

Abstract
Long non-coding RNAs (lncRNAs) have been recognized as critical components of a broad genomic regulatory network and play pivotal roles in physiological and pathological processes. Identification of disease-associated lncRNAs is becoming increasingly crucial for fundamentally improving our understanding of molecular mechanisms of disease and developing novel biomarkers and therapeutic targets. Considering lower efficiency and higher time and labor cost of biological experiments, computer-aided inference of disease-associated RNAs has become a promising avenue for facilitating the study of lncRNA functions and provides complementary value for experimental studies. In this study, we first summarize data and knowledge resources publicly available for the study of lncRNA-disease associations. Then, we present an updated systematic overview of dozens of computational methods and models for inferring lncRNA-disease associations proposed in recent years. Finally, we explore the perspectives and challenges for further studies. Our study provides a guide for biologists and medical scientists to look for dedicated resources and more competent tools for accelerating the unraveling of disease-associated lncRNAs.
Keywords: long non-coding RNAs, disease, lncRNA-disease association, computational method, bioinformatics
Graphical Abstract
Identification of disease-associated lncRNAs is a significant and challenging task in biomedical research. This study presents an overview of computer-aided inference of lncRNA-disease associations and provides a guide for biologists and medical scientists to look for dedicated resources and more competent tools for accelerating the unraveling of disease-associated lncRNAs.
Introduction
Advances in genomic and transcriptional analyses have markedly expanded our knowledge of the genomic dark matter and revealed that only about 2% of the human genome encodes protein-coding genes, and the vast majority are transcribed into non-coding RNAs (ncRNAs).1,2 Long ncRNAs (lncRNAs), constituting the biggest class of ncRNAs, were arbitrarily defined as ncRNAs with more than 200 nt in length.3,4 There is increasing evidence that lncRNAs are hidden critical components of a broad genomic regulatory network involved in gene transcription, epigenetic regulation, and post-transcriptional regulation, and they thus play pivotal roles in a wide variety of biological processes.5,6 A large number of lncRNAs with oncogenic or tumor-suppressor function have been found, highlighting the emerging role of lncRNAs in complex diseases.7, 8, 9
Identification of disease genes is a significant and challenging task in biomedical research. Systematic identification of disease-associated lncRNAs not only contributes to our understanding of the underlying molecular mechanisms of complex diseases, but it also has been shown to have the intrinsic advantage over protein-coding genes in disease diagnosis, prognosis, and treatment.10, 11, 12, 13, 14, 15, 16, 17 Despite increasing efforts that have been taken to explore the function of lncRNAs and their implications in diseases, the vast majority of lncRNAs are not functionally well characterized, and their associations with diseases remain unknown. Low-throughput biological experiments in vivo or in vitro have been extensively used to dissect disease-related lncRNAs. Although the exact association between disease and lncRNAs, as well as the pathogenic mechanism of lncRNAs, could be elucidated through in vivo or in vitro experiments, these low-throughput biological experiments tend to be time-consuming, expensive, and inefficient when faced with tens of thousands of lncRNAs with unknown function. With the application of high-throughput technologies (e.g., microarray and next-generation sequencing) to disease transcriptomes, a large number of dysregulated lncRNAs were identified to be associated with disease. However, results of high-throughput technologies contained much noise, and most of the dysregulated lncRNAs tend to be unrelated rather than causal lncRNAs because the aberrant expression in disease is not sufficient evidence to confirm the causal association between lncRNAs and diseases. With large-scale available heterogeneous data resources of lncRNAs and diseases, great efforts have been devoted to system-level inference of lncRNA-disease association through computational or bioinformatics approaches, which constitute a complement to wet-lab experiments.18,19
In this study, we present an overview of the computer-aided inference of the lncRNA-disease association. First, data sources accessible to the lncRNA-disease association study are introduced in detail. Second, novel computational methods and software tools, as well as their application in lncRNA-disease association prediction, are summarized and reviewed. Finally, we explore the future perspectives and challenges in this field.
Results
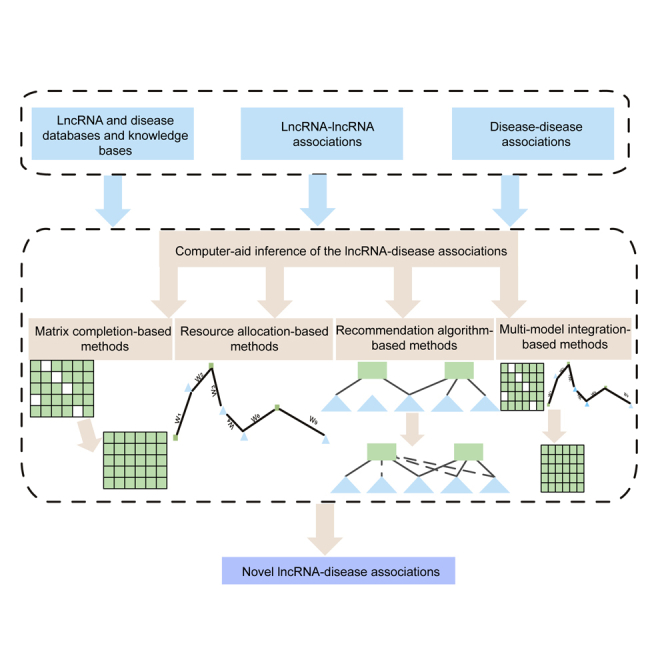
In this section, we reviewed dozens of novel computational methods in inferring the lncRNA-disease association proposed in recent years. Based on the core idea of the algorithm, these computational methods could be divided into four categories: (1) matrix completion-based methods, (2) recommendation algorithm-based methods, (3) resource allocation-based methods, and (4) integration-based methods.
Matrix Completion-Based Methods
The universal characteristic of the matrix completion-based methods is to complete the data set with missing values in the form of a matrix. As shown in Figure 1, three matrices, including the lncRNA-disease association matrix, lncRNA-lncRNA matrix, and disease-disease matrix, were obtained. Then feature extraction was accomplished based on the above three matrices to obtain lncRNA feature vectors and disease feature vectors. Finally, matrix completion methods were conducted on the lncRNA-disease association matrix to acquire the lncRNA-disease scoring matrix based on lncRNA feature vectors and disease feature vectors (Table 1).
Figure 1.

Schematic Workflow of Matrix Completion-Based Methods
Three matrices (including the lncRNA-disease association matrix, lncRNA-lncRNA matrix, and disease-disease matrix) were first obtained as the input data. Then, feature extraction was accomplished based on the above three matrices to obtain lncRNA feature vectors and disease feature vectors. Finally, matrix completion methods were performed on the lncRNA-disease association matrix to acquire the lncRNA-disease association.
Table 1.
Overview of Categories and Corresponding Method/Tool for Acquiring lncRNA-lncRNA Association
| Categories | Method/Tool | Data Types | Data Resources | References |
|---|---|---|---|---|
| Sequence similarity | EMBOSS Needle tool | lncRNA sequence | LncRNADisease, UCSC, LNCipedia | Needleman and Wunsch34 |
| Functional similarity | LNCSIM | lncRNA-disease association, MeSH descriptors | LncRNADisease, Lnc2Cancer, MNDR, MeSH | Chen et al.35 |
| Functional similarity | ILNCSIM | lncRNA-disease association, MeSH descriptors | MNDR, Lnc2Cancer, MeSH | Huang et al.36 |
| Functional similarity | NA | lncRNA-gene association, protein-protein interaction | LncRNA2Target, StarBase, HPRD | Paik et al.37 |
| Functional similarity | NA | lncRNA-miRNA association | StarBase | Zhao et al.40 |
| Expression similarity | Spearman/Pearson correlation | lncRNA expression profiles | Array Express, UCSC Genome Bioinformatics | Chen and Yan41 |
| Cosine similarity | cosine similarity | lncRNA-disease association | MNDR, Lnc2Cancer, LncRNADisease | Cheng et al.42 |
NA, not applicable.
Li et al.19 developed a computational model of faster randomized matrix completion for latent disease lncRNA association (named FRMCLDA) that used the faster singular value threshold (fSVT) algorithm to predict lncRNA-disease associations based on the idea of matrix completion. FRMCLDA uses the disease similarity matrix, lncRNA similarity matrix, lncRNA-disease association matrix, and transpose matrix of the association matrix to construct the adjacency matrix, which improves the prediction performance by fitting the adjacency matrix. LDAPM and SIMCLDA also use a matrix completion approach, but the difference is that LDAPM denotes the approximated matrix as the multiplication of the two matrices.20,21 TSSR exploits learned representation matrices as feature matrices to reconstruct the original matrix.22
Resource-Allocation-Based Methods
Resource allocation is the allocation of available resources to each node. To predict the lncRNA-disease association, resource allocation is based on the initial value of the multi-data source matrices as a possible value for the relationship between nodes. The process is demonstrated in Figure 2. Resource allocation-based methods are built on data from multiple sources, such as lncRNA-disease association, miRNA-disease association, miRNA-lncRNA association, and so on. The heterogeneous multilayer network is constructed, and the edges are weighted according to the corresponding values of the matrix. The lncRNA-disease scoring matrix was produced by post-processing resource allocation on the heterogeneous network (Table 2).
Figure 2.

Schematic Workflow of Resource Allocation-Based Methods
Multi-type data source matrices were first obtained as the input data. Then, a heterogeneous multilayer network is constructed, and the edges are weighted by the corresponding values of the matrix. Finally, the lncRNA-disease scoring matrix was produced by post-processing resource allocation on the heterogeneous network.
Table 2.
Overview of Categories and Corresponding Method/Tool for Acquiring Disease-Disease Association
| Categories | Method/Tool | Data Types | Data Resources | References |
|---|---|---|---|---|
| Semantic similarity | R package DOSE | MeSH descriptor | Disease Ontology, MeSH | Yu and Wang43 |
| Semantic similarity | NA | MeSH descriptor, Disease Ontology terms | MeSH, DincRNA | Chen et al.35 |
| Functional similarity | Jaccard coefficient | disease-gene association, gene-Gene Ontology terms association | Ensembl, DisGeNET | Mathur and Dinakarpandian44 |
| Functional similarity | NA | disease-miRNA association | HMDD | Zhao et al.40 |
| Gaussian interaction profile kernel similarity | Gaussian interaction profile kernel similarity/radial basis function (RBF) kernel similarity | disease-miRNA association, disease-gene association, lncRNA-disease association, sequence, expression | DisGeNet, HMDD, MNDR, Lnc2Cancer, LncRNADisease | Chen and Yan41 |
| Cosine similarity | Cosine similarity | lncRNA-disease association | MNDR, Lnc2Cancer, LncRNADisease | Hamaneh and Yu45 |
NA, not applicable.
Resource allocation has been implemented in more than a dozen methods for predicting the lncRNA-disease association. Fan et al.23 proposed a computational model of IDHI-MIRW by integrating diverse heterogeneous information sources with positive pointwise mutual information and random walk with restart algorithm. Xiao et al.24 developed BPLLDA to predict lncRNA-disease associations based on simple paths with limited lengths in a heterogeneous network. Zhang et al.25 proposed a rule-based inference method on the linked tripartite network, which was constructed by integrating heterogeneous data with deep learning algorithms. Some other information had also been introduced to allocate resources for improving prediction performance. For example, LION and DislncRF introduced protein information and genome-wide tissue expression profiles, which are aided by protein-coding genes.26,27 NBLDA and LLCLPLDA both constructed four matrices and used the label propagation algorithm for resource allocation.28,29 By constructing a disease weight matrix based on the similarity between the lncRNA disease set and the specified disease, IIRWR introduced the concept of disease clique and added the weight of disease linkages to the traveling network.30 Lap-BiRWRHLDA and BiWalkLDA both used laplacian normalization and bi-random walk algorithm on similarity networks.31,32 The other two methods, TPGLDA and ncPred, allocated resources from the disease to lncRNAs and other nodes, respectively, but the difference is that the resources were returned to the initial nodes.33,34
Recommendation Algorithm-Based Methods
The common characteristic of the recommendation algorithm-based methods is to recommend a node that may be related to another node. It mainly includes content-based recommendation, collaborative filtering, and matrix factorization. The process is depicted in Figure 3. After applying a recommendation system algorithm to multi-data matrices, recommendation matrices at multiple levels (such as lncRNA, miRNA, etc.) were obtained. Finally, the possibility of the potential relationship between lncRNA and disease was measured through the combination of the recommendation matrices (Table 3).
Figure 3.

Schematic Workflow of Recommendation Algorithm-Based Methods
Multi-type data source matrices were first obtained as the input data. Then, recommendation matrices at multiple levels (e.g., lncRNAs, miRNAs) are obtained by applying a recommendation system algorithm. Finally, the possibility of the potential relationship between lncRNA and disease is measured through the combination of the recommendation matrices.
Table 3.
Overview of Matrix Completion-Based Computational Methods for Inferring lncRNA-Disease Association
| Method Name | Computational Principle | Data Types | Available Tool (Package or Code) | References |
|---|---|---|---|---|
| SIMCLDA | inductive matrix completion, singular value decomposition | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | code (https://github.com//bioinfomaticsCSU/SIMCLDA) | Lu et al.47 |
| LDAPM | inductive matrix completion, singular value decomposition | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | NA | Fraidouni and Zaruba48 |
| FRMCLDA | faster randomized matrix completion, faster singular value threshold | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | code (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6749816/bin/Table_7.docx) | Li et al.46 |
| TSSR | sparse self-representation | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | code (https://github.com/Oyl-CityU/TSSR) | Ou-Yang et al.49 |
NA, not applicable.
The first category of recommendation algorithm-based methods is the content-based recommendation. Content-based recommendation refers to the recommendation of similar nodes of previous related nodes for this node. For example, NCPLDA, proposed by Li et al.,35 measured the lncRNA-disease association score based on network consistency projection. The second category of recommendation algorithm-based methods is based on collaborative filtering. Collaborative filtering refers to adding other nodes similar to the nodes and using the information of these nodes to make inferences. CFNBC, developed by Yu et al.,36 and NBCLDA, proposed by Yu et al.,37 both used collaborative filtering on multi-data matrices to uncover a new relationship between lncRNA and disease and then took advantage of a naive Bayesian classifier to determine whether there is an association between lncRNA and disease in the set of the lncRNA-associated node and disease-associated node. A similarity correlation fusion method introduced neighbor information and then was used to predict the association by making the original matrix fit as well as possible in ILDMSF and SKF-LDA.38,39 BLM-NPAI, developed by Cui et al.,40 introduced the nearest profile to get the final prediction results after constructing the local model of lncRNA and disease. Another method, proposed by Ping et al.,41 measured the one-step neighbor of a node based on simrank measure when there was no common neighbor. DCSLDA, proposed by Zhao et al.,42 calculated the shortest path between lncRNA and disease and the distance correlation coefficient to construct the final matrix. The third category of recommendation algorithm-based methods is based on matrix factorization, including MFLDA, WMFLDA, and PMFILDA. The three methods above are based on matrix factorization, and the difference is that the latter two add weight and probability separately.43, 44, 45 Li et al.46 developed a computational method, DNILMF-LDA, that is anchored in the neighborhood regularized logistic matrix factorization and optimizes the above parameters to predict interaction probabilities. NNLDA, determined by Hu et al.,47 solved some of the disadvantages of traditional matrix factorization by changing the training method and the loss function and adding a fully connected layer. DSCMF combines matrix factorization and collaborative filtering to predict associations efficiently by introducing neighbor information.48
Multi-model Integration-Based Methods
Multi-model integration methods have also been proposed to overcome the shortcomings of the single model and improve prediction performance (Table 4). A combination of matrix completion ideas and recommendation system ideas was applied in three models to predict potential lncRNA-disease associations, including LDASR, ECLDA, and weighted bagging LightGBM model.49, 50, 51 Three methods (CNNLDA, CNNDLP, and GCNLDA) were developed by Xuan et al.52, 53, 54 to construct the final module through the integration of the convolutional module and attention module. LDAPred, proposed by Xuan et al.,55 introduced the convolutional neural network based on the integration of resource allocation and matrix completion.
Table 4.
Overview of Resource Allocation-Based Computational Methods for Inferring lncRNA-Disease Association
| Method Name | Computational Principle | Data Types | Available Tool (Package or Code) | References |
|---|---|---|---|---|
| BPLLDA | paths together with a decay function | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | NA | Xiao et al.51 |
| TPGLDA | resource allocation | disease-gene association, lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | code (https://github.com/USTC-HIlab/TPGLDA) | Ding et al.62 |
| IDHI-MIRW | positive pointwise mutual information, random walk with restart algorithm | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | IDHI-MIRW (https://github.com/NWPU-903PR/IDHI-MIRW) | Fan et al.50 |
| Lap-BiRWRHLDA | Laplacian normalization, random walks | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | NA | Wen et al.60 |
| IIRWR | random walk with restart algorithm | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | code (https://github.com/xiaoyubin123/code) | Wang et al.59 |
| LLCLPLDA | label propagation algorithm, locality-constrained linear coding | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | NA | Xie et al.55 |
| LION | network diffusion approach | lncRNA-protein interaction, protein-protein interaction, protein-disease interaction | NA | Sumathipala et al.53 |
| NBLDA | label propagation algorithm | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | NA | Liu et al.58 |
| DislncRF | random forest | RNA sequencing data, disease-protein coding gene association, lncRNA-disease association | code (https://github.com/xypan1232/DislncRF) | Pan et al.54 |
| NA | DeepWalk and a rule-based inference method | lncRNA-disease association, lncRNA-miRNA association, miRNA-disease association | code (https://github.com/Pengeace/lncRNA-disease-link) | Zhang et al.52 |
| NA | ncPred | disease-target association, target-ncRNA association, ncRNA-ncRNA association, target-target association | NA | Mori et al.63 |
| BiWalkLDA | Laplacian normalization, random walks | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | code (https://github.com/screamer/BiwalkLDA) | Gao et al.61 |
NA, not applicable.
Discussion
During the past decade, it has been well documented that lncRNAs play a critical role in nearly all biological processes and have become an emerging paradigm of human disease research.56,57 Identification of disease-associated lncRNAs is becoming increasingly important for fundamentally improving our understanding of molecular mechanisms and developing novel therapeutic targets, and thus has attracted more and more attention in the scientific community and is becoming one of the hotspots in medical research. Although current experimental studies in vitro and in vivo could directly link identified lncRNAs with disease phenotypes, they are affected by the limitation of lower efficiency and higher time and labor cost. Taking into account the limitations of experimental studies, high-throughput technologies were then implemented, leading to exponential growth in the number of dysregulated lncRNAs in diseases. However, the aberrant expression of lncRNAs is not sufficient evidence for ascribing to them a functional role in disease.7 Therefore, efficient and accurate identification and functional elucidation of disease-associated lncRNAs are in their infancy and remain a major challenge. With the rapidly increasing quantity and quality of bioinformatics databases and resources in lncRNAs and diseases, computer-aided inference of disease-associated RNAs has become a promising avenue for facilitating the unraveling of the functional role of lncRNAs in diseases and provides complementary value for experimental studies. A large number of computational models, algorithms, and tools have been developed and proposed, compensating for this dearth.
In this work, we first summarize data and knowledge sources available for the lncRNA-disease association study, which contains databases of lncRNAs, diseases, and known lncRNA-disease associations. We then present a detailed overview of previously proposed computational methods for inferring lncRNA-disease associations. Based on the core idea implemented, these computational methods can be divided into four categories: (1) matrix completion-based methods, (2) recommendation algorithm-based methods, (3) resource allocation-based methods, and (4) multi-model integration-based methods. Despite that the performance of each computational method is very great according to the reports in their own studies, one emerging critical issue is that most of these methods used different data sources as their training dataset and carried out cross-validation on their dataset, lacking benchmark performance evaluation. These computational methods have distinct limitations and weaknesses, which are noted at follows. First, matrix completion-based methods considered the feature vectors of lncRNA and disease to improve the accuracy of the prediction. However, these algorithms hold the disadvantage of poor robustness. The ranks of diverse datasets are likely to vary widely. Second, because experimentally verified lncRNA-disease associations are still too incomplete, resource allocation-based methods need to consider the prediction of separate nodes or integrate additional biological information. However, although this can improve prediction accuracy, some interactions from other databases may contain some noise to interfere with prediction results. In addition, recommendation algorithm-based methods are stated separately. Content-based recommendation models only need node prior knowledge, but new levels of disease-lncRNA associations cannot be recognized. Although collaborative filtering-based recommendation methods complement this shortcoming, the spare lncRNA-disease association matrix is harmful to the recommendation, and the complexity and time cost of the algorithm will sharply increase when the amount of data is too large. Additionally, matrix factorization-based recommendation methods reduce space complexity by mapping a matrix to a product of low-dimensional matrices. These methods also make it easy to add additional data from different sources and use the intrinsic structure. Matrix factorization-based methods also have the same weaknesses as collaborative filtering-based recommendation methods. The above computational approaches can thus complement each other. Therefore, multi-model integration-based methods were proposed to achieve better performance when investigating the association between lncRNAs and diseases. Finally, only several computational approaches have been developed as online web tools, and most are still theoretical studies that hampered their use for biologists and medical scientists.
With the rapidly increasing knowledge for the functional mechanism of lncRNAs, several challenges that would be helpful to improve the accuracy and practicality of the predictors could be highlighted. It is well known that the majority of lncRNAs exhibited precise subcellular localization, thus performing regulatory roles in a spatiotemporal manner.5,6 Therefore, some interactions between lncRNAs and other biological molecules (DNA, RNA, and proteins) used in previous predictors are derived from prediction and do not exist in the real biological world. Therefore, co-localization information of lncRNAs and other biological molecules should be considered. Additionally, it has also been observed that lncRNAs were expressed in highly cell type-specific, tissue-specific, and disease-specific manners. Therefore, more molecular information in the appropriate biological contexts should be introduced into predictors that are more suitable for the specific disease. Finally, the prediction results of these computational approaches are only descriptive associations, and the specific association type (e.g., casual or non-causal association of lncRNA with the disease) is still a challenging task and needs to be answered. The implementation of efficient and reliable computational predictions, together with systematic biological experiments, will greatly accelerate the study of lncRNA functions and mechanisms in physiological and pathological conditions.
Materials and Methods
Databases and Knowledge Bases
NONCODE (http://www.noncode.org/) collects and integrates data from PubMed and other resources via text mining.20 Users can use CNCI to predict their protein-encoding potential and display the results of functional annotation and enrichment through the ncFANs online website. The current version covers expression, function, sequence, structure, disease relevance of lncRNA, and other factors. Compared with other lncRNA databases, NONCODE stores more information about lncRNA transcripts and unique annotations.
LncRBase (http://bicresources.jcbose.ac.in/zhumur/lncrbase) is an annotation database resource for analyzing lncRNA functions based on feature sequences.21 It has recorded transcript information about 133,361 human lncRNA entries and 83,201 mouse lncRNA entries. Information about the lncRNA subtypes and small ncRNA-lncRNA associations is included. The database also provides microarray probes mapped to specific lncRNAs and expression in tissues.
LncBook (http://bigd.big.ac.cn/lncbook) collects information on 268,848 experimentally verified and predicted lncRNAs (including 1,867 functional lncRNAs) and includes information on related functions, diseases, expressions, methylation, mutations, and miRNA interactions (via software prediction).22 The team developed a database called LncRNAWiki, which is an integrated database.23 LncRNAWiki has set up a model of collaborative annotation. Then LncBook has been established to organize large-scale annotations systematically as a complement to LncRNAWiki.
MONOCLdb (https://www.monocldb.org/) contains 20,728 lncRNAs from the sequencing of virus-infected lungs of eight respiratory-infected mice, of which 5,329 were differentially expressed.24 These differentially expressed lncRNAs are annotated by different methods (enrichment methods, as well as module-based and rank-based methods). The correlation score of lncRNA expression profiles and six phenotypic data were determined as pathogenic associations.
lncRNome (http://genome.igib.res.in/lncRNome) is a comprehensive database of human lncRNAs, which collects information on annotation, sequence, structure, interacting proteins, genomic variations, conservation, and epigenetic modifications for more than 18,000 lncRNAs.25 Annotation is manually curated from literature and databases, including associated diseases, related literature, and the mapping of disease-associated variation in lncRNA gene loci.
LncRNASNP (http://bioinfo.life.hust.edu.cn/lncRNASNP/) mainly sorts information on the single nucleotide polymorphism (SNP) loci located on the lncRNA gene in humans and mice.26 The cancer mutations in lncRNA transcripts and lncRNA expression in cancer, the predicted interactions of miRNAs and associated diseases, and the impact of variations on lncRNA structures were integrated into LncRNASNP. This database also collects experimentally verified and predicted disease-lncRNA associations in humans.
LncRNADisease (http://www.rnanut.net/lncrnadisease/) is an open-access database that has been updated to version 2.0.27 As one of the more commonly used databases, LncRNADisease 2.0 provides 10,564 experimental lncRNA-disease associations and 195,395 computational lncRNA-disease associations in four species. Additionally, a confidence score was obtained for each pair of lncRNA-disease associations based on known experimental information. LncRNADisease also collets lncRNA regulatory networks.
Lnc2Cancer (http://www.bio-bigdata.net/lnc2cancer) collected 4,989 comprehensive experimentally supported associations between 1,614 lncRNAs and 165 human cancers.28,29 These records were built through text mining on the PubMed database. The database consists of three classifications of relationships between lncRNAs and cancers: circulating, drug-resistant, and prognostic-related lncRNAs. Additionally, it collects transcription factor (TF), mircroRNA (miRNA), variant, and methylation molecular information on the regulation of lncRNAs.
MNDR (http://www.rna-society.org/mndr/) is built through manual curation of scientific literature.30 The current release (MNDR v2.0) has recorded 261,042 entries including six species and 1,416 diseases from 26,600 studies. Detailed and comprehensive annotations for lncRNA-disease associations are presented at the bottom of each record, including the data from articles and evidence support.
EVLncRNAs (http://biophy.dzu.edu.cn/EVLncRNAs) is a database that intends to include all lncRNA-disease associations that are validated by low-throughput experiments.31 The database includes 1,543 lncRNAs from 77 species, 886 of which are associated with 338 diseases, along with experimental information. For other lncRNAs that are not associated with diseases, their functional information is collected.
NSDNA (http://www.bio-bigdata.net/nsdna/) is an online knowledge base of ncRNA-nervous system disease (NSD) associations.32 It contains 24,713 entries of associations covering 142 NSDs and 8,593 ncRNAs from more than 1,300 articles, of which 4,608 lncRNAs-NSDs are included. Users can browse by ncRNAs, diseases, species, or tissue name. Also, if searching for data, users can select low-throughput or high-throughput experimental data or both.
Nc2Eye (http://nc2eye.bio-data.cn/) is the first high-quality manually curated ncRNAomics knowledge base associated with eye disease and includes 1,147 lncRNA-associated entries.33
Computational Methods for Acquiring lncRNA-lncRNA Associations
Most of the computational methods for inferring lncRNA-disease association are based on lncRNA-lncRNA association data. Therefore, acquiring a high-quality lncRNA-lncRNA association is critical for improving performance in predicting the lncRNA-disease association. We have summarized the currently available computational methods for acquiring the lncRNA-lncRNA association. In general, these methods could be basically divided into four categories (Table 1): sequence similarity-based methods, functional similarity-based methods, cosine similarity-based methods, and expression-based methods.
Sequence Similarity-Based Methods
Due to plentiful information about the lncRNA sequence, the similarity between two lncRNAs was measured by comparing the sequence features of lncRNAs. Needleman and Wunsch34 first proposed the Needleman-Wunsch global alignment algorithm in 1970. Then, researchers developed it as a web tool, which calculates the optimum alignment and best score of two sequences in the order of sequence steps along their entire length. The alignment score SW(li,lj) was obtained from EMBOSS Needle, and the sequence similarity is defined as follows:
| (1) |
Functional Similarity-Based Methods
Based on the assumption that if lncRNA-related molecules have a similar function, lncRNA functions are identical, several functional similarity-based computational methods were developed. Chen et al.35 proposed a method, named LNCSIM, to measure the semantic similarity of lncRNAs’ associated two groups of diseases. They integrated two semantic similarity models to achieve better performance. Two models both collected diseases’ MeSH descriptors and constructed a directed acyclic graph (DAG). Then, the contribution of disease term t to disease A was calculated, which is also the difference between these two models. In the first model, it was calculated as Equation 2. Since the contribution of other diseases to the semantic value of the disease decreases with the increase of the distance between this disease and disease A, the decay factor is added. For the second model, diseases that appear in DAG(A) and are less common in other diseases, DAGs have a more significant contribution that can be calculated in Equation 3:
| (2) |
| (3) |
Therefore, the semantic value of disease A is defined as the sum of contributions from ancestral diseases and disease A itself:
| (4) |
Thus, the semantic similarity between two diseases A and B is calculated based on the common nodes of DAG(A) and DAG(B):
| (5) |
Finally, lncRNA functional similarity was obtained by calculating the average of the similarities between the two groups of diseases.
Huang et al.36 proposed an improved model called ILNCSIM, which introduced information content (IC) and focused on the hierarchical structure of disease DAGs. First, the information content value was calculated. Information content of disease term a is defined as the negative log-likelihood of each term:
| (6) |
Second, IC-based distances were used to calculate the most informative common ancestors (MICAs) and the most informative leaf (MIL):
| (7) |
Third, components α, to measure the specificity of MICA, β, to measure the generality of two disease terms, and γ, to estimate the total IC-based distances between two terms and their MICA, are computed:
| (8) |
| (9) |
| (10) |
Fourth, based on the above three equations, to compute the semantic similarity of two diseases:
| (11) |
Finally, lncRNA functional similarity was measured by calculating the average of the similarities between the two groups of diseases.
ICod measures disease similarity by scoring disease-related gene similarity, and researchers applied this idea to lncRNAs.37, 38, 39 The similarity between lncRNAs i and j was calculated based on the shortest path between each pair of lncRNA-related genes in the integrated human protein-protein interaction (PPI) network. The shortest distance between two proteins in the PPI network is indicated as d(pm,pn). D(pm,pn) denotes the transformed distance between the networks. t is the threshold of d(pm,pn). NETi and NETj represent the networks related to two lncRNAs, respectively. E and H are freely adjustable parameters:
| (12) |
| (13) |
Zhao et al.40 proposed a computational model to infer lncRNA-lncRNA association, which introduced lncRNA-miRNA associations and was defined as for Equation 14 and 15. The process is the sum of contributions of commonly associated miRNA divided by the number of miRNAs associated with two lncRNAs. The contribution value of each miRNA for lncRNA is computed by Equation 14. D(i) and D(j) are the number of lncRNAi-related edges and lncRNAj-related edges, respectively:
| (14) |
| (15) |
Expression Similarity-Based Methods
lncRNAs are expressed in highly cell type-specific, tissue-specific, and disease-specific manners. Consequently, the expression similarity between two lncRNAs is an important point. The co-expression relationship between lncRNAs measured by a Pearson or Spearman correlation coefficient was commonly used to infer the lncRNA-lncRNA association.41
Cosine Similarity-Based Methods
The concept of cosine is the origin of mathematics. Cheng et al.42 proposed a computational method called IntNetLncSim, which linked cosine similarity with lncRNA similarity. The similarity between two lncRNAs, lnc1 and lnc2, was calculated as follows:
| (18) |
where w1,i represents the vector values of lnc1 on the ith dimension.
Computational Methods for Acquiring Disease-Disease Associations
Disease-disease association data are critical data used in most of the computational methods for inferring lncRNA-disease association. In general, these methods for obtaining disease-disease association can be divided into four categories (Table 2): semantic similarity-based methods, functional similarity-based methods, Gaussian interaction profile kernel similarity-based methods, and cosine similarity-based methods.
Semantic Similarity-Based Methods
Semantic similarity between diseases is one of the commonly used methods that use mesh descriptors or disease ontology terms and determine the similarity of two disease terms based on the information content of common ancestral terms. A package named DOSE developed by Yu and Wang43 can calculate disease semantic similarity. Other methods also obtained the same results by mathematical formulas. The detailed algorithm is described in LNCSIM of lncRNA functional similarity.
Functional Similarity-Based Methods
Functional similarity-based methods were achieved by using the Jaccard coefficient to measure the similarity and difference of disease-related gene ontology.44 Disease-gene interaction and gene-gene ontology interaction were used and calculated as Equation 19. GOi represents the gene ontology terms related to disease i:
| (19) |
Zhao et al.40 proposed a model to infer the disease-disease association in their computational method, which introduced disease-miRNA associations and was defined as Equation 20 and 21. The process is the sum of contributions of commonly associated miRNA divided by the number of miRNAs associated with two diseases. The contribution value of each miRNA for a disease is computed by Equation 20. D(i) and D(j) are the number of diseasei-related edges and diseasej-related edges, respectively:
| (20) |
| (21) |
Gaussian Interaction Profile Kernel Similarity-Based Methods
Based on the assumption that genes with similar functions tend to be associated with a similar disease, Chen and Yan41 applied the Gaussian interaction profile kernel (also called the radial basis function kernel) similarity to measure disease-disease association as in Equation 22. IP(i) indicates the row of diseasei in the disease-lncRNA association matrix:
| (22) |
The parameter γd controls the kernel bandwidth, which is defined as follows:
| (23) |
where nd denotes the number of the contained diseases. γd′ is a novel bandwidth parameter by the average number of associations with lncRNAs per disease.
Cosine Similarity-Based Methods
In a previous study, Hamaneh and Yu45 linked cosine similarity with disease similarity. We used the lncRNA-disease association matrix to replace the original matrix for display. The similarity between the two diseases, dis1 and dis2, was calculated as follows:
| (24) |
where wl,i represents the vector values of dis1 on the ith dimension.
Computational Methods for Inferring lncRNA-Disease Associations
In this section, we review dozens of novel computational methods in inferring the lncRNA-disease associations proposed in recent years. Based on the core idea of the algorithm, these computational methods can be divided into four categories: matrix completion-based methods, recommendation algorithm-based methods, resource allocation-based methods, and multi-model integration-based methods.
Matrix Completion-Based Methods
The universal characteristic of the matrix completion-based methods is to complete the dataset with missing values in the form of a matrix. As shown in Figure 1, three matrices, including the lncRNA-disease association matrix, lncRNA-lncRNA matrix, and disease-disease matrix, were obtained. Then, feature extraction is accomplished based on the above three matrices to obtain lncRNA feature vectors and disease feature vectors. Finally, matrix completion methods were conducted on the lncRNA-disease association matrix to acquire the lncRNA-disease scoring matrix based on lncRNA feature vectors and disease feature vectors (Table 3).
Li et al.46 developed a computational model of faster randomized matrix completion for latent disease-lncRNA association (named FRMCLDA) that used the fSVT algorithm to predict lncRNA-disease associations based on the idea of matrix completion. FRMCLDA uses the disease similarity matrix, lncRNA similarity matrix, lncRNA-disease association matrix, and transpose matrix of the association matrix to construct the adjacency matrix, which improves the prediction performance by fitting the adjacency matrix. LDAPM and SIMCLDA also use a matrix completion approach, but the difference is that LDAPM denotes the approximated matrix as the multiplication of the two matrices.47,48 Also, TSSR exploits learned representation matrices as feature matrices to reconstruct the original matrix.49
Resource Allocation-Based Methods
Resource allocation is used to allocate available resources to each node. To predict the lncRNA-disease association, resource allocation is based on the initial value of the multi-data source matrices as a possible value for the relationship between nodes. The process is demonstrated in Figure 2. Resource allocation-based methods are built on data from multiple sources, such as lncRNA-disease association, miRNA-disease association, miRNA-lncRNA association, and so forth. The heterogeneous multilayer network is constructed, and the edges are weighted according to the corresponding values of the matrix. The lncRNA-disease scoring matrix was produced by post-processing resource allocation on the heterogeneous network (Table 4).
Resource allocation has been implemented in more than a dozen methods for predicting the lncRNA-disease association. Fan et al.50 proposed a computational model of IDHI-MIRW by integrating diverse heterogeneous information sources with positive pointwise mutual information and random walk with a restart algorithm. Xiao et al.51 developed BPLLDA to predict lncRNA-disease associations based on simple paths with limited lengths in a heterogeneous network. Zhang et al.52 proposed a rule-based inference method on the linked tripartite network, which was constructed by integrating heterogeneous data with deep-learning algorithms. Some other information had also been introduced to allocate resources for improving prediction performance. For example, LION and DislncRF introduced protein information and genome-wide tissue expression profiles, which are aided by protein-coding genes.53,54 NBLDA and LLCLPLDA both constructed four matrices and used the label propagation algorithm to resource allocation.55,58 By constructing a disease weight matrix based on the similarity between the lncRNA disease set and the specified disease, IIRWR introduced the concept of disease clique and added the weight of disease linkages to the traveling network.59 Lap-BiRWRHLDA and BiWalkLDA both used Laplacian normalization and a bi-random walk algorithm on similarity networks.60,61 The other two methods, TPGLDA and ncPred, allocated resources from the disease to lncRNAs and other nodes, respectively, but the difference is that the resources are returned to the initial nodes.62,63
Recommendation Algorithm-Based Methods
The common characteristic of the recommendation algorithm-based methods is to recommend a node that may be related to another node. It mainly includes content-based recommendation, collaborative filtering, and matrix factorization. The process is depicted in Figure 3. After applying a recommendation system algorithm to multi-data matrices, recommendation matrices at multiple levels (e.g., such as lncRNA, miRNA) are obtained. Finally, the possibility of the potential relationship between lncRNA and disease is measured through the combination of the recommendation matrices (Table 5).
Table 5.
Overview of Recommendation Algorithm-Based Computational Methods for Inferring lncRNA-Disease Association
| Method Name | Computational Principle | Data Types | Available Tool (Package or Code) | References |
|---|---|---|---|---|
| ILDMSF | similarity network fusion | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | NA | Chen et al.38 |
| NBCLDA | naive Bayesian, collaborative filtering | miRNA-disease association, miRNA-lncRNA association, lncRNA-disease association, disease-disease association | NA | Yu et al.66 |
| NCPLDA | network consistency projection | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | code (https://github.com/ghli16/NCPLDA) | Li et al.64 |
| MFLDA | matrix factorization | lncRNA-miRNA association, lncRNA-gene association, lncRNA-Gene Ontology (GO) association, lncRNA-disease association, miRNA-gene association, miRNA-disease association, gene-disease association, gene-gene association, gene-drug association, drug-drug association, gene-GO association | code (http://mlda.swu.edu.cn/codes.php?name=MFLDA) | Fu et al.70 |
| WMFLDA | matrix factorization | lncRNA-miRNA association, lncRNA-gene association, lncRNA-GO association, lncRNA-disease association, miRNA-gene association, miRNA-disease association, gene-disease association, gene-gene association, gene-drug association, drug-drug association, gene-GO association | code (http://mlda.swu.edu.cn/codes.php?name=WMFLDA) | Wang et al.83 |
| PMFILDA | probabilities matrix factorization | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association, miRNA-disease association, miRNA-lncRNA association | NA | Xuan et al.71 |
| DNILMF-LDA | logistic matrix factorization, Bayesian optimization | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | NA | Li et al.73 |
| DSCMF | collaborative matrix factorization | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | NA | Gao et al.75 |
| NNLDA | matrix factorization | lncRNA-disease association | code (https://github.com/gao793583308/NNLDA) | Hu et al.74 |
| NA | SimRank measure, common neighbor-based | lncRNA-disease association | NA | Ping et al.69 |
| CFNBC | naive Bayesian, collaborative filtering | miRNA-disease association, miRNA-lncRNA association, lncRNA-disease association, disease-disease association | code (https://github.com/jingwenyu18/CFNBC) | Yu et al.65 |
| DCSLDA | distance correlation set | disease-disease association, lncRNA-disease association, miRNA-disease association, miRNA-LncRNA association, lncRNA-lncRNA association | NA | Zhao et al.40 |
| SKF-LDA | similarity kernel fusion | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | NA | Xie et al.67 |
| BLM-NPAI | bipartite local model | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | NA | Cui et al.84 |
NA, not applicable.
The first category of recommendation algorithm-based methods is the content-based recommendation. Content-based recommendation refers to recommending similar nodes of previous related nodes for this node. For example, NCPLDA, proposed by Li et al.,64 measured the lncRNA-disease association score based on network consistency projection. The second category of recommendation algorithm-based methods is based on collaborative filtering. Collaborative filtering refers to adding other nodes similar to the nodes and using the information of these nodes to make inferences. CFNBC developed by Yu et al.65 and NBCLDA proposed by Yu et al.66 both used collaborative filtering on multi-data matrices to uncover a new relationship between lncRNA and disease and then took advantage of a naive Bayesian classifier to determine whether there is an association between lncRNA and disease in the set of lncRNA-associated node and disease-associated node. A similarity correlation fusion method introduced neighbor information and then was used to predict the association by making the original matrix fit as good as possible in ILDMSF and SKF-LDA.38,67 BLM-NPAI, developed by Cui et al.,68 introduced the nearest profile to get the final prediction results after constructing the local model of lncRNA and disease. Another method proposed by Ping et al.69 measured the one-step neighbor of a node based on SimRank measure when there is no common neighbor. DCSLDA, proposed by Zhao et al.,40 calculated the shortest path between lncRNA and disease and distance correlation coefficient to construct the final matrix. The third category of recommendation algorithm-based methods is based on matrix factorization, including MFLDA, WMFLDA, and PMFILDA. These three methods are based on matrix factorization, and the difference is that the latter two add weight and probability separately.70, 71, 72 Li et al. developed a computational method, DNILMF-LDA, which is anchored in the neighborhood-regularized logistic matrix factorization and optimizes the above parameters to predict interaction probabilities.73 NNLDA, determined by Hu et al.,74 solved some of the disadvantages of traditional matrix factorization by changing the training method and the loss function and adding a fully connected layer. Additionally, DSCMF combines matrix factorization and collaborative filtering to predict associations efficiently by introducing neighbor information.75
Multi-Model Integration-Based Methods
Multi-model integration methods have also been proposed to overcome the shortcomings of the single model and improve prediction performance (Table 6). A combination of matrix completion ideas and recommendation system ideas was applied in three models to predict potential lncRNA-disease associations, including LDASR, ECLDA, and the weighted bagging LightGBM model.76, 77, 78 Three methods (CNNLDA, CNNDLP, and GCNLDA) were developed by Xuan et al.79, 80, 81 to construct the final module through the integration of the convolutional module and attention module. LDAPred, proposed by Xuan et al.,82 introduced the convolutional neural network based on the integration of resource allocation and matrix completion.
Table 6.
Overview of Multi-Model Integration-Based Computational Methods for Inferring lncRNA-Disease Association
| Method Name | Computational Principle | Data Types | Available Tool (Package or Code) | References |
|---|---|---|---|---|
| NA | weighted bagging lightGBM model | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | NA | Chen and Liu77 |
| LDASR | rotation forest | lncRNA-disease association | NA | Guo et al.76 |
| ECLDA | extreme learning machine, convolutional neural networks | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association | NA | Guo et al.78 |
| CNNLDA | convolutional neural networks, attention mechanisms | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association, miRNA-disease association, miRNA-lncRNA association, miRNA-miRNA association | NA | Xuan et al.79 |
| CNNDLP | convolutional neural networks, attention mechanisms | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association, miRNA-disease association, miRNA-lncRNA association, miRNA-miRNA association | NA | Xuan et al.80 |
| GCNLDA | convolutional neural networks, graph convolutional network | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association, miRNA-disease association, miRNA-lncRNA association, miRNA-miRNA association | NA | Xuan et al.81 |
| LDAPred | convolutional neural networks, information flow propagation | lncRNA-disease association, lncRNA-lncRNA association, disease-disease association, miRNA-disease association, miRNA-lncRNA association, miRNA-miRNA association | NA | Xuan et al.82 |
NA, not applicable.
Discussion
During the past decade, it has been well documented that lncRNAs play a critical role in nearly all biological processes and have become an emerging paradigm of human disease research.56,57 Identification of disease-associated lncRNAs is becoming increasingly important for fundamentally improving our understanding of molecular mechanisms and developing novel therapeutic targets, and thus has attracted more and more attention in the scientific community and is becoming one of the hotspots in medical research. Although current experimental studies in vitro and in vivo could directly link identified lncRNAs with disease phenotypes, they are affected by the limitation of lower efficiency and higher time and labor cost. Taking into account the limitations of experimental studies, high-throughput technologies were then implemented, leading to exponential growth in the number of dysregulated lncRNAs in diseases. However, the aberrant expression of lncRNAs is not sufficient evidence for ascribing to them a functional role in disease.7 Therefore, efficient and accurate identification and functional elucidation of disease-associated lncRNAs are in their infancy and remain a major challenge. With the rapidly increasing quantity and quality of bioinformatics databases and resources in lncRNAs and diseases, computer-aided inference of disease-associated RNAs has become a promising avenue for facilitating the unraveling of the functional role of lncRNAs in diseases and provides complementary value for experimental studies. A large number of computational models, algorithms, and tools have been developed and proposed, compensating for this dearth.
In this work, we first summarize data and knowledge sources available for the lncRNA-disease association study, which contains databases of lncRNAs, diseases, and known lncRNA-disease associations. We then present a detailed overview of previously proposed computational methods for inferring lncRNA-disease associations. Based on the core idea implemented, these computational methods can be divided into four categories: (1) matrix completion-based methods, (2) recommendation algorithm-based methods, (3) resource allocation-based methods, and (4) multi-model integration-based methods. Despite that the performance of each computational method is very great according to the reports in their own studies, one emerging critical issue is that most of these methods used different data sources as their training dataset and carried out cross-validation on their dataset, lacking benchmark performance evaluation. These computational methods have distinct limitations and weaknesses, which are noted at follows. First, matrix completion-based methods considered the feature vectors of lncRNA and disease to improve the accuracy of the prediction. However, these algorithms hold the disadvantage of poor robustness. The ranks of diverse datasets are likely to vary widely. Second, because experimentally verified lncRNA-disease associations are still too incomplete, resource allocation-based methods need to consider the prediction of separate nodes or integrate additional biological information. However, although this can improve prediction accuracy, some interactions from other databases may contain some noise to interfere with prediction results. In addition, recommendation algorithm-based methods are stated separately. Content-based recommendation models only need node prior knowledge, but new levels of disease-lncRNA associations cannot be recognized. Although collaborative filtering-based recommendation methods complement this shortcoming, the spare lncRNA-disease association matrix is harmful to the recommendation, and the complexity and time cost of the algorithm will sharply increase when the amount of data is too large. Additionally, matrix factorization-based recommendation methods reduce space complexity by mapping a matrix to a product of low-dimensional matrices. These methods also make it easy to add additional data from different sources and use the intrinsic structure. Matrix factorization-based methods also have the same weaknesses as collaborative filtering-based recommendation methods. The above computational approaches can thus complement each other. Therefore, multi-model integration-based methods were proposed to achieve better performance when investigating the association between lncRNAs and diseases. Finally, only several computational approaches have been developed as online web tools, and most are still theoretical studies that hampered their use for biologists and medical scientists.
With the rapidly increasing knowledge for the functional mechanism of lncRNAs, several challenges that would be helpful to improve the accuracy and practicality of the predictors could be highlighted. It is well known that the majority of lncRNAs exhibited precise subcellular localization, thus performing regulatory roles in a spatiotemporal manner.5,6 Therefore, some interactions between lncRNAs and other biological molecules (DNA, RNA, and proteins) used in previous predictors are derived from prediction and do not exist in the real biological world. Therefore, co-localization information of lncRNAs and other biological molecules should be considered. Additionally, it has also been observed that lncRNAs were expressed in highly cell type-specific, tissue-specific, and disease-specific manners. Therefore, more molecular information in the appropriate biological contexts should be introduced into predictors that are more suitable for the specific disease. Finally, the prediction results of these computational approaches are only descriptive associations, and the specific association type (e.g., casual or non-causal association of lncRNA with the disease) is still a challenging task and needs to be answered. The implementation of efficient and reliable computational predictions, together with systematic biological experiments, will greatly accelerate the study of lncRNA functions and mechanisms in physiological and pathological conditions.
Authors Contributions
J.S., C.X., and M.Z. designed the study. C.Y., Z.Z., S.B., and P.H. collected and reviewed literature. J.S., M.Z., and C.Y. drafted the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grant 61973240) and the Scientific Research Foundation for Talents of Wenzhou Medical University (QTJ18029, and QTJ18024). The funders had no roles in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Contributor Information
Chongyong Xu, Email: stony693100@163.com.
Jie Sun, Email: suncarajie@wmu.edu.cn.
References
- 1.Johnson J.M., Edwards S., Shoemaker D., Schadt E.E. Dark matter in the genome: evidence of widespread transcription detected by microarray tiling experiments. Trends Genet. 2005;21:93–102. doi: 10.1016/j.tig.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 2.Dinger M.E., Pang K.C., Mercer T.R., Mattick J.S. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput. Biol. 2008;4:e1000176. doi: 10.1371/journal.pcbi.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ingolia N.T., Lareau L.F., Weissman J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mercer T.R., Dinger M.E., Mattick J.S. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 5.Fang Y., Fullwood M.J. Roles, functions, and mechanisms of long non-coding RNAs in cancer. Genomics Proteomics Bioinformatics. 2016;14:42–54. doi: 10.1016/j.gpb.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kapranov P., Cheng J., Dike S., Nix D.A., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermüller J., Hofacker I.L. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–1488. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- 7.Huarte M. The emerging role of lncRNAs in cancer. Nat. Med. 2015;21:1253–1261. doi: 10.1038/nm.3981. [DOI] [PubMed] [Google Scholar]
- 8.Zeng W., Wang F., Ma Y., Liang X.C., Chen P. Dysfunctional mechanism of liver cancer mediated by transcription factor and non-coding RNA. Curr. Bioinform. 2019;14:100–107. [Google Scholar]
- 9.Tang W., Wan S., Yang Z., Teschendorff A.E., Zou Q. Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics. 2018;34:398–406. doi: 10.1093/bioinformatics/btx622. [DOI] [PubMed] [Google Scholar]
- 10.Bao S., Zhao H., Yuan J., Fan D., Zhang Z., Su J., Zhou M. Computational identification of mutator-derived lncRNA signatures of genome instability for improving the clinical outcome of cancers: a case study in breast cancer. Brief. Bioinform. 2019:bbz118. doi: 10.1093/bib/bbz118. [DOI] [PubMed] [Google Scholar]
- 11.Cheetham S.W., Gruhl F., Mattick J.S., Dinger M.E. Long noncoding RNAs and the genetics of cancer. Br. J. Cancer. 2013;108:2419–2425. doi: 10.1038/bjc.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou M., Hu L., Zhang Z., Wu N., Sun J., Su J. Recurrence-associated long non-coding RNA signature for determining the risk of recurrence in patients with colon cancer. Mol. Ther. Nucleic Acids. 2018;12:518–529. doi: 10.1016/j.omtn.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou M., Zhang Z., Zhao H., Bao S., Cheng L., Sun J. An immune-related six-lncRNA signature to improve prognosis prediction of glioblastoma multiforme. Mol. Neurobiol. 2018;55:3684–3697. doi: 10.1007/s12035-017-0572-9. [DOI] [PubMed] [Google Scholar]
- 14.Zhou M., Zhao H., Xu W., Bao S., Cheng L., Sun J. Discovery and validation of immune-associated long non-coding RNA biomarkers associated with clinically molecular subtype and prognosis in diffuse large B cell lymphoma. Mol. Cancer. 2017;16:16. doi: 10.1186/s12943-017-0580-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zou Q., Ma Q. The application of machine learning to disease diagnosis and treatment. Math. Biosci. 2020;320:108305. doi: 10.1016/j.mbs.2019.108305. [DOI] [PubMed] [Google Scholar]
- 16.Liao Z.J., Li D.P., Wang X.R., Li L.S., Zou Q. Cancer diagnosis through isomiR expression with machine learning method. Curr. Bioinform. 2018;13:57–63. [Google Scholar]
- 17.Sun J., Zhang Z., Bao S., Yan C., Hou P., Wu N., Su J., Xu L., Zhou M. Identification of tumor immune infiltration-associated lncRNAs for improving prognosis and immunotherapy response of patients with non-small cell lung cancer. J. Immunother. Cancer. 2020;8:e000110. doi: 10.1136/jitc-2019-000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou M., Wang X., Li J., Hao D., Wang Z., Shi H., Han L., Zhou H., Sun J. Prioritizing candidate disease-related long non-coding RNAs by walking on the heterogeneous lncRNA and disease network. Mol. Biosyst. 2015;11:760–769. doi: 10.1039/c4mb00511b. [DOI] [PubMed] [Google Scholar]
- 19.Sun J., Shi H., Wang Z., Zhang C., Liu L., Wang L., He W., Hao D., Liu S., Zhou M. Inferring novel lncRNA-disease associations based on a random walk model of a lncRNA functional similarity network. Mol. Biosyst. 2014;10:2074–2081. doi: 10.1039/c3mb70608g. [DOI] [PubMed] [Google Scholar]
- 20.Fang S., Zhang L., Guo J., Niu Y., Wu Y., Li H., Zhao L., Li X., Teng X., Sun X. NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2018;46(D1):D308–D314. doi: 10.1093/nar/gkx1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chakraborty S., Deb A., Maji R.K., Saha S., Ghosh Z. LncRBase: an enriched resource for lncRNA information. PLoS ONE. 2014;9:e108010. doi: 10.1371/journal.pone.0108010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma L., Cao J., Liu L., Du Q., Li Z., Zou D., Bajic V.B., Zhang Z. LncBook: a curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2019;47(D1):D128–D134. doi: 10.1093/nar/gky960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma L., Li A., Zou D., Xu X., Xia L., Yu J., Bajic V.B., Zhang Z. LncRNAWiki: harnessing community knowledge in collaborative curation of human long non-coding RNAs. Nucleic Acids Res. 2015;43:D187–D192. doi: 10.1093/nar/gku1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Josset L., Tchitchek N., Gralinski L.E., Ferris M.T., Eisfeld A.J., Green R.R., Thomas M.J., Tisoncik-Go J., Schroth G.P., Kawaoka Y. Annotation of long non-coding RNAs expressed in collaborative cross founder mice in response to respiratory virus infection reveals a new class of interferon-stimulated transcripts. RNA Biol. 2014;11:875–890. doi: 10.4161/rna.29442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhartiya D., Pal K., Ghosh S., Kapoor S., Jalali S., Panwar B., Jain S., Sati S., Sengupta S., Sachidanandan C. LncRNome: a comprehensive knowledgebase of human long noncoding RNAs. Database (Oxford) 2013;2013:bat034. doi: 10.1093/database/bat034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miao Y.-R., Liu W., Zhang Q., Guo A.-Y. lncRNASNP2: an updated database of functional SNPs and mutations in human and mouse lncRNAs. Nucleic Acids Res. 2018;46(D1):D276–D280. doi: 10.1093/nar/gkx1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bao Z., Yang Z., Huang Z., Zhou Y., Cui Q., Dong D. LncRNADisease 2.0: an updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019;47(D1):D1034–D1037. doi: 10.1093/nar/gky905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao Y., Wang P., Wang Y., Ma X., Zhi H., Zhou D., Li X., Fang Y., Shen W., Xu Y. Lnc2Cancer v2.0: updated database of experimentally supported long non-coding RNAs in human cancers. Nucleic Acids Res. 2019;47(D1):D1028–D1033. doi: 10.1093/nar/gky1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ning S., Zhang J., Wang P., Zhi H., Wang J., Liu Y., Gao Y., Guo M., Yue M., Wang L., Li X. Lnc2Cancer: a manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 2016;44(D1):D980–D985. doi: 10.1093/nar/gkv1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui T., Zhang L., Huang Y., Yi Y., Tan P., Zhao Y., Hu Y., Xu L., Li E., Wang D. MNDR v2.0: an updated resource of ncRNA-disease associations in mammals. Nucleic Acids Res. 2018;46(D1):D371–D374. doi: 10.1093/nar/gkx1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou B., Zhao H., Yu J., Guo C., Dou X., Song F. EVLncRNAs: a manually curated database for long non-coding RNAs validated by low-throughput experiments. Nucleic. Acids Res. 2018;46:D100–D105. doi: 10.1093/nar/gkx677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J., Cao Y., Zhang H., Wang T., Tian Q., Lu X., Lu X., Kong X., Liu Z., Wang N. NSDNA: a manually curated database of experimentally supported ncRNAs associated with nervous system diseases. Nucleic Acids Res. 2017;45(D1):D902–D907. doi: 10.1093/nar/gkw1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y., Xue Z., Guo F., Yu F., Xu L., Chen H. Nc2Eye: a curated ncRNAomics knowledgebase for bridging basic and clinical research in eye diseases. Front. Cell. Devel. Biol. 2020;8:75. doi: 10.3389/fcell.2020.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Needleman S.B., Wunsch C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 35.Chen X., Yan C.C., Luo C., Ji W., Zhang Y., Dai Q. Constructing lncRNA functional similarity network based on lncRNA-disease associations and disease semantic similarity. Sci. Rep. 2015;5:11338. doi: 10.1038/srep11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang Y.-A., Chen X., You Z.-H., Huang D.-S., Chan K.C. ILNCSIM: improved lncRNA functional similarity calculation model. Oncotarget. 2016;7:25902–25914. doi: 10.18632/oncotarget.8296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paik H., Heo H.-S., Ban H.J., Cho S.B. Unraveling human protein interaction networks underlying co-occurrences of diseases and pathological conditions. J. Transl. Med. 2014;12:99. doi: 10.1186/1479-5876-12-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Q., Lai D., Lan W., Wu X., Chen B., Chen Y.P., Wang J. ILDMSF: inferring associations between long non-coding RNA and disease based on multi-similarity fusion. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019 doi: 10.1109/TCBB.2019.2936476. Published online August 20, 2019. [DOI] [PubMed] [Google Scholar]
- 39.Zou Q., Li J., Song L., Zeng X., Wang G. Similarity computation strategies in the microRNA-disease network: a survey. Brief. Funct. Genomics. 2016;15:55–64. doi: 10.1093/bfgp/elv024. [DOI] [PubMed] [Google Scholar]
- 40.Zhao H., Kuang L., Wang L., Xuan Z. A novel approach for predicting disease-lncRNA associations based on the distance correlation set and information of the miRNAs. Comput. Math. Methods Med. 2018;2018:6747453. doi: 10.1155/2018/6747453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen X., Yan G.-Y. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics. 2013;29:2617–2624. doi: 10.1093/bioinformatics/btt426. [DOI] [PubMed] [Google Scholar]
- 42.Cheng L., Shi H., Wang Z., Hu Y., Yang H., Zhou C., Sun J., Zhou M. IntNetLncSim: an integrative network analysis method to infer human lncRNA functional similarity. Oncotarget. 2016;7:47864–47874. doi: 10.18632/oncotarget.10012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu G., Wang L.G., Yan G.R., He Q.Y. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics. 2015;31:608–609. doi: 10.1093/bioinformatics/btu684. [DOI] [PubMed] [Google Scholar]
- 44.Mathur S., Dinakarpandian D. Finding disease similarity based on implicit semantic similarity. J. Biomed. Inform. 2012;45:363–371. doi: 10.1016/j.jbi.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 45.Hamaneh M.B., Yu Y.-K. Relating diseases by integrating gene associations and information flow through protein interaction network. PLoS ONE. 2014;9:e110936. doi: 10.1371/journal.pone.0110936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li W., Wang S., Xu J., Mao G., Tian G., Yang J. Inferring latent disease-lncRNA associations by faster matrix completion on a heterogeneous network. Front. Genet. 2019;10:769. doi: 10.3389/fgene.2019.00769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu C., Yang M., Luo F., Wu F.-X., Li M., Pan Y., Li Y., Wang J. Prediction of lncRNA-disease associations based on inductive matrix completion. Bioinformatics. 2018;34:3357–3364. doi: 10.1093/bioinformatics/bty327. [DOI] [PubMed] [Google Scholar]
- 48.Fraidouni N., Zaruba G. Proceedings of the International Conference on Bioinformatics & Computational Biology (BIOCOMP) CSREA Press; 2019. A Matrix Completion Approach for Predicting lncRNA-disease association; pp. 61–66.https://csce.ucmss.com/cr/books/2019/LFS/CSREA2019/BIC4058.pdf [Google Scholar]
- 49.Ou-Yang L., Huang J., Zhang X.-F., Li Y.-R., Sun Y., He S., Zhu Z. lncRNA-disease association prediction using two-side sparse self-representation. Front. Genet. 2019;10:476. doi: 10.3389/fgene.2019.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fan X.-N., Zhang S.-W., Zhang S.-Y., Zhu K., Lu S. Prediction of lncRNA-disease associations by integrating diverse heterogeneous information sources with RWR algorithm and positive pointwise mutual information. BMC Bioinformatics. 2019;20:87. doi: 10.1186/s12859-019-2675-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao X., Zhu W., Liao B., Xu J., Gu C., Ji B., Yao Y., Peng L., Yang J. BPLLDA: predicting lncRNA-disease associations based on simple paths with limited lengths in a heterogeneous network. Front. Genet. 2018;9:411. doi: 10.3389/fgene.2018.00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang H., Liang Y., Peng C., Han S., Du W., Li Y. Predicting lncRNA-disease associations using network topological similarity based on deep mining heterogeneous networks. Math. Biosci. 2019;315:108229. doi: 10.1016/j.mbs.2019.108229. [DOI] [PubMed] [Google Scholar]
- 53.Sumathipala M., Maiorino E., Weiss S.T., Sharma A. Network Diffusion Approach to Predict LncRNA disease associations using multi-type biological networks: LION. Front. Physiol. 2019;10:888. doi: 10.3389/fphys.2019.00888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pan X., Jensen L.J., Gorodkin J. Inferring disease-associated long non-coding RNAs using genome-wide tissue expression profiles. Bioinformatics. 2019;35:1494–1502. doi: 10.1093/bioinformatics/bty859. [DOI] [PubMed] [Google Scholar]
- 55.Xie G., Huang S., Luo Y., Ma L., Lin Z., Sun Y. LLCLPLDA: a novel model for predicting lncRNA-disease associations. Mol. Genet. Genomics. 2019;294:1477–1486. doi: 10.1007/s00438-019-01590-8. [DOI] [PubMed] [Google Scholar]
- 56.Li J., Xuan Z., Liu C. Long non-coding RNAs and complex human diseases. Int. J. Mol. Sci. 2013;14:18790–18808. doi: 10.3390/ijms140918790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DiStefano J.K. The emerging role of long noncoding RNAs in human disease. Methods Mol. Biol. 2018;1706:91–110. doi: 10.1007/978-1-4939-7471-9_6. [DOI] [PubMed] [Google Scholar]
- 58.Liu Y., Feng X., Zhao H., Xuan Z., Wang L. A novel network-based computational model for prediction of potential lncRNA-disease association. Int. J. Mol. Sci. 2019;20:1549. doi: 10.3390/ijms20071549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang L., Xiao Y., Li J., Feng X., Li Q., Yang J. IIRWR: internal inclined random walk with restart for lncRNA-disease association prediction. IEEE Access. 2019;7:54034–54041. [Google Scholar]
- 60.Wen Y., Han G., Anh V.V. Laplacian normalization and bi-random walks on heterogeneous networks for predicting lncRNA-disease associations. BMC Syst. Biol. 2018;12(Suppl 9):122. doi: 10.1186/s12918-018-0660-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao Y., Hu J., Shang X. BIBM; 2018. Identification of lncRNA-disease association using bi-random walks. 2018 IEEE International Conference on Bioinformatics and Biomedicine; pp. 1249–1255. [Google Scholar]
- 62.Ding L., Wang M., Sun D., Li A. TPGLDA: novel prediction of associations between lncRNAs and diseases via lncRNA-disease-gene tripartite graph. Sci. Rep. 2018;8:1065. doi: 10.1038/s41598-018-19357-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mori T., Ngouv H., Hayashida M., Akutsu T., Nacher J.C. ncRNA-disease association prediction based on sequence information and tripartite network. BMC Syst. Biol. 2018;12(Suppl 1):37. doi: 10.1186/s12918-018-0527-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li G., Luo J., Liang C., Xiao Q., Ding P., Zhang Y. Prediction of lncRNA-disease associations based on network consistency projection. IEEE Access. 2019;7:58849–58856. [Google Scholar]
- 65.Yu J., Xuan Z., Feng X., Zou Q., Wang L. A novel collaborative filtering model for lncRNA-disease association prediction based on the naïve Bayesian classifier. BMC Bioinformatics. 2019;20:396. doi: 10.1186/s12859-019-2985-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu J., Ping P., Wang L., Kuang L., Li X., Wu Z. A novel probability model for lncRNA–disease association prediction based on the Naïve Bayesian Classifier. Genes (Basel) 2018;9:345. doi: 10.3390/genes9070345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie G., Meng T., Luo Y., Liu Z. SKF-LDA: Similarity Kernel Fusion for Predicting lncRNA-Disease Association. Mol. Ther. Nucleic AcidCs. 2019;18:45–55. doi: 10.1016/j.omtn.2019.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cui Z., Liu J.-X., Gao Y.-L., Zhu R., Yuan S.S. lncRNA-disease associations prediction using bipartite local model with nearest profile-based association inferring. IEEE J. Biomed. Health Inform. 2019;24:1519–1527. doi: 10.1109/JBHI.2019.2937827. [DOI] [PubMed] [Google Scholar]
- 69.Ping P., Wang L., Kuang L., Ye S., Iqbal M.F.B., Pei T. A novel method for lncRNA-disease association prediction based on an lncRNA-disease association network. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2019;16:688–693. doi: 10.1109/TCBB.2018.2827373. [DOI] [PubMed] [Google Scholar]
- 70.Fu G., Wang J., Domeniconi C., Yu G. Matrix factorization-based data fusion for the prediction of lncRNA-disease associations. Bioinformatics. 2018;34:1529–1537. doi: 10.1093/bioinformatics/btx794. [DOI] [PubMed] [Google Scholar]
- 71.Xuan Z., Li J., Yu J., Feng X., Zhao B., Wang L. A probabilistic matrix factorization method for identifying lncRNA-disease associations. Genes (Basel) 2019;10:126. doi: 10.3390/genes10020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Y., Yu G., Wang J., Fu G., Guo M., Domeniconi C.J.M. Weighted matrix factorization on multi-relational data for lncRNA-disease association prediction. Methods. 2020;173:32–43. doi: 10.1016/j.ymeth.2019.06.015. [DOI] [PubMed] [Google Scholar]
- 73.Li Y., Li J., Bian N. DNILMF-LDA: prediction of lncRNA-disease associations by dual-network integrated logistic matrix factorization and Bayesian optimization. Genes (Basel) 2019;10:608. doi: 10.3390/genes10080608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hu J., Gao Y., Li J., Shang X. Deep learning enables accurate prediction of interplay between lncRNA and disease. Front. Genet. 2019;10:937. doi: 10.3389/fgene.2019.00937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao M.-M., Cui Z., Gao Y.-L., Li F., Liu J.-X. Dual sparse collaborative matrix factorization method based on Gaussian kernel function for predicting lncRNA-disease associations. In: Huang D.S., Huang Z.K., Hussain A., editors. Intelligent Computing Methodologies. ICIC 2019. Lecture Notes in Computer Science. volume 11645. Springer; 2019. pp. 318–326. [Google Scholar]
- 76.Guo Z.-H., You Z.-H., Wang Y.-B., Yi H.-C., Chen Z.-H. A Learning-Based Method for LncRNA-Disease Association Identification Combing Similarity Information and Rotation Forest. iScience. 2019;19:786–795. doi: 10.1016/j.isci.2019.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen X., Liu X. A weighted bagging LightGBM model for potential lncRNA-disease association identification. In: Qiao J., Zhao X., Pan L., Zuo X., Zhang X., Zhang Q., Huang S., editors. Bio-Inspired Computing: Theories and Applications. BIC-TA 2018. Communications in Computer and Information Science. volume 951. Springer; 2018. pp. 307–314. [Google Scholar]
- 78.Guo Z.-H., You Z.-H., Li L.-P., Wang Y.-B., Chen Z.-H. Combining high speed ELM with a CNN feature encoding to predict lncRNA-disease associations. In: Huang D.S., Jo K.H., Huang Z.K., editors. Intelligent Computing Theories and Application. ICIC 2019. Lecture Notes in Computer Science. volume 11644. Springer; 2019. pp. 406–417. [Google Scholar]
- 79.Xuan P., Cao Y., Zhang T., Kong R., Zhang Z. Dual convolutional neural networks with attention mechanisms based method for predicting disease-related lncRNA genes. Front. Genet. 2019;10:416. doi: 10.3389/fgene.2019.00416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xuan P., Sheng N., Zhang T., Liu Y., Guo Y. CNNDLP: a method based on convolutional autoencoder and convolutional neural network with adjacent edge attention for predicting lncRNA-disease associations. Int. J. Mol. Sci. 2019;20:4260. doi: 10.3390/ijms20174260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xuan P., Pan S., Zhang T., Liu Y., Sun H. Graph convolutional network and convolutional neural network based method for predicting lncRNA-disease associations. Cells. 2019;8:1012. doi: 10.3390/cells8091012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xuan P., Jia L., Zhang T., Sheng N., Li X., Li J. LDAPred: a method based on information flow propagation and a convolutional neural network for the prediction of disease-associated lncRNAs. Int. J. Mol. Sci. 2019;20:4458. doi: 10.3390/ijms20184458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Y., Yu G., Wang J., Fu G., Guo M., Domeniconi C. Weighted matrix factorization on multi-relational data for lncRNA-disease association prediction. Methods. 2020;173:32–43. doi: 10.1016/j.ymeth.2019.06.015. [DOI] [PubMed] [Google Scholar]
- 84.Cui Z., Liu J.-X., Gao Y.-L., Zhu R., Yuan S.-S. lncRNA-disease associations prediction using bipartite local model with nearest profile-based association inferring. IEEE J. Biomed. Health Inform. 2020;24:1519–1527. doi: 10.1109/JBHI.2019.2937827. [DOI] [PubMed] [Google Scholar]