

Abstract
The amyloid-based yeast prions are folded in-register parallel beta sheet polymers. Each prion can exist in a wide array of variants, with different biological properties resulting from different self-propagating amyloid conformations. Yeast has several anti-prion systems, acting in normal cells (without protein overexpression or deficiency). Some anti-prion proteins partially block prion formation (Ssb1,2p - ribosome-associated Hsp70s), others cure a large portion of prion variants that arise (Btn2p, Cur1p, Hsp104 - a disaggregase, Siw14p, Upf1,2,3p - nonsense-mediated decay proteins) and others prevent prion-induced pathology (Sis1p - essential cytoplasmic Hsp40). Study of the anti-prion activity of Siw14p, a pyrophosphatase specific for 5-diphosphoinositol-pentakisphosphate (5PP-IP5), led to the discovery that inositol polyphosphates, signal transduction molecules, are involved in [PSI+] prion propagation. Either inositol hexakisphosphate or 5PP-IP4 (or 5PP-IP5) can supply a function that is needed by nearlyall [PSI+] variants. Because yeast prions are informative models for mammalian prion diseases and other amyloidoses, detailed examination of the anti-prion systems, some of which have close mammalian homologs, will be important for development of therapeutic measures.
“Prion” has come to mean “infectious protein” without the need for an essential nucleic acid component. Most yeast prions are based on amyloid forms of normally soluble proteins in which the amyloid form cannot carry out the normal function of the protein, and so a phenotype appears that is similar to mutants in the gene for the prion protein (1, 2)). However, some yeast prions have novel toxicity not seen for the chromosomal gene mutants (3). Amyloid is a filamentous polymer of hundreds to thousands of molecules, largely of a single protein, a sort of linear crystal. Beta sheet dominates amyloid structures, with the beta strands perpendicular to the long axis of the filaments (reviewed in (4)). There are about 10 yeast prions known, most of them based on self-propagating amyloid formation (Table 1). The exception is the [β] prion, which is vacuolar protease B that can proteolytically activate its own inactive precursor (5).
Table 1.
Yeast and fungal prions
| Priori | Priori protein | Normal form function | Ref. |
|---|---|---|---|
| [URE3] | Ure2p | Nitrogen catabolite repression | (1) |
| [PSI+] | Sup35p | Translation termination | (1) |
| [PIN+]/[RNQ+] | Rnq1p | unknown | (83–85) |
| [Het-s] | HET-s | Heterokaryon incompatibility. The prion form carries out the normal function. | (86) |
| [BETA] | Prb1p | Vacuolar protease B. Activated (prion) form needed for sporulation and survival in stationary phase. | (5) |
| [SWI+] | Swi1p | Chromatin remodeling | (87) |
| [OCT+] | Cyc8p | Transcription co-repressor | (88) |
| [MOT+] | Mot3p | Transcription factor | (89) |
| [MOD+] | Mod5p | tRNA isopentenyltransferase | (90) |
The most studied yeast prions, each amyloid-based, are [PSI+], a prion of Sup35p, which is a subunit of the translation termination apparatus; [URE3], a prion of Ure2p, a regulator of nitrogen catabolism; and [PIN+]/[RNQ+], a prion of Rnq1p, a protein of unknown function (Table 1). Each prion protein has a domain that largely determines the prion properties, and roughly comprises the part of the protein that actually forms the beta sheets of the amyloid. Of course, these ‘prion domains’ have functions in the normal form of the protein, so the cell cannot so easily dispense with the risk of prions (see below) by losing these domains.
Yeast prions are non-chromosomal genes, and, like chromosomal genes, can have ‘alleles’ or ‘prion variants’, which are a variety of self-propagating amyloid forms, all amyloids of the same protein sequence, but with differing amyloid conformations (6, 7). These prion variants are each rather stable, implying there must be a mechanism by which protein conformation can be templated. The folded in-register parallel beta sheet architecture that has been shown for the infectious amyloid of the prion domains of Sup35p, Ure2p and Rnq1p (8–11) has led to a model (Fig. 1) explaining the conformational templating of prion variant information (2).
Figure 1.
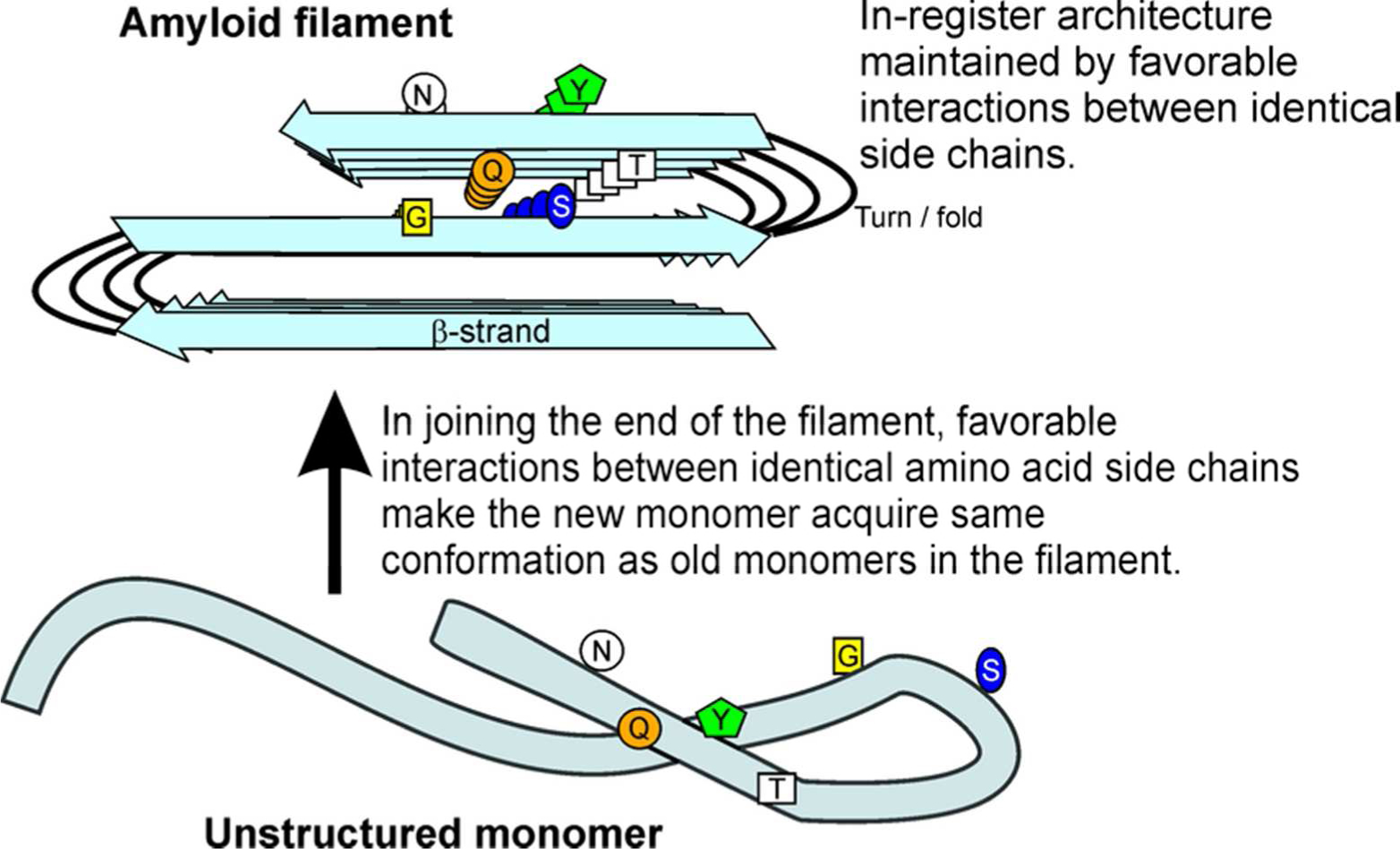
End-on view of an amyloid filament. The folded, parallel in-register β-sheet architecture of infectious amyloids of the prion domains of Sup35p, Ure2p and Rnq1p may explain the ability of prion variants to propagate the amyloid conformation. Prion variants are proposed to differ in the location of the folds of the β-sheet. Favorable interactions among identical amino acid side chains (H-bonding or hydrophobic interactions) require that adjacent molecules be aligned (in-register). This forces molecules joining the ends of the filaments to have their turns (= folds of the sheet) in the same locations as molecules already in the sheet. This constitutes templating of conformation, and can explain how each prion variant is stably propagated (2). Modified from ref. (2).
The classic mammalian prion, the cause of the transmissible spongiform encephalopathies (TSEs, Creutzfeldt-Jakob disease in humans, scrapie in sheep, Mad Cow disease, Chronic wasting disease of deer) is based on amyloid formation by the PrP protein, a cell surface GPI-anchored protein (12). Although the TSEs are uniformly lethal diseases, deletion of the gene encoding PrP produces, at most, a subtle phenotype (13). Thus, the amyloid is not simply inactivating the protein, but is positively toxic.
The common human amyloidoses, including Alzheimer’s disease, Parkinson’s disease, type 2 diabetes and amyotrophic lateral sclerosis, have, increasingly, shown signs of being prions. Animal models of Alzheimer’s disease show infectious properties (14, 15), and patients who succumbed to CJD due to accidental infection with pituitary-derived growth hormone injections often showed postmortem evidence of Alzheimer’s disease at a very early age (16).
Prions [PSI+] and [URE3] are diseases of yeast.
A majority of the variants of [PSI+] or [URE3] arising are severely toxic or lethal ((3), reviewed in (2)). Even the mildest variants of these prions are rare in wild strains (17, 18). Evidently, the loss of translation termination or of proper nitrogen regulation is detrimental, but yeast prion toxicities not due to loss of the normal prion protein’s function are also common among a random collection of variants (3). To prevent these diseases, one expects to find ‘anti-prion’ systems, cellular components that prevent prion formation, cure prions as they arise or prevent their toxicity once they have arisen. Evidently, such systems are not completely effective, as toxic prions do arise and propagate, but we shall see that an array of proteins are devoted to this task.
What is an ‘anti-prion system’?
Overproduction or deficiency of a variety of cellular components, particularly chaperones, can lead to the curing of yeast prions. Examples include overproduced Hsp104 (19), Hsp40s (20, 21), transcription factors and ribosomal protein Rrp0 (21), Sse1p (22), Hsp42 (23) and the ribosome-associated Hsp70s Ssb1,2 (24). Deficiency of Hsp104 (19), Ssa1p (25) or Ssa2p (26), Sis1p (27, 28), Cpr7p (29), Swa2p (30), Fes1p or Sse1p (22) can also cure prions. These studies have revealed many of the requirements for prion propagation, conditions that affect prion stability, and have led to the discovery of anti-prion systems. However, not all such manipulations reflect the action of anti-prion systems, as the artificial amplification or elimination of protein expression may not naturally occur. By ‘anti-prion system’ we mean components that cure prions in a normal cell, a cell in which no proteins are deleted or overproduced. This phenomenon has been shown by allowing prions to arise in a strain lacking the component being tested, and then replacing the missing gene. If prions that arose in its absence are cured by restoring the normal level of the protein, this protein is assumed to be part of an ‘anti-prion system’.
Anti-prion action of Btn2p and Cur1p.
In a screen for proteins whose overexpression cures the [URE3] prion, paralogs Btn2p and Cur1p were detected (31). Overexpressed Btn2p was found to collect the prion aggregates of Ure2p in one place in the cell, a place that coincided with Btn2p itself. It was suggested that Btn2p sequestered the Ure2p prion aggregates in one place so that on cell division, one of the daughter cells would get the lump of Ure2p amyloid and the other would often get no amyloid and so be cured of the prion (31)(Fig. 2). Subsequent work showed that overexpressed Btn2p could also cure an artificial prion (32), and that Btn2p’s aggregate-sequestering abilities were not limited to prion aggregagtes, but worked on a variety of non-prion aggregates as well (32, 33).
Figure 2.
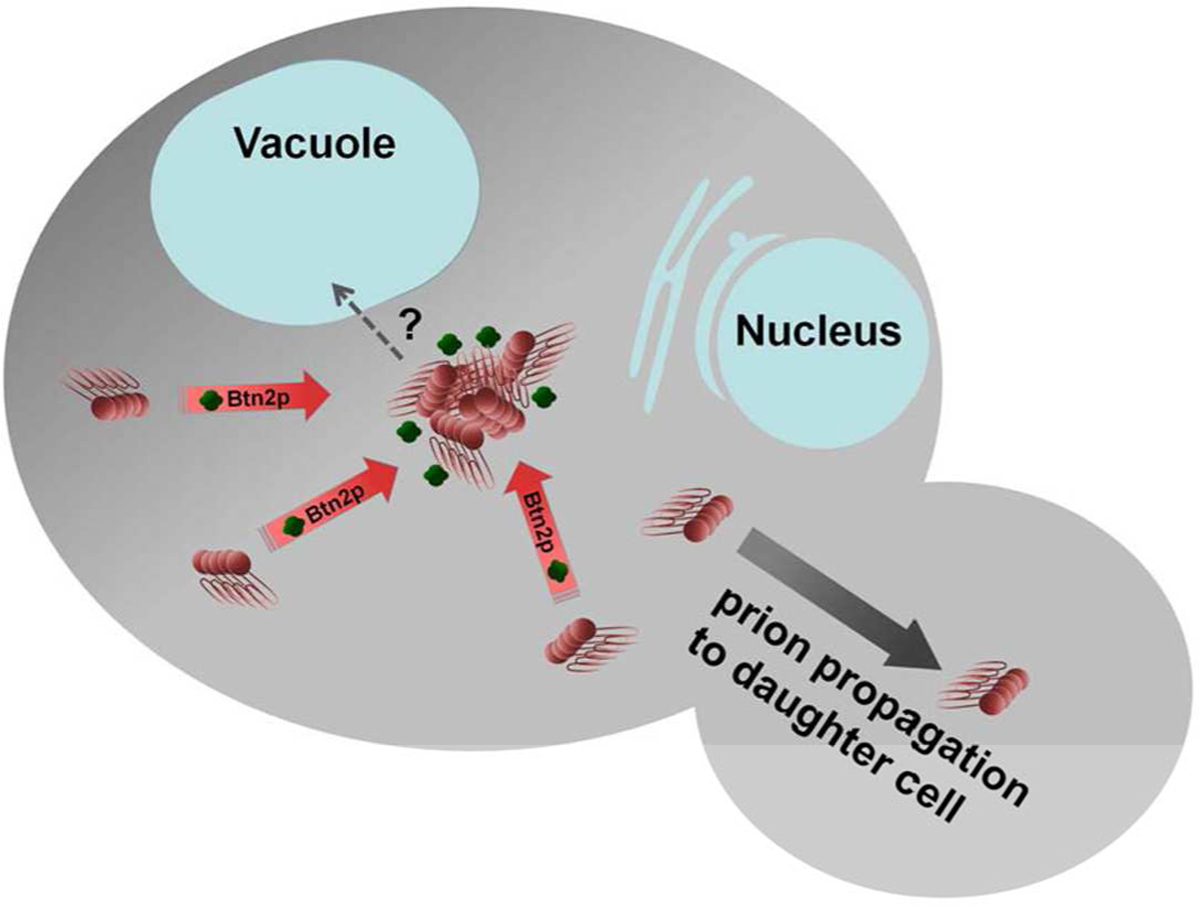
The sequestration model for Btn2p curing of [URE3] (31). The amyloid aggregates of Ure2p are collected at one place in the cell so that on cell division, the chance of one of the daughter cells receiving no aggregate is enhanced. Modified from (31).
Btn2p also acts as a protein-sorting gene, involved in retrieval of specific ‘cargo proteins’ from the late endosomes to the Golgi (34). However the relation of this activity to its role in collecting aggregates is, at this time, obscure.
Cur1p overproduction was as effective as Btn2p in curing [URE3], but no co-localization of Cur1p with Ure2p aggregates could be observed (31). Btn2p was found to be located adjacent to the nucleus (the latter marked by the tRNA:pseudouridine synthase Pus1-GFP), while Cur1p was located inside and throughout the nucleus. Btn2p did not need Cur1p to cure [URE3], nor did Cur1p need Btn2p. However, curing by Btn2p requires Hsp42 (23), a small heat shock protein previously shown to capture non-prion aggregates (35). Hsp42 overproduction also cures [URE3], in a process requiring Cur1p (23). Whether normal levels of Hsp42 cure some [URE3] variants has not yet been reported. Btn2p also collects non-prion aggregates to a non-nuclear site (33). Another group reports Btn2p is in the nucleus, to which it attracts non-prion aggregates (36). Hsp42 also collects non-prion aggregates to a peripheral site (35, 36).
To determine whether normal levels of Btn2p or Cur1p could cure some [URE3] variants, we isolated [URE3]s in a btn2Δ cur1Δ double mutant, and restored normal levels of both proteins by mating with an isogenic wild-type strain. Surprisingly, nearly all of the [URE3] variants were cured in the diploids, and the frequency of spontaneous [URE3] generation is elevated five-fold in the btn2Δ cur1Δ double mutant (23). The few variants not cured by restoring Btn2p and Cur1p were those with a high prion seed/propagon number, consistent with Btn2p’s sequestration mechanism (Fig. 2) (23). Normal levels of either Btn2p or Cur1p could cure these low propagon [URE3] variants, but overexpression was needed to cure variants with a high number of amyloid lumps.
Because overproduction of Sis1p, an Hsp40 chaperone required for propagation of [URE3] and [PIN+] (among others) (27, 28), suppresses Btn2p and Cur1p overproduction curing, and both Btn2p and Cur1p associate with Sis1p in vivo, it was proposed that Btn2p and Cur1p overproduction cure [URE3] by sequestering Sis1p (32, 37). This proposal for Btn2p does not explain the association of Btn2p with Ure2p aggregates in the course of curing (31, 38), the specificity for low-propagon variants of [URE3], and the fact that the very low abundance Btn2p or Cur1p (together or individually) cure many [URE3] variants without overproduction in the face of vastly higher amounts of Sis1p (23). Nor does it account for the failure of overproduced Btn2p or Cur1p to cure [PIN+] (31), whose requirement for Sis1p resembles that of [URE3] (27). Deletion of the nuclear localization signal of Btn2p is reported to prevent its curing [URE3] (32), but other deletions eliminating the same NLS were reported by others not to prevent [URE3] curing (31). Rather, the overproduced Sis1p may be sequestering Btn2p (or Cur1p) and preventing their prion curing action, the reverse of the proposed mechanism. Btn3p is a Btn2p-binding protein that has just this action, inhibiting curing of [URE3] by overproduced Btn2p as well as the protein-sorting actions of Btn2p (38).
Thus, Btn2p and Cur1p are anti-prion components, acting at their normal levels to cure the [URE3] prion. Neither protein cures the [PSI+] prion, even if overproduced (31).
An anti-[PSI+] activity of Hsp104.
Deficiency of the disaggregating chaperone Hsp104 results in loss of all amyloid-based yeast prions (19, 39). Hsp104, working with Hsp70s and Hsp40s (40, 41), cleaves filaments by removing a monomer from the middle of the fiber (42, 43), thereby generating new prion seeds (reviewed by (39)). Overproduction of Hsp104 also cures the [PSI+] prion (19).
The prion-curing activity of Hsp104 is distinct from its prion – propagating activity in several ways. First, the overproduction-curing activity only works on [PSI+], not on any other yeast amyloid-based prions, all of which require Hsp104 for propagation. Second, deletion of or mutations (such as hsp104T160M) in the N-terminal domain of Hsp104 completely eliminate the prion-curing activity without affecting the [PSI+]-propagation activity of the protein (42). Third, the overproduction curing activity requires the activity of the co-chaperone Sti1p (44, 45), a TPR repeat containing protein that interacts with the EEVD or DDLD C-terminal sites on the Hsp70, Hsp90 or Hsp104 chaperones, but Sti1p is not needed for [PSI+] propagation (46). Partial inhibition of Hsp90 by radicicol or mutation also block curing of [PSI+] by overproduction of Hsp104, but did not affect propagation of [PSI+] (44).
Using an approach similar to that developed to study Btn2p and Cur1p curing, [PSI+] variants were isolated in an hsp104T160M host and transferred to isogenic wild type or hsp104T160M strains by cytoduction (cytoplasmic mixing). Over half of the [PSI+] variants were efficiently transferred to the hsp104T160M recipient and propagated stably, but were quickly lost in a wild type host (47). Detailed analysis showed that normal levels of wild type Hsp104 cured a majority of [PSI+] variants arising in the hsp104T160M mutant (47). Like Hsp104 overproduction curing, curing by normal levels of the protein required Sti1p and Hsp90 activity. The frequency of [PSI+] arising in the hsp104T160M mutant was ~13 fold higher than in the isogenic wild type strain (47). Remarkably, Hsp104, whose filament cleaving activity is indispensable for [PSI+] propagation, removes most of the [PSI+] prions arising in a normal cell by a different activity of the same protein.
Siw14p and inositol polyphosphate control of [PSI+] prion propagation.
A general screen was devised that did not depend on overproduction curing, but could detect components which at their normal levels could cure some [PSI+] variant (48). This screen detected SIW14, and detailed analysis showed that about half of [PSI+] variants arising in an siw14Δ strain were cured by replacing the normal gene, expressed from its own promoter. Thus, SIW14 qualifies as an anti-prion gene, which in normal cells cures a portion of [PSI+] variants arising (48).
SIW14 encodes a pyrophosphatase specifically attacking 5- pyrophosphate inositol pentakisphosphate (5PP-IP5) (49). The inositol polyphosphates (see Fig.3) are important signalling molecules, affecting polyphosphate accumulation, vesicle trafficking, telomere shortening, DNA repair, phosphatase regulation, response to certain stress conditions, and other functions (50, 51). In siw14Δ mutants, the amount of 5PP-IP5 is substantially elevated (49), suggesting that elevation of this and/or other inositol polyphosphates is favorable for [PSI+] propagation. Confirming this hypothesis, it was found that loss of Arg82p, encoding the inositol multikinase that phosphorylates the 6 and 3 positions (52, 53), was needed for propagation of all of 24 random [PSI+] variants tested (48). Detailed examination of various mutants blocked in the inositol polyphosphate pathway (Fig. 3) showed that IP6, 5PP-IP4 and presumably 5PP-IP5 are each sufficient to support [PSI+] propagation (48). Evidence suggesting that 1PP-IP5 inhibits [PSI+] propagation was also reported. The loss of [PSI+] in arg82Δ strains was not abrogated by a mutation of Hsp104 eliminating its [PSI+]-curing activity, implying that these are two distinct systems (48). [URE3] was also not affected by arg82Δ.
Figure 3.
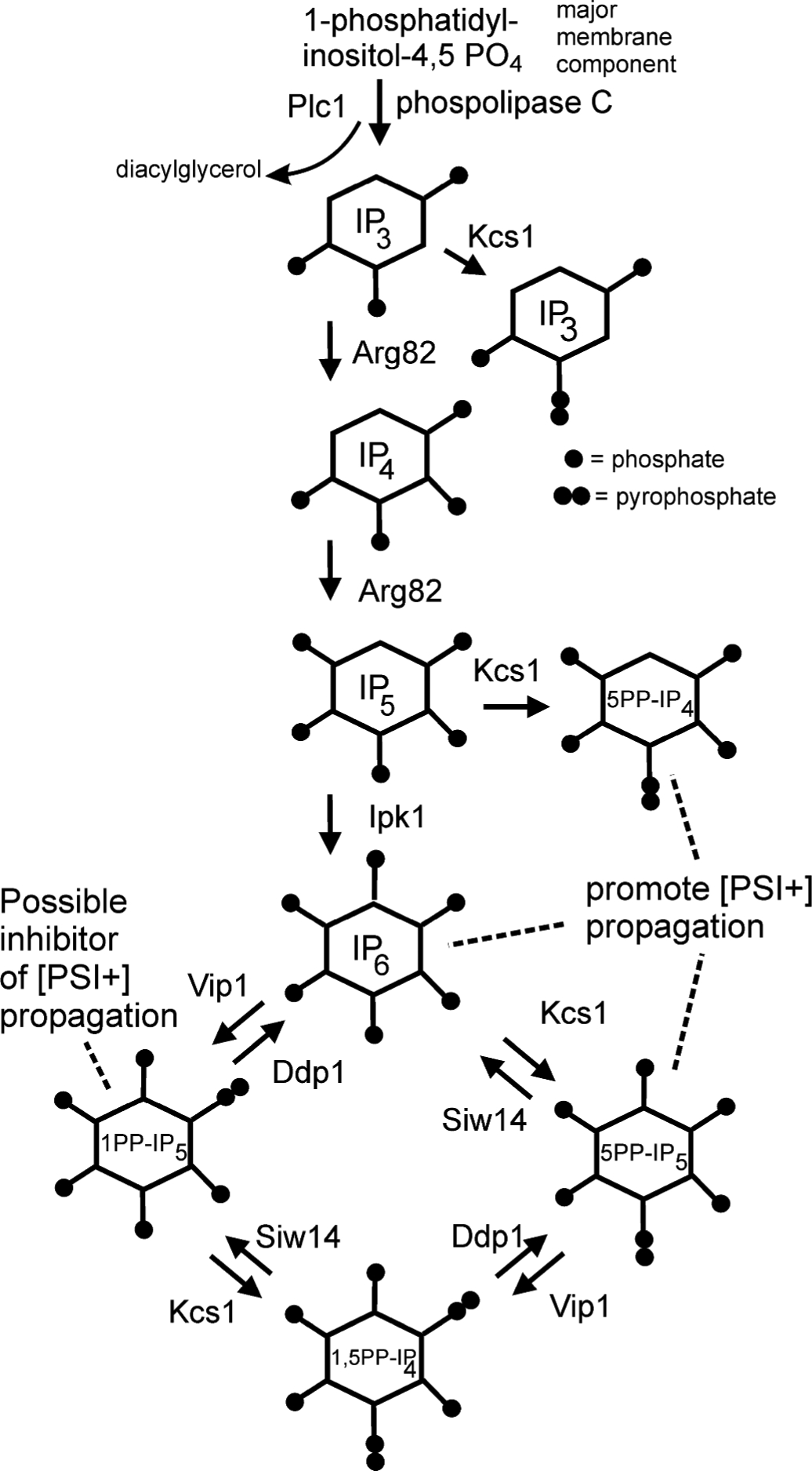
Pathways of biosynthesis and degradation of the soluble inositol poly-/pyro-phosphates (49–51). Myo-inositol is an isomer of cyclohexane-1,2,3,4,5,6 hexol. Some species help [PSI+] propagate, while one other species may block [PSI+] propagation (48). Modified from (48).
The “environmental stress response” is a transcriptional program induced by exposure to heat, oxidation or hyperosmotic conditions, and requiring inositol pyrophosphates (54). However, this system is inoperative in kcs1Δ vip1Δ double mutants, but [PSI+] propagates well in such strains. Moreover, the requirement for inositol polyphosphates for [PSI+] propagation was observed without any of the known stress conditions that induce this transcriptional response (48).
The mechanism by which inositol polyphosphates control [PSI+] prion propagation remains unclear at this time. Possible targets identified as binding specifically to affinity columns for IP6 and 5PP-IP5 included Ssb1p, Ssb2p, Hsp26 and Sse1p (55). While each of these proteins are known to affect [PSI+], there is as yet no clear evidence for a role in inositol polyphosphate effect.
Nonsense-mediated mRNA decay proteins cure [PSI+] prions arising in their absence.
The same screen that found the inositol pyrophosphate cleaving pyrophosphatase had an anti – prion action, selected the UPF genes, encoding components of the nonsense-mediated mRNA decay pathway (NMD) (56). Upf1p, Upf2p and Upf3p form a complex that binds to Sup35p and Sup45p, and screens mRNAs during translation, inducing rapid degradation of those whose termination codon is located early in the open reading frame (57, 58). The upf mutants show increased frequencies of spontaneous and induced [PSI+] formation, and nearly all of the [PSI+] variants arising are cured by merely restoring normal levels of these proteins (56). A [PSI+] variant isolated in a upf1Δ strain (called [PSI+u1s] for Upf1p sensitive) is stable in a upf2Δ or upf3Δ strain, suggesting that it is the Upf1-2-3 complex that is important here. Detailed mutation analysis of UPF1 and UPF2 show that there is no clear correlation of ability to cure [PSI+u1s] and either NMD activity or translation termination efficiency, or any of the ATPase, helicase or other activities of the encoded proteins, but that the ability of the proteins to interact with Sup35p and to form the Upf1-2-3 complex is crucial (56).
Upf1p is known to associate with Sup35p in extracts of wild-type or [PSI+] strains (59), and using Upf1-RFP and Sup35-GFP, the association was confirmed in vivo (56). The prion aggregates appear to deplete the rest of the cytoplasm of Upf1p, suggesting that some of the nonsense-suppression phenotype of [PSI+] cells is a result of preventing NMD and the known nonsense-suppression effect of Upf protein deficiency. In addition, purified Upf1p at decinormal concentrations inhibited amyloid formation by Sup35p in vitro, but had no effect on amyloid formation by Ure2p at ten-fold levels (56).
The normal direct binding of Upf monomers and the Upf1-2-3 complex to Sup35p may compete with the amyloid filaments for Sup35p monomers sufficiently well that many prion variants fail to propagate. Alternatively, the binding of the Upf1-2-3 complex to the amyloid filaments may block the fiber growth points sufficiently to allow other cellular systems to dismantle the prions (56) (Fig. 4). The Upf proteins are evolved primarily to carry out nonsense-mediated mRNA decay, but their clear anti-prion activity, presumably specific for [PSI+], may reflect a general notion, namely, that normal protein-protein interactions (between Upfs and Sup35p) can be expected to inhibit (or even reverse) the pathologic protein-protein interactions between Sup35p monomers in forming amyloid. Seeking ways to strengthen these normal interactions may provide an approach to treatment of amyloid/prion diseases.
Figure 4.
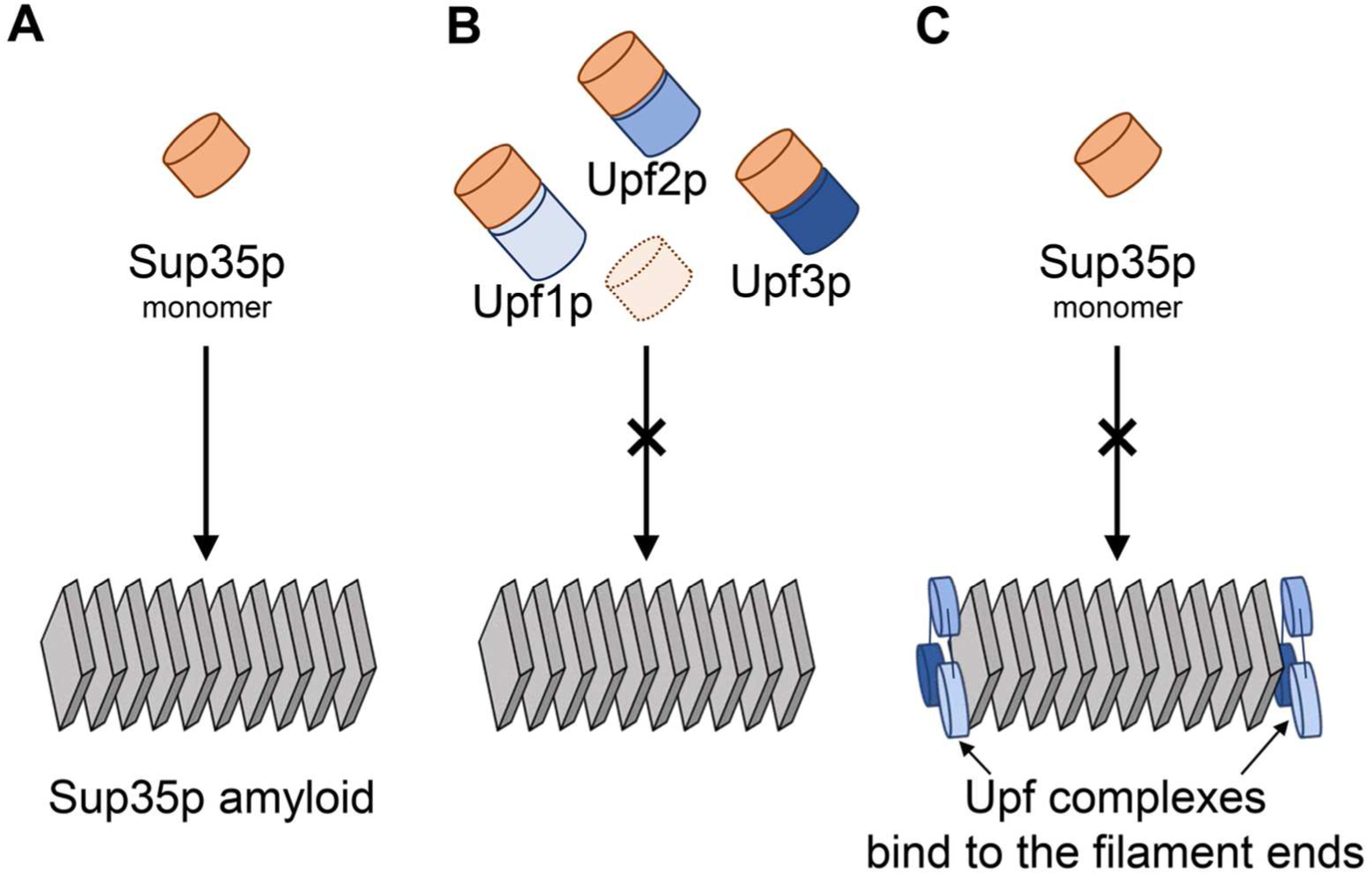
Two models for Upf proteins curing of [PSI+u1s] variants: A. In cells missing a Upf protein, Sup35p monomers add to the ends of amyloid filaments. B. Upf proteins and the Upf1,2,3 complex compete with amyloid filaments for Sup35p monomers slowing filament growth. B. The Upf1,2,3 complex binds to the ends/growing points of amyloid filaments of Sup35p blocking addition of Sup35 monomers (56).
Like the Upf effected discussed above, the elevated frequency of [PSI+] generation induced by overproduction of Sup35p (60) is prevented by overproduction of the other subunit of the translation termination factor, Sup45p (61). However, it is not clear that Sup35p is ever naturally overexpressed, and it would be difficult to test whether normal levels of Sup45p can cure any [PSI+] prions because Sup45 is essential.
Ssb chaperones of the Hsp70 family inhibit [PSI+] prion generation.
Ssb1p and Ssb2p are paralogous ribosome-associated chaperones that assist the proper folding of nascent proteins (62, 63). Deletion of SSB1 and SSB2, (or ZUO1 or SSZ1, other components of the ribosome-associated complex) results in a ten-fold increase in the frequency with which [PSI+] arises spontaneously, suggesting that Ssb1p and Ssb2p play a role for proteins analogous to DNA repair proteins (64–67). Restoration of Ssb proteins to a ssb1 ssb2 double mutant does not cure the [PSI+]s formed in their absence, unlike other systems discussed above. However, overexpression of Ssb1p can cure [PSI+] (68). The Ssb’s apparently act at normal levels to prevent the generation of [PSI+], presumably by their normal role in facilitating proper folding of nascent proteins.
Do proteases inhibit [PSI+] prion generation?
It has recently been reported that mutants lacking protease B (or protease A, needed for protease B maturation) have 2–3 fold higher rates of [PSI+] generation (69). This effect is correlated with the appearance in extracts of wild type strains of a shortened form of Sup35p, lacking the first 38 residues of the prion domain. This could be viewed as an anti-prion system. However, it remains to be shown that the Sup35p cleavage happens inside the cells, and that the small difference in [PSI+] generation is not due to a chance difference in [PIN+] variants between the strains (70).
Sis1p chaperone of the Hsp40 family prevents lethality of otherwise mild [PSI+] variants.
Over half of the [PSI+] variants arising in a wild type cell are toxic or lethal, as discussed below, but many do not have any obvious effect on growth (‘mild’ variants) (3). Sis1p is an abundant essential Hsp40 that has been shown to be more required for propagation of [URE3], [PIN+], [PSI+] and [SWI+] than it is for cell growth (27, 28). Sis1p has a role in Hsp104-overproduction curing of [PSI+], but it also is important in preventing the toxicity of [PSI+] variants that are mild in wild type strains (71). The C-terminal domain of Sis1p is not essential for cell growth, or for the propagation of [PSI+], but its deletion makes a ‘mild’ [PSI+] variant (of strong phenotype but not very pathogenic in a wild-type host) be lethal (71). The experiments clearly showed that the prion was not lost in the host with deleted Sis1p C-terminal domain, but when [PSI+] was present, the cells could not grow. The mechanism of the toxicity is not yet clear.
Multiple anti-prion systems are (another) sign that yeast prions are diseases.
There have been several reports of special growth conditions under which yeast prions, particularly [PSI+], were advantageous (18, 72, 73), but these have not been reproducible (2, 73, 74). However, even if a reproducible advantage of some prion were shown, it would be essential to show that the yeast ecological niche included the corresponding growth condition to an extent that it would justify the risks of developing a lethal yeast prion. Moreover, the reported tests considered only the mildest variants of these prions, not the more common toxic or lethal variants. The contention that yeast prions contribute to evolution of yeast by enhancing the diversity of phenotypes is not tenable if none of the phenotypes are reproducibly advantageous.
Sup35p is an essential protein and, suspecting that some [PSI+] variants would so efficiently convert the protein to amyloid that lethality would ensue, [PSI+] variants were isolated in a strain expressing a minimal amount of the essential part of the molecule, Sup35C, lacking the prion domain, from a counter-selectable plasmid (3). In fact over half of the [PSI+] isolates grew extremely slowly on loss of the source of Sup35C, and some were lethal (3). In a strain in which deletion of URE2 resulted in no slowing of growth, a majority of [URE3] variants were extremely slow growing, with loss of the prion relieving the growth defect. These are toxic [URE3]s, harmful because of damage done by the Ure2p amyloid rather than by the absence of Ure2p (3). It is likely that similarly toxic [PSI+] variants or lethal [URE3]s exist, but that the right permissive condition has not yet been devised.
The rules of meiosis serve evolution of organisms because they demand that an allele become more common only because it benefits the host, improving its survival or reproductive ability. Infectious agents violate these rules, and can become widespread even if they are lethal. Chronic wasting disease, the uniformly lethal prion disease of elk and deer is prevalent in many areas of the west because it is infectious. If an infectious element were beneficial, it would spread more rapidly because selective advantage and infectivity would be working in the same direction instead of in opposition (17). Indeed, [URE3] and [PSI+] are rare in wild strains, showing that even the most advantageous [PSI+] or [URE3] variants are not advantageous at all (17).
Prion-forming ability of Ure2p and Sup35p is not conserved, but is sporadically distributed among species (75, 76), suggesting it is an unavoidable rare side-effect of maintaining the known functions of the prion domains of these proteins (77–79). Yeast cells show a stress reaction on acquiring even a mild [PSI+] or [URE3] prion (25, 80), suggesting the cells do not consider these prions an advantage. The disadvantages of acquiring the [URE3] or [PSI+] prion are manifested, in part, by the more rapid evolution of the prion domains of Ure2p and Sup35p than the rest of the molecule, producing barriers to transmission of the prions, even within cerevisiae in the case of [PSI+] (81, 82).
All these facts argue that the known [PSI+] and [URE3] variants are detrimental to cells. The existence of multiple anti-prion systems reinforces this argument by bringing testimony from the cell itself. Arguments based on the advantages for evolution of more diversity of phenotype ignore the fact that there is no shortage of diversity among yeast or any other organism. Chromosomal DNA is constantly under assault from without (cosmic rays, X-rays, UV light, etc.) and from within (transposons, spontaneous C to U change, chemical mutagens, polymerase errors, etc.), and the myriad of repair systems are inevitably imperfect. Even without new mutations, most populations already contain a vast array of alleles that are constantly being reshuffled by recombination, providing new combinations for natural selection to act on.
The Ssb proteins lower [PSI+] formation by about 10-fold by affecting generation, Hsp104 by about 13-fold by curing, Siw14p by about 2-fold by curing, and the Upf proteins about 5 to 10-fold by curing. If these systems are working independently, the true frequency of [PSI+] formation must be dramatically higher than is apparent in a wild type strain. Further, this work reveals a scope of possible prion variants much wider than is usually met with. Looking for curing in normal cells identifies systems that are working all the time. Of course, Hsp104, Btn2p, and Cur1p were found by overproduction curing screens, and later proven to work without overproduction, but Siw14p and the Upf’s cannot be found this way as their overproduction does not cure the usual [PSI+] variants. It is possible that other components whose overproduction cures yeast prions will be found to be anti-prion elements in the sense used here. It is further noteworthy that the extensive studies of [PSI+] and [URE3] have revealed mostly non-overlapping sets of proteins involved in their propagation and elimination. It would doubtless be useful if a similar level of effort were applied to other yeast prions.
With the increasing recognition of the close relation between the human prion diseases, and the traditional amyloidoses, the characterization of anti-prion systems is of great importance, and may lead to applications in therapy. Just as we harness the cellular and humoral and RNAi, etc. systems to prevent human infections, we should try to harness the natural anti-prion / anti-amyloid systems to combat human prions and amyloidoses. One important distinction between the anti-viral/anti-bacterial systems and anti-prion/anti-amyloid systems is that the former deal with an external invader, while the latterdeal with a truly endogenous danger. Even infections with prions of external origin are using the same potential for amyloid formation that is the basis of the spontaneous (endogenous) prion formation cases. The elucidation of the anti-prion systems has proceeded largely through studies in yeast, but remains in an early stage of understanding.
Table 2.
Anti-prion systems in yeast
| Anti-prion protein | Target priori | Action | Mechanism | Ref. |
|---|---|---|---|---|
| Btn2p | [URE3] | curing | Sequester amyloid | (23, 31) |
| Cur1p | [URE3] | curing | unkown | (23, 31) |
| Ssb1,2p | [PSI+] | block generation | assist proper folding | (64, 91) |
| Sis1p | [PSI+] | block lethality | unknown | (71) |
| Hsp104p | [PSI+] | curing | controversial | (47) |
| Siw14p | [PSI+] | curing | lowering inositol 5-pyrophosphates | (48) |
| Upf1,2,3p | [PSI+] | curing | binding Sup35p or blocking amyloid elongation | (56) |
Acknowledgements:
This work was supported by the Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health.
References:
- 1.Wickner RB (1994) [URE3] as an altered URE2 protein: evidence for a prion analog in S. cerevisiae, Science 264, 566–569. [DOI] [PubMed] [Google Scholar]
- 2.Wickner RB, Shewmaker F, Bateman DA, Edskes HE, Gorkovskiy A, Dayani Y, and Bezsonov EE (2015) Yeast prions: structure, biology and prion-handling systems, Microbiol. Mol. Biol. Rev 79, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGlinchey R, Kryndushkin D, and Wickner RB (2011) Suicidal [PSI+] is a lethal yeast prion, Proc. Natl. Acad. Sci. USA 108, 5337–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tycko R (2014) Physical and structural basis for polymorphism in amyloid fibrils, Protein Sci. 23, 1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts BT, and Wickner RB (2003) A class of prions that propagate via covalent autoactivation, Genes Dev. 17, 2083–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, and Liebman SW (1996) Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae, Genetics 144, 1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tanaka M, Chien P, Naber N, Cooke R, and Weissman JS (2004) Conformational variations in an infectious protein determine prion strain differences, Nature 428, 323–328. [DOI] [PubMed] [Google Scholar]
- 8.Shewmaker F, Wickner RB, and Tycko R (2006) Amyloid of the prion domain of Sup35p has an in-register parallel β-sheet structure, Proc. Natl. Acad. Sci. USA 103, 19754–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baxa U, Wickner RB, Steven AC, Anderson D, Marekov L, Yau W-M, and Tycko R (2007) Characterization of β-sheet structure in Ure2p1–89 yeast prion fibrils by solid state nuclear magnetic resonance, Biochemistry 46, 13149–13162. [DOI] [PubMed] [Google Scholar]
- 10.Wickner RB, Dyda F, and Tycko R (2008) Amyloid of Rnq1p, the basis of the [PIN+] prion, has a parallel in-register β-sheet structure, Proc Natl Acad Sci U S A 105, 2403–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorkovskiy A, Thurber KR, Tycko R, and Wickner RB (2014) Locating folds of the in- register parallel β-sheet of the Sup35p prion domain infectious amyloid, Proc. Natl. Acad. Sci. USA 111, E4615–E4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collinge J (2016) Mammalian prions and their wider relevance in neurodegenerative diseases, Nature 539, 217–226. [DOI] [PubMed] [Google Scholar]
- 13.Wulf MA, Senatore A, and Aguzzi A (2017) The biological function of the cellular prion protein: an update, BMC Biol. 15, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jucker M, and Walker LC (2013) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases, Nature 501, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watts JC, Condello C, Stohr J, Oehler A, Lee J, DeArmond SJ, Lannfelt L, Ingelsson M, Giles K, and Prusiner SB (2014) Serial propagation of distinct strains of Aβ prions from Alzheimer’s disease patients, Proc. Natl. Acad. Sci. USA 111, 10323–10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaunmuktane Z, Mead S, Ellis M, Wadsworth JD, Nicoll AJ, Kenny J, Launchbury F, Linehan J, Richard-Loendt A, Walker AS, Rudge P, Collinge J, and Brandner S (2015) Evidence for human transmission of amyloid- β pathology and cerebran amyloid angiopathy, Nature 525, 247–250. [DOI] [PubMed] [Google Scholar]
- 17.Nakayashiki T, Kurtzman CP, Edskes HK, and Wickner RB (2005) Yeast prions [URE3] and [PSI+] are diseases, Proc Natl Acad Sci U S A 102, 10575–10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halfmann R, Jarosz DF, Jones SK, Chang A, Lancster AK, and Lindquist S (2012) Prions are a common mechanism for phenotypic inheritance in wild yeasts, Nature 482, 363–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chernoff YO, Lindquist SL, Ono B-I, Inge-Vechtomov SG, and Liebman SW (1995) Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+], Science 268, 880–884. [DOI] [PubMed] [Google Scholar]
- 20.Moriyama H, Edskes HK, and Wickner RB (2000) [URE3] prion propagation in Saccharomyces cerevisiae: requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p, Mol. Cell. Biol 20, 8916–8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kryndushkin D, Smirnov VN, Ter-Avanesyan MD, and Kushnirov VV (2002) Increased expression of Hsp40 chaperones, transcriptional factors, and ribosomal protein Rpp0 can cure yeast prions, J. Biol. Chem 277, 23702–23708. [DOI] [PubMed] [Google Scholar]
- 22.Kryndushkin D, and Wickner RB (2007) Nucleotide exchange factors for Hsp70s are required for [URE3] prion propagation in Saccharomyces cerevisiae, Mol. Biol. Cell 18, 2149–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wickner RB, Beszonov E, and Bateman DA (2014) Normal levels of the antiprion proteins Btn2 and Cur1 cure most newly formed [URE3] prion variants, Proc. Natl. Acad. Sci. USA 111, E2711–E2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chacinska A, Szczesniak B, Kochneva-Pervukhova NV, Kushnirov VV, Ter-Avanesyan MD, and Boguta M (2001) Ssb1 chaperone is a [PSI+] prion-curing factor, Curr Genet 39, 62–67. [DOI] [PubMed] [Google Scholar]
- 25.Jung G, Jones G, Wegrzyn RD, and Masison DC (2000) A role for cytosolic Hsp70 in yeast [PSI+] prion propagation and [PSI+] as a cellular stress, Genetics 156, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts BT, Moriyama H, and Wickner RB (2004) [URE3] prion propagation is abolished by a mutation of the primary cytosolic Hsp70 of budding yeast, Yeast 21, 107–117. [DOI] [PubMed] [Google Scholar]
- 27.Higurashi T, Hines JK, Sahi C, Aron R, and Craig EA (2008) Specificity of the J-protein Sis1 in the propagation of 3 yeast prions, Proc. Natl. Acad. Sci. USA 105, 16596–16601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hines JK, Li X, Du Z, Higurashi T, Li L, and Craig EA (2011) [SWI], the prion formed by the chromatin remodeling factor Swi1, is highly sensitive to alterations in Hsp70 chaperone system activity, PLOS Genet. 7, e1001309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar N, Gaur D, Gupta A, Puri A, and Sharma D (2015) Hsp90-associated immunophilin homolog Cpr7 is required for the mitotic stability of [URE3] prion in Saccharomyces cerevisiae, PLOS Genet. 11, e1005567. doi:1005510.1001371/journal.pgen.1005567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Troisi EM, Rockman ME, Nguyen PP, Oliver EE, and Hines JK (2015) Swa2, the yeast homolog of mammalian auxilin, is specifically required for the propagation of the prion variant [URE3–1], Mol. Microbiol 97, 926–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kryndushkin D, Shewmaker F, and Wickner RB (2008) Curing of the [URE3] prion by Btn2p, a Batten disease-related protein, EMBO J. 27, 2725–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malinovska L, Kroschwald S, Munder MC, Richter D, and Alberti S (2012) Molecular chaperones and stress-inducible protein-sorting factors coordinate the spaciotemporal distribution of protein aggregates, Mol. Biol. Cell 23, 3041–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kryndushkin D, Ihrke G, Piermartiri TC, and Shewmaker F (2012) A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity, Mol. Microbiol 86, 1531–1547. [DOI] [PubMed] [Google Scholar]
- 34.Kama R, Robinson M, and Gerst JE (2007) Btn2, a Hook1 ortholog and potential Batten disease-related protein, mediates late endosome-Golgi protein sorting in yeast, Mol. Cell. Biol 27, 605–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Specht S, Miller SBM, Mogk A, and Bukau B (2011) Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae, J. Cell. Biol 195, 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller SB, Ho CT, Winkler J, Khokhrina M, Neuner A, Mohamed MY, Guilbride DL, Richter K, Lisby M, Scheibel E, Mogk A, and Bukau B (2015) Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition, EMBO J 34, 778–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barbitoff YA, Matveenko AG, Moskalnko SE, Zemlyanko OM, Newnam GP, Patel A, Chernova TA, Chernoff YO, and Zhouravleva GA (2017) To CURe or not to CURe? Differential effects of the chaperone sorting factor Cur1 on yeast prions are mediated by the chaperone Sis1, Mol. Microbiol 105, 242–257. [DOI] [PubMed] [Google Scholar]
- 38.Kanneganti V, Kama R, and Gerst JE (2011) Btn3 is a negative regulator of Btn2-mediated endosomal protein trafficking and prion curing in yeast, Mol. Biol. Cell 22, 1648–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liebman SW, and Chernoff YO (2012) Prions in yeast, Genetics 191, 1041–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glover JR, and Lindquist S (1998) Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins, Cell 94, 73–82. [DOI] [PubMed] [Google Scholar]
- 41.Reidy M, and Masison DC (2012) Prokaryotic chaperones support yeast prions and thermotolerance and define disaggregation machinery interactions, Genetics 192, 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hung GC, and Masison DC (2006) N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression, Genetics 173, 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tessarz P, Mogk A, and Bukau B (2008) Substrate threading through the central pore of the Hsp104 chaperone as a common mechanism for protein disaggregation and prion propagation, Mol. Microbiol 68, 87–97. [DOI] [PubMed] [Google Scholar]
- 44.Reidy M, and Masison DC (2010) Sti1 regulation of Hsp70 and Hsp90 is critical for curing of Saccharomyces cerevisiae [PSI+] prions by Hsp104, Mol. Cell. Biol 30, 3542–3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moosavi B, Wongwigkam J, and Tuite MF (2010) Hsp70/Hsp90 co-chaperones are required for efficient Hsp104-mediated elimination of the yeast [PSI+] prion but not for prion propagation, Yeast 27, 167–179. [DOI] [PubMed] [Google Scholar]
- 46.Jones G, Song Y, Chung S, and Masison DC (2004) Propagation of yeast [PSI+] prion impaired by factors that regulate Hsp70 substrate binding, Mol Cell Biol 24, 3928–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gorkovskiy A, Reidy M, Masison DC, and Wickner RB (2017) Hsp104 at normal levels cures many [PSI+] variants in a process promoted by Sti1p, Hsp90 and Sis1p, Proc. Natl. Acad. Sci. USA 114, E4193–E4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wickner RB, Kelly AC, Bezsonov EE, and Edskes HE (2017) Prion propagation is controlled by inositol polyphosphates, Proc. Natl. Acad. Sci. USA 114, E8402–E8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steidle EA, Chong LS, Wu M, Crooke E, Fiedler D, Resnick AC, and Rolfes RJ (2016) A novel inositol pyrophosphate phosphatase in Saccharomyces cerevisiae: Siw14 protein selectively cleaves the β-phosphate from 5-diphosphoinositol pentakisphosphate (5PP-IP5), J. Biol. Chem 291, 6772–6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shears SB (2017) Initimate connections: inositol pyrophosphates at the interface of metabolic regulation and cell signaling, J. Cell. Physiol DOI: 10.1002/jcp.26017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hatch AJ, Odom AR, and York JD (2017) Inositol phosphate multikinase dependent transcriptional control, Adv. Biol. Regul 64, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saiardi A, Erdjument-Bromage H, Snowman AM, Tempst P, and Snyder SH (1999) Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases, Curr. Biol 9, 1323–1326. [DOI] [PubMed] [Google Scholar]
- 53.York JD, Odom AR, Murphy R, Ives EB, and Wente SR (1999) A phospholipase Cdependent inositol polyphosphate kinase pathway required for efficient messenger RNA export, Science 285, 96–100. [DOI] [PubMed] [Google Scholar]
- 54.Worley J, Luo X, and Capaldi AP (2013) Inositol pyrophosphates regulate cell growth and the environmental stress response by activating the HDAC Rpd3L, Cell Reports 3, 1476–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu M, Chong LS, Perlman DH, Resnick AC, and Fiedler D (2016) Inositol polyphosphates intersect with signaling and metabolic networks via two distinct mechanisms, Proc. Natl. Acad. Sci. USA 113, E6757–E6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Son M, and Wickner RB (in press) Yeast prion [PSI+] is under surveillance by nonsense-mediated mRNA decay factors, Proc. Natl. Acad. Sci. USA [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He F, and Jacobson A (2015) Nonsense-mediated mRNA decay: degradation of defective transcripts is only part of the story, Ann, Rev. Genet 49, 339–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilusz CJ, Wormington M, and Peltz SW (2001) The cap-to-tail guide to mRNA turnover, Nat. Rev. Mol. Cell Biol 2, 237–246. [DOI] [PubMed] [Google Scholar]
- 59.Czaplinski K, Ruiz-Echevarria MJ, Paushkin SV, Han X, Weng Y, Perlick HA, Dietz HC, Ter-Avanesyan MD, and Peltz SW (1998) The surveillance complex interacts with the translation release factors to enhance termination and degrade aberrant mRNAs, Genes Dev. 12, 1665–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chernoff YO, Derkach IL, and Inge-Vechtomov SG (1993) Multicopy SUP35 gene induces de-novo appearance of psi-like factors in the yeast Saccharomyces cerevisiae, Curr. Genet 24, 268–270. [DOI] [PubMed] [Google Scholar]
- 61.Derkatch IL, Bradley ME, and Liebman SW (1998) Overexpression of the SUP45 gene encoding a Sup35p-binding protein inhibits the induction of the de novo appearance of the [PSI+] prion, Proc Natl Acad Sci U S A 95, 2400–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nelson RJ, Ziegilhoffer T, Nicolet C, Werner-Washburne M, and Craig EA (1992) The translation machinery and 70 kDal heat shock protein cooperate in protein synthesis, Cell 71, 97–105. [DOI] [PubMed] [Google Scholar]
- 63.Pfund C, Lopez-Hoyo N, Ziegelhoffer T, Schilke BA, Lopez-Buesa P, Walter WA, Wiedmann M, and Craig EA (1998) The molecular chaperone Ssb from Saccharomyces cerevisiae is a component of the ribosome-nascent chain complex, Embo J 17, 3981–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chernoff YO, Newnam GP, Kumar J, Allen K, and Zink AD (1999) Evidence for a protein mutator in yeast: role of the Hsp70-related chaperone Ssb in formation, stability and toxicity of the [PSI+] prion, Mol. Cell. Biol 19, 8103–8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chernoff YO, and Kiktev DA (2016) Dual role of ribosome-associated chaperones in prion formation and propagation, Curr. Genet DOI 10.1007/s00294-016-0586-2. [DOI] [PubMed] [Google Scholar]
- 66.Kiktev DA, Melomed MM, Lu CD, Newnam GP, and Chernoff YO (2015) Feedback control of prion formation and propagation by the ribosome-associated chaperone complex, Mol. Microbiol 96, 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Amor AJ, Castanzo DT, Delany SP, Selechnik DM, van Ooy A, and Cameron DM (2015) The ribosome-associated complex antagonizes prion formation in yeast, Prion 9, 144–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cachinska A, Szczesniak B, Kochneva-Pervukhova NV, Kushnirov VV, Ter-Avanesyan MD, and Boguta M (2001) Ssb1 chaperone is a [PSI+] prion-curing factor, Curr Genet 39, 62–67. [DOI] [PubMed] [Google Scholar]
- 69.Okamoto A, Hosoda N, Tanaka A, Newnam GP, Chernoff YO, and Hoshino S-I (2017) Proteolysis suppresses spontaneous prion generation in yeast, J. Biol. Chem 292, 20113–20124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bradley ME, Edskes HK, Hong JY, Wickner RB, and Liebman SW (2002) Interactions among prions and prion “strains” in yeast, Proc Natl Acad Sci U S A 99 (Suppl. 4), 16392–16399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kirkland PA, Reidy M, and Masison DC (2011) Functions of yeast Hsp40 chaperone Sis1p dispensable for prion propagation but important for prion curing and protection from prion toxicity, Genetics 188, 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eaglestone SS, Cox BS, and Tuite MF (1999) Translation termination efficiency can be regulated in Saccharomyces cerevisiae by environmental stress through a prion-mediated mechanism, EMBO J. 18, 1974–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.True HL, and Lindquist SL (2000) A yeast prion provides a mechanism for genetic variation and phenotypic diversity, Nature 407, 477–483. [DOI] [PubMed] [Google Scholar]
- 74.Namy O, Galopier A, Martini C, Matsufuji S, Fabret C, and Rousset C (2008) Epigenetic control of polyamines by the prion [PSI+], Nat. Cell. Biol 10, 1069–1075. [DOI] [PubMed] [Google Scholar]
- 75.Edskes HK, Engel A, McCann LM, Brachmann A, Tsai H-F, and Wickner RB (2011) Prion-forming ability of Ure2 of yeasts is not evolutionarily conserved, Genetics 188, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Edskes HE, Khamar HJ, Winchester C-L, Greenler AJ, Zhou A, McGlinchey RP, Gorkovskiy A, and Wickner RB (2014) Sporadic distribution of prion-forming ability of Sup35p from yeasts and fungi, Genetics 198, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shewmaker F, Mull L, Nakayashiki T, Masison DC, and Wickner RB (2007) Ure2p function is enhanced by its prion domain in Saccharomyces cerevisiae, Genetics 176, 1557–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hoshino S, Imai M, Kobayashi T, Uchida N, and Katada T (1999) The eukaryotic polypeptide chain releasing factor (eRF3/GSPT) carrying the translation termination signal to the 3’-poly(A) tail of mRNA, J. Biol. Chem 274, 16677–16680. [DOI] [PubMed] [Google Scholar]
- 79.Li X, Kandel ER, and Derkatch IL (2014) Functional role of Tia1/Pub1 and Sup35 prion domains: directing protein synthesis machinery to the tubulin cytoskeleton, Mol. Cell 55, 305–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schwimmer C, and Masison DC (2002) Antagonistic interactions between yeast [PSI+] and [URE3] prions and curing of [URE3] by Hsp70 protein chaperone Ssa1p but not by Ssa2p, Mol. Cell.Biol 22, 3590–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Edskes HK, and Wickner RB (2002) Conservation of a portion of the S. cerevisiae Ure2p prion domain that interacts with the full - length protein, Proc. Natl. Acad. Sci. USA 99 (Suppl. 4), 16384–16391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bateman DA, and Wickner RB (2012) [PSI+] prion transmission barriers protect Saccharomyces cerevisiae from infection: intraspecies ‘species barriers’, Genetics 190, 569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Derkatch IL, Bradley ME, Zhou P, Chernoff YO, and Liebman SW (1997) Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae, Genetics 147, 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sondheimer N, and Lindquist S (2000) Rnq1: An epigenetic modifier of protein function in yeast, Mol. Cell 5, 163–172. [DOI] [PubMed] [Google Scholar]
- 85.Derkatch IL, Bradley ME, Hong JY, and Liebman SW (2001) Prions affect the appearance of other prions: the story of [PIN], Cell 106, 171–182. [DOI] [PubMed] [Google Scholar]
- 86.Coustou V, Deleu C, Saupe S, and Begueret J (1997) The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog, Proc. Natl. Acad. Sci. USA 94, 9773–9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Du Z, Park K-W, Yu H, Fan Q, and Li L (2008) Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae, Nat. Genet 40, 460–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Patel BK, Gavin-Smyth J, and Liebman SW (2009) The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion, Nat. Cell Biol 11, 344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Alberti S, Halfmann R, King O, Kapila A, and Lindquist S (2009) A systematic survey identifies prions and illuminates sequence features of prionogenic proteins, Cell 137, 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Suzuki G, Shimazu N, and Tanaka M (2012) A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress, Science 336, 355–359. [DOI] [PubMed] [Google Scholar]
- 91.Allen KD, Wegrzyn RD, Chernova TA, Muller S, Newnam GP, Winslett PA, Wittich KB, Wilkinson KD, and Chernoff YO (2005) Hsp70 chaperones as modulators of prion life cycle: novel effects of Ssa and Ssb on the Saccharomyces cerevisiae prion [PSI+], Genetics 169, 1227–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]