CTG repeat (guide 1, 2, and 3). This region was amplified by PCR using the primers flanking the target sequence. (B) HEK293 cells were co-transfected with guide RNAs designed at the 5′ and 3′ regions with Cas9 nuclease, and genomic PCR was performed. By mock transfection, i.e., 5′ guide (–), 3′ guide (–), a PCR product of 400 bp was observed (arrow). Upon transfection with each combination of guide RNA, lower molecular weight bands were observed (asterisk). (C) Genomic PCR of DM1 patient-derived fibroblasts GM03991, harboring 50–80 CTG repeats, showed two distinct bands with an apparent molecular mass of 400 and 700 bp, corresponding to the wild-type and the mutant allele with an expanded CTG repeat, respectively. By genome editing using 5′ guide 2/3′ guide 2 and 5′ guide 3/3′ guide 2, the bands with a lower molecular mass above 200 bp were observed. (D) Sanger sequencing revealed that the lower molecular weight bands consisted of amplicons in which the CTG repeat region was excised. A typical junctional sequence is shown here. The blue, green, and red characters represent the position of the 5′ guide 2, 3′ guide 2, and CTG repeat, respectively. Arrowheads indicate the position of the expected DSBs. (E) Genome editing was repeated five times with an interval of 3 days to HEK293 cells (upper panel) and three times with an interval of 7 days to DM1-derived fibroblasts (lower panel). The CTG repeat excision was facilitated by repeating the genome editing (arrow).
We investigated whether similar events could be observed in the fibroblasts of DM1, denoted as GM03991, which contains 50–80 CTG repeats. Using PCR amplification, the control fibroblasts (mock electroporation) exhibited two distinct bands at 400 and 700 bp, corresponding to the wild-type and mutant allele, respectively. Based on the CRISPR design tool score, we chose two combinations of sgRNA pairs: (1) 5′ guide 2/3′ guide 2 and (2) 5′ guide 3/3′ guide 2. These showed the lowest likelihood of off-target digestion. By electroporation with these sgRNAs, the lower molecular weight bands appeared above 200 bp (Figure 1C). Sanger sequencing confirmed that these bands consisted of amplicons lacking the CTG repeat (Figures 1D, S2A, and S2B). Although several junctional sequences were obtained, they were mostly homogeneous (57.1% for type deletion [del] 2 in HEK293 cells and 83.3% for type del 1 in fibroblasts; Figure S2C). In the following experiments, we used the sgRNA pair 5′ guide 3/3′ guide 2 for Cas9 nuclease cleavage. The T7 endonuclease 1 assay did not reveal any obvious off-target mutations at the top five sites predicted by the CRISPR design tool in fibroblasts (Figure S3).
When designing in vivo gene therapy for DM1 using CRISPR-Cas9 technology, it is of particular importance to remove the CTG repeats from as many cells as possible. To this purpose, we investigated whether repeated genome editing may facilitate the excision. The repeated genome editing of HEK293 cells and fibroblasts using Cas9 nuclease increased the intensity of the lower bands gradually, indicating it facilitated repeat excision (Figure 1E).
Excision of DMPK CTG Repeat by Cas9 Nickase
In order to adapt the CRISPR-Cas9 technology for clinical usage, it is critical to reduce the potential for off-target effects. For this purpose, we tested whether the CTG repeat was excised using the CRISPR-Cas9 double nicking strategy. We designed seven (Nick 1, 2, 5, 6, 7, 8, and 9) and two (Nick 3 and 4) nicking pairs of sgRNAs on the 5′ and 3′ regions of the CTG repeat, respectively (Figure 2A). Upon transfection of these nicking pairs into HEK293 cells, a T7 endonuclease 1 assay demonstrated indel frequencies ranging from 30% to 40% or more (Figure S4). We then co-transfected HEK293 cells with Cas9 nickase and two pairs of sgRNAs, one targeting the 5′ region and another the 3′ region of the repeat. Genomic PCR showed lower molecular weight bands in all of the combinations of sgRNA pairs (Figure 2B). Among them, we chose the nicking pairs of Nick 1 and Nick 3 for the subsequent experiments, since the lower band was robust and the decrease in molecular weight was small, and the latter may only minimally affect the 3′ UTR structure of the DMPK gene. Sanger sequencing demonstrated that the lower band consisted of amplicons lacking the CTG repeat (Figures 2C, S5A, and S5B). By aligning the sequence data, it was evident that the junctional sequences were heterogeneous, compared to those observed using the conventional Cas9 nuclease (Figures S2B and S5B). No identical junctional sequence was observed among the clones we tested (Figure S5C). Repeated genome editing of HEK293 cells using the double nicking strategy gradually increased the intensity of the lower bands, indicating that it facilitated the excision of the repeat (Figures 2D and 2E).
Figure 2.

Excision of DMPK CTG Repeat Using Cas9 Nickase
(A) Schematic representation of the 3′ UTR of the DMPK gene. The CTG repeat sequence is indicated in red characters. Seven (Nick 1, 2, 5, 6, 7, 8, and 9) and two (Nick 1 and 2) nicking pairs of guide RNAs were designed at the 5′ and 3′ regions of the CTG repeat, respectively. This region was amplified by PCR using the primers flanking the target sequence. (B) HEK293 cells were co-transfected with two pairs of guide RNAs, one against the 5′ region and another against the 3′ region of the repeat together with Cas9 nickase. Genomic PCR showed a PCR product of 400 bp by mock transfection i.e., 5′ Nick (–), 3′ Nick (–) (arrow). Upon transfection with each combination of nicking pairs of guide RNAs, lower molecular weight bands were observed (asterisk). (C) Sanger sequencing of the lower molecular weight band observed in Figure 3B by 5′ Nick 1 and 3′ Nick 3 revealed that it consisted of amplicons lacking the CTG repeat. A few junctional sequences are shown here. The blue, green, and red characters represent the position of Nick 1, Nick 3, and the CTG repeat, respectively. Arrowheads indicate the position of expected nicks. (D) Genome editing with Cas9 nickase was repeated five times with an interval of 3 days to HEK293 cells. The CTG repeat excision was facilitated by repeating the genome editing. (E) Quantitative densitometric analyses of the lower bands in (B) confirmed that the repeated genome editing increases the efficiency of repeat excision. The results are expressed as the mean ± SEM (n = 3).
Suppression of RNA Foci Formation by CRISPR-Cas9 Genome Editing
As described above, the fibroblast GM03991, harboring 50–80 CTG repeats, was found to contain a wild-type allele at 400 bp and a mutant allele at 700 bp, according to the PCR results. In contrast, the fibroblast GM05163, harboring 400 repeats, showed only a wild-type allele, suggesting that the PCR failed to amplify the large mutant allele (Figure 3A, mock). Upon the genome editing of these fibroblasts using conventional Cas9 nuclease or double nicking with Cas9 nickase, the lower molecular weight bands appeared above 200 bp, indicating the successful excision of the CTG repeat (Figure 3A).
Figure 3.

Suppression of RNA Foci Formation by the Repeat Excision
(A) Fibroblast GM03991, harboring 50–80 CTG repeats, was found to consist of a wild-type allele at 400 bp and a mutant allele at 700 bp by PCR. However, fibroblast GM05163, with 400 CTG repeats, exhibited only the wild-type allele, presumably because the PCR failed to amplify the highly repetitive sequence of the mutant allele. Genome editing was performed to these cells using conventional Cas9 nuclease and two sgRNAs (5′ guide 3 and 3′ guide 2) or the double nicking strategy with Cas9 nickase and four sgRNAs (Nick 1 and Nick 3 pairs). Using both of the procedures, lower molecular weight bands were observed above 200 bp (arrow), indicating the successful excision of the CTG repeat. (B) Fibroblast GM05163 was co-transfected with GFP-tagged Cas9 nuclease or Cas9 nickase together with sgRNAs. The RNA-FISH image shows intense RNA foci in the GFP-negative fibroblast (left) but not in the GFP-positive Cas9 nuclease-expressing cell (right). (C) In the nuclei of the control fibroblasts (no plasmids), several RNA foci were consistently observed. However, the RNA foci were mostly undetectable in fibroblasts expressing Cas9 nuclease or nickase. (D) Quantitative analyses of the number and the total intensity of intranuclear RNA foci were performed. Histograms of both the number (upper panel) and the intensity (lower panel) with a left-side skew were obtained for Cas9 nuclease and nickase. (E) The average number of RNA foci was significantly reduced by both Cas9 nuclease and nickase. When compared between Cas9 nuclease and nickase, the nuclease-treated cells exhibited significantly fewer foci (upper panel). The average intensity of RNA foci was significantly decreased by both Cas9 nuclease and nickase. There was no significant difference between the nuclease and nickase by this parameter (lower panel). The results are expressed as the mean ± SEM.
RNA foci were not detected in GM03991 using the RNA-fluorescence in situ hybridization (FISH) assay. As such, we studied GM05163 to evaluate the formation of RNA foci, one of the major pathological hallmarks of DM1 (Figure S6). To determine whether the removal of the CTG repeat affected the formation of foci, fibroblasts were co-transfected with GFP-tagged Cas9 nuclease or Cas9 nickase together with sgRNAs targeting the 5′ and 3′ regions flanking the CTG repeat. A typical RNA-FISH image 3 days after genome editing is provided in Figure 3B. In the nucleus of the GFP-negative fibroblasts (Figure 3B, left), several intense dot-shaped RNA foci were detected, which were not observed in GFP-positive Cas9-expressing fibroblasts (Figure 3B, right). In the nuclei of the control fibroblasts (mock electroporation), several RNA foci were consistently observed. However, the RNA foci were mostly undetectable in the nuclei of fibroblasts expressing Cas9 nuclease and nickase (Figure 3C). Furthermore, to ascertain the sequestration of MBNL1 to the RNA foci, an RNA-FISH assay was performed, followed by immunofluorescent analysis using anti-MBNL1 antibody. As a result, we found that MBNL1 colocalized with RNA foci in the DM1 patient-derived fibroblasts. Upon genome editing using Cas9 nuclease, both of the signals of the RNA foci and MBNL1 were abolished, indicating that the trapped MBNL1 was released from the foci (Figure S7).
Subsequently, we obtained more than 100 images of the nuclei from each group to quantitatively analyze the formation of the RNA foci. The number of RNA foci and the total intensity of RNA foci in each nucleus were measured. Histograms of both the number (Figure 3D, upper panel) and the total intensity (Figure 3D, lower panel) showed a left-sided skew resulting from Cas9 nuclease and nickase compared to the control. These data demonstrated that both genome editing strategies suppressed the formation of RNA foci. Furthermore, the average number of RNA foci was significantly reduced by both Cas9 nuclease and nickase. When comparing Cas9 nuclease and nickase, the nuclease-treated cells exhibited significantly fewer foci (Figure 3E, upper panel). The average intensity of the RNA foci was also significantly decreased by these procedures. There was no significant difference between the nuclease and nickase with regard to this parameter (Figure 3E, lower panel).
Unbiased Genome-wide Detection of On- and Off-Target Mutations
To evaluate the unexpected mutations caused by CRISPR-Cas9, we performed LAM-HTGTS. Generally, DSBs generated by genome editing are rejoined via the classic NHEJ pathway, with occasional indels at the break site, and rarely by HDR. However, some DSBs, which are not rejoined immediately at the original loci, are fused with the separate cut ends when the other DSBs occur due to off-target cleavage. This leads to translocations to other chromosomes or intra-chromosomal deletions. By designing the sequence-specific primers at the fixed “bait” (DMPK locus in this case), LAM-HTGTS analyzes the genome-wide off-target “prey” DSBs captured by the on-target bait DSBs using next-generation sequencing (Figure 4A).
Figure 4.

Unbiased Genome-wide Detection of On- and Off-Target Mutations
(A) A diagram of LAM-HTGTS for the detection of unbiased genome-wide off-target effects is shown. The broken end of the DMPK locus generated by on-target DSB works as a “bait” and captures genome-wide “prey” induced by unintended DSBs in other loci. Following PCR using a biotinylated primer, enrichment with avidin beads, and nested PCR, the bait-prey hybrid sequences are read using next-generation sequencing. (B) Using Cas9 nuclease and one sgRNA targeting the RAG1A site, a single DSB was generated in the HEK293 cells as a positive control. With RAG1 as a bait, LAM-HTGTS was used to reveal several translocation hotspots. The chromosomal location is indicated in the outermost part of the Circos plot. Split reads binned into 50-Mb regions (orange bars) have been plotted on a log scale. Colored lines (orange and red) connect the RAG1 bait site on chromosome 11 to the genome-wide prey hotspots binned into 100-bp regions. The orange and red line colors indicate a split-reads frequency of 3–9 and of more than 10, respectively. (C) LAM-HTGTS was performed without genome editing using DMPK as a bait, a negative control for Cas9 nuclease. No translocation hotspot was detected. (D) Using Cas9 nuclease and two sgRNAs targeting the DMPK locus, dual DSBs were generated. Using DMPK as a bait, six translocation hotspots were identified. (E) LAM-HTGTS was performed without genome editing using DMPK as a bait, a negative control for the double nicking strategy with Cas9 nickase. (E) differs from (C) in the filtering process of split reads since the deduced on-target breakpoint is different between them (details are found in Materials and Methods). (F) Using Cas9 nickase and four sgRNAs, dual DSBs were generated. Using DMPK as a bait, as many as 24 translocation hotspots were identified. (G) Split reads obtained by LAM-HTGTS were mapped in integrative genomics viewer. Possible deletions, jugged from the orientation of the paired reads and their intervals, were extracted and the top 20 were sorted according to their size. By generating a single DSB on the RAG1 locus using Cas9 nuclease, possible on-target deletions ranging from several hundred bp to several kb were observed (upper panel). By generating dual DSBs on the DMPK locus using Cas9 nuclease and two sgRNAs, larger deletions with an estimated size ranging from several to 20 kb were observed (middle panel). By generating dual DSBs on the DMPK locus using Cas9 nickase and four sgRNA, larger deletions with an estimated size ranging from 20 to 60 kb were observed (lower panel). Arrows indicate the position of sgRNA.
First, as a positive control, we induced a DSB using Cas9 nuclease and one sgRNA at the RAG1 locus in HEK293 cells. Using this single DSB, the translocation hotspots were identified in chromosomes 7, 12, 15, and 19, as shown in a Circos plot (Figure 4B). This pattern of hotspots was consistent with that reported in a previous study.37 Next, we performed genome editing to the DMPK 3′ UTR using Cas9 nuclease and two sgRNAs to excise the CTG repeat, and detected translocations using DMPK as a bait. Although no hotspots were found in the negative control (no editing) (Figure 4C), six translocation hotspots were observed in chromosomes 1, 14, 15, 17, 19, and X by the dual DSBs (Figure 4D). Furthermore, we attempted the double nicking strategy using Cas9 nickase and four sgRNAs to generate dual DSBs in the DMPK locus. We anticipated that off-target effects would be strikingly reduced by this procedure. However, contrary to our expectation, as many as 25 translocation hotspots were identified using double nicking genome editing (Figures 4E and 4F). The frequency of translocation to each hotspot is shown in Table S1. The total frequency of these translocations in total mapped reads by a DSB at the RAG1 locus with Cas9 nuclease was 0.002625%. Comparably, the total frequencies by DSBs at the DMPK locus using Cas9 nuclease and Cas9 nickase were 0.001592% and 0.002169%, respectively.
Furthermore, to evaluate the on-target mutations neighboring the cutting sites, we mapped the paired reads obtained by LAM-HTGTS using integrative genomics viewer software.39 Upon generation of a single DSB at the RAG1 locus, the left-sided reads of some of the paired reads were mapped several kb apart from the breakpoint (Figure S8A). The non-edited samples using DMPK as a bait, which were used as negative controls for Cas9 nuclease and Cas9 nickase, exhibited only minimal gaps between the paired reads (Figures S8B and S8C). However, dual DSBs, generated by both Cas9 nuclease and Cas9 nickase, resulted in much larger gaps between the paired reads. These large gaps indicate that genomic rearrangements occurred including large deletions. Among them, paired reads with opposite orientations facing each other and with inferred insert sizes larger than expected, such as several hundred bp or larger, represent possible deletions. We extracted the paired reads with these possible deletions and ranked the top 20 according to their size (Figure 4G; Table S2). Using RAG1 cleavage with a single sgRNA, potential on-target deletions ranging from several hundred bp to several kb were observed. However, the dual digestion at DMPK by Cas9 nuclease and nickase led to the generation of much larger deletions spanning beyond 20 and 60 kb, respectively.
Suppression of RNA Foci by CRISPRi
The generation of unexpected mutagenesis is of great concern when DSBs are generated using CRISPR-Cas9, as described above. Therefore, we examined whether the RNA foci were suppressed by the downregulation of DMPK transcription using CRISPRi, a DSB-free method. We searched the TSS of the human DMPK gene using the FANTOM5 database and identified the most enriched peak at chromosome 19 (ch19), 46,285,748 (hg19). Then, three sgRNAs neighboring the peak were designed (Figure 5A). Three days after the transfection of HEK293 cells with dCas9-KRAB and each sgRNA, the RNA was extracted from the whole cell population without selection. Quantitative RT-PCR revealed a significant reduction in the level of DMPK mRNA by guide 2 (Figure 5B). Next, to evaluate the formation of RNA foci, an RNA-FISH assay was performed using the fibroblast GM05163. Several RNA foci were clearly observed in the control fibroblasts, whereas RNA foci were mostly inconspicuous in the transfected cells with the guide 2, according to the red fluorescent protein (RFP) fluorescence of EF1-RFP-U6-gRNA plasmid (Figure 5C). Finally, the number and total intensity of the RNA foci were quantitatively analyzed. Histograms of both the number (Figure 5D, upper panel) and the total intensity (Figure 5D, lower panel) of RNA foci with a left-sided skew were obtained using CRISPRi. Although the average foci number was not significantly different (Figure 5E, upper panel), the average intensity of foci was significantly decreased by CRISPRi (Figure 5E, lower panel).
Figure 5.
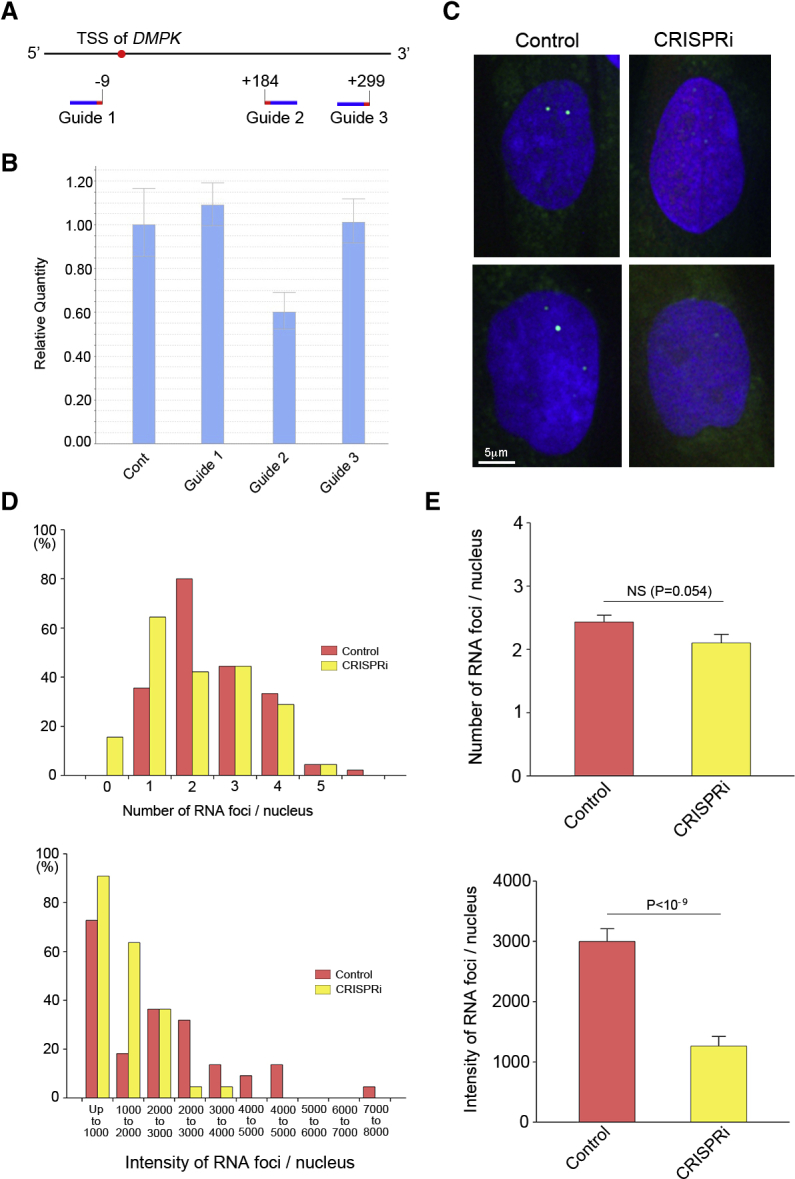
Suppression of RNA Foci by CRISPRi
(A) To suppress the transcription of the DMPK gene by CRISPRi, three sgRNAs, one upstream and two downstream of the TSS, were designed. (B) After the transfection of the HEK293 cells, total RNA was extracted from the whole cell population without selection. Quantitative RT-PCR revealed a significant reduction in the level of DMPK mRNA by guide 2. The results are expressed as the mean ± SEM (N = 3). (C) An RNA-FISH assay was performed using fibroblast GM05163 after CRISPRi using guide 2. Several RNA foci were clearly observed in the control fibroblasts, but they were mostly inconspicuous in the transfected cells. (D) Quantitative analysis of the number and total intensity of the intranuclear RNA foci was performed. Histograms of both the number (upper panel) and the total intensity (lower panel) shifted toward the left side by CRISPRi compared to control. (E) Although the fibroblasts transfected for CRISPRi tended to show fewer foci, the difference in the average foci number was not statistically significant (upper panel). The average intensity of the RNA foci was significantly decreased by CRISPRi (lower panel). The results are expressed as the mean ± SEM.
Discussion
Thus far, no therapeutic strategy has been successfully applied in clinical use for the treatment of DM1, although many experimental approaches have been attempted in the past two decades. These include small molecular therapeutic strategies and posttranscriptional silencing using nucleotide sequences. For example, small molecules, such as erythromycin, and several designer small molecular compounds have been reported to block the interaction between CUG repeat and MBNL1.40,41 It was also demonstrated that RNase-H active gapmer antisense oligonucleotides (ASOs) modified by 2′-O-methoxyethyl and 2′-4′-constrained ethyl effectively corrected the phenotype of DM1 in model mice.42,43 Note that a gapmer ASO developed by Ionis Pharmaceuticals (IONIS-DMPKRX) entered a phase I/IIa trial, but enough concentration of the drug was not achieved in skeletal muscle to provide a therapeutic benefit (ClinicalTrials.gov: NCT02312011). Recently, genome editing technologies based on CRISPR-Cas9 have been found to have a robust applicability in a variety of research fields, including medical therapeutics. Several groups have reported successful excision of the CTG repeat in DM1 with conventional Cas9 nuclease by designing sgRNAs at the 5′ and 3′ region flanking the repeats.27, 28, 29, 30 In alternative approaches, polyadenylation signals were inserted in the 3′ UTR upstream of the CTG repeat using CRISPR-Cas9 or transcription activator-like effector nuclease (TALEN).44,45 This insertion led to the premature termination of transcription and the reversal of aberrant splicing. In terms of the delivery method for in vivo gene targeting, one of the most attractive vectors is the adeno-associated virus (AAV).46 AAV does not integrate into the genome and produces high levels of long-term gene expression. A variety of serotypes are available that provide increased delivery efficiencies for the specific cell/tissue types. However, AAV has some disadvantages, including (1) a prolonged and uncontrollable expression that potentially increases the off-target mutagenesis, (2) a production of antibodies against AAV that may reduce the therapeutic effectiveness, and (3) a limited packing capacity that sometimes requires separate vector systems. Alternatively, CRISPR-Cas9 components can be delivered as a ribonucleoprotein (RNP) complex, which is generally considered to be safe since it is rapidly degraded. Although a local injection of RNP itself was shown to successfully induce genome editing,47 many non-viral delivery systems are currently under development for more efficient RNP delivery. These include lipid nanoparticles,48 gold nanoparticles,49 cell-penetrating peptides,50 and extracellular nanovesicles.51
In this study, we tried to validate the excision of expanded CTG repeats using a conventional CRISPR-Cas9 system. By designing sgRNAs at the 5′ and 3′ region of the repeats, Cas9 nuclease successfully removed the targeted region. Upon this procedure, RNA foci were suppressed, and trapped MBNL1 was released from the foci. Although we have not examined the correction of the mis-splicing of mRNAs, including CLCN1, BIN1, or IRα, it is presumed to be difficult to validate this using fibroblasts.21 CRISPR-Cas9 is an innovative tool that has potential applications in clinical use in the future; however, it can lead to unintended mutagenesis. To reduce this possibility, we excised the CTG repeats using the double nicking strategy, by which the off-target cuttings are considered to be strictly inhibited. We found that the repeat sequence was successfully deleted in HEK293 cells and DM1 fibroblasts using this procedure. Note that on-target junctional sequences were found to be heterogeneous using Sanger sequencing after double nicking. The breakpoints spanned widely, from the upstream of 5′ nicking sgRNAs to the downstream of 3′ sgRNAs. This is in contrast to Cas9 nuclease, which mostly generated the expected junctions. It is difficult to predict the exact on-target cutting sites when the double nicking strategy is employed, which is a potential disadvantage of this procedure. In addition to the accuracy, a degree of effectiveness is required to apply the genome editing to clinical use, especially for disorders in which the pathogenic mechanism is based on the gain of toxic functions, including DM1. We found that the excision of CTG repeats was facilitated by genome editing. Thus, several rounds of repeating genome editing therapies could be used in severe cases to obtain an additive efficacy, such as in chemotherapy for cancer or immunosuppressive therapies for autoimmune-related disorders.52
Although both the conventional Cas9 nuclease and the double nicking strategy with Cas9 nickase significantly reduced the RNA foci, Cas9 nuclease seems to have a subtle advantage over Cas9 nickase in terms of its efficiency. One possible reason for this difference may be that four sgRNAs are necessary to exert an effect simultaneously at each targeting loci in the double nicking strategy, in contrast to Cas9 nuclease, which requires two sgRNAs. Alternatively, the efficiency of the DSB formation by paired nickase may be lower than that by Cas9 nuclease. The former is more likely since D10A Cas9 nickase used in this study was recently reported to have a higher cutting efficiency than Cas9 nuclease.53
So far, many methodologies for the detection of unintended mutations by genome editing have been reported, and they are grouped into two categories: “biased” methods and “unbiased” methods.54 In the biased methods, potential off-target sites are predicted using in silico homology searches, and the resulting sites are analyzed by a T7 endonuclease 1 assay or deep sequencing.26 However, these methods have only a limited capability to detect off-target mutations and cannot reveal unpredicted genomic alterations. In contrast, the unbiased methods directly detect mutations at the genome-wide level independent of in silico prediction tools.54 Therefore, to develop medical therapeutics for human diseases using genome editing, on- and off-target mutations need to be thoroughly assessed using the unbiased method, although most studies in the context of disease treatment do not use them. Using LAM-HTGTS, we clearly demonstrated a significant translocation hotspot, even in the case of the double nicking strategy, against our expectations.37 One possible explanation for the off-target cleavages is that the Cas9 nickase may exert residual nuclease activity. Indeed, D10A Cas9 nickase with a single sgRNA (not a double nicking) exhibited low levels of on-target indels.24,55 The sgRNAs we used for the double nicking strategy may have relatively frequent homologous sequences along the genome, thereby generating DSBs individually without pairing. Another possibility is that the single-strand breaks (SSBs; nicks) generated by Cas9 nickase led to the formation of DSBs during the replication of the chromosome. SSBs are mainly repaired via the base excision repair pathway with a high degree of fidelity.56 However, if they are left unrepaired, replicative polymerases encounter these SSBs, which can result in the collapse of the replication fork, and subsequently in the formation of DSBs.57, 58, 59 In the presence of Cas9 nickase, the cycles of nick formation and their repair will be continuously repeated until the enzyme is inactivated. This may result in the formation of frequent DSBs, especially in the actively dividing cells, such as HEK293 cells.
Apart from the off-target effects, LAM-HTGTS revealed on-target genomic rearrangements, including possible large deletions. The largest deletion size estimated was several kb, resulting from a single DSB at the RAG1 locus. However, this was exceeded by 20 and 60 kb by dual DSBs at the DMPK locus as a result of Cas9 nuclease and Cas9 nickase, respectively. Recently, on-target large deletions caused by CRISPR-Cas9 have been reported by several research groups. They have found deletions ranging from several hundred bp up to several kb in mouse embryonic stem cells (ESCs) or cultured cell lines using long PCR and Sanger sequencing.60, 61, 62, 63 However, larger deletions, such as those identified in our study, are difficult to detect using these strategies, since PCR primer binding sites are lost. In another report, an unbiased mutation detection methodology, UDiTaS (uni-directional targeted sequencing), was used to detect on-target mutations.64 They generated dual DSBs using Cas9 nuclease at the DMD locus and found large deletions of up to several kb in mice.65 The difference in the deletion size between these studies and our own study (several kb versus several 10s of kb) may be attributed to the differences in the targeting species, locus, or the procedures used. In our study, the dual DSBs rather than the single DSB, and the double nicking rather than the conventional Cas9 nuclease, induced larger deletions. However, no general conclusions can be currently drawn due to the limited number of experiments. As such, further investigations will be required to confirm our findings.
As discussed above, DSBs can exert potential deleterious effects on genomic structure, even when the double nicking strategy is used. In addition, recent reports found that DSBs induced by CRISPR-Cas9 trigger a p53-mediated DNA damage response and cell cycle arrest.66,67 Prompted by these observations, we sought to clarify the effects of CRISPRi, a methodology independent of a DSB formation, using dCas9. Since it was demonstrated that the FANTOM5 promoter atlas represented the most reliable source of TSS annotations, we used this online database for the prediction of TSSs.68 Although the position of sgRNA spanning −50 to +150 relative to the TSS is generally recommended, its functionality also depends on the chromatin accessibility of the target site.68 In our case, using the sgRNA designed at +184 relative to the DMPK TSS, the transcription and the formation of RNA foci were successfully suppressed. Although this procedure inhibits the transcription of both the normal and mutant allele of DMPK, serious undesired effects were not presumed since a targeted deletion of DMPK exhibited no significant phenotypic alteration in mice.69 So far, two studies have reported on the use of dCas9 to inhibit the pathogenic pathway of DM1. The strategies reported in these studies differ from our own in that the CTG or CUG repeat sequence on the genome or mRNA was directly targeted by sgRNAs.70,71 These approaches allow for the selective reduction of mutant DMPK mRNA, while non-selectively affecting the CTG repeat tracts found in the form of microsatellite sequences on the human genome.
In conclusion, we demonstrated that both a conventional Cas9 nuclease method and a double nicking strategy using Cas9 nickase can be used to successfully excise CTG repeats in the DM1 locus and suppress the formation of RNA foci. However, contrary to our expectations, off-target cleavage and on-target genomic rearrangements were observed as a result of using the double nicking strategy, to a comparable degree as that observed when using Cas9 nuclease excision. By reducing the transcription of DMPK using CRISPRi, a DSB-free procedure, the formation of RNA foci was significantly inhibited. We propose that this alternative approach should be used for the development of a safer therapeutic strategy for the treatment of DM1 in the future.
Materials and Methods
Plasmid Construction
The expression plasmids for Cas9 nuclease (pSpCas9(BB)-2A-GFP) and Cas9 nickase, a D10A mutant version of Cas9 nuclease (pSpCas9n(BB)-2A-GFP), were gifts from Dr. Feng Zhang (Addgene, plasmid nos. 48138 [http://addgene.org/48138] and 48140 [http://addgene.org/48140]). The target sites of sgRNA were selected using the online CRISPR design tool (http://zlab.bio/guide-design-resources). Based on the input of the sequences flanking the 5′ and 3′ regions of the CTG repeat of the DMPK gene, several targeting sites were chosen to generate the fewest number of off-target sites as close to the CTG repeat as possible. The sgRNAs were cloned into the plasmids as described previously.26 Briefly, the top and bottom strands of oligonucleotides with BbsI restriction sites on their 5′ termini were chemically synthesized at FASMAC (Atsugi, Japan). The oligonucleotides were phosphorylated and annealed in a thermal cycler using the following program: 37°C for 30 min; 95°C for 5 min; ramp down to 25°C at 5°C/min. Then, pSpCas9(BB)-2A-GFP and pSpCas9n(BB)-2A-GFP were digested by BbsI (NEB, Ipswich, MA, USA) and ligated with each sgRNA. The insertions of the sgRNAs were confirmed by Sanger sequencing. For the CRISPRi experiments, AAV CMV-dSaCas9-KRAB-bGHpA, gifted by Dr. Charles Gersbach (Addgene, plasmid no. 106219 [http://addgene.org/106219]) and EF1-RFP-U6-gRNA (System Biosciences, Palo Alto, CA, USA) were used to express dCas9 and sgRNA, respectively.72 The TSS of the human DMPK gene was identified using the online database FANTOM5 (https://fantom.gsc.riken.jp/5/).73 sgRNAs with a PAM sequence of NNGRRT or NNGRR were designed in the vicinity using Benchling software (https://www.benchling.com). The corresponding oligonucleotides were synthesized, annealed, and ligated to the EF1-RFP-U6-gRNA plasmid according to the manufacturer’s instructions. The sgRNA sequences used are listed in Table S3.
Cell Culture, Transfection, and Electroporation
HEK293 cells and DM1 patient-derived fibroblasts were obtained from RIKEN BioResource Research Center (BRC) (Tsukuba, Japan) and the Coriell Institute (Camden, NJ, USA), respectively. The fibroblast GM03991, harboring 50–80 CTG repeats, was obtained from a mildly affected patient, while the fibroblast GM05163, with 400 CTG repeats, was obtained from a moderately affected individual. HEK293 cells were plated on plastic culture dishes or six-well plates (Corning Life Sciences, Oneonta, NY, USA) and grown in the Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum and antibiotic-antimycotic (Thermo Fisher Scientific, Waltham, MA, USA). Fibroblasts were plated on six-well plates and grown in minimal essential medium (Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum and antibiotic-antimycotic. All cells were grown in a humidified 37°C incubator with 5% CO2 and 95% air. The HEK293 cells were transfected with the expression plasmids using Effectene (QIAGEN, Hilden, Germany). The fibroblasts were transfected using electroporation with an Amaxa 4D-Nucleofector (Lonza, Basel, Switzerland) by condition CA-137 according to the manufacturer’s instructions. The cells were harvested 3 days after the transfection.
PCR Amplification and T7 Endonuclease 1 Assay
Genomic DNA was extracted from the cultured cells using a DNeasy Blood & Tissue kit (QIAGEN, Hilden, Germany) and quantified using a Qubit 2.0 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). The PCR amplification of the on-target DMPK locus was performed using the extracted genomic DAN as a template with KOD FX (Toyobo, Osaka, Japan). The PCR primers used are indicated in Table S3. The following program was used: an initial denaturation at 94°C for 2 min; 35 cycles at 98°C for 10 s, 68°C for 3 min. After the separation of the PCR products in 2% agarose gel, the DNA was extracted using a QIAquick gel extraction kit (QIAGEN, Hilden, Germany) and analyzed by Sanger sequencing. A T7 endonuclease 1 assay was performed using a Surveyor mutation detection kit (Integrated DNA Technologies, Coralville, IA, USA), according to the manufacturer’s protocol. Briefly, after genome editing with Cas9 nuclease or Cas9 nickase, the genomic DNA was extracted from HEK293 cells or fibroblasts. On- and off-target loci were amplified by PCR using LA Taq DNA polymerase (Takara Bio, Shiga, Japan) and the primers listed in Table S3. The PCR products were heat-denaturated and then re-annealed according to the following program in a thermal cycler: 95°C for 10 min; ramp down to 25°C at 1°C/15 s; hold at 4°C. Heteroduplex PCR products comprised of wild-type and mutant alleles were digested by incubating with Surveyor nuclease at 42°C for 60 min. The PCR products were separated by 4%–20% gradient polyacrylamide gel electrophoresis, and the band intensities were measured using ImageJ software. Indel occurrence (indel %) was estimated as previously described.26
Quantitative RT-PCR
The RNA was extracted from cultured cells using an RNeasy mini kit (QIAGEN, Hilden, Germany) and quantified using a Qubit 2.0 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). Then, the cDNA was reverse transcribed using ReverTra Ace (Toyobo, Osaka, Japan) according to the manufacturer’s instructions. Quantitative RT-PCR was conducted with a 7500 Fast real-time PCR system (Thermo Fisher Scientific, Waltham, MA, USA) using the cDNAs as templates. The gene expression was quantitatively analyzed by TaqMan assay using TaqMan probes Hs01094329_m1 and Hs02786624_g1 for DMPK and GAPDH, where the latter was used as an internal control.
RNA-FISH and Quantitative Analysis of RNA Foci
For the RNA-FISH assay, a DNA/locked nucleic acid (LNA) chimeric oligonucleotide labeled with Cy3 or Alexa Fluor 488 was synthesized as follows (GeneDesign, Osaka, Japan): 5′-Cy3/Alexa 488-CAGCAGCAGCAGCAGCAGCA-3′, where underlining denotes LNAs. The fibroblasts were grown on non-coated coverslips. The cells were fixed with cold 4% paraformaldehyde for 30 min at 4°C and permeabilized for 5 min at 4°C with 2% acetone in PBS pre-chilled at ‒20°C. Hybridization was performed with 2 ng/μL of DNA/LNA probe in hybridization buffer (30% formamide, 2× SSC, 200 ng/mL single-stranded DNA [ssDNA], 0.02% BSA, 10% dextran sulfate, and 2 mM vanadyl ribonucleoside) overnight at 37°C. Then, the cells were washed with 30% formamide in 2× SSC for 30 min at 45°C and 30% formamide in 1× SSC for 30 min at 37°C. For the immunofluorescence detection of the transfected cells, fibroblasts were blocked with 3% BSA in PBS for 1 h and incubated with anti-GFP (Thermo Fisher Scientific, Waltham, MA, USA) or anti-RFP (MBL International, Nagoya, Japan) antibody. After washing, the coverslips were mounted with ProLong Diamond antifade mountant with DAPI (Thermo Fisher Scientific, Waltham, MA, USA), and fluorescent images were taken using an FSX100 fluorescence microscope (Olympus, Tokyo, Japan). For the quantitative analysis of the RNA foci, images of more than 100 nuclei were captured from each group. The numbers of RNA foci in the nuclei were counted by visual observation, and the intensities of the RNA foci in the nuclei were measured using ImageJ software. Statistical differences were evaluated using Student’s t test. p values <0.05 were considered statistically significant. For the detection of MBNL1, the RNA-FISH assay was followed by immunofluorescent staining with anti-MBNL1 MB1a (Developmental Studies Hybridoma Bank, Iowa City, IA, USA), according to the standard protocol.
Unbiased Genome-wide Detection of Off-target DSBs
LAM-HTGTS was performed as previously reported with some modifications.37,38 Briefly, HEK293 cells (6 × 106 cells for each group) were transfected with Cas9 nuclease or Cas9 nickase with the respective sgRNAs (5′ guide 3 and 3′ guide 2 for Cas9 nuclease, Nick 1 and Nick 3 for Cas9 nickase). As a positive control of the off-target effects, the cells were transfected with Cas9 nuclease and sgRNA targeting RAG1 gene site A (RAG1A) (Table S3).37 Three days after the transfection, genomic DNA was extracted from the cells with a DNeasy Blood & Tissue kit (QIAGEN, Hilden, Germany) and incubated overnight at 56°C. Then, the genomic DNA was sheared using a Covaris Focused-ultrasonicator (Covaris, Woburn, MA, USA). After purification using Agencourt AMPure XP beads (Beckman Coulter, Brea, CA, USA), the sheared DNA was amplified by LAM-PCR using KOD FX (Toyobo, Osaka, Japan) and sequence-specific biotinylated primers for DMPK and RAG1 (Table S4) under the following conditions: an initial denaturation at 98°C for 2 min; 80 cycles at 95°C for 30 s, 58°C for 30 s, 72°C for 90 s; a final extension at 72°C for 2 min. The PCR products were incubated with Dynabeads MyONE streptavidin C1 beads (Thermo Fisher Scientific, Waltham, MA, USA) for 4 h. The DNA-beads complex was then captured by the magnetic stand. After annealing the upper and lower strands of the oligonucleotides for the bridge adaptor (Table S4), this was ligated to the DNA-beads complex using T4 DNA ligase (Promega, Madison, WI, USA) by incubating at 25°C for 1 h, 92°C for 2 h, and 16°C for 1 h. The on-beads ligation products were again captured by the magnetic stand and washed twice. Nested PCR was performed using the on-beads ligation products as templates with the primers indicated in Table S4. The PCR conditions were as follows: an initial denaturation at 95°C for 5 min; 15 cycles at 95°C for 60 s, 60°C for 30 s, 72°C for 60 s; a final extension at 72°C for 6 min. The PCR products were centrifuged at 15,000 × g for 5 min and the supernatants were concentrated using a QIAquick gel extraction kit (QIAGEN, Hilden, Germany). The DNA samples were barcoded for multiplexing using dual index primer sets (Illumina, San Diego, CA, USA) (Table S4) using the following conditions: an initial denaturation at 95°C for 3 min; 16 cycles at 95°C for 30 s, 62°C for 30 s, 72°C for 60 s; a final extension at 72°C for 6 min. The PCR products were run on a 1% agarose gel and the DNA was extracted from an area ranging from 500 to 1,000 bp using a QIAquick gel extraction kit. After barcoding, the samples were pooled in equal mass ratios. A denatured and diluted library pool of 600 μL of PhiX control was sequenced with the 300-bp paired-end sequencing reactions on the MiSeq sequencer using the a V3 600 cycle kit (Illumina, San Diego, CA, USA). After trimming the adaptor sequences, the sequence reads were mapped to the hg19 reference genome, and any PCR duplicates were removed. The reads, which contain 20 bp of an adjacent to on-target breakpoint locus and at least 10 bp of uncertain nucleotides beyond the breakpoint, were specifically extracted for subsequent analysis. Off-target digestion and genomic rearrangements were detected as split reads, which mapped more than two different genomic loci in the same reads. Split reads were counted in a series of 100-bp equal-sized bins of entire genome. Off-target sites and the number of split reads were drawn using a Circos plot.74
Author Contributions
F.S. conducted the study and wrote the manuscript. M.I. performed most of the experiments. M.T.-I. and T.K. performed deep sequencing. Y.S. performed bioinformatics analysis. S.T., H.H., N.S., and T.M. analyzed the experiments. K.M., M.S., and H.K. supervised the project.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
F.S. was supported by an Intramural Research Grant (29-4) for Neurological and Psychiatric Disorders of NCNP from the Ministry of Health, Labour and Welfare of Japan and a Grant-in-Aid for Scientific Research C (19K07981) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. F.S. received research support from Eisai, Takeda Pharmaceutical, Daiichi Sankyo, and Novartis. K.M. was supported by a Grant-in-Aid for Scientific Research C (19K07406) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. H.H. was supported by a Grant-in-Aid for Scientific Research C (19K11359) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. T.M was supported by a Grant-in-Aid for Scientific Research C (18K10693) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. M.T.I. was supported by the Naito Foundation and the Hoansya Foundation.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.05.024.
Supplemental Information
References
- 1.Turner C., Hilton-Jones D. The myotonic dystrophies: diagnosis and management. J. Neurol. Neurosurg. Psychiatry. 2010;81:358–367. doi: 10.1136/jnnp.2008.158261. [DOI] [PubMed] [Google Scholar]
- 2.Mahadevan M., Tsilfidis C., Sabourin L., Shutler G., Amemiya C., Jansen G., Neville C., Narang M., Barceló J., O’Hoy K. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 3.Fu Y.H., Pizzuti A., Fenwick R.G., Jr., King J., Rajnarayan S., Dunne P.W., Dubel J., Nasser G.A., Ashizawa T., de Jong P. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–1258. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- 4.Zerylnick C., Torroni A., Sherman S.L., Warren S.T. Normal variation at the myotonic dystrophy locus in global human populations. Am. J. Hum. Genet. 1995;56:123–130. [PMC free article] [PubMed] [Google Scholar]
- 5.Harley H.G., Rundle S.A., MacMillan J.C., Myring J., Brook J.D., Crow S., Reardon W., Fenton I., Shaw D.J., Harper P.S. Size of the unstable CTG repeat sequence in relation to phenotype and parental transmission in myotonic dystrophy. Am. J. Hum. Genet. 1993;52:1164–1174. [PMC free article] [PubMed] [Google Scholar]
- 6.Tsilfidis C., MacKenzie A.E., Mettler G., Barceló J., Korneluk R.G. Correlation between CTG trinucleotide repeat length and frequency of severe congenital myotonic dystrophy. Nat. Genet. 1992;1:192–195. doi: 10.1038/ng0692-192. [DOI] [PubMed] [Google Scholar]
- 7.Lavedan C., Hofmann-Radvanyi H., Shelbourne P., Rabes J.P., Duros C., Savoy D., Dehaupas I., Luce S., Johnson K., Junien C. Myotonic dystrophy: size- and sex-dependent dynamics of CTG meiotic instability, and somatic mosaicism. Am. J. Hum. Genet. 1993;52:875–883. [PMC free article] [PubMed] [Google Scholar]
- 8.Ashizawa T., Anvret M., Baiget M., Barceló J.M., Brunner H., Cobo A.M., Dallapiccola B., Fenwick R.G., Jr., Grandell U., Harley H. Characteristics of intergenerational contractions of the CTG repeat in myotonic dystrophy. Am. J. Hum. Genet. 1994;54:414–423. [PMC free article] [PubMed] [Google Scholar]
- 9.Davis B.M., McCurrach M.E., Taneja K.L., Singer R.H., Housman D.E. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc. Natl. Acad. Sci. USA. 1997;94:7388–7393. doi: 10.1073/pnas.94.14.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michalowski S., Miller J.W., Urbinati C.R., Paliouras M., Swanson M.S., Griffith J. Visualization of double-stranded RNAs from the myotonic dystrophy protein kinase gene and interactions with CUG-binding protein. Nucleic Acids Res. 1999;27:3534–3542. doi: 10.1093/nar/27.17.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mooers B.H., Logue J.S., Berglund J.A. The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc. Natl. Acad. Sci. USA. 2005;102:16626–16631. doi: 10.1073/pnas.0505873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller J.W., Urbinati C.R., Teng-Umnuay P., Stenberg M.G., Byrne B.J., Thornton C.A., Swanson M.S. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mankodi A., Urbinati C.R., Yuan Q.P., Moxley R.T., Sansone V., Krym M., Henderson D., Schalling M., Swanson M.S., Thornton C.A. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 2001;10:2165–2170. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- 14.Fardaei M., Rogers M.T., Thorpe H.M., Larkin K., Hamshere M.G., Harper P.S., Brook J.D. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 2002;11:805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 15.Kanadia R.N., Johnstone K.A., Mankodi A., Lungu C., Thornton C.A., Esson D., Timmers A.M., Hauswirth W.W., Swanson M.S. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 16.Kuyumcu-Martinez N.M., Wang G.S., Cooper T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalsotra A., Singh R.K., Gurha P., Ward A.J., Creighton C.J., Cooper T.A. The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell Rep. 2014;6:336–345. doi: 10.1016/j.celrep.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mankodi A., Takahashi M.P., Jiang H., Beck C.L., Bowers W.J., Moxley R.T., Cannon S.C., Thornton C.A. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 19.Charlet-B N., Savkur R.S., Singh G., Philips A.V., Grice E.A., Cooper T.A. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 20.Fugier C., Klein A.F., Hammer C., Vassilopoulos S., Ivarsson Y., Toussaint A., Tosch V., Vignaud A., Ferry A., Messaddeq N. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat. Med. 2011;17:720–725. doi: 10.1038/nm.2374. [DOI] [PubMed] [Google Scholar]
- 21.Savkur R.S., Philips A.V., Cooper T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 22.Garneau J.E., Dupuis M.È., Villion M., Romero D.A., Barrangou R., Boyaval P., Fremaux C., Horvath P., Magadán A.H., Moineau S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 23.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Komor A.C., Badran A.H., Liu D.R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017;168:20–36. doi: 10.1016/j.cell.2016.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Agtmaal E.L., André L.M., Willemse M., Cumming S.A., van Kessel I.D.G., van den Broek W.J.A.A., Gourdon G., Furling D., Mouly V., Monckton D.G. CRISPR/Cas9-induced (CTG⋅CAG)n repeat instability in the myotonic dystrophy type 1 locus: implications for therapeutic genome editing. Mol. Ther. 2017;25:24–43. doi: 10.1016/j.ymthe.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Provenzano C., Cappella M., Valaperta R., Cardani R., Meola G., Martelli F., Cardinali B., Falcone G. CRISPR/Cas9-mediated deletion of CTG expansions recovers normal phenotype in myogenic cells derived from myotonic dystrophy 1 patients. Mol. Ther. Nucleic Acids. 2017;9:337–348. doi: 10.1016/j.omtn.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dastidar S., Ardui S., Singh K., Majumdar D., Nair N., Fu Y., Reyon D., Samara E., Gerli M.F.M., Klein A.F. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 2018;46:8275–8298. doi: 10.1093/nar/gky548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lo Scrudato M., Poulard K., Sourd C., Tomé S., Klein A.F., Corre G., Huguet A., Furling D., Gourdon G., Buj-Bello A. Genome editing of expanded CTG repeats within the human DMPK gene reduces nuclear RNA foci in the muscle of DM1 mice. Mol. Ther. 2019;27:1372–1388. doi: 10.1016/j.ymthe.2019.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X.H., Tee L.Y., Wang X.G., Huang Q.S., Yang S.H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther. Nucleic Acids. 2015;4:e264. doi: 10.1038/mtna.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ran F.A., Hsu P.D., Lin C.Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y., Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen B., Zhang W., Zhang J., Zhou J., Wang J., Chen L., Wang L., Hodgkins A., Iyer V., Huang X., Skarnes W.C. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods. 2014;11:399–402. doi: 10.1038/nmeth.2857. [DOI] [PubMed] [Google Scholar]
- 34.Canver M.C., Bauer D.E., Dass A., Yien Y.Y., Chung J., Masuda T., Maeda T., Paw B.H., Orkin S.H. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J. Biol. Chem. 2014;289:21312–21324. doi: 10.1074/jbc.M114.564625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frock R.L., Hu J., Meyers R.M., Ho Y.J., Kii E., Alt F.W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015;33:179–186. doi: 10.1038/nbt.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu J., Meyers R.M., Dong J., Panchakshari R.A., Alt F.W., Frock R.L. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat. Protoc. 2016;11:853–871. doi: 10.1038/nprot.2016.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakamori M., Taylor K., Mochizuki H., Sobczak K., Takahashi M.P. Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy. Ann. Clin. Transl. Neurol. 2015;3:42–54. doi: 10.1002/acn3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rzuczek S.G., Colgan L.A., Nakai Y., Cameron M.D., Furling D., Yasuda R., Disney M.D. Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol. 2017;13:188–193. doi: 10.1038/nchembio.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wheeler T.M., Leger A.J., Pandey S.K., MacLeod A.R., Nakamori M., Cheng S.H., Wentworth B.M., Bennett C.F., Thornton C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. 2012;488:111–115. doi: 10.1038/nature11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jauvin D., Chrétien J., Pandey S.K., Martineau L., Revillod L., Bassez G., Lachon A., MacLeod A.R., Gourdon G., Wheeler T.M. Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol. Ther. Nucleic Acids. 2017;7:465–474. doi: 10.1016/j.omtn.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao Y., Guo X., Santostefano K., Wang Y., Reid T., Zeng D., Terada N., Ashizawa T., Xia G. Genome therapy of myotonic dystrophy type 1 iPS cells for development of autologous stem cell therapy. Mol. Ther. 2016;24:1378–1387. doi: 10.1038/mt.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y., Hao L., Wang H., Santostefano K., Thapa A., Cleary J., Li H., Guo X., Terada N., Ashizawa T., Xia G. Therapeutic genome editing for myotonic dystrophy type 1 using CRISPR/Cas9. Mol. Ther. 2018;26:2617–2630. doi: 10.1016/j.ymthe.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang D., Zhang F., Gao G. CRISPR-based therapeutic genome editing: strategies and in vivo delivery by AAV vectors. Cell. 2020;181:136–150. doi: 10.1016/j.cell.2020.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Staahl B.T., Benekareddy M., Coulon-Bainier C., Banfal A.A., Floor S.N., Sabo J.K., Urnes C., Munares G.A., Ghosh A., Doudna J.A. Efficient genome editing in the mouse brain by local delivery of engineered Cas9 ribonucleoprotein complexes. Nat. Biotechnol. 2017;35:431–434. doi: 10.1038/nbt.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finn J.D., Smith A.R., Patel M.C., Shaw L., Youniss M.R., van Heteren J., Dirstine T., Ciullo C., Lescarbeau R., Seitzer J. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 2018;22:2227–2235. doi: 10.1016/j.celrep.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 49.Lee K., Conboy M., Park H.M., Jiang F., Kim H.J., Dewitt M.A., Mackley V.A., Chang K., Rao A., Skinner C. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 2017;1:889–901. doi: 10.1038/s41551-017-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramakrishna S., Kwaku Dad A.B., Beloor J., Gopalappa R., Lee S.K., Kim H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 2014;24:1020–1027. doi: 10.1101/gr.171264.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gee P., Lung M.S.Y., Okuzaki Y., Sasakawa N., Iguchi T., Makita Y., Hozumi H., Miura Y., Yang L.F., Iwasaki M. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat. Commun. 2020;11:1334. doi: 10.1038/s41467-020-14957-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwab I., Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat. Rev. Immunol. 2013;13:176–189. doi: 10.1038/nri3401. [DOI] [PubMed] [Google Scholar]
- 53.Gopalappa R., Suresh B., Ramakrishna S., Kim H.H. Paired D10A Cas9 nickases are sometimes more efficient than individual nucleases for gene disruption. Nucleic Acids Res. 2018;46:e71. doi: 10.1093/nar/gky222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yee J.K. Off-target effects of engineered nucleases. FEBS J. 2016;283:3239–3248. doi: 10.1111/febs.13760. [DOI] [PubMed] [Google Scholar]
- 55.Alateeq S., Ovchinnikov D., Tracey T., Whitworth D., Al-Rubaish A., Al-Ali A., Wolvetang E. Identification of on-target mutagenesis during correction of a β-thalassemia splice mutation in iPS cells with optimised CRISPR/Cas9-double nickase reveals potential safety concerns. APL Bioeng. 2018;2:046103. doi: 10.1063/1.5048625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caldecott K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008;9:619–631. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 57.Kuzminov A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA. 2001;98:8241–8246. doi: 10.1073/pnas.131009198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ensminger M., Iloff L., Ebel C., Nikolova T., Kaina B., Lӧbrich M. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 2014;206:29–43. doi: 10.1083/jcb.201312078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cannan W.J., Pederson D.S. Mechanisms and consequences of double-strand DNA break formation in chromatin. J. Cell. Physiol. 2016;231:3–14. doi: 10.1002/jcp.25048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adikusuma F., Piltz S., Corbett M.A., Turvey M., McColl S.R., Helbig K.J., Beard M.R., Hughes J., Pomerantz R.T., Thomas P.Q. Large deletions induced by Cas9 cleavage. Nature. 2018;560:E8–E9. doi: 10.1038/s41586-018-0380-z. [DOI] [PubMed] [Google Scholar]
- 61.Shin H.Y., Wang C., Lee H.K., Yoo K.H., Zeng X., Kuhns T., Yang C.M., Mohr T., Liu C., Hennighausen L. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat. Commun. 2017;8:15464. doi: 10.1038/ncomms15464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Owens D.D.G., Caulder A., Frontera V., Harman J.R., Allan A.J., Bucakci A., Greder L., Codner G.F., Hublitz P., McHugh P.J. Microhomologies are prevalent at Cas9-induced larger deletions. Nucleic Acids Res. 2019;47:7402–7417. doi: 10.1093/nar/gkz459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Giannoukos G., Ciulla D.M., Marco E., Abdulkerim H.S., Barrera L.A., Bothmer A., Dhanapal V., Gloskowski S.W., Jayaram H., Maeder M.L. UDiTaS™, a genome editing detection method for indels and genome rearrangements. BMC Genomics. 2018;19:212. doi: 10.1186/s12864-018-4561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 67.Ihry R.J., Worringer K.A., Salick M.R., Frias E., Ho D., Theriault K., Kommineni S., Chen J., Sondey M., Ye C. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018;24:939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 68.Radzisheuskaya A., Shlyueva D., Müller I., Helin K. Optimizing sgRNA position markedly improves the efficiency of CRISPR/dCas9-mediated transcriptional repression. Nucleic Acids Res. 2016;44:e141. doi: 10.1093/nar/gkw583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carrell S.T., Carrell E.M., Auerbach D., Pandey S.K., Bennett C.F., Dirksen R.T., Thornton C.A. Dmpk gene deletion or antisense knockdown does not compromise cardiac or skeletal muscle function in mice. Hum. Mol. Genet. 2016;25:4328–4338. doi: 10.1093/hmg/ddw266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pinto B.S., Saxena T., Oliveira R., Méndez-Gómez H.R., Cleary J.D., Denes L.T., McConnell O., Arboleda J., Xia G., Swanson M.S., Wang E.T. Impeding transcription of expanded microsatellite repeats by deactivated Cas9. Mol. Cell. 2017;68:479–490.e5. doi: 10.1016/j.molcel.2017.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Batra R., Nelles D.A., Pirie E., Blue S.M., Marina R.J., Wang H., Chaim I.A., Thomas J.D., Zhang N., Nguyen V. Elimination of toxic microsatellite repeat expansion RNA by RNA-targeting Cas9. Cell. 2017;170:899–912.e10. doi: 10.1016/j.cell.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thakore P.I., Kwon J.B., Nelson C.E., Rouse D.C., Gemberling M.P., Oliver M.L., Gersbach C.A. RNA-guided transcriptional silencing in vivo with S. aureus CRISPR-Cas9 repressors. Nat. Commun. 2018;9:1674. doi: 10.1038/s41467-018-04048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Noguchi S., Arakawa T., Fukuda S., Furuno M., Hasegawa A., Hori F., Ishikawa-Kato S., Kaida K., Kaiho A., Kanamori-Katayama M. FANTOM5 CAGE profiles of human and mouse samples. Sci. Data. 2017;4:170112. doi: 10.1038/sdata.2017.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hu Y., Yan C., Hsu C.H., Chen Q.R., Niu K., Komatsoulis G.A., Meerzaman D. OmicCircos: a simple-to-use R package for the circular visualization of multidimensional omics data. Cancer Inform. 2014;13:13–20. doi: 10.4137/CIN.S13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.