

SUMMARY
Deregulation of MYC plays an essential role in T cell acute lymphoblastic leukemia (T-ALL), yet the mechanisms underlying its deregulation remain elusive. Herein, we identify a molecular mechanism responsible for reciprocal activation between Aurora B kinase (AURKB) and MYC. AURKB directly phosphorylates MYC at serine 67, counteracting GSK3β-directed threonine 58 phosphorylation and subsequent FBXW7- mediated proteasomal degradation. Stabilized MYC, in concert with T cell acute lymphoblastic leukemia 1 (TAL1), directly activates AURKB transcription, constituting a positive feedforward loop that reinforces MYC-regulated oncogenic programs. Therefore, inhibitors of AURKB induce prominent MYC degradation concomitant with robust leukemia cell death. These findings reveal an AURKB-MYC regulatory circuit that underlies T cell leukemogenesis, and provide a rationale for therapeutic targeting of oncogenic MYC via AURKB inhibition.
Graphical Abstract
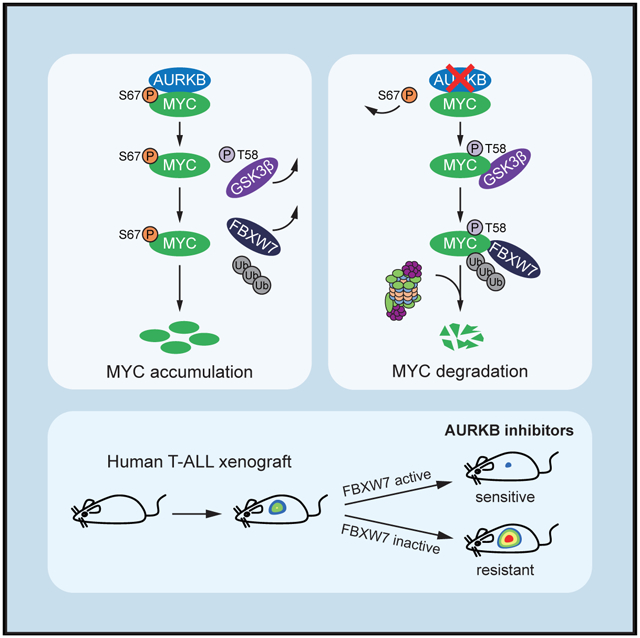
In Brief
Jiang et al. describe how MYC is stabilized by AURKB-mediated phosphorylation to prevent its degradation by FBXW7 in T cell acute lymphoblastic leukemia (T-ALL) and show that MYC induces the transcription of AURKB to support leukemogenesis. Wild-type FBXW7 T-ALL is sensitive to AURKB inhibition.
INTRODUCTION
MYC (also termed c-MYC) is broadly deregulated in human cancers (Dang, 2012). The unleashed MYC oncogene frequently produces abundant MYC protein, which mediates a transcriptional response involved in a variety of biological processes, contributing to almost every aspect of tumorigenesis (Meyer and Penn, 2008). The significance of MYC deregulation has been recognized in T cell acute lymphoblastic leukemia (T-ALL) (Dang, 2012; Sanchez-Martin and Ferrando, 2017), a life-threatening hematological malignancy with dismal outcome due to disease relapse and drug resistance (Inaba et al., 2013). Specific expression of MYC under the control of lymphatic-specific rag2 promoter induces T-ALL in zebrafish (Langenau et al., 2003). Moreover, MYC is linked to the leukemia-initiating cell activity, and suppression of MYC inhibits T-ALL progression in murine models (King et al., 2013; Roderick et al., 2014). Its involvement in both T-ALL initiation and maintenance suggests that therapeutic efforts aimed at inhibiting MYC expression or activity should have an important clinical relevance. However, attempts to directly disrupt MYC function have met with limited success, in part due to its “undruggable” protein structure (Chen et al., 2018).
Rapid protein degradation by the ubiquitin-proteasome system is an essential mechanism responsible for tight control of physiological levels of MYC (Farrell and Sears, 2014). A well-defined event in MYC degradation involves sequential phosphorylations of two critical residues, serine 62 (S62) and threonine 58 (T58), respectively. MYC is stabilized upon phosphorylation of S62 by ERK and/or CDKs (Bachireddy et al., 2005; Sears, 2004). S62 phosphorylation primes subsequent phosphorylation at T58 by GSK3β (Gregory et al., 2003), and MYC with phosphorylated T58 is recognized by the E3 ubiquitin ligase FBXW7 and degraded by the 26S proteasome (Welcker et al., 2004; Yada et al., 2004). Highlighting the importance of this degradation pathway in cancer, many of the signaling proteins implicated in the MYC S62/T58 phosphorylation are often deregulated in tumor cells, resulting in altered MYC phosphorylation and increased MYC protein stability (Sears et al., 2000; Yeh et al., 2004). Therefore, impairment of the pathway that regulates MYC degradation represents an important mechanism for oncogenic activation of MYC in human cancers, and a focus for therapeutic targeting.
Aurora kinases, a multi-genic family of serine/threonine kinases, comprise Aurora A (AURKA), Aurora B (AURKB), and Aurora C (AURKC) (Willems et al., 2018), which are well-characterized to play integral roles in the regulation of cell division. Amplification or overexpression of Aurora kinases is frequently found in human cancers with clear evidence of oncogenic potential, implicating Aurora kinases as rational anti-tumor targets (Falchook et al., 2015; Tang et al., 2017). AURKB is the catalytic subunit of the chromosomal passenger complex, which regulates multiple facets of cell division, including the spindle checkpoint, chromosome segregation, and cytokinesis (Vader et al., 2006). Overexpression of AURKB has been reported in a variety of cancers and predicts poor overall survival (Hayama et al., 2007; Takeshita et al., 2013). It appears that MYC-overex-pressing tumor cells are susceptible to AURKB inhibitors, such as AZD1152 (Topham et al., 2015; Yang et al., 2010). AZD1152 is a highly potent and selective inhibitor of Aurora B; preclinical evidence of anti-tumor efficacy with AZD1152 has extended to the clinical setting and demonstrated tolerable toxicity (Collins et al., 2015; Löwenberg et al., 2011). Despite considerable studies, it remains largely unclear how expression of AURKB is elevated and, in particular, how elevated levels of AURKB reprogram cells to promote the cancer progression.
In this study, we unravel a molecular mechanism responsible for reciprocal activation between AURKB and MYC, and delineate the functional importance of this AURKB-MYC axis in T cell leukemogenesis. Moreover, we highlight a mechanism-based therapeutic strategy of targeting oncogenic MYC via AURKB inhibition.
RESULTS
Identification of AURKB as a MYC Binding Partner
Besides transcriptional activation by NOTCH1 as a mechanism underlying deregulated MYC expression in T-ALL (Sanchez-Martin and Ferrando, 2017), binding partners that sustain MYC stability and transcriptional activity remain to be determined. To identify such factors, we generated a Jurkat stable cell line ectopically expressing Flag-tagged MYC. Flag-bound immunoprecipitates from these cells were resolved by SDS-PAGE and visualized by silver staining (Figure 1A). Flag-tagged MYC and associated proteins were subjected to liquid chromatography-tandem mass spectrometry analysis, which led to the identification of AURKB as a MYC binding partner (Figures 1A and 1B and Table S1). To validate the physical interaction, we enforced the expression of Flag-AURKB and HA-MYC in 293T cells for reciprocal immunoprecipitation and confirmed associations between both proteins (Figure 1C). Moreover, co-immunoprecipitation using CUTLL1 and HPB-ALL cell lysates validated the specific interaction between endogenous AURKB and MYC in T-ALL cells (Figure 1D). To define the precise region(s) in MYC for this interaction, we expressed full-length Flag-tagged AURKB in combination with respective HA-tagged fragments of MYC in 293T cells (Figure 1E). The N-terminal region of MYC (amino acids 1–221) containing the first three MYC box (MB) domains did not interact with AURKB, whereas the C-terminal region (amino acids 220–439) showed significant interaction (Figure 1E, lanes 3 and 4). As the N-terminal region of MYC extended further to cover a peptide sequence that is rich in proline (P), glutamic acid (E), serine (S), and threonine (T) (PEST), the MB IV domain, and nuclear localization signal (amino acids 1–345), the truncated protein gained the ability to interact with AURKB (Figure 1E, lane 5), suggesting that the interacting module likely resides in amino acids 221–345. We then expressed the truncated fragments (221–345) and determined their contribution to the association between the two proteins. Indeed, this small portion of MYC protein is capable of binding to AURKB (Figure 1E, Lane 6). Amino acids 221–345 within MYC contain the PEST domain (Figure 1E); we therefore narrowed down this interacting module from amino acids 221–345 to PEST (226–270) and performed co-immunoprecipitation. PEST domain alone, fused with streptavidin-binding peptide, was sufficient to pull down AURKB (Figure 1F, left). More importantly, PEST deletion (MYCΔPEST) disrupted the AURKB-MYC interaction (Figure 1F, right), supporting the PEST domain as a minimal AURKB-binding motif. These results thereby provide strong evidence supporting that AURKB is a bona fide MYC interacting partner.
Figure 1. MYC Interacts with AURKB.
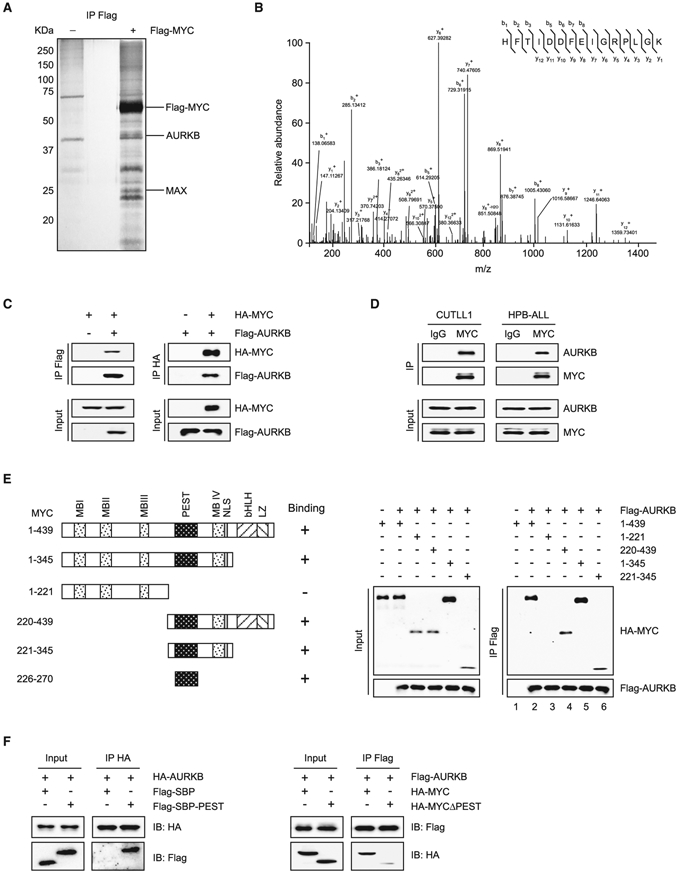
(A) Purification of MYC and its interacting partners. Total protein extract from Jurkat cells expressing Flag-tagged MYC or vector was subjected to immunoprecipitation using anti-Flag beads. Eluted proteins were resolved by SDS-PAGE and visualized by silver staining.
(B) Liquid chromatography-tandem mass spectrometry analysis of the Flag-MYC-associated peptides corresponding to AURKB.
(C) Lysates of 293T cells overexpressing Flag-AURKB and/or HA-MYC were subjected to reciprocal co-immunoprecipitation (co-IP) to detect protein interaction.
(D) CUTLL1 and HPB-ALL cell lysates were subjected to co-IP and immunoblot to detect endogenous MYC and AURKB interaction.
(E) Schematic presentation of various human MYC truncations used in AURKB-binding assays (left). Lysates from 293T cells overexpressing Flag-AURKB and/or various HA-MYC truncations were subjected to co-IP and immunoblot (right).
(F) Co-IP of HA-AURKB and Flag-tagged streptavidin-binding peptide (SBP)-fused MYC PEST domain (left), or Flag-AURKB and HA-MYCΔPEST (right) from lysates of 293T cells overexpressing respective tagged proteins.
See also Table S1.
AURKB Inhibition Destabilizes MYC and Impairs the Transcriptional Activity
To assess the potential role of AURKB in regulating MYC, we knocked down AURKB in CUTLL1 and HPB-ALL cells by expressing specific short hairpin RNAs (shRNAs). Depletion of AURKB reduced the steady-state levels of MYC protein (Figure 2A). However, knockdown of AURKA or AURKC failed to do so (Figure S1A). Enforced expression of a shRNA-resistant AURKB efficiently rescued the MYC depletion due to endogenous AURKB silencing (Figure S1B), arguing the specific role of AURKB in regulation of MYC in T-ALL. Pharmacologic targeting of AURKB by AZD1152, which specifically suppresses AURKB autophosphorylation (Figure S1C), diminished MYC expression as well in multiple T-ALL cells (Figure 2B). Inactivation of Aurora B or depletion of AURKB failed to induce a substantial decrease in MYC mRNA levels (Figures S1D and S1E), suggesting that Aurora B regulates MYC via a post-transcriptional mechanism. Indeed, time-course experiments revealed that knockdown of AURKB significantly reduced the half-life of endogenous MYC in CUTLL1 cells from 30 to 15 min (Figure 2C). Similarly, administration of AZD1152 accelerated MYC protein turnover with a half-life from 40 to 18 min (Figure S1F). Decrease in MYC protein abundance was efficiently rescued by the proteasome inhibitor MG132 (Figures 2D and S1G), suggesting that ablation of AURKB stimulates proteasomal degradation of MYC. Of particular interest, the N-MYC protein accumulation and stability in neuroblastoma cells was barely changed upon AURKB inhibition (Figures S1H-S1K), indicating a specific regulation of (c-)MYC, but not N-MYC, by Aurora B kinase.
Figure 2. AURKB Inhibition Attenuates the MYC Protein Accumulation and Transcriptional Activity.

(A) AURKB was depleted by specific shRNAs in T-ALL cells as indicated. AURKB and MYC proteins were analyzed by immunoblot, with ACTIN as a loading control. shGFP, control shRNA targeting GFP.
(B) Immunoblots of MYC and ACTIN in T-ALL cells treated with 10 nM AZD1152 for 24 h.
(C) Time-course analysis of MYC protein levels in AURKB-depleted CUTLL1 cells. MYC proteins were quantified and plotted on the right.
(D) AURKB-depleted CUTLL1 and KOPTK1 cells were treated with MG132 (10 μM) for 6 h before harvest. AURKB and MYC were analyzed by immunoblot, with ACTIN as a loading control.
(E) A heatmap depicting top 30 down- and upregulated genes upon AURKB depletion in CUTLL1 cells (Q < 0.05).
(F) qRT-PCR analysis of representative MYC-induced target genes in CUTLL1 cells upon AURKB depletion. Data shown represent the means (±SD) of biological triplicates. *p < 0.05, **p < 0.01, unpaired two-tailed Student’s t test.
(G) Gene set enrichment analysis of MYC target gene sets in the expression profiles of CUTLL1 cells expressing AURKB shRNA (http://software.broadinstitute.org/gsea).
See also Figure S1.
We predicted that inactivation of AURKB would affect global MYC transcriptional program. To test this notion, RNA sequencing was performed using AURKB or control depleted CUTLL1 cells. In the list of top 30 differentially expressed genes, previously reported MYC transcriptional targets, such as ULBP1 (Nanbakhsh et al., 2014) and PSAT1 (Hsieh et al., 2015; Vazquez et al., 2011), emerged as responsive genes sensitive to AURKB depletion (Figure 2E). Comparison with the database of MYC target genes by qRT-PCR confirmed that depletion of Aurora B reduced expression of many such genes (Figure 2F). We further interrogated our gene expression profile with published, validated MYC-regulated gene signatures (Kim et al., 2005) for statistically significant enrichment by gene set enrichment analysis (Subramanian et al., 2005). Again, AURKB depletion resulted in significant downregulation of MYC-induced gene expression programs (Figure 2G), corroborating an essential role of AURKB in sustaining MYC transcriptional activity.
AURKB-Mediated MYC Phosphorylation Contributes to MYC Stability
Regulation of MYC degradation by the ubiquitin-proteasome system is dependent on MYC phosphorylation. Initiation of MYC turnover requires GSK3β-mediated phosphorylation of T58 (p-T58) (Gregory et al., 2003). We examined whether AURKB inhibition affected MYC p-T58. Time-course analysis showed that treatment with AZD1152 in both CUTLL1 and HPB-ALL cells resulted in a notable increase in MYC p-T58 (Figure 3A), suggesting that inactivation of AURKB enhances GSK3β-mediated T58 phosphorylation. Conversely, increasing doses of AURKB expression abolished MYC and GSK3β interaction (Figure 3B). AURKB overexpression significantly extended the half-life of exogenous MYC from 31 to >100 min (Figure 3C) and counteracted FBXW7-mediated acceleration of MYC degradation (Figure S2A). Of note, AURKB kinase inactive mutant (K106R) (Honda et al., 2003), which is capable of binding with MYC (Figure S2B), failed to prolong its half-life (Figure 3C), suggesting that MYC stabilization requires the kinase activity of AURKB.
Figure 3. AURKB Enhances MYC Protein Stability through the S67 Phosphorylation.

(A) Time-course analysis of MYC p-T58 by immunoblot upon AZD1152 (50 nM) treatment in CUTLL1 and HPB-ALL cells, with ACTIN as a loading control.
(B) Protein interaction between GSK3β and MYC in the presence of AURKB. pCDH-Flag-GSK3β (1 μg), pCDH-HA-MYC (1 μg), and/or increasing doses of pCDH-AURKB (0.5,1, and 2 μg) were transfected into 293T cells for 48 h as indicated. Cell lysates were then subjected to co-IP and immunoblot. The star (*) designates immunoglobulin G heavy chain.
(C) Time-course analysis of HA-MYC levels in 293T cells expressing ectopic HA-MYC, Flag-AURKB WT, or mutant (K106R) as indicated. MYC proteins were quantified and plotted on the right. Data shown are means (±SD) of three independent experiments.
(D) Peptide sequence alignment of MYC (amino acids 58–74) in various species. The S67 site resides in the AURKB phosphorylation consensus motif.
(E) Time-course analysisofHA-tagged MYC(WT ormutants) byimmunoblot in 293T cells(left). MYC proteinswerequantified and plotted ontheright. Datashown are means (±SD) of three independent experiments.
(F) Co-IP of Flag-GSK3β and HA-MYC (WT, S67D, or S67A) from lysates of 293T cells overexpressing respective tagged proteins.
(G) In vitro kinase analysis of recombinant GST-MYC. Active human AURKB proteins were incubated with GST-MYC for kinase reaction. Phosphorylated proteins were separated by SDS-PAGE and visualized by autoradiography. Loading controls were shown as Coomassie blue staining in the bottom panels.
(H) Detection of endogenous MYC p-S67. CUTLL1 and primary T-ALL cells were treated with Nocodazole(Noco, 1 μg/mL) for 4 h or AZD1152 (10 nM) for 24 h as indicated. Noco-treated cell lysates were treated with calf-intestinal alkaline phosphatase (CIP) for 30 min before immunoblot. Histone 3 p-S10 (p-S10 H3) levels were shown as a positive control reflecting AURKB activity.
See also Figure S2.
We next sought to explore whether AURKB directly phosphorylated MYC. We examined the MYC protein sequence and identified a potential site matching AURKB phosphorylation consensus motif (Figure 3D). The potential phosphorylation residue serine 67 (S67) and the surrounding amino acids are highly conserved among vertebrates (Figure 3D), indicating an evolutionarily conserved role in regulation of the MYC protein. We then constructed MYC non-phosphorylatable alanine mutants (S67A) and phosphomimetic aspartic acid mutants (S67D), respectively, and performed a series of cycloheximide chase experiments to assess the half-life of these mutant proteins. In comparison with the wild-type (WT) counterpart, the mutation of S67D increased the half-life of MYC from 33 to >100 min, whereas S67A further decreased the MYC half-life to 17 min (Figure 3E). Consistent with these results, the S67D mutant diminished, whereas the S67A mutant enhanced the binding with GSK3β (Figure 3F). To further determine the critical role of p-S67 in affecting the interaction between MYC and GSK3β, we synthesized peptides (amino acids 51–69 within MYC) that contain p-S62, p-S67, or both. Co-immunoprecipitation demonstrated that S62 phosphorylation alone was sufffcient to enable binding with GSK3β; however, inclusion of p-S67 significantly disrupted the interaction, demonstrating that S67 phosphorylation antagonized GSK3β-MYC interaction (Figure S2C). Most likely, S67 phosphorylation prevents GSK3β from recognizing the MYC p-S62 and blocks the subsequent ubiquitin-mediated proteasomal degradation. In support of this notion, the S67D mutation impaired the interaction between MYC and FBXW7, and subsequently reduced the MYC polyubiquitylation (Figures S2D and S2E). Taken together, these data suggest that p-S67 dissociates MYC with GSK3β and blocks the MYC degradation pathway.
To determine whether MYC is directly phosphorylated by Aurora B, we performed in vitro kinase assays and found that AURKB phosphorylated glutathione S-transferase (GST)-MYC (Figure 3G, lane 4) but not GST alone (Figure 3G, lane 2). Mutagenesis of S67 to alanine abolished the phosphorylation signal (Figure 3G, lane 6), demonstrating that this residue within MYC is the major site subjected to AURKB phosphorylation. Consistently, mass spectrometry analysis manifested the S67 phosphorylation of MYC resulting from the in vitro kinase reaction (Figure S2F), and this modification was further validated by a rabbit polyclonal antibody against p-S67 (Figures S2G-S2I). We observed endogenous S67 phosphorylation signals in both CUTLL1 and primary T-ALL#1 cells. AZD1152 treatment in these cells diminished the phosphorylation, concomitantly with decrease in MYC protein levels (Figure 3H). Notably, the endogenous phosphorylation signals were enhanced by Nocodazole (Noco) treatments through full activation of AURKB (Yang et al., 2011), and calf-intestinal alkaline phosphatase treatment in cell lysates rapidly erased the phosphorylation signal (Figure 3H). Altogether, these results corroborate the notion that AURKB phosphorylation of MYC at S67 is an important mechanism in regulation of MYC stability in T-ALL. Interestingly, polypeptide sequence alignments failed to reveal S67 in N-MYC (Figure S2J), which may explain non-regulatory role of AURKB in N-MYC (Figures S1H-S1K).
Previous studies show that PLK1 contributes to MYC protein stability through phosphorylation of MYC in colorectal cancer (Popov et al., 2010; Tan et al., 2013) or phosphorylation of FBXW7 in neuroblastoma and small-cell lung carcinoma (Xiao et al., 2016). AURKB was capable of activating PLK1 in the M phase of the cell cycle (Archambault and Carmena, 2012), raising the possibility that AURKB regulation of MYC could be indirectly through PLK1. To evaluate this, we respectively treated KOPTK1 cells with PLK1 inhibitor BI6727 or AURKB inhibitor AZD1152. BI6727 decreased p-S62 and increased p-T58 as expected, whereas AZD1152 decreased p-S67 and increased p-T58 with p-S62 largely unchanged at the tested time point (Figure S2K). These results suggest that, most likely, stabilization of MYC by Aurora B is through direct S67 phosphorylation, but not mediated by PLK1. Nevertheless, we would not rule out the possibility that the indirect regulation of MYC through the AURKB-PLK1-MYC axis, to some extent, contributes to MYC protein stabilization and MYC-mediated leukemogenesis.
MYC and T Cell Acute Lymphoblastic Leukemia 1 Cooperate to Activate AURKB Transcription
We analyzed the Cancer Cell Line Encyclopedia and revealed the second highest expression of AURKB in T-ALL among 1,457 human cancer cell lines from 40 tumor types (Barretina et al., 2012) (Figure 4A). The AURKB protein is also more abundant in human T-ALL than normal thymocytes (Figure 4B), suggesting its crucial role in transformed T cells. To decipher the molecular mechanisms underlying AURKB expression, we performed in silico analysis in the University of California Santa Cruz genome browser for transcription factor binding cis-elements in the AURKB locus and identified MYC and T cell acute lymphoblastic leukemia 1 (TAL1) binding consensus sequences (data not shown). Depletion of MYC or TAL1 led to a significant decrease in AURKB expression at mRNA and protein levels, comparable with the well-characterized MYC or TAL1 target gene (Figures 4C and 4D). We next identified putative MYC E-box response elements (RE1, −1,287 bp and RE2, −2,969 bp) and TAL1 binding site (RE3, +110 bp) (Figure 4E). Chromatin immunoprecipitation (ChIP) assay revealed a marked increase in MYC or TAL1 recruitment to respective sites (Figure 4F). Luciferase reporter assays confirmed that MYC significantly activated RE1 and RE2 as compared with vector control or RE mutants (RE1 or RE2 Mut). Similar results were observed in TAL1-driven reporter activity (Figure 4G). Although MYC or TAL1 depletion affected cell-cycle progression to some extent (Figures S3A and S3B), the corresponding changes in AURKB expression should not be a consequence of cell-cycle arrest as treatment of the CDK4/6 inhibitor Palbociclib (Palbo) induced G1 arrest but failed to change the AURKB expression (Figures S3C and S3D). Moreover, expression of an inducible MYC transgene in Jurkat cells significantly activated AURKB transcription (Figure S3E). Taken together, these findings support the notion that MYC, in concert with TAL1, directly activates the AURKB transcription in T-ALL, thus constituting a reciprocal regulation between MYC and AURKB. Interestingly, previous studies suggested that AURKB expression was dependent on MYC as well in B cell lymphoma (den Hollander et al., 2010), suggesting a general regulation of AURKB by MYC in various tumor contexts.
Figure 4. MYC and TAL1 Activate AURKB Transcription.

(A) AURKB mRNA expression was analyzed among 1,457 human cancer cell lines in CCLE database (https://portals.broadinstitute.org/ccle). The distributions of AURKB mRNA expression are presented as log2 median-centered intensity and shown in box-and-whisker plots, which depict the first and third quartiles, with the median shown as a solid line inside the box and whiskers extending to 1.5 interquartile range from first and third quartiles.
(B) AURKB proteins were analyzed by immunoblot in normal thymuses from healthy donors, primary T-ALL samples and T-ALL cell lines, respectively.
(C) AURKB mRNA and protein were analyzed in MYC-depleted CUTLL1 cells by qRT-PCR and immunoblot. MYC target gene NCL was used as a positive control.
(D) AURKB mRNA and protein were analyzed in TAL1-depleted Jurkat cells by qRT-PCR and immunoblot. TAL1 target gene NKX3.1 was used as a positive control.
(E) Schematic presentation of MYC and TAL1 binding sites on the AURKB locus. MYC response elements, RE1 and RE2; TAL1 responsive element, RE3. Consensus sequence mutations are shown as RE Mut.
(F) ChIP analysis of MYC binding to the AURKB locus in CUTLL1 cells (left) orTAL1 binding in Jurkat cells (right). Binding signals to NCL and NKX3.1 are shown as positive controls, ACTIN as a negative control.
(G) Luciferase reporter activities were assessed using MYC3×RE constructs in the presence of exogenous MYC in 293T cells (left) or when co-expressing TAL1 3×RE reporter constructs with TAL1 (right).
Data shown represent the means (±SD) of three biological replicates, **p < 0.01; significance was determined by unpaired two-tailed Student’s t test (F) or one-way ANOVA test followed by Tukey’s correction (C, D, and G). See also Figure S3.
AURKB-Mediated MYC Phosphorylation Accelerates Leukemogenesis
Myc-induced T-ALL zebrafish model is created by expressing mouse Myc and EGFP fusion protein, which was placed under the control of zebrafish lymphatic-specific rag2 promoter (Langenau et al., 2003). This model allows putative genes to be assessed for their ability collaborating with MYC in leukemogenesis. We thus co-injected the one-cell-stage embryos with the rag2:EGFP-Myc construct alone or together with rag2:AURKB followed by fluorescent microscopy analysis. As shown in Figures 5A and S4A, co-expression of AURKB with Myc led to a significant increase in tumor dissemination and penetrance, with a 4-fold increase of GFP fluorescence signals as compared with Myc expressed alone. We next used an inducible MYC transgenic fish Tg (rag2:Loxp-dsRED2-Loxp-EGFP-Myc;hsp70:Cre) to analyze tumor cell propagation (Feng et al., 2007). After heat shock treatment at 3 days post-fertilization (dpf), over 80% of these fish developed T-ALL. The transgenic fish were further bred with the stable transgenic line simultaneously expressing human AURKB and mCherry (Figure S4B). Again, AURKB expression induced more pronounced distant dissemination of lymphoblasts at 80 dpf as demonstrated by increased GFP and mCherry fluorescence signals (Figure 5B).
Figure 5. AURKB Promotes Murine Myc-Induced T-ALL.

(A) Zebrafish embryos were injected respectively with the rag2:EGFP-Myc construct alone or in combination with rag2:AURKB. Representative images of GFP fluorescent microscopy analysis at 8 dpf are shown on the left. GFP fluorescence from the thymuses of EGFP-Myc (n = 13) and EGFP-Myc;AURKB fish (n = 14) were qualified and plotted on the right. Scale bar, 1 mm. Data are means ± SD, **p < 0.01, unpaired two-tailed Student’s t test.
(B) Representative images of GFP or mCherry dissemination at 80 dpf in EGFP-Myc;mCherty (n = 10) and EGFP-Myc;mCherry;AURKB (n = 12) transgenic fish receiving heat shock treatments. Scale bar, 1 mm.
(C) Representative GFP images of zebrafish injected with indicated constructs (left). Scale bar, 1 mm. Quantifications of the GFP fluorescence are shown on the right (n = 6 in each group). Data are mean ± SD, **p < 0.01, two-way ANOVA test followed by Tukey’s correction.
(D) Time-course analysis of exogenous murine Myc from zebrafish expressing various transgenes as indicated. The arrow denotes human AURKB transgene expression and the bands below (*) are endogenous zebrafish Aurkb. Zebrafish Actin was used as a loading control (left). Myc protein levels at each point were quantified and plotted on the right.
See also Figure S4.
We next determined whether the enhanced leukemogenic effect of AURKB is dependent on Myc p-S67 by microinjecting constructs expressing Myc S67D or S67A mutant into one-cell-stage embryos. Time-course analysis revealed that expression of Myc S67D yielded greater GFP signals at each time point as compared with WT Myc, highly resembling the phenotype of Myc;AURKB co-expression. In contrast, expression of Myc S67A diminished the accelerated leukemogenesis induced by Myc S67D (Figure 5C). Protein stability assays, performed in lymphoblasts dissected from zebrafish thymuses, revealed that overexpression of AURKB significantly extended the half-life of Myc from 25 to ~200 min. Again, S67D mutation increased Myc protein half-life to 185 min, whereas the S67A mutation slightly reduced the half-life to 19 min (Figure 5D). These findings provide strong evidence demonstrating that the S67 phosphorylation of MYC is associated with the tumor-promoting effect of AURKB and is functionally important for MYC stabilization and T cell leukemogenesis in vivo.
AURKB Inhibitor Synergizes with Vincristine to Induce T-ALL Cell Death
AURKB depletion led to significant KOPTK1 cell death associated with MYC downregulation and enforced expression of a shRNA-resistant AURKB efficiently rescued both cell death and MYC depletion (Figures S1B and S5A). Similarly, pharmacological inhibition of AURKB by AZD1152 induced marked cell death in KOPTK1, CUTLL1, and HPB-ALL cells concomitant with the decline in MYC expression (Figures 6A and 6B). Ectopically expressed MYC S67D significantly rescued cell death and reverted MYC target gene downregulation (Figures 6C left, S5B-S5D), although had minimal effect on cell-cycle progression (Figure S5E), suggesting that MYC is a crucial downstream effector of AURKB mediating the pro-survival signals. In contrast, Jurkat and PEER cells were resistant to AZD1152 associated with unchanged MYC abundance (Figure 6A and 6B). We reasoned that the resistance is due to the presence of FBXW7 loss-of-function mutant alleles in these cells. In support of this notion, primary T-ALL#1 cells with WT FBXW7 alleles, but not those (T-ALL#4) harboring FBXW7 R465C mutant alleles, were responsive to AZD1152 (Figure 6A). We surmised that loss-of-function FBXW7 mutant protein failed to elicit efficient MYC polyubiquitylation and proteasomal degradation, conferring intrinsic drug resistance. Indeed, knockdown of MYC sensitized Jurkat cells to AURKB inhibition (Figures 6C, middle and S5F, left); AZD1152-sensitive CUTLL1 cells became refractory upon FBXW7 depletion concomitant with elevated MYC levels (Figures 6C, right and S5F, right). These data suggest that decrease in MYC expression is required for T-ALL cells to respond to AZD1152 and expression of functional FBXW7 predicts anti-T-ALL efficacy. Of note, NOTCH1 activating mutations are the most prevailing genetic lesions in T-ALL and NOTCH1 plays a crucial role in MYC transcriptional activation (Liu et al., 2010; Weng et al., 2004). However, both NOTCH1 WT (SUP-T1) and mutant (KOPTK1) cells were sensitive to AURKB inhibition, suggesting that NOTCH1 mutational status may not predict therapeutic consequences of AZD1152 (Figure S5G). In principle, AURKB inhibition would cause MYC protein degradation even though mutant NOTCH1 enhances MYC transcription.
Figure 6. AURKB Inhibition Induces Apoptosis in T-ALL Cells Expressing WT FBXW7.
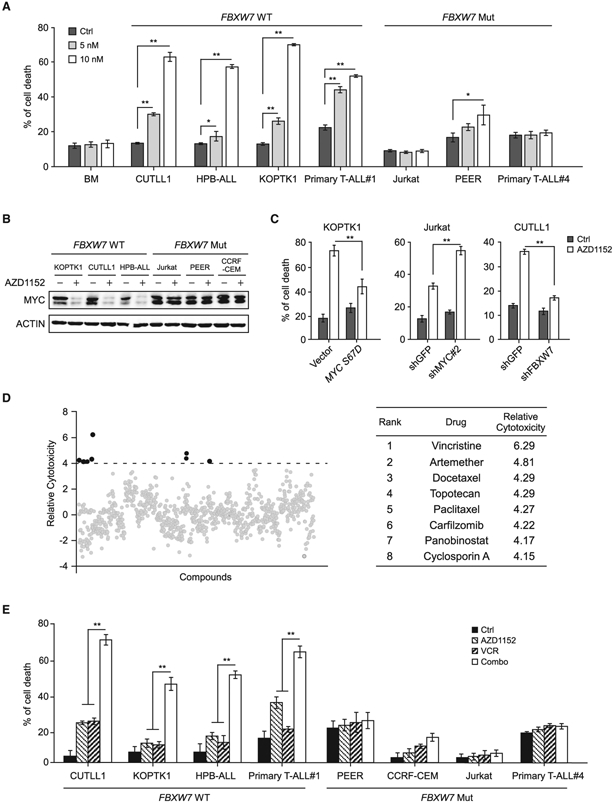
(A) Analysis of apoptotic cell death by Annexin V/PI staining in normal bone marrow cells and a panel of T-ALL cell lines, harboring WT or mutant FBXW7, treated with AZD1152 for 48 h.
(B) Immunoblots of MYC in 10 nM AZD1152-treated T-ALL cells.
(C) Cell death analysis in MYC S67D expressing KOPTK1 cells (left), MYC-depleted Jurkat cells (middle), or FBXW7-depleted CUTLL1 cells (right), which were subjected to 5, 25, or 5 nM AZD1152 treatment, respectively.
(D) High-throughput screening of US Food and Drug Administration-approved drugs in KOPTK1 cells to identify small molecules synergistic with AZD1152. Top hits are listed on the right.
(E) Assessment of cell death in T-ALL cells treated with AZD1152 (5 nM) and/or vincristine (1 nM) for 48 h. Combo, combination treatment.
Data shown represent the means (±SD) of three biological replicates, *p < 0.05, **p < 0.01; significance was determined by one-way (A and E) or two-way (C) ANOVA test followed by Tukey’s correction. See also Figures S5 and S6 and Table S2.
To test the effect of AURKB inhibition in a wider tumor context, we treated RS4; 11 (B cell acute lymphoblastic leukemia, B-ALL), Daudi (Burkitts’ lymphoma), and NB4 (acute myeloid leukemia, -AML-) cells with the same doses of AZD1152 as used in T-ALL. AZD1152 induced robust cell death in RS4; 11 cells associated with decrease in MYC protein levels. In contrast, it caused minimal effects on MYC protein abundance and cell death in Daudi and NB4 cells (Figures S5H and S5I), suggesting a tumor context-dependent effect of AURKB inhibition. These data also raise the possibility that ALL cells could be more responsive to AZD1152 treatment.
To improve AZD1152 anti-leukemia activity and durability, we performed a drug combination screen from a collection of Food and Drug Administration-approved drugs to identify potential candidates capable of eliciting synergistic lethality in KOPTK1 cells. In this screen, we observed various drug combination effects that are additive, antagonistic, or synergistic (Figure S6). To pinpoint the most synergistic molecules, positive hits were defined as compounds exhibiting more than 4-fold toxic effects in combination treatment as compared with single treatment (Figure 6D, left). Vincristine, the first-line ALL therapeutics (Inaba et al., 2013; Jackson et al., 2007), emerged as the top hit to elicit remarkable nanomolar synergistic lethal effect (Figure 6D, right; Table S2). Again, dual-compound treatments manifested robust synergism in T-ALL cells expressing WT FBXW7, but not in those harboring FBXW7 loss-of-function mutations (Figures 6E and S6B). We therefore identify FBXW7 mutational status as a biomarker predicting sensitivity to AZD1152 treatment, alone or in combination with vincristine.
AZD1152 and Vincristine Synergistically Suppress Tumor Progression in T-ALL
To evaluate the therapeutic efficacy of AZD1152 and vincristine combination in vivo, we established human xenografts using CUTLL1 (WT FBXW7) and Jurkat (mutant FBXW7) cells, respectively, which were subjected to AZD1152/vincristine single or combination treatment. To visualize leukemia cell expansion in vivo, lentiviruses expressing firefly luciferase were infected to these cells before intravenous injection into immunodeficient NPG mice (Figure 7A). Administration of either AZD1152 or vincristine alone inhibited CUTLL1 engraftment, and AZD1152/vincristine combination resulted in a synergistic and remarkable suppression of leukemogenesis (Figure 7B, left). However, neither single nor dual administration efficiently inhibited tumor progression in Jurkat xenografts (Figure 7B, right).
Figure 7. AZD1152 and Vincristine Synergistically Inhibit Xenograft Tumor Growth.

(A) Schematic representation of in vivo imaging of T-ALL xenografts. CUTLL1 (FBXW7 WT) or Jurkat (FBXW7 Mut) cells, expressing both luciferase and GFP markers (Luc-GFP), were injected into NPG mice and subjected to treatments, followed by in vivo bioimaging to assess therapeutic responses.
(B) Representative images of tumor burden assessed by bioimaging in mice xenografted with CUTLL1 or Jurkat cells upon single or combination treatments (n = 3 per group). Drug administrations started at day 0.
(C) Anti-leukemia effects of drug combination in a patient-derived xenograft (PDX). Human CD45+ cells from bone marrow (BM) and spleen were analyzed by flow cytometry.
(D) Representative spleen and bone images of mice in each group at day 25 after engraftment.
(E) Representative immunohistological images of MYC, proliferating cell nuclear antigen (PCNA), and cleaved caspase-3 (c-Cas-3) in the spleen sections from mice receiving indicated treatments. Scale bar, 50 μm. Quantifications of immunohistochemistry are shown on the right. Data are mean ± SD, *p < 0.05, **p < 0.01, one-way ANOVA test followed by Tukey’s correction.
(F) Kaplan-Meier survival curves of T-ALL PDX treated with AZD1152 and/or vincristine (n = 5 in each group). Significance was determined by logrank test, **p < 0.01.
(G) Model depicting the regulation of (c-)MYC or N-MYC by Aurora kinases through kinase-dependent or independent mechanism. See text for more details.
To further explore the clinical relevance of Aurora B inhibition therapies in combination with vincristine, we evaluated the efficacy of dual treatments in xenografts from primary T-ALL#1 cells expressing WT FBXW7 alleles. Again, the combination treatment induced synergistic tumor growth inhibition compared with the administration of either single agent, as demonstrated by much less CD45+ tumor cell burden (Figure 7C), more reddish bones and smaller spleen sizes (Figure 7D). Immunohistochemistry staining confirmed that administration of AZD1152 depleted MYC expression. Again, AZD1152 and vincristine in combination further inhibited cell proliferation and induced intratumoral apoptosis, as quantified by proliferating cell nuclear antigen and cleaved caspase-3 staining, respectively (Figure 7E). As a consequence, dual treatments significantly prolonged animal survival as compared with single treatment (Figure 7F). All of these results support the clinical potential of the combination of AURKB inhibitors and vincristine as T-ALL therapeutics.
DISCUSSION
Aberrant high levels of MYC expression are broadly present in T-ALL (Bonnet et al., 2011). Herein, we reveal that AURKB directly phosphorylates and stabilizes MYC, which in turn activates AURKB transcription. This reciprocal activation between AURKB and MYC is essential for sustaining mutual high expression, leading to MYC-dependent transcriptional amplification and aggressive leukemogenic phenotype. Multiple previous elegant studies show that the NOTCH1 signaling pathway activates MYC at the transcriptional and post-transcriptional levels (Herranz et al., 2014; Liu et al., 2010; Su et al., 2018; Weng et al., 2006; Yashiro-Ohtani et al., 2014). In principle, all these regulatory mechanisms would cooperate in MYC deregulation and malignant transformation.
In our experimental setting, we demonstrate that AURKB plays a prominent role in regulating (c-)MYC stability in T-ALL through a kinase activity-dependent mechanism. However, AURKB seems to have marginal effect on N-MYC stability in MYCN-amplified neuroblastoma cells. Interestingly, AURKA was shown to bind N-MYC in neuroblastoma and counteract FBXW7-mediated N-MYC degradation through a kinase-independent mechanism (Otto et al., 2009; Richards et al., 2016). Therefore, conformation-changing AURKA inhibitors, which disrupted AURKA and N-MYC interaction, induced proteolytic N-MYC protein turnover (Gustafson et al., 2014). A follow-up study suggested the anti-tumor efficacy of these inhibitors in p53-altered liver cancer via dissociation of AURKA and MYC (Dauch et al., 2016). All these findings argue that Aurora kinases sustain the expression of MYC protein family via multiple mechanisms in a context-dependent manner (see a simplified model in Figure 7G).
High levels of MYC were shown to sensitize cancer cells to antimitotic drugs by upregulating the expression of an apoptotic network (Topham et al., 2015), and mitotic regulators are synthetic lethal targets in MYC-overexpressing tumors, including Aurora B (Yang et al., 2010). Alternatively, approaches to inhibit MYC expression/activity have proven potent anti-tumor efficacy (McKeown and Bradner, 2014). Therefore, either maintaining high MYC expression or reducing MYC expression in tumor cells could achieve remarkable anti-cancer effects. Most likely, MYC functions as a double-edged sword in regulating cellular activities, that is activation of MYC promotes survival or apoptosis depending on expression thresholds and cellular context (Murphy et al., 2008). In particular, MYC inhibition leads to T-ALL remission in part through eliminating leukemia-initiating cell activity (King et al., 2013; Roderick et al., 2014), arguing inhibition of MYC is a potentially effective anti-T-ALL strategy. Therefore, our findings argue that applications of AURKB inhibitors hold great promise as a regimen for MYC-targeted therapy in T-ALL.
AZD1152, a highly specific and potent AURKB inhibitor, is under multiple clinical trials. As monotherapy, AZD1152 has manifested short-lived therapeutic response in diffuse large B cell lymphoma patients (Collins et al., 2015) and resulted in an overall hematologic response rate of 25% in AML patients (Löwenberg et al., 2011). These clinical studies suggest that AZD1152 may not achieve durable clinical responses as a single treatment and optimization of patient populations is of immediate emergence. Our findings show that ALL patients may be more responsive to this regimen, particularly in combination with vincristine. We found that leukemia cell expressing functionally intact FBXW7 were selectively responsive to AZD1152 treatment. Because FBXW7 loss-of-function mutations occur in ~20% of T-ALL cases (O’Neil et al., 2007; Thompson et al., 2007), this mutational status may serve as a genetic biomarker predicting drug sensitivity. These findings encourage an immediate translation of AZD1152-based therapeutics in T-ALL treatment, which currently lacks effective targeted therapies.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hudan Liu (hudanliu@whu.edu.cn).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
Human T-ALL cell lines were grown in complete RPMI-1640 (Hyclone) supplemented with 10% fetal bovine serum (FBS, Hyclone), 1% penicillin/streptomycin (Hyclone), 1% non-essential amino acids (Gibco), 2 mM L-glutamine (Sigma-Aldrich), 1 mM sodium pyruvate (Sigma-Aldrich), and 55 μM β-mercaptoethanol (Sigma-Aldrich). Human primary T-ALL cells were obtained with informed consents from Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou, China, and Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences, Tianjin, China; these cells were co-cultured in OP9-DL1 feeder cells as described (Su et al., 2018). 293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone) containing 10% FBS and 1% penicillin/streptomycin. All cell lines were authenticated with short tandem repeats (STR) profiling, cultured for fewer than 6 months after resuscitation, and tested for mycoplasma contamination every 3 months using MycoAlert (Lonza).
Animals
Immunocompromised NPG mice (NOD.Cg-PrkdcscidIl2rgtm1Vst/Vst, Beijing Vitalstar Biotechnology) were used for human T-ALL xenografts. Mice were maintained in Specific Pathogen Free (SPF) animal facility of Medical Research Institute, Wuhan University. All animal experiments were performed under animal ethical regulations and the study protocol was approved by the Institutional Animal Care and Use Committee of Wuhan University.
Zebrafish of the AB background strain were maintained in a recirculating filtration system, according to standards and approved protocols of the Institutional Animal Care and Use Committee of the China Zebrafish Resource Center, Institute of Hydrobiology, Chinese Academy of Sciences.
Microbe Strains
E. coli DH5α or BL21 (DE3) competent cells were transformed and grown on plates of LB Agar with Ampicillin or Kanamycin 100 μg/mL for plasmid selection at 37°C.
METHOD DETAILS
Immunoblot and Immunoprecipitation
Cells were lysed in RIPA buffer (Su et al., 2018) containing protease inhibitor cocktail for immunoblot and protein concentrations were determined using BCA assay kit (Thermo Fisher Scientific). 30-50 μg total cellular proteins were subjected to SDS-PAGE gel and transferred to polyvinylidene difluoride membrane (Bio-Rad). After being blocked with 5% fat free milk, blots were generally incubated with primary antibodies at 4°C overnight. Appropriate horseradish peroxidase conjugated secondary antibodies were applied for 1 hr at room temperature before detection with SuperSignal Chemiluminescent Substrate (Bio-Rad). Densitometric analyses of protein abundance were determined by ImageJ software. For immunoprecipitation, cells were lysed in buffer (50 mM Tri-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1 mM DTT and protease inhibitor cocktail). 1 mg of lysate was incubated with corresponding antibodies conjugated to Protein G beads (Thermo Fisher Scientific) overnight at 4°C. Antibodies used in this study are listed in the Key Resources Table. Phospho-MYC S67 antibody was generated by immunizing rabbits with phosphorylated S67 peptides (CSPSRR-(phospho)S-GLASPSY) conjugated with carrier protein keyhole limpet hemacyanin (PTM Biolabs).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ANTI-FLAG® M2 Affinity Gel | Sigma-Aldrich | Cat# A2220; RRID: AB_10063035 |
| AURKA | Cell Signaling Technology | Cat# 14475; RRID: AB_2665504 |
| AURKB | Cell Signaling Technology | Cat# 3094; RRID: AB_10695307 |
| AURKC | ABclonal | Cat# A7930; RRID: AB_2768528 |
| β-ACTIN | ABclonal | Cat# AC026; RRID: AB_2768234 |
| cleaved Caspase-3 | ABclonal | Cat# A11021; RRID: AB_2758369 |
| FBXW7 | Abcam | Cat# 109617; RRID: AB_2687519 |
| FLAG-tag | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| HA-tag | ABclonal | Cat# AE008; RRID: AB_2770404 |
| HA-tag (peroxidase conjugate) | Roche | Cat# 12013819001; RRID: AB_390917 |
| Histone H3 | Cell Signaling Technology | Cat# 3638; RRID: AB_1642229 |
| MYC (N-262) | Santa Cruz Biotechnology | Cat# sc-764; RRID: AB_631276 |
| MYC (9E10) | Santa Cruz Biotechnology | Cat# sc-40; RRID: AB_627268 |
| MYC/N-MYC (D3N8F) | Cell Signaling Technology | Cat# 13987; RRID: AB_2631168 |
| Myc-tag | ABclonal | Cat# AE010; RRID: AB_2770408 |
| NCL | ABclonal | Cat# A5904; RRID: AB_2766650 |
| NKX3.1 | Sigma-Aldrich | Cat# SAB1412056 |
| p53 | Cell Signaling Technology | Cat# 9282; RRID: AB_331476 |
| PCNA | Santa Cruz Biotechnology | Cat# sc-56; RRID: AB_628110 |
| Phospho-Aurora A(Thr288)/Aurora B (Thr232)/Aurora C (Thr198) |
Cell Signaling Technology | Cat# 2914; RRID: AB_2061631 |
| Phospho-Histone H3 Ser10 | Millipore | Cat# 06-570; RRID: AB_310177 |
| Phospho-MYC S62 | Cell Signaling Technology | Cat# 13748; RRID: AB_2687518 |
| Phospho-MYC S67 | PTM BioLabs | NA |
| Phospho-MYC T58 | Santa Cruz Biotechnology | Cat# sc-135647; RRID: AB_2148600 |
| TAL1 | Santa Cruz Biotechnology | Cat# sc-393287 X |
| Biological Samples | ||
| Primary human T-ALL cells | This paper | NA |
| Bacterial and Virus Strains | ||
| BL21 (DE3) Competent E. coli | Thermo Fisher Scientific | Cat# C601003 |
| DH5α Competent E. coli | Thermo Fisher Scientific | Cat# 12297016 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ATP, [γ-32P] | PerkinElmer | Cat# NEG502A |
| ATP | Thermo Fisher Scientific | Cat# 18330019 |
| Aurora B Kinase, GST-tag, His-tag | BPS Bioscience | Cat# 40002 |
| Barasertib (AZD1152-HQPA) | Selleck | Cat# S1147 |
| Cycloheximide | Sigma-Aldrich | Cat# C7698 |
| FDA-approved drug library | Selleck | Cat# L1300 |
| MG132 | Selleck | Cat# S2619 |
| Nocodazole | Selleck | Cat# S2775 |
| Vincristine | Selleck | Cat# S1241 |
| Commercial Kits | ||
| Annexin V-FITC Apoptosis Kit | BioVision | Cat# K201 |
| CellTox™ Green Cytotoxicity Assay kit | Promega | Cat# G8743 |
| Chromatin Immunoprecipitation (ChIP) Assay Kit | Sigma-Aldrich | Cat# 17-295 |
| DAB substrate kit | Vector Laboratories | Cat# SK-4100 |
| Dual Luciferase Reporter Assay System | Promega | Cat# E1980 |
| In vitro protein kinase buffer | Cell Signaling Technology | Cat# 9802S |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat# 1725124 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 11668027 |
| ReverTra Ace qPCR RT kit | TOYOBO | Cat# FSQ-101 |
| Experimental Models: Cell Lines | ||
| 293T | ATCC | Cat# ACS-4500 |
| CCRF-CEM | ATCC | Cat# CCL-119 |
| CUTLL1 | (Weng et al., 2006) | NA |
| HPB-ALL | (Weng et al., 2006) | NA |
| Jurkat | ATCC | Cat# TIB-152 |
| KOPTK1 | (Weng et al., 2006) | NA |
| PEER | (O’Neil et al., 2007) | NA |
| SUP-T1 | ATCC | Cat# CRL-1942 |
| Experimental Models: Organisms | ||
| Mouse: NPG (NOD.Cg-PrkdcscidIl2rgtm1Vst/Vst) | Beijing Vitalstar Biotechnology | Cat# VS000187 |
| Zebrafish: Tg(rag2:Loxp-dsRED2-Loxp-EGFP-Myc; hsp70:Cre) | (Feng et al., 2007) | NA |
| Zebrafish: Tg(rag2:Loxp-dsRED2-Loxp-EGFP-Myc; hsp70:Cre; rag2:mCherry) | This paper | NA |
| Zebrafish: Tg(rag2:Loxp-dsRED2-Loxp-EGFP-Myc; hsp70:Cre; rag2:mCherry; rag2:AURKB) | This paper | NA |
| Oligonucleotides | ||
| See Table S3 | This paper | NA |
| Software and Algorithms | ||
| CalcuSyn | Biosoft | http://www.biosoft.com/ |
| FlowJo | Tree Star | https://www.flowjo.com/ |
| Gene Set Enrichment Analysis | (Subramanian et al., 2005) | http://software.broadinstitute.org/gsea/index.jsp |
| Graphpad Prism 7.0 | Graphpad software | https://www.graphpad.com/ |
| ImageJ | ImageJ: Image Processing and Analysis in Java |
https://imagej.nih.gov/ij/ |
| Mev | My BioSoftware | http://www.mybiosoftware.com/mev-4-6-2-multiple-experiment-viewer.html |
| Deposited Data | ||
| CUTLL1 RNA-seq | This paper | GSE140739 |
Time-course Analysis of MYC Degradation
Cells undergoing AZD1152 treatment or AURKB depletion/overexpression were subjected to cycloheximide (CHX, 50 μg/mL) treatment and harvested at specific time-points. Cells were then lysed in RIPA buffer containing protease inhibitor cocktail and subjected to immunoblot. MYC protein band densities were quantified by ImageJ software and normalized to ACTIN.
RNA Isolation and Real-time Quantitative PCR
Total cellular RNA was extracted using TRIzol (Thermo Fisher Scientific). Complementary DNA (cDNA) synthesis was performed using the ReverTra Ace qPCR RT Kit according to the manufacturer’s instructions (TOYOBO). Quantitative PCR was conducted using FAST SYBR Green Master Mixon CFX Connect Real-Time PCR System (Bio-Rad). Relative expression of the mRNA was calculated by 2−ΔΔCt method and normalized to 18S or ACTIN.
RNA Sequencing
Total RNA was extracted using TRIzol Reagent following the manufacturer’s instructions, and subjected to quality control to inspect RNA integrity by Agilent Bioanalyzer 2100 (Agilent technologies). Total RNAs were further purified by RNAClean XP Kit (Beckman Coulter) and RNase-Free DNase Set (QIAGEN). RNA libraries were constructed using TruSeq RNA Library Prep Kit v2 (Illumina) and sequenced as 150 bp paired-end reads by Illumina HiSeq X-Ten (Shanghai Biotechnology Corporation). RNA-seq reads quality was evaluated using FastQC and mapped to the human genome reference assembly (hg38) using Bowtie 2. Significant differential expressed genes (DEGs) were identified as those with a False Discovery Rate (FDR) value above the threshold (Q<0.05), fold-change>1.2 and FPKM>5 using edgeR software. The heatmap was generated using Mev software.
Lentiviral Transduction
Lentiviral production and transduction were performed as described (Su et al., 2018). Briefly, lentiviral constructs (pLKO.1 for shRNA and pCDH for overexpression) were used for plasmid construction and transfected into 293T cells together with helper plasmids (pMD2.G and psPAX2) using Lipofectamine 2000 (Thermo Fisher Scientific). Viral supernatants were generally collected 48 hr post transfection. One million T-ALL cells were incubated with 0.5 mL viral supernatant and 8 μg/mL polybrene (Sigma-Aldrich) in a final volume of 2 mL for 30 min, and subjected to centrifugation at 1000×g for 90 min at room temperature. Cells were then supplemented with 3 mL fresh medium and continued culture for at least 48 hr to achieve efficient gene silencing or overexpression. Cell viabilities or gene expression changes were assessed 48 hr post-infection.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed according to the manufacture’s instruction. Ten million cells were fixed with 1% formaldehyde for 10 min, quenched with 0.125 M glycine for 5 min at 37°C and lysed in SDS Lysis Buffer. Cell lysate was sonicated by Bioruptor Pico Sonifier to shear chromatin DNA to a size range of 200-1000 bp. The supernatant was diluted 10 fold in ChIP Dilution Buffer and precleared with 60 μL agarose beads for 30 min. The supernatant fraction was immunoprecipitated with indicated antibodies (2 μg) against MYC or TAL1 overnight at 4°C. Antibody-chromatin complexes were pulled down with protein A agarose/salmon sperm DNA beads (Sigma-Aldrich) for 1 hr at 4°C. De-crosslinked DNA was subjected to qPCR analysis using specific primers listed in Table S3.
Luciferase Reporter Assay
0.5 μg pGL3 vector expressing 3×MYC-RE or TAL1-RE (or indicated mutant), along with 0.5 μg pCDH-MYC or TAL1, 50 ng Renilla luciferase reporter was co-transfected in triplicates into 293T cells using Lipofectamine 2000. Luciferase activities were measured 24 hr later using Dual Luciferase Reporter Assay System (Promega). Firefly luciferase activities were normalized to Renilla luciferase control values and shown as an average of triplicates.
In Vitro Protein Kinase Assay
Flag-tagged human AURKB were purified from 293T cells treated with Nocodazole (1 μg/mL) for 4 hr using Protein G agarose beads. GST-MYC and MYC S67A mutant were expressed in the E. coli BL21 (DE3) strain. 2 μg of the indicated GST fusion proteins were incubated with AURKB in the kinase buffer (Cell Signaling Technologies) together with 5 μCi [γ-32P] ATP (PerkinElmer) and 200 μM cold ATP (Thermo Fisher Scientific) for 1 hr at 30°C. Reactions were stopped by addition of SDS loading buffer, and samples were then heated for 5 min at 95°C before analysis by SDS-PAGE and autoradiography.
Mass Spectrometry Analysis
Sample preparations: In Figure 1B, total protein extract from Jurkat cells expressing Flag-tagged MYC or vector was subjected to immunoprecipitation using ANTI-FLAG® M2 Affinity Gel (Sigma-Aldrich). Flag-MYC and associated proteins eluted by Flag peptides (100 μg/mL) were resolved by SDS-PAGE. Gel slices including all proteins were excised. In Figure S2F, His/GST-tagged AURKB was purchased from BPS Bioscience and GST-tagged MYC was expressed and purified from E.coli BL21 (DE3) strain. Recombinant AURKB (1 μg) and MYC (10 μg) were incubated in kinase buffer for 1 hr at 30°C. Reaction was stopped by addition of SDS loading buffer, followed by SDS-PAGE and Coomassie blue staining. Gel bands corresponding to GST-MYC were excised.
LC-MS/MS analysis was performed in the Protein Chemistry Facility, Center of Biomedical Analysis at Tsinghua University (Beijing, China) according to previous publications (Herold et al., 2019; Liu et al., 2014). Briefly, proteins extracted from gels were digested with trypsin in 50 mM ammonium bicarbonate at 37°C overnight. After treatment with 5 mM dithiotreitol and 11 mM iodoacetamide, the resulting peptides were separated by silica capillary column and eluted at a flow rate of 0.3 μL/min with the UltiMate 3000 HPLC system (Thermo Fisher Scientific) coupled with the Q Exactive mass spectrometer (Thermo Fisher Scientific), which was set in the data-dependent acquisition mode using Xcalibur 2.2 software.
Zebrafish Studies
Human AURKB coding sequences were ligated into the Rag2/pBKS plasmid (Langenau et al., 2003) through BamHI and ClaI sites to generate the rag2:AURKB construct. rag2:EGFP-Myc alone (Langenau et al., 2003), or in combination with rag2:AURKB, prepared in the microinjection solution [30 ng/μL DNA, 0.5×I-SceI buffer and 0.5 U/μL I-SceI meganuclease (NEB)] was injected into embryos of zebrafish at the one-cell stage of development. AURKB expression was analyzed by immunoblot. Thymus enlargement and T-ALL onset were detected by GFP fluorescence using fluorescent microscopy (Leica).
To generate Tg(rag2:AURKB) stable transgenic line, mosaic F0 fish simultaneously injected with rag2:AURKB and rag2:mCherry constructs in single-cell-stage embryos were bred with the WT fish, and the resultant F1 offsprings were assessed for the presence of transgene by PCR. Stable transgenic Tg(rag2:AURKB;rag2:mCherry) fish were bred with double transgenic fish Tg(rag2:Loxp-dsRED2-Loxp-EGFP-Myc;hsp70:Cre) and subjected to heat-shock treatments as described (Feng et al., 2007). Specifically, the embryos were heat-shocked at 3 dpf for 45 min at 37°C and recovered in a 28°C incubator. Genomic DNA was extracted from the tail fin of adult fish and analyzed by gene-specific PCR to obtain the triple transgenic fish. T-ALL onset and progression were assessed by imaging at both GFP and mCherry channels using fluorescent microscopy. Fish genotyping was performed using gene-specific primers listed in Table S3.
Drug Screening and Drug Combination Analysis
To identify small molecules synergistic with AZD1152, an FDA-approved drug library was purchased from Selleck Chemicals for a high-throughput drug screening. KOPTK1 cells were seeded at a density of 20000 cells per well in 96-well plates. These cells were then treated with AZD1152 (5 nM) alone or in combination with each compound (10 nM) for 24 hr, and cell death was assessed by the CellToxTM-Green Cytotoxicity Assay kit (Promega). Fluorescence readings were collected by automatic microplate reader (Molecular devices). Synergy analysis for AZD1152 drug combinations were performed using the Chou-Talalay method based on the median-effect principle (Chou, 2010). Combination index values were generated using CalcuSyn software (Biosoft).
Cell Line-based Xenograft
CUTLL1 (FBXW7 WT) and Jurkat (FBXW7 Mut) xenografts were carried out as previously described (Hu et al., 2016; Wang et al., 2017). 4-6 week-old NPG mice were irradiated (2 Gray) before tail vein injection of two million cells infected with pWPXLd-Luciferase-GFP lentiviruses. Treatment started eight days post engraftment; AZD1152 (5 mg/kg) was intraperitoneally injected into mice three times a week, and Vincristine (0.2 mg/kg) twice a week for consecutive two weeks. Disease progression and therapeutic response were assessed by in vivo bioimaging (IVIS Lumina II) through injection of D-Luciferin (150 mg/kg).
Patient-derived Xenografts (PDX)
Ten million primary T-ALL#1 cells were injected into each NPG mouse after irradiation (2 Gray). Treatment of T-ALL PDX started eight days post engraftment. AZD1152 (5 mg/kg) was injected into mice twice a week, Vincristine (0.2 mg/kg) once a week for continuing three weeks. Mock-treated mice were euthanized when demonstrating characteristic disease symptoms and becoming moribund. Cells were then isolated from spleens by mechanical disaggregation and red blood cell lysis, and collected from bone marrow by flushing of femurs with Phosphate-buffered saline (PBS). Leukemia burden in vivo was assessed by FACS analysis of human CD45+ cell distribution in bone marrow and spleen cells. Spleen sections were prepared for immunohistochemistry staining. To evaluate therapeutic response, two or three mice in treatment groups were simultaneously sacrificed and assessed for leukemia burden to compare disease progression. Remaining mice in treatment groups were kept to monitor overall survival.
Flow Cytometry Analysis
Cells with GFP fluorescence, stained with indicated antibodies or Annexin-V/PI were resuspended in PBS. Acquisition was performed on an Accuri C6 (BD Biosciences) and live cells were gated based on FSC-A and SSC-A characteristics. Data were analyzed with FlowJo software (TreeStar). Flow cytometry sorting was conducted using a FACS Aria (BD Biosciences).
Immunohistochemistry (IHC)
The IHC analysis was performed in paraffin-embedded sections using specific antibodies against MYC (Santa Cruz Biotechnology), PCNA (Santa Cruz Biotechnology) or cleaved Caspase-3 (ABclonal). These slides were then subjected to horseradish peroxidase-linked secondary antibodies for 1 hr at room temperature. Staining was visualized by the DAB substrate kit (Vector Laboratories). Representative IHC images were captured at ×400 magnification. ImageJ software was used to quantify the staining results.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis were carried out using GraphPad Prism 7. Statistical significance was calculated by unpaired two-tailed Student’s t-test between two groups or by one-way or two-way ANOVA with Tukey’s corrections when comparing three or more groups. Differences were considered significant when p<0.05. Survival in mouse experiments was represented with Kaplan-Meier curves and significance was estimated by log-rank test.
DATA AND CODE AVAILABILITY
The accession number for the RNA-Seq data reported in this paper is deposited in Gene Expression Omnibus (GEO: GSE140739).
Supplementary Material
Highlights.
Aurora B kinase phosphorylates MYC at serine 67 and promotes its protein stability
MYC directly activates AURKB transcription
AURKB and MYC constitute a feedforward circuit promoting T cell leukemogenesis
Targeting AURKB destabilizes MYC and induces apoptosis in FBXW7-active T-ALL cells
Significance.
Aberrant activation of MYC is one of the most common features in human cancers, yet direct targeting of MYC remains unsuccessful. Using T-ALL, a MYC-mediated cancer lacking targeted therapeutics, as a model system, we unravel a regulatory mechanism of MYC involving its serine 67 phosphorylation by AURKB, which promotes MYC protein stability. Treatments with AZD1152, a selective AURKB inhibitor currently being evaluated in clinical trials, result in pronounced MYC degradation and T-ALL cell death. Notably, T-ALL cells expressing wild-type FBXW7, but not loss-of-function mutant, are selectively responsive to AZD1152, suggesting that FBXW7 mutational status may serve as a genetic biomarker predicting drug sensitivity. Our findings provide strong preclinical evidence supporting the trial of AZD1152-based targeted therapeutics in T-ALL patients.
ACKNOWLEDGMENTS
We thank members of the Liu and Qing Laboratories for helpful suggestions, and the Core Facility of Medical Research Institute at Wuhan University for flow sorting and histological analysis. . We also thank Dr Hongling Peng ( the Second Xiangya Hospital of Central South University) for providing transgenic zebrafish lines. This study was supported by grants from National Key R&D Program of China (2017YFA0505600 to G.Q.), National Natural Science Foundation of China (81970152, 81770177 to H.L., 81830084 to G.Q., and 81803003 to M.Y.), National Science Foundation for Distinguished Young Scholar (81725013 to G.Q.), Hubei Provincial Natural Science Fund for Distinguished Young Scholars (2017CFA072 to H.L.), Hubei Provincial Natural Science Fund for Creative Research Groups (2018CFA018 to G.Q.), Medical Science Advancement Program (Basic Medical Sciences) of Wuhan University (TFJC2018005 to G.Q.), H.F. acknowledges grant supports from National Institutes of Health (CA215059) and American Cancer Society (RSG-17-204-01-TBG).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.ccell.2020.01.001.
REFERENCES
- Archambault V, and Carmena M (2012). Polo-like kinase-activating kinases: Aurora A, Aurora B and what else? Cell Cycle 11, 1490–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachireddy P, Bendapudi PK, and Felsher DW (2005). Getting at MYC through RAS. Clin. Cancer Res 11, 4278–4281. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancerdrug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet M, Loosveld M, Montpellier B, Navarro J-M, Quilichini B, Picard, , Di Cristofaro J, Bagnis C, Fossat C, Hernandez L, et al. (2011). Posttranscriptional deregulation of MYC via PTEN constitutes a majoralternative pathway of MYC activation in T-cell acute lymphoblastic leukemia. Blood 117, 6650–6659. [DOI] [PubMed] [Google Scholar]
- Chen H, Liu H, and Qing G (2018). Targeting oncogenic Myc as a strategy for cancer treatment. Signal. Transduct. Target Ther 3, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T-C (2010). Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 70, 440–446. [DOI] [PubMed] [Google Scholar]
- Collins GP, Eyre TA, Linton KM, Radford J, Vallance GD, Soilleux E, and Hatton C (2015). A phase II trial ofAZD1152 in relapsed/refractory diffuse large B-cell lymphoma. Br. J. Haematol 170, 886–890. [DOI] [PubMed] [Google Scholar]
- Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauch D, Rudalska R, Cossa G, Nault JC, Kang TW, Wuestefeld T, Hohmeyer A, Imbeaud S, Yevsa T, Hoenicke L, et al. (2016). A MYC-aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med 22, 744–753. [DOI] [PubMed] [Google Scholar]
- den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, Hoellein A, Kremer M, Graf N, Scheerer M, Hall MA, et al. (2010). Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 116, 1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchook GS, Bastida CC, and Kurzrock R (2015). Aurora kinase inhibitors in oncology clinical trials: current state of the progress. Semin. Oncol 42, 832–848. [DOI] [PubMed] [Google Scholar]
- Farrell AS, and Sears RC (2014). MYC degradation. Cold Spring Harb Perspect. Med 4, a014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Langenau DM, Madge JA, Quinkertz A, Gutierrez A, Neuberg DS, Kanki JP, and Thomas Look A (2007). Heat-shock induction ofT-cell lymphoma/leukaemia in conditional Cre/lox-regulated transgenic zebrafish. Br. J. Haematol 138, 169–175. [DOI] [PubMed] [Google Scholar]
- Gregory MA, Qi Y, and Hann SR (2003). Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J. Biol. Chem 278, 51606–51612. [DOI] [PubMed] [Google Scholar]
- Gustafson WC, Meyerowitz JG, Nekritz EA, Chen J, Benes C, Charron E, Simonds EF, Seeger R, Matthay KK, Hertz NT, et al. (2014). Drugging MYCN through an allosteric transition in aurora kinase A. Cancer Cell 26, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayama S, Daigo Y, Yamabuki T, Hirata D, Kato T, Miyamoto M, Ito T, Tsuchiya E, Kondo S, and Nakamura Y (2007). Phosphorylation and activation of cell division cycle associated 8 by aurora kinase B plays a significant role in human lung carcinogenesis. Cancer Res. 67, 4113–4122. [DOI] [PubMed] [Google Scholar]
- Herold S, Kalb J, BGchel G, Ade CP, Baluapuri A, Xu J, Koster J, Solvie D, Carstensen A, Klotz C, et al. (2019). Recruitment of BRCA1 limits MYCN-driven accumulation of stalled RNA polymerase. Nature 567, 545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz D, Ambesi-Impiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, Xu L, Castillo-Martin M, Llobet-Navas D, Cordon-Cardo C, et al. (2014). A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med 20, 1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Körner R, and Nigg EA (2003). Exploring the functional interactions between aurora B, INCENP, and survivin in mitosis. Mol. Biol. Cell 14, 3325–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AL, Walton ZE, Altman BJ, Stine ZE, and Dang CV (2015). MYC and metabolism on the path to cancer. Semin. Cell Dev. Biol 43, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Su H, Liu C, Wang Z, Huang L, Wang Q, Liu S, Chen S, Zhou J, Li P, et al. (2016). DEPTOR is a direct NOTCH1 target that promotes cell proliferation and survival in T-cell leukemia. Oncogene 36, 1038–1047. [DOI] [PubMed] [Google Scholar]
- Inaba H, Greaves M, and Mullighan CG (2013). Acute lymphoblastic leukaemia. Lancet 381, 1943–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JR, Patrick DR, Dar MM, and Huang PS (2007). Targeted antimitotic therapies: can we improve on tubulin agents? Nat. Rev. Cancer 7, 107–117. [DOI] [PubMed] [Google Scholar]
- Kim YH, Girard L, Giacomini CP, Wang P, Hernandez-Boussard T, Tibshirani R, Minna JD, and Pollack JR (2005). Combined microarray analysisofsmall cell lung cancerrevealsaltered apoptotic balanceand distinct expression signatures of MYC family gene amplification. Oncogene 25, 130–138. [DOI] [PubMed] [Google Scholar]
- King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P, Aranda-Orgilles B, Perez-Garcia A, Shi J, Vakoc C, et al. (2013). The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 153, 1552–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löwenberg B, Muus P, Ossenkoppele G, Rousselot P, Cahn J-Y, Ifrah N, Martinelli G, Amadori S, Berman E, Sonneveld P, et al. (2011). Phase 1/2 study to assess the safety, efficacy, and pharmacokinetics of barasertib (AZD1152) in patients with advanced acute myeloid leukemia. Blood 118, 6030–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenau DM, Traver D, Ferrando AA, Kutok JL, Aster JC, Kanki JP, Lin S, Prochownik E, Trede NS, Zon LI, and Look AT (2003). Myc-induced T cell leukemia in transgenic zebrafish. Science 299, 887–890. [DOI] [PubMed] [Google Scholar]
- Liu H, Chi AW, Arnett KL, Chiang MY, Xu L, Shestova O, Wang H, Li YM, Bhandoola A, Aster JC, et al. (2010). Notch dimerization isrequired for leukemogenesis and T-cell development. Genes Dev. 24, 2395–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, et al. (2014). Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 508, 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown MR, and Bradner JE (2014). Therapeutic strategies to inhibit MYC. Cold Spring Harb. Perspect. Med 4, a014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer N, and Penn LZ (2008). Reflecting on 25 years with MYC. Nat. Rev. Cancer 8, 976–990. [DOI] [PubMed] [Google Scholar]
- Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, Brown-Swigart L, Johnson L, and Evan GI (2008). Distinct thresholds govern Myc’s biological output in vivo. Cancer Cell 14, 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanbakhsh A, Pochon C, Mallavialle A, Amsellem S, Bourhis JH, and Chouaib S (2014). c-Myc regulates expression of NKG2D ligands ULBP1/2/3 in AML and modulates their susceptibility to NK-mediated lysis. Blood 123, 3585–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, et al. (2007). FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J. Exp. Med 204, 1813–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R, Christiansen H, et al. (2009). Stabilization of N-Myc is a critical function ofAurora A in human neuroblastoma. Cancer Cell 15, 67–78. [DOI] [PubMed] [Google Scholar]
- Popov N, Schulein C, Jaenicke LA, and Eilers M (2010). Ubiquitylation of the amino terminus of Myc by SCF(beta-TrCP) antagonizes SCF(Fbw7)-mediated turnover. Nat. Cell Biol 12, 973–981. [DOI] [PubMed] [Google Scholar]
- Richards MW, Burgess SG, Poon E, Carstensen A, Eilers M, Chesler L, and Bayliss R (2016). Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc. Natl. Acad. Sci. USA 113, 13726–13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roderick JE, Tesell J, Shultz LD, Brehm MA, Greiner DL, Harris MH, Silverman LB, Sallan SE, Gutierrez A, Look AT, et al. (2014). c-Myc inhibition preventsleukemia initiation in miceand impairsthegrowth ofrelapsed and induction failure pediatric T-ALL cells. Blood 123, 1040–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Martin M, and Ferrando A (2017). The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 129, 1124–1133. [DOI] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, and Nevins JR (2000). Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 14, 2501–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears RC (2004). The life cycle ofC-myc: from synthesistodegradation. Cell Cycle 3, 1131–1135. [PubMed] [Google Scholar]
- Su H, Hu J, Huang L, Yang Y, Thenoz M, Kuchmiy A, Hu Y, Li P, Feng H, Zhou Y, et al. (2018). SHQ1 regulation of RNA splicing is required for T-lymphoblastic leukemia cell survival. Nat. Commun 9, 4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita M, Koga T, Takayama K, Ijichi K, Yano T, Maehara Y, Nakanishi Y, and Sueishi K (2013). Aurora-B overexpression is correlated with aneuploidy and poor prognosis in non-small cell lung cancer. Lung Cancer 80, 85–90. [DOI] [PubMed] [Google Scholar]
- Tan J, Li Z, Lee PL, Guan P, Aau MY, Lee ST, Feng M, Lim CZ, Lee EY, Wee ZN, et al. (2013). PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov. 3, 1156–1171. [DOI] [PubMed] [Google Scholar]
- Tang A, Gao K, Chu L, Zhang R, Yang J, and Zheng J (2017). Aurora kinases: novel therapy targets in cancers. Oncotarget 8, 23937–23954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, Ferrando A, and Aifantis I (2007). The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J. Exp. Med 204, 1825–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topham C, Tighe A, Ly P, Bennett A, Sloss O, Nelson L, Ridgway RA, Huels D, Littler S, Schandl C, et al. (2015). MYC is a major determinant of mitotic cell fate. Cancer Cell 28, 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vader G, Medema RH, and Lens SMA (2006). The chromosomal passenger complex: guiding Aurora-B through mitosis. J. Cell Biol 173, 833–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez A, Markert EK, and Oltvai ZN (2011). Serine biosynthesis with one carbon catabolism and the glycine cleavage system represents a novel pathway for ATP generation. PLoS One 6, e25881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Hu Y, Xiao D,Wang J, Liu C, Xu Y, Shi X, Jiang P, Huang L, Li P, et al. (2017). Stabilization of Notch1 by the Hsp90 chaperone is crucial for T-cell leukemogenesis. Clin. Cancer Res 23, 3834–3846. [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, and Clurman BE (2004). The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 101, 9085–9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, and Aster JC (2004). Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271. [DOI] [PubMed] [Google Scholar]
- Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, Del Bianco C, Rodriguez CG, Sai H, Tobias J, et al. (2006). c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 20, 2096–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems E, Dedobbeleer M, Digregorio M, Lombard A, Lumapat PN, and Rogister B (2018). The functional diversity of Aurora kinases: a comprehensive review. Cell Div. 13, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao D, Yue M, Su H, Ren P, Jiang J, Li F, Hu Y, Du H, Liu H, and Qing G (2016). Polo-like kinase-1 regulates myc stabilization and activates a feedforward circuit promoting tumor cell survival. Mol. Cell 64, 493–506. [DOI] [PubMed] [Google Scholar]
- Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, and Nakayama KI (2004). Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBOJ. 23, 2116–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Tang X, Guo X, Niikura Y, Kitagawa K, Cui K, Wong STC, Fu L, and Xu B (2011). Aurora-B mediated ATM serine 1403 phosphorylation is required for mitotic ATM activation and the spindle checkpoint. Mol. Cell 44, 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Liu H, Goga A, Kim S, Yuneva M, and Bishop JM (2010). Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc. Natl. Acad. Sci. U S A 107, 13836–13841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro-Ohtani Y, Wang H, Zang C, Arnett KL, Bailis W, Ho Y, Knoechel B, Lanauze C, Louis L, Forsyth KS, et al. (2014). Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc. Natl. Acad. Sci. USA 111, E4946–E4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, et al. (2004). A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol 6, 308–318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-Seq data reported in this paper is deposited in Gene Expression Omnibus (GEO: GSE140739).